

Cofactors are very special because they reveal the needs of catalysts that cannot be satisfied by the myriad abilities of proteins. Cofactors are often the most easily replicated components of a biocatalyst. Of course, cofactors are important because they shed light on biosynthetic pathways that often reveal novel reaction mechanisms. Some of the most spectacular scientific achievements are the result of the enduring affinity of chemists for cofactors, for example, the B12 story. It is intriguing that cofactors are potential keys in this new era of energy-driven research: How exactly does nature design catalysts that process important gases, such as H2, CO, and CO2?[1] The answers are being found, but it has been hard work.
The hydrogenases have proven to be a particularly rich source of cofactors. The active site of the fastest hydrogenases, the [FeFe]-hydrogenases, consists solely of cofactors—the diiron core is coordinated to seven cofactors: an 4Fe–4S cluster, an aminedithiolate cofactor,[2] three CO ligands, and two cyanide ligands (Scheme 1). The biosyntheses of the ligands, aside from the 4Fe–4S cluster, are unknown. This situation is changing rapidly now with the elucidation of the pathway to the cyanide ligands.[3,4]
Scheme 1.
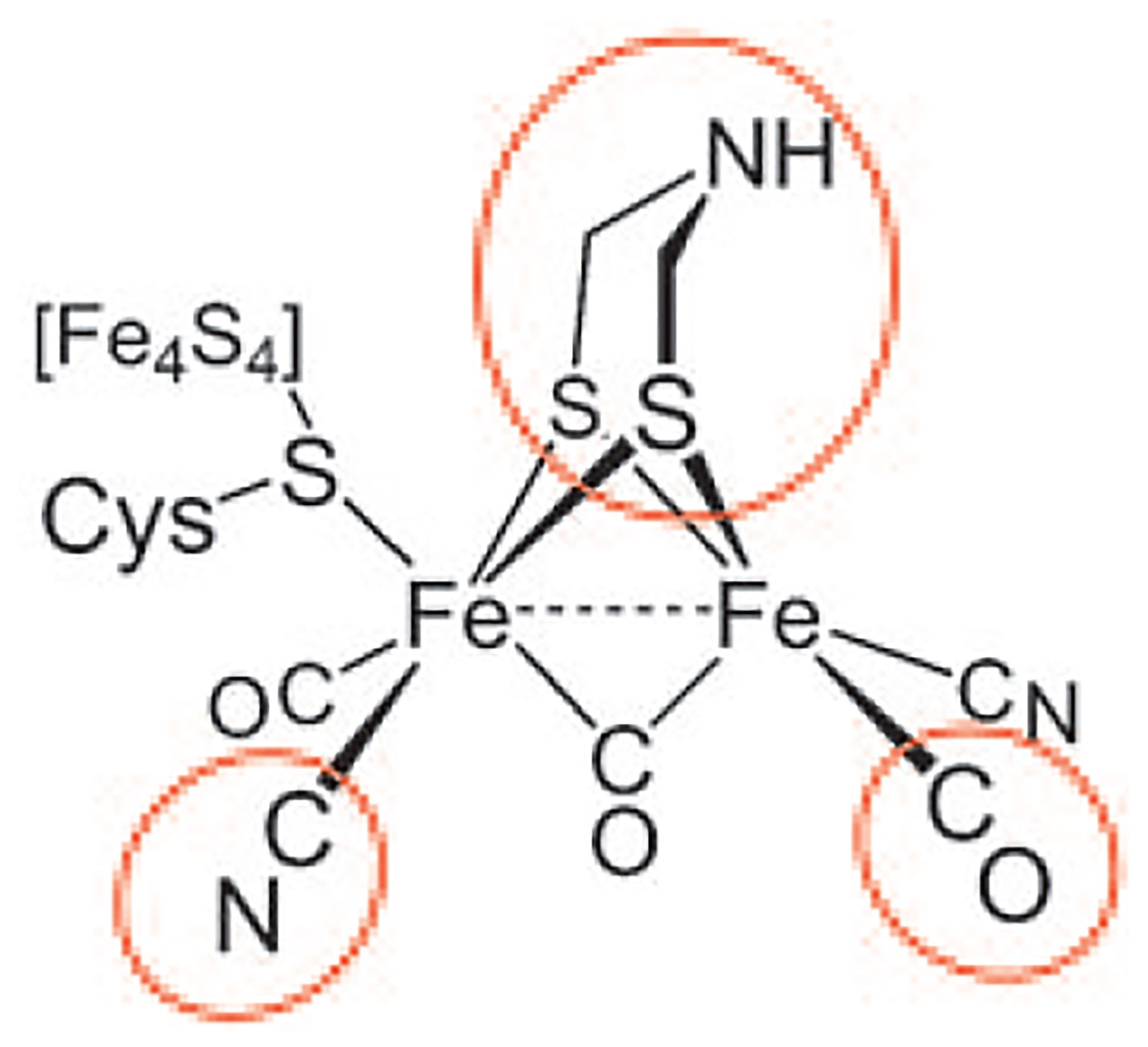
Active site (“H cluster”) of the [FeFe]-hydrogenases. The three types of cofactors of unknown origin are highlighted. Roach and co-workers recently showed that at least one cyanide ligand is derived from tyrosine via the intermediacy of dehydroglycine.[4]
Cyanide is widely considered by inorganic chemists as the ultimate “strong ligand”. It is featured in the first synthetic coordination compound, Prussian Blue. As a ligand, cyanide is at or near the top of many lists, notably for the stability constants of its complexes and the spectrochemical series. For related reasons, it is a potent poison which disables key respiratory metalloenzymes.
Industrial routes to cyanide begin with the platinum catalyzed reaction of ammonia–methane–oxygen mixtures. In the main variant of this process, the aza analogue of steam reforming, oxygen is excluded, and H2 is also generated. By these two processes, more than a billion kilograms of HCN are produced annually.[5]
Many routes are known to metal cyanides,[6,7] not surprisingly, since such complexes are highly stable. Typically, metal cyanides are made from cyanide salts. Metal–cyano complexes also arise from many CN-containing precursors: hydrogen cyanide, cyanogen, cyanogen halides and thiocyanates, and even nitriles[8] and isonitriles.[9] More interesting and more obscure routes to metal cyanides do not start with a preformed C N bond. For example, [Fe(CO)5] reacts with ammonia (and its equivalents[10]) to give cyano complexes. Such reactions appear to occur via carbamoyl intermediates.[7,11] Metal complexes of methylamines are susceptible to oxidative dehydrogenation to the corresponding cyanide complexes (Scheme 2).[12]
Scheme 2.
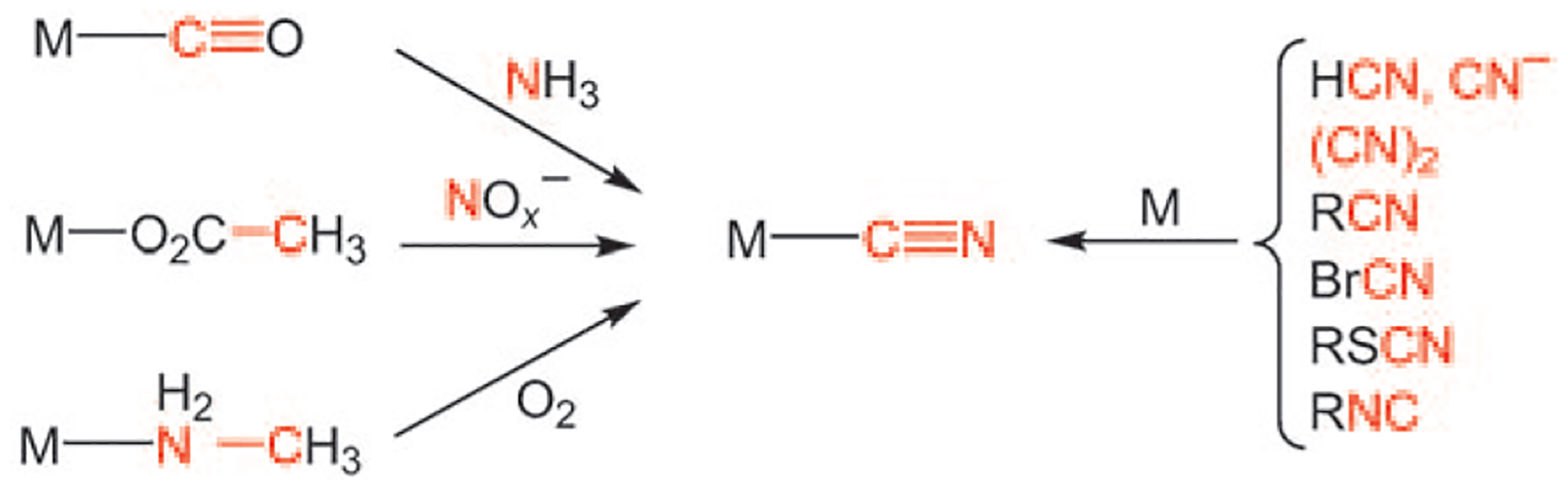
Routes to metal cyanides.
In plants and arthropods, cyanide is produced from any of several amino acids, but not from glycine. The oxidation of the amine is catalyzed by two different cytochrome P450 enzymes and results in α-hydroxynitriles. These compounds or their derivatives are utilized by their hosts as precursors to HCN as a defensive antifeedant response.[13] Bacteria, on the other hand, commonly generate cyanide from glycine [Eq. (1)].
| (1) |
The biosynthetic precursor of the cyanide in the [NiFe] hydrogenases is the metabolite carbamoyl phosphate (H2NC(O)OPO3H−). In the Böck pathway, the C(O)NH2 unit is transferred to the C-terminal cysteinyl thiol of the protein HypE.[14] The dehydration of HypE–SC(O)NH2 requires adenosine triphosphate hydrolysis and affords HypE–SCN, which is the precursor to at least one and probably both of the Fe–CN subunits of this hydrogenase. [NiFe]- and [FeFe]-hydrogenases are, however, unrelated genetically,[15] so the biosynthesis of the cyanide ligands in the [FeFe] enzyme was expected to differ.
Following the development of a heterologous expression system for the Chlamydomonas reinhardtii [FeFe]-hydrogenase, three proteins, HydE, HydF, and HydG, were implicated in the biosynthesis of the CO, cyanide, and dithiolate ligands of the H cluster.[16] HydE and HydG are members of the radical S-adenosylmethionine (SAM) family,[16] the class of enzymes that are responsible for generating the C–S bonds in lipoic acid and biotin. By using a cell-free approach, Roach and co-workers demonstrated that HydG converts tyrosine into the cyanide needed for the biosynthesis of the H cluster.[4] In a further illustration of the special capabilities of radical SAM enzymes, tyrosine was degraded into p-cresol and dehydroglycine, which released CN− into the medium, where it was detected by conversion into a fluorescent derivative. Dehydroglycine is also involved in the formation of thiamin in some bacteria, where it is also generated by radical SAM enzymes (Scheme 3). In Escherichia coli, the enzyme is called ThiH and shares significant sequence homology with HydG.[3,17]
Scheme 3.
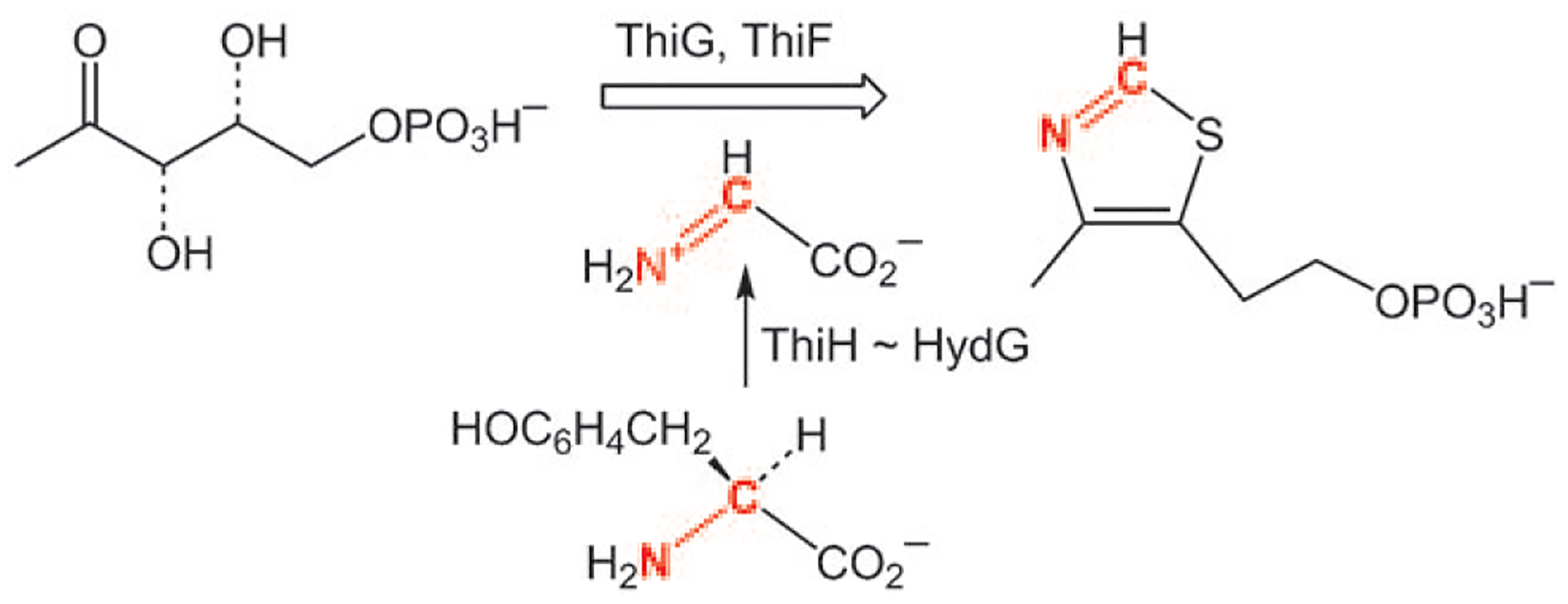
Role of dehydroglycine in the biosynthesis of the thiazole in thiamin.
The details of the process by which cyanide becomes attached to the diiron center remain unknown. Being a promiscuous ligand, CN− probably needs to be guided to the targeted Fe2 center, not simply released in its vicinity. The Fe center that receives the cyanide is bound to a scaffold protein,[18] which may solve this problem. For the coordination chemist, the role of the dehydroglycine is especially intriguing. Several metal complexes of dehydroglycine are known; they are mainly prepared by oxidative dehydrogenation of glycinate complexes (Scheme 4).[19] Thus, it is reasonable to hypothesize that cyanide is formed by oxidative degradation of an iron–dehydroglycinate complex. The conversion would be reminiscent of ethylene biosynthesis, which occurs through the iron(II/III)-mediated release of HCN from aminocyclopropanecarboxylic acid.[20] Glyoxalic acid, obtained upon the hydrolysis of dehydroglycine, is a potential source of CO. In contrast to this unfolding (and still unconfirmed) biosynthetic scenario, the CO and CN− ligands in the [NiFe] hydrogenase do not originate from the same precursor.[21]
Scheme 4.

Formation and acid–base reaction of a cobalt(III) complex of dehydroglycine.
Regarding the biosynthesis of the dithiolate cofactor, Pilet et al. have suggested that this subunit arises through the condensation of two equivalents of dehydroglycine with a 2Fe–2S center.[3] This route is reminiscent of the known aminomethylation of iron sulfides (Scheme 5).[22]
Scheme 5.
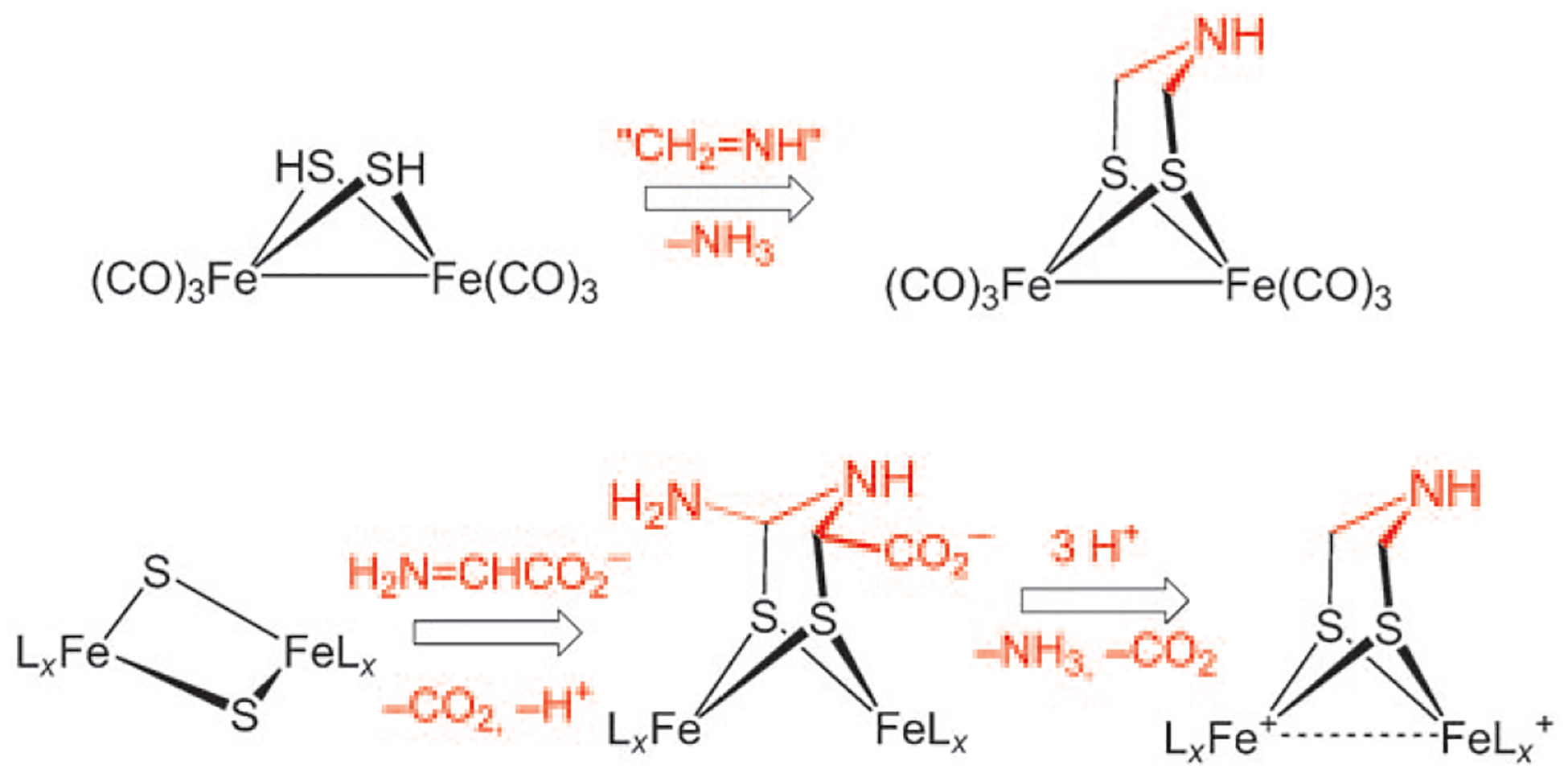
Organometallic route to the azadithiolate cofactor and the pathway proposed by Pilet et al.[3]
The elucidation of the routes to the dithiolate as well as the three CO ligands will form the basis of exciting further research. Overall, the hydrogenases have proven to be a particularly rich source of novel chemistry, not only in the realm of catalysis, but also for new synthetic routes that might be more broadly applicable.[23]
Acknowledgments
The NIH funds our research in this area. Prof. Richard S. Glass and Dr. David Schilter provided helpful advice.
References
- [1].Fontecilla-Camps JC, Amara P, Cavazza C, Nicolet Y, Volbeda A, Nature 2009, 460, 814. [DOI] [PubMed] [Google Scholar]
- [2].Silakov A, Wenk B, Reijerse E, Lubitz W, Phys. Chem. Chem. Phys 2009, 11, 6592. [DOI] [PubMed] [Google Scholar]
- [3].Pilet E, Nicolet Y, Mathevon C, Douki T, Fontecilla Camps JC, Fontecave M, FEBS Lett. 2009, 583, 506. [DOI] [PubMed] [Google Scholar]
- [4].Driesener RC, Challand MR, McGlynn SE, Shepard EM,Boyd ES, Broderick JB, Peters JW, Roach PL, Angew. Chem 2010, 122, 1731; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2010, 49, 1687 [Google Scholar]
- [5].Gail E, Gos S, Kulzer R, Lor sch J, Rubo A, Sauer M in Ullmann s Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2004. [Google Scholar]
- [6].Fehlhammer WP, Fritz M, Chem. Rev 1993, 93, 1243. [Google Scholar]
- [7].Glass RS, Paschos A, Reissmann S, Singh MS, Wang H, Böck A, Phosphorus Sulfur Silicon Relat. Elem 2005, 180, 1183. [Google Scholar]
- [8].Hiroshi N, Masumi I, Kouji K, Kensuke U, Chem. Asian J 2007, 2, 882.17534995 [Google Scholar]
- [9].Bassett JM, Barker GK, Green M, Howard JAK, Stone FGA, Wolsey WC, J. Chem. Soc. Dalton Trans 1981, 219; [Google Scholar]; Halbauer K, Dönnecke D, Görls H, Imhof W, Anorg Z. Allg. Chem 2006, 632, 1477 [Google Scholar]
- [10].Lyon EJ, Georgakaki IP, Reibenspies JH, Darensbourg MY, J. Am. Chem. Soc 2001, 123, 3268. [DOI] [PubMed] [Google Scholar]
- [11].Uchida H, Bando K, J. Chem. Soc. Jpn 1956, 29, 40; [Google Scholar]; Behrens H, Adv. Organomet. Chem 1980, 18, 1. [Google Scholar]
- [12].Diamond SE, Tom GM, Taube H, J. Am. Chem. Soc 1975, 97, 2661; [DOI] [PubMed] [Google Scholar]; McWhinnie WR, Miller JD, Watts JB, Waddan DY, Inorg. Chim. Acta 1973, 7, 461. [Google Scholar]
- [13].Zagrobelny M, Bak S, Møller BL, Phytochemistry 2008, 69, 1457. [DOI] [PubMed] [Google Scholar]
- [14].Reissmann S, Hochleitner E, Wang H, Paschos A, Lott speich F, Glass RS, Böck A, Science 2003, 299, 1067. [DOI] [PubMed] [Google Scholar]
- [15].Fontecilla-Camps JC, Volbeda A, Cavazza C, Nicolet Y, Chem. Rev 2007, 107, 4273. [DOI] [PubMed] [Google Scholar]
- [16].Posewitz MC, King PW, Smolinski SL, Zhang L, Seibert M,Ghirardi ML, J. Biol. Chem 2004, 279, 25711. [DOI] [PubMed] [Google Scholar]
- [17].Kriek M, Martins F, Challand MR, Croft A, Roach PL, Angew. Chem 2007, 119, 9383; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2007, 46, 9223; [Google Scholar]; Begley TP, Chatterjee A, Hanes JW, Hazra A, Ealick SE, Curr. Opin. Chem. Biol 2008, 12, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].McGlynn SE, Shepard EM, Winslow MA, Naumov AV,Duschene KS, Posewitz MC, Broderick WE, Broderick JB, Peters JW, FEBS Lett. 2008, 582, 2183–2187. [DOI] [PubMed] [Google Scholar]
- [19].Mori T, Yamaguchi M, Sato M, Yamagishi T, Inorg. Chim. Acta 1998, 267, 329; [Google Scholar]; Bendahl L, Hammershøi A, Jensen DK,Larsen S, Riisager A, Sargeson AM, Sørensen HO, J. Chem. Soc. Dalton Trans 2002, 3054;; Siebenlist R, Fruehauf H-W, Vrieze K, Kooijman H, Smeets WJJ, Spek AL, Organometallics 2000, 19, 3016. [Google Scholar]
- [20].Lin Z, Zhong S, Grierson D, J. Exp. Bot 2009, 60, 3311; [DOI] [PubMed] [Google Scholar]; Tierney DL, Rocklin AM, Lipscomb JD, Que L Jr., Hoffman BM, J. Am. Chem. Soc 2005, 127, 7005. [DOI] [PubMed] [Google Scholar]
- [21].Forzi L, Hellwig P, Thauer RK, Sawers RG, FEBS Lett. 2007, 581, 3317; [DOI] [PubMed] [Google Scholar]; Lenz O, Zebger I, Hamann J, Hildebrandt P,Friedrich B, FEBS Lett. 2007, 581, 3322. [DOI] [PubMed] [Google Scholar]
- [22].Li H, Rauchfuss TB, J. Am. Chem. Soc 2002, 124, 726; [DOI] [PubMed] [Google Scholar]; Stanley JL, Rauchfuss TB, Wilson SR, Organometallics 2007, 26, 1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gloaguen F, Rauchfuss TB, Chem. Soc. Rev 2009, 38, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]