

Highlights
► Hipposideridae is underestimated in the study of coronaviruses. ► Hipposideridae harbor Alphacoronavirus and Betacoronavirus in South-East Asia. ► Hipposideridae colony can host Betacoronavirus close to SARS-CoV over long period. ► BetaCoV (SARS) spill-over chain: Hipposideridae, Rhinolophidae, Carnivora, Human. ► Hipposideridae and Rhinolophidae coevolved with independent Betacoronavirus lineages.
Keywords: Betacoronavirus, Phylogeny, Hipposideridae, SARS-CoV, Thailand, Emergence
Abstract
One of the great challenges in the ecology of infectious diseases is to understand what drives the emergence of new pathogens including the relationship between viruses and their hosts. In the case of the emergence of SevereAcute Respiratory Syndrome Coronavirus (SARS-CoV), several studies have shown coronavirus diversity in bats as well as the existence of SARS-CoV infection in apparently healthy bats, suggesting that bats may be a crucial host in the genesis of this disease. To elucidate the biogeographic origin of SARS-CoV and investigate the role that bats played in its emergence, we amplified coronavirus sequences from bat species captured throughout Thailand and assessed the phylogenetic relationships to each other and to other published coronavirus sequences. To this end, RdRp sequence of Coronavirinae was targeted by RT-PCR in non-invasive samples from bats collected in Thailand. Two new coronaviruses were detected in two bat species: one Betacoronavirus in Hipposideros larvatus and one Alphacoronavirus in Hipposiderosarmiger. Interestingly, these viruses from South-East Asia are related to those previously detected in Africa (Betacoronavirus-b) or in Europe (Alphacoronavirus & Betacoronavirus-b). These findings illuminate the origin and the evolutionary history of the SARS-CoV group found in bats by pushing forward the hypothesis of a Betacoronavirus spill-over from Hipposideridae to Rhinolophidae and then from Rhinolophidae to civets and Human. All reported Betacoronaviruses-b (SARS-CoV group) of Hipposideridae and Rhinolophidae respectively cluster in two groups despite their broad geographic distribution and the sympatry of their hosts, which is in favor of an ancient and genetically independent evolution of Betacoronavirus-b clusters in these families. Moreover, despite its probable pathogenicity, we found that a Betacoronavirus-b can persistently infect a medium-sized hipposiderid bat colony. These findings illustrate the importance of the host phylogeny and the host/pathogen ecological interactions in the description and the understanding of pathogen emergence. The host’s phylogeny, biogeography and behaviour, combined with already described roles of pathogen plasticity and anthropic changes are likely to be co-factors of disease emergence. Elucidating the common ancestor of Hipposideridae and Rhinolophidae is key to understanding the evolutionary history of actual betacoronaviruses and therefore to get an insight of the deep origin of SARS-CoV.
1. Introduction
Pathogens mostly emerge in densely human populated areas as did the Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV), which had the potential to cause the first viral pandemic of the XXIst century (WHO, 2003, Jones et al., 2008). SARS-CoV most likely originated from bats through an intermediate species that could be the masked palm civet Paguma larvata (Viverridae) or another bridge-host, itself very probably contaminated by a SARS-like CoV from horseshoe bats (Song et al., 2005, Li et al., 2006, Cui et al., 2007). Several elements indicate that this bridge-host could presumably be a Carnivora as suggested by both detections in wild animals from markets or experimental infections (Guan et al., 2003, Van den Brand et al., 2008). Several other mammalian or avian species are susceptible to coronaviruses infection (displaying enteric, hepatic, neurological, or respiratory tropism), but little is known about their viral ecology and host interactions. Coronaviruses are members of the order Nidovirales which contains three families of single stranded positive RNA viruses: Arteriviridae, Coronaviridae and Roniviridae. Coronaviridae are divided in two subfamilies: Coronavirinae and Torovirinae. Because of serological and genotypic differences, coronaviruses were traditionally divided into three groups labeled group I, II and III (González et al., 2003, Gorbalenya et al., 2004). These three groups have recently been elevated at the genus level and named respectively Alphacoronavirus, Betacoronavirus and Gammacoronavirus by the coronavirus Study Group of the International Committee for Taxonomy of Viruses, thus invalidating the “Coronavirus” genus name (http://talk.ictvonline.org/media/p/1230.aspx). As other Nidovirales, Coronaviridae such as SARS-CoV possess a mutation prone RNA-dependent RNA-polymerase (RdRp) associated with high genome mutability (Castro et al., 2005). Furthermore, they frequently recombine in vivo (Lai et al., 1985, Makino et al., 1986, Keck et al., 1987, Jia et al., 1995, Herrewegh et al., 1998, Lee and Jackwood, 2000, Stadler et al., 2003). This genetic variability confers to coronaviruses a high potential of evolution that allows them to sometimes overcome species barriers and host specificity (Baric et al., 1995, Baric et al., 1997) (Thackray and Holmes, 2004). From a more general viewpoint, these characteristics are known to account for the frequent involvement of RNA viruses in disease emergence (Domingo and Holland, 1997, Chomel, 1998, Daszak et al., 2000, Cleavel et al., 2001, Woolhouse and Gowtage-Sequeria, 2005).
Until now, Asian SARS-related coronavirus (SARSr-CoV, from Betacoronavirus-b group – Fig. 1 A) RNA has been detected in only one bat family: the Rhinolophidae Gray, 1825 (Table 2). Therefore, this monogeneric bat family is considered as the host at the origin of the deadly SARS-CoV outbreak (Lau et al., 2005). However, other bat families are involved in the maintenance and the diversification of a wide range of coronavirus species belonging to both Alphacoronavirus and Betacoronavirus genera (i.e. Vespertillionidae, Phyllostomidae, Pteropodidae, Molossidae, and Hipposideridae families have been positively tested for coronavirus nucleic acid) (Chu et al., 2006, Tang et al., 2006, Dominguez et al., 2007, Carrington, 2008, Brandao et al., 2008, Tong et al., 2009, Pfefferle et al., 2009). Whether or not another bat family could be involved (in addition to Rhinolophidae) in the natural history of betacornaviruses would have been an interesting question but since the discovery of SARSr-CoV in wildlife, most of the attention and sampling efforts have been focusing on Rhinolophidae (Table 2). Several species from this family were shown to carry different SARSr-CoV such as SARSr Rhinolophidae Bat Coronavirus strain Rp3 in Rhinolophus pearsoni and SARSr Rhinolophidae Bat Coronavirus strain Rf1 in Rhinolophus ferrumequinum in the Chinese provinces of Guangxi and Hubei, respectively (Lau et al., 2005, Li et al., 2005, Tang et al., 2006). The sampling pressure on both Rhinolophidae (1148 samples in 12 species – Table 2) and Vespertilionidae (900 samples in 17 species – Table 2) has induced an over representation of these families compared to others (Lau et al., 2005, Li et al., 2005, Tang et al., 2006). By contrast, Hipposideridae (206 samples in 6 species) were under-represented in previous studies (Poon et al., 2005, Woo et al., 2006). The higher northern distribution limit of Rhinolophidae (versus Hipposideridae) may partially explain the sampling bias in non-Southern Chinese provinces (Fig. 1B). This bias illustrates the need for testing other sympatric species for betacoronaviruses to study their natural history and emergence process.
Fig. 1.
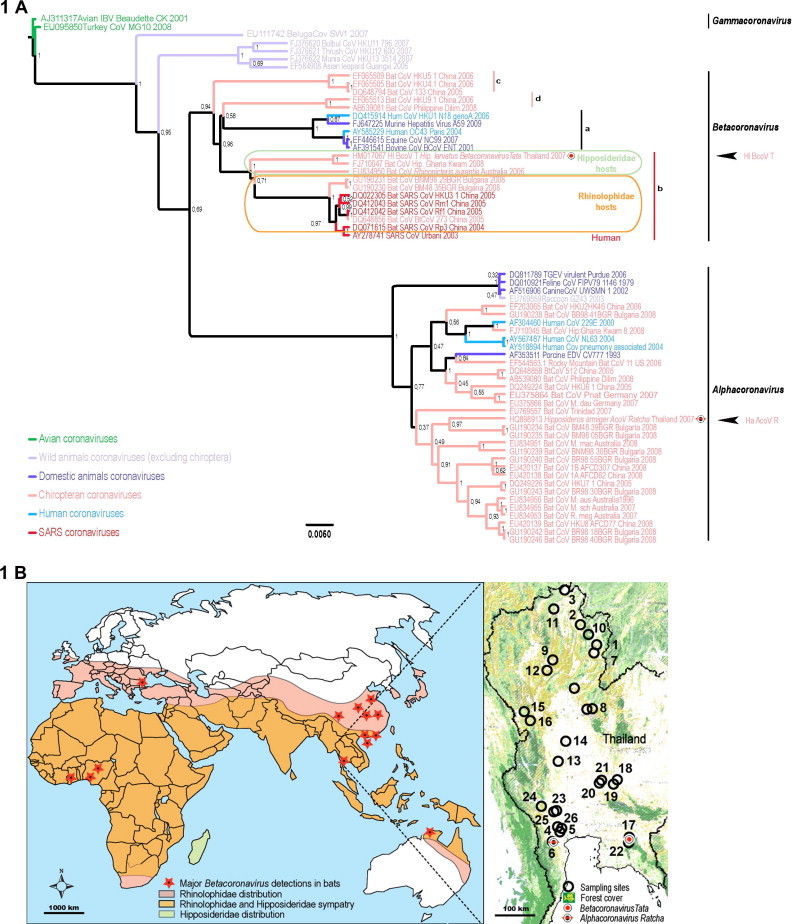
Phylogenetic reconstruction of main coronaviruses based on the analysis ofof 333 bases of the RNA-dependent RNA polymerase protein (partial nsp12) and geographical localization of Hipposideros larvatus Betacoronavirus Tata and Hipposideros larvatus Alphacoronavirus Ratcha. (A) Bayesian phylogeny of coronaviruses showing host taxonomy of Betacoronavirus-b group and reporting two new coronaviruses (posterior probabilities are shown for each node). (B) Repartition of hosts families (Hutson 2003), sampling sites and Hl BCoV T & Ha BCoV R spatial position.
Table 2.
Asian bat species sampled in coronavirus studies. Some values should be considered cautiously because the related data were not always available and were in some cases extrapolated: (∗) Details not available, supposedly two samples per bat (respiratory and fecal numbers extrapolated here). (†) Expressed as number of bats in original publication, details not available.
|
The biogeography and the natural history of the Betacoronavirus-b species, among which are SARSr-CoVs, remain poorly described. However, Betacoronavirus have been detected in China, Africa, Europe and even Australia (Guan et al., 2003, Lau et al., 2005, Li et al., 2005, Müller et al., 2007, Tong et al., 2009, Quan et al., 2010, Drexler et al., 2010) (Fig. 1B). The biogeographical origin of the ancestors of SARSr-CoVs (from Betacoronavirus-b sub-group) is therefore far more extended than the epicentre of the human epidemic (Southern China). Understanding the biogeography of this group would be essential for assessing the risk for emergence of another SARS-CoV like agent. South-China and its Pearl River delta combined various ecological and anthropic factors that have led the SARS-CoV to emerge but the distribution of the viral clade at the origin of SARS-CoV is much wider than this region and many locations still remain to be investigated.
Emergence results from a combination of numerous factors. Environmental changes (ecological, climatic and agricultural), whether they are man-driven or not (Schrag and Wiener, 1995, Kuiken et al., 2003), globalization (trade or travel) and societal changes (land-use, demography), are among the most reported ones (Reperant, 2010), but others have been highlighted recently like the hosts’ phylogenetic relationships (Daszak, 2010, Streicker et al., 2010), the variation of biodiversity (Keesing et al., 2010) and species interactions in ecological communities whether between hosts or between hosts and pathogens (Cleavel et al., 2001, Woolhouse and Gowtage-Sequeria, 2005, Reperant, 2010). Consequently, the biogeography and host-pathogen relationships of Betacoronavirus-b are pivotal to understanding their radiation. The diversity of the SARSr-CoV has been described by an increasing number of published studies and the number of discovered and sequenced viral taxa has improved the accuracy of the phylogenetic reconstruction of this group.
In this study we explored the biodiversity and the ecology of Coronavirinae, enriched the current phylogeny and challenged the biogeography of this clade. More particularly, we focused on the ecology of the Betacoronavirus-b sub-group (Fig. 1A) by testing hypotheses on the origination of these viruses not only in Rhinolophidae but also in their sister taxa Hipposideridae. Considering that Hipposideridae might host a diverse community of coronaviruses comparable to that of Rhinolophidae because of their phylogenetic proximity and comparable diversity (Nunn et al., 2004, Simmons, 2007), it is very probable that the prevalence and diversity of coronaviruses hosted by this family has been underestimated because of biased sampling (Table 2 – Lau et al., 2005, Li et al., 2005, Tang et al., 2006). We hypothesize also that some bat taxa could play a refuge role for betacoronaviruses and therefore constitute a source of infection for other species, consistently with the recent application of the theory of island biogeography (MacArthur and Wilson, 1967) to emerging pathogens (Reperant, 2010). This study is particularly focused on, but not restricted to, Betacoronavirus in Hipposideridae in South-East Asia (Table 1 ).
Table 1.
Detection of coronaviruses in saliva (Oral Swab), feces and urine in bats from Thaïland.
 |
2. Materials and methods
To test our hypotheses, we targeted, by RT-PCR, the Coronavirinae RdRp RNA sequence (more precisely a coding fragment of a subunit of the RdRp, the non-structural protein 12 – nsp12 –) in bat samples and compare it with the published sequences downloaded from public online databases. The SRAS-CoV nsp12 and nsp13 exhibit the highest conservation profiles among proteins from all coronaviruses (Stadler et al., 2003). Moreover, as RdRps (nsp12) are involved in replication and transcription processes and therefore play a central role in the virion offspring genome synthesis (the vertical information transmission), its relatively conserved ribonucleic acid sequence is appropriate for both viral detection and phylogeny reconstruction.
2.1. Sampling
From 2006–2008, samples (feces, urine and oral swabs) were collected from bats at 26 sites in Thailand (Fig. 1B and Table 1). Sampling locations were selected considering land cover and geological data as forests are known to enhance biodiversity (Harvey et al., 2006, Trisurat et al., 2010) and limestone hills harbour numerous caves. The rocky forest-covered hills surrounded by cultivated lowlands, considered as potential refuge areas, were particularly sought. Sampling sessions covered an area of approximately 800 km from North to South Thailand by 300 km from West to East. Dates and locations are described in Table 1 and Fig. 1B. Sites were sometimes sampled several times to confirm the results, to screen another population or species of bats or to assess a putative long-lasting infection of a given colony (Table 1). Samples were collected from 25 species of bats and were divided and preserved in two different media: (i) modified Minimum Essential Medium antibiotic supplemented (viral transportation medium) to preserve entire virus particles integrity and (ii) RNAlater (RNA preservation medium) to limit RNA degradation, both kept on ice before and immediately after collection (prior to liquid nitrogen conservation in the field). Each sample was immediately aliquoted into four tubes (including absolute backup) and kept at −80 °C before RNA extraction. Traceability was assured by assigning a unique number to each sample. Handling of animals was conducted in accordance with the American Society of Mammalogists Guidelines for the capture, Handling and Care of Mammals (Gannon and Sikes, 2007). Trapping sessions were performed using harp-traps, flip-nets and hand-nets. Bats were transferred to individual clean cotton bags immediately after being captured and kept in a dark, cool and quiet place for one to a few hours. Immediately after sample collection, bats were released where they were trapped. Particular attention was paid: (i) to avoid disturbance inside the cave and near the colony; (ii) to disinfect material and shoes between sampling sessions and study sites; and, (iii) to preserve the surroundings as first encountered.
2.2. RNA extraction
Qiamp viral RNA mini kit (Qiagen) was used for RNA extraction following the manufacturer’s instructions. RNA was eluted in 60 μl RNAse-free saline buffer, 7 μl were immediately used for the reverse transcription reaction and the remaining 53 μl were aliquoted and kept at −80 °C for subsequent detection assays. The amount and quality of the extracted RNA was measured using Nanodrop and Agilent techniques prior to reverse transcription (Mueller et al., 2004, Gallagher and Desjardins, 2006).
2.3. Reverse transcription (RT), amplification by polymerase chain reaction (PCR) and sequencing
Complement DNA synthesis was performed with M-MLV Reverse Transcriptase (Bio-Active) and random hexamer primers. Random hexamer primers were used at a final ratio of 0.15 μg for 1 μg of RNA. Then, 5 μl of RT products were used in 25 μl final volume PCR reaction targeting a 438 nucleotide fragment of the RdRp coding region. PCR primers were designed from the RdRp coding region by selecting a highly conserved region (nsp12) among all Coronavirinae sequences available from online public databases in August 2007. Assuming a higher mutation rate for the third codon position and according to the published sequence variations, degenerate primers were designed: BatCoV pol 15197 (forward primer: 5′-GGTTGGGAYTAYCCWAARTGTGA-3′) and BatCoV pol 15635 (reverse primer: 5′-CCATCRTCMGAHARAATCATCATA-3′), respectively at position 15197 to 15219 and 15635 to 15614 on Bat SARS-CoV Rf1 reference sequence (DQ412042). In order to increase both the sensitivity and the specificity of the detection, a third primer were designed and combined with the previous ones in a semi-nested PCR: BatCoV pol nested 15419 (forward nested primer: 5′-GCNAATWSTGTNTTTAACAT-3′), at the position 15419 to 15438 on the same reference sequence. The first PCR produced a fragment of 438 nucleotides and the semi-nested PCR confirmed the detection and enhanced significantly the detection limit of the semi-nested PCR by amplifying a 216 base pairs fragment from the first amplicon. Contaminations were avoided by observing strict lab procedures, carrying out at each step (mix, PCRI, PCRII) in a UV cabinet and using bleached plastic-ware. Manipulations included extraction negative controls at the beginning and at the end of each sample series as well as several PCR negative controls per run. The sequencing of the 438 nucleotides PCR products was performed in both directions using the external primers.
2.4. Cell culture and inoculation assay
Fifty microliters of sample preserved in modified Minimum Essential Medium antibiotic supplemented (viral transportation medium) were inoculated into 12 wells plates where subconfluent cell monolayers were grown. Madin–Darby Canine Kidney (MDCK), Vero and Vero clone E6 cells were used in these assays. MDCK were chosen because Carnivora are highly suspected to have efficiently contributed to the transmission from bats to Humans (Paguma larvata, Nyctereutes procyonides, Melogale moschata) (Guan et al., 2003) and several species are susceptible to SARS-CoV infection (Martina et al., 2003). The Vero cell line is known to efficiently replicate SARS-CoV (Qinfen et al., 2004). Cells were cultivated in Minimum Essential Medium supplemented with 120 μg/ml of streptomycin, 120 units/ml of penicillin and 10% of Fœtal Calf Serum (FCS). Once confluence was nearly reached, the supernatant was removed and samples were added on the cell monolayer and left at 20 °C on a rocking agitator for 60 min. Then, 2 ml of the same medium (2% FCS) were added to each well. Plates were then incubated at 37 °C and daily checked on a phase contrast inverted microscope for CPE occurrence.
2.5. Phylogenetic reconstruction
A 304 or 333 nucleotide region was selected within the sequences from both amplicons (Ha AcoV R for 304 or Hl Bcov T, respectively). The corresponding region from 60 taxa available in databases were added to the matrix used in the phylogenetic reconstructions.
1/ Firstly a dynamic homology approach implemented in Poy v.4.1.2 was used (Wheeler, 1996, Janies and Wheeler, 2002, Varón et al., 2010). This method did not need any prior alignment because this step was performed during the cladogram diagnosis and therefore established the putative homology statements during the tree reconstruction. Poy was used under parsimony as the optimality criterion (Wagner algorithm), complemented with the tree bisection reconnection swapping algorithm (TBR), perturbation injection, ratcheting and fuse iterations steps (Nixon, 1999). These combined steps provided a reasonably fast and accurate way to better explore the tree space by escaping from local optima. Although RdRp is a conserved gene and the matrix included a great number of taxa, resampling taxa tests were used to evaluate whether this matrix was subjected to the long branch attraction phenomenon which could constitute a limitation of some phylogenetic reconstructions. This limitation could be particularly true in the case of high evolution rate and very limited number of taxa when combined with static homology approach of molecular characters. The over-representation of sub-groups by several similar sequences was avoided as it could artificially report changes on branches located between internal nodes, leading to the long branch attraction issue.
2/ Secondly, a probabilistic approach (Bayesian) implemented in BEAST (Drummond and Rambaut, 2007) was applied to the same data set. Sequences were previously aligned using Sea-View (Gouy et al., 2010) under the Muscle algorithm (Edgar, 2004) and resulted in a frank border matrix showing a unique gap of three codons for four sequences (Bulbul, Trush, Munia and Asian leopard CoVs, position 33 to 36). Then the adequacy of the matrix to 88 substitution models was evaluated using jModeltest (Posada, 2008). The three codons partition model of evolution was specified (under HYU substitution model with a gamma site heterogeneity distribution), so that each of the three codon position could follow its own evolution rate. Yule speciation model showed similar estimates as the constant population size coalescent model. Thus, the Yule speciation model was chosen as tree prior. The MCMC chain was set to 10,000,000 states to get an adequate posterior ESS value superior to 300.
The support of the nodes was evaluated via a bootstrap analysis (Felsenstein, 1985) for the direct optimization method and posterior probabilities we assessed for the Bayesian method (Fig. 1 and S1). Except for the sequences of Hipposideros larvatus Betacoronavirus Tata (Hl BcoV T) and Hipposideros armiger Alphacoronavirus Ratcha (Ha AcoV R) which were newly identified in this study, all other sequences used in phylogenetic reconstructions were downloaded from the National Center for Biotechnology Information database (http://www.ncbi.nlm.nih.gov/nucleotide/). Each Genbank sequence identification number is indicated directly on trees (Fig. 1 and S1).
2.6. Rooting the trees
In the case of coronavirus phylogeny, rooting with one member of the sister group (Bafinivirus or Torovirus, the two genus of Torovirinae sub-family) would have been a good choice to study the relation within the in-group Coronavirinae. Unfortunately, the region that we used here was too divergent and hardly comparable to Torovirinae sequence. However, as Torovirinae sequence can not be used to root the trees in this case, we used the results provided by a published study (Snijder et al., 2003), that used Torovirinae as root. This study, based on an amino acid matrix, inferred a Coronaviridae phylogeny, and verified the monophyly of Coronavirus and the basal branching of Gammacoronavirus clade. According to these conclusions, we chose an avian Coronavirus taxa to root our trees and therefore study the Alphacoronavirus and Betacoronavirus genera phylogeny. An additional taxon, from the same clade of the taxa used as root (Gammacoronavirus – Avian Coronavirus Beaudette), was included in the analysis to allow the polarization of the characters (ancestral versus apomorphic) along the tree.
3. Theory
3.1. Identifying the phylogenetic signal of filiation in Betacoronavirus metagenome
A genome comprises ribonucleic acid fragments that can independently be submitted to various selection pressures of different intensity. The history of these fragments can be deeply shaped by the function they encode, eventually resulting in incongruent phylogenetic signals. The only way to fully recover and clearly exploit the phylogenetic signal of a given fragment (by comparison to the putative homolog fragment of other taxa) is to analyze it independently. Therefore, the combination of several fragments in a same analysis should be carefully thought and documented as mixing the information they provide can produce a blurred signal and therefore can lead to an inaccurate phylogeny. Here we focused on a fragment of the polymerase sequence only for phylogenetic reconstruction of coronaviruses because: (i) the key function of this protein involved in vertical transmission and consequently in viruses filiation is likely to regulate variation and provide a phylogenetic signal that reflects the filiation and, (ii) its relatively short length, genome position and associated transcription characteristics make it unlikely to be a putative recombination site. Moreover, the analysis of this region by the RDP3 package (Martin et al., 2010) did not detect any putative recombination sites.
During the inference of a species tree, the identification of the appropriate vertical transmission of information, is one of the classical pitfalls of phylogenetic reconstruction. Therefore, careful attention must be paid in the choice of a genetic fragment that reflects the species phylogeny and not only the gene history. A given genetic fragment, coding for a positively selected function which appeared in one species, may benefit another one by horizontal transfer (recombination), increasing the fitness of the recombinant virus and its progeny. This phenomenon have happened also between viruses that are actually recognized as members of separated families as described for the two independent recombination events that occurred between a Torovirus (Coronaviridae, Torovirinae), a Coronavirus and an Orthomyxoviridae (Influenzavirus C) (Snijder et al., 1991). These particular events did not involve the RdRp sequence. All these reasons lead us to rely on the analysis of this RdRp fragment for the phylogenetic reconstruction of Coronavirus.
3.2. Biodiversity, host phylogeny and micro hot-spots
According to the distribution of both the known mammalian host families, the Rhinolophidae Gray, 1825, and its sister family the Hipposideridae Lydekker, 1891, Betacoronavirus-b might be widespread over Asia and most of the old world. Moreover, the biodiversity of these two mammalian families is surely higher than what has been currently described (S.J. Puechmaille, unpublished data), which in turn induces a potential greater coronavirus diversity than is currently described. In discordance to what has been reported to date and based on the phylogenetic proximity of these sister families, Hipposideridae might harbour an equal or richer diversity of coronaviruses (at least of the Betacoronavirus genus) compared to the Rhinolophidae. Furthermore, the fragmentation of the forested habitat, produced by human activities such as land use practices and agriculture, leads to the isolation of preserved biodiversity spots (surrounded by cultivated fields) that have the potential to maintain both host and viral populations, preserving by this way the footprints of the natural history of coronaviruses in wildlife.
4. Results
Twenty-five species of bats belonging to the Rhinolophidae (N = 8), Hipposideridae (N = 8), Emballonuridae (N = 1), Molossidae (N = 1), Megadermatidae (N = 2), Pteropodidae (N = 2), Miniopteridae (N = 1) (Table 1), Craseonycteridae (N = 1) and Vespertilionidae (N = 1) families were sampled. Species were identified using morphological and acoustic characters.
A total of seven samples among 552 (265 feces, 97 urine collections and 190 oral swabs), were positive using the RT-PCR on the RdRp region described in the material and methods section. This amount rose to 28 when the results from the second sampling at the study site 17 were included (Table 1 and Fig. 1B). All of the positive samples came from two species: Hipposideros larvatus (Fig. 2 A) and Hipposideros armiger (Fig. 2B). Two new viruses were detected in this study but the isolation trials on cell cultures remained unsuccessful: no cytopathic effect was observed after three passages on cell cultures and no viral material was amplified from the third passage supernatants with the semi-nested PCR described in material and methods.
Fig. 2.

Colonies of Hipposideros at roost, showing the scattered distribution of individuals, with a zoom on the face of one specimen (bar is equal to 1 cm). (A) Hipposideroslarvatus (B) Hipposideros armiger.
4.1. Two coronaviruses from two Hipposideridae: Hipposideros armiger Alphacoronavirus Ratcha (Ha AcoV R) and Hipposideros larvatus Betacoronavirus Tata (Hl BcoV T)
The Ha ACoV R was detected in bat feces from a unique colony (Hipposideros armiger Hodgson, 1835). Among 10 bats tested, four were positive (36% – study site 4 – Table 1). The BLAST 78% identity value indicated that the sequence was related to a bat Alphacoronavirus reported to infect Rhinolophus ferrumequinum and Rhinolophus pearsoni in the Chinese Shandong region in 2005 (Table 2) and (Tang et al., 2006). For this sequence, 30 bases from the 5′ side of the PCR products were ambiguous and therefore removed from the analysis because the sequencing with one degenerated primer gave low quality results.
Concerning Hl BcoV T, the sequences were free of ambiguous sites and identical among the molecular isolates amplified from feces collected from 22 specimens of bats from the same colony (Table 1). The 391 nucleotides long sequences obtained showed that this new virus was related to the SARS-CoV clade and shared 75% nucleotide identity (online BLAST analysis) with the Bat SARSr Coronavirus Rf1 from Rhinolophus ferrumequinum. Hl BcoV T was detected in fecal samples from one colony of Hipposideros larvatus (Hipposideridae Lydekker, 1891), in one sampling site among 26. In this colony (estimated to 250 individuals), 3/11 (27%) bats tested were positive, then, eighteen months later, 21/40 (52%). This is the first evidence of SARSr-CoV long-lasting infection in a bat colony (Table 1).
4.2. Other species and study sites
Besides the detection of Ha ACoV-R in one site, no other Alphacoronavirus was detected from the other study sites. Hipposideros armiger individuals positive for Ha ACoV-R shared their roost with Hipposideros larvatus individuals, which were negative for the detection of Ha ACoV-R. Similarly, besides Hl BcoV-T detected in one site, no other Betacoronavirus-b was detected from the other study sites even if they were populated by the same species as the positive site and in two instances (cave 3 and 14 – Table 1), with comparable sample sizes. The other species sampled, Taphozous longimanus and Hipposideros armiger, that were sharing the same roost with Hipposideros larvatus (cave 17 – Table 1), were all negative for the detection of Hl BcoV T.
4.3. Phylogenetic reconstruction
The same global topology was obtained from both direct optimization (Poy) and probabilistic methods (Beast) as apparent differences mostly concerned nodes that are not supported by respective bootstrap or posterior probability values (inferior to 95%) (Fig. 1A and S1). Indeed, direct optimization supported Betacoronavirus paraphyly but basal nodes exhibited low bootstrap support values. Accordingly, the separation between Alphacoronavirus and Betacoronavirus is not significantly supported in the Bayesian phylogeny (posterior probablility value: 0.69 < 0.95). Several other nodes remained weakly supported by the data in each method. Moreover, the HKU4/HKU5/BtCoV 133 group significantly segregated in basal position of the Betacoronavirus in both reconstructions. Alphacoronavirus exhibited mainly nodes with low support in both methods of reconstruction despite comparable topologies. The Alphacoronavirus detected in this study, Ha AcoV R, rooted two alphacoronaviruses reported to infect bats in Bulgaria (Drexler et al., 2010). Concerning Betacoronavirus-b topology, most of the nodes were well supported. Our RdRp gene based phylogenetic reconstructions placed Hl BCoV-T as a sister taxon of an african Hipposideros Betacoronavirus-b (Fig. 1A and S1). These taxa formed together the sister group of all other betacoronaviruses-b, which comprised the monophyletic group of Rhinolophidae betacoronaviruses-b. These were rooted by one Hipposideridae Betacoronavirus-b: the Australian Bat Coronavirus R. aur, detected in Rhinonicteris aurantia. Even paraphyletic, Hipposideridae betacoronaviruses mainly grouped together and rooted the Rhinolophidae betacoronaviruses among which is the SARS-CoV (Fig. 1A and S1).
5. Discussion
The species demarcation criteria proposed to the ICTV by the coronavirus study group (http://talk.ictvonline.org/media/p/1230.aspx) recommend a minimal 90% amino acid sequence identity threshold over seven domains of the replicase (nsps 3, 5, 12, 13, 14, 15, 16). Only one domain is available in our data (nsp12) but it is located in one of the three most conserved and contiguous regions of the replicase (nsp11, 12 and 13) (Stadler et al., 2003). The relatively low identity values obtained by the BLAST analyses (78% for the Ha AcoV-R, and 75% for the Hl BcoV-T) in the most conserved region of the coronavirus genome suggests that Hl BcoV T and Ha AcoV R are new species, even though there was only one fragment available, instead of the ad hoc set of seven regions proposed by the ICTV working group. More investigations are necessary to fully characterize these coronaviruses and to get the identity values of the seven replicase domains to rule on these two taxa description in complete accordance with the ICTV recommendations. For clarity and non-ambiguous taxonomy, these two putative new species are proposed to be named Hipposideros larvatus Betacoronavirus Tata (Hl BcoV-T) and Hipposideros armiger Alphacoronavirus Ratcha (Ha AcoV R). These Betacoronavirus and Alphacoronavirus were detected from the same bat genus Hipposideros (super-family: Rhinolophoidea sensu Teeling et al. (2005), family: Hipposideridae Lydekker, 1891). Considering the diversity of both Chiroptera and Coronavirinae, we explicitly implied the host species and the virus species within the name, thus avoiding further possible confusion once other coronaviruses will be discovered and described within these genera (Betacoronavirus and Hipposideros).
5.1. Sampling pressure and host species involved
Sampling bias could explain the uneven distribution of coronavirus detection among bat families, more specifically concerning SARSr-CoV in Rhinolophidae versus Hipposideridae. In contrast with other studies, the Hipposideridae family in our study was the most sampled family with five times more samples than those collected from Rhinolophidae. Only two sampling sites among 28 (site 17 for Hl BcoV-T and site 4 for Ha AcoV-R) explored with various sampling intensity, gave positive results. For some site, the low level of sampling might be insufficient to detect any Coronavirinae (Table 1). We cannot exclude that betacoronaviruses are hosted by others hipposiderid bat colonies and could circulate around Thailand or South-East Asia. Moreover, it is likely that other yet unknown betacoronaviruses circulate in Hipposideridae and even in other bat families around South-East Asia. An appropriate sampling would be able to test these hypotheses.
5.2. Long-lasting infection in colony, roost-sharing behaviour and host phylogenetic affinities are suspected to be species-jumping promoting factors
Chu and collaborators have repeatedly detected alphacoronaviruses in Miniopterus (Miniopteridae) colonies from Hong Kong. Positive detections in bat colonies were obtained over a period ranging from 3 months in a Miniopterus pusillus colony to 6 months in a Miniopterus magnater colony (Chu et al., 2006). These results, suggested that alphacoronaviruses were able to persist through time in bat colonies. Another study on Alphacoronavirus (and Betacoronavirus – SARSr-Rh-BatCoV –) infected Rhinolophus sinicus bats modulated this conclusion. No significant persistence was observed in Rhinolophus sinicus bat colonies and the viral clearance was estimated between 2 weeks and 4 months (Lau et al., 2010). Moreover, the authors reported a significant loss of body weight in bats infected by Betacoronavirus (SARSr-CoVs) that was not observed in Alphacoronavirus infected bats. This loss of weight could be the sign of a loss of fitness associated with Betacoronavirus infection in Rhinolophus sinicus, without presuming that it would be a promoting factor or a consequence of Betacoronavirus infection. Even though: (i) Betacoronavirus persistence was not observed in Rhinolophus sinicus colonies and, (ii) the loss of body mass correlated with Betacoronavirus infection might reduce the fitness of Rhinolophus, we report here that a Betacoronavirus was persisting for 18 months in a Hipposideridae bat colony. This suggests that a Hipposideridae colony might be more tolerant to Betacoronavirus than Rhinolophidae over a long period of time or that Betacoronavirus of Rhinolophidae might have acquired virulence factor limiting long-lasting infection in this host. Further investigations are needed to confirm and describe this suspected difference. This Betacoronavirus detected so far in Hipposideros larvatus in Thaïland is restricted to a single cave. Thus, Betacoronavirus long-lasting infection in a bat colony might be a rare event. Although rare, long-lasting infection can promote cross-species transmission by the emergence of new properties in viral populations (Baric et al., 1999). Despite the coincidence of several promoting factors in study site 17 (the long-lasting infection – Table 1 –, the sharing of the same small-sized cave by three bat species and the phylogenetic proximity of the two Hipposideros species) none of the other species were detected positive for Betacoronavirus (Hipposideros armiger and Taphozous longimanus were negative – Table 1). Interestingly, we noted that the Taphozous longimanus (negative) colony was roosting separately from the Hipposideros larvatus (positive) cluster whereas the Hipposideros armiger (negative) colony occupied also another specific area of the cave. Similarly, in site 4 (Table 1) the Hipposideros armiger colony, infected by ACoV-Ha, was roosting separately from the other (negative) species. Again, this typical spatial segregation within a roosting site (e.g. cave) limits direct contacts between species and is suspected to account for a behavioral barrier to cross-species transmission in the wild, as previously described for Lentivirus and Felidae (Troyer et al., 2008, VandeWoude et al., 2010). Nevertheless, we mentioned these observations here but these mechanisms still remain to be precisely described in the complex relationship between Rhinolophoidea and betacoronaviruses.
By contrast, previous studies of Asian coronaviruses that focused on the species level, suggested that there was no strict association between bat species and Alphacoronavirus species (Chu et al., 2006, Tang et al., 2006), and other studies on Betacoronavirus in Africa reached similar conclusions (Quan et al., 2010). Species-barrier crossing has been reported occasionally for Betacoronavirus in Asian, European and African Rhinolophidae or Hipposideridae but more frequently in Miniopteridae (Chu et al., 2006, Tang et al., 2006, Tong et al., 2009, Drexler et al., 2010). Moreover, Miniopterus species exhibit an aggregative behaviour, and mixed species colonies are commonly observed as described in literature (Benda et al., 2003). Mixed species roosting, inducing direct contact between species and therefore promoting transmission, in synergy with viral persistence in a colony and the phylogenetic proximity of the hosts, is likely to promote cross species transmission of alphacoronaviruses. As a result, a non-specific host-virus association for Alphacoronavirus in Miniopterus follows the same model as what has been described for these bats and their ectoparasites (Bruyndonckx et al., 2009). The apparent higher frequency of cross-species transmission (or the absence of strict virus-host association) in Miniopteridae compared to Hipposideridae and Rhinolphidae might partially be explained by their different roosting behaviour.
5.3. Phylogeny and biogeography: Relation between betacoronaviruses filiation and host distribution
The two methods of phylogenetic reconstructions employed here supported comparable relationships within Coronavirinae (Fig. 1 and S1). The uncertainty associated with some relationships was often similarly reported by each method. These phylogenetic issues might be reduced by increasing the size of the analysed fragment, that would increase the number of informative characters and therefore provide more information to support nodes. Adding new taxa would also improve the phylogeny by providing probable currently lacking information, within or around groups that are not yet well supported: (i) Betacoronavirus sub-groups relationships might be improved by looking for new taxa related to the group HKU9/Bat CoV Philippine Dilim (weak posterior probability: 0.59 and bootstrap value: 0.24 are reported for this node); (ii) Other sequences from new Betacoronavirus (or taxa from a new group) might help to clarify the relationship between Betacoronavirus, Alphacoronavirus and putative new groups as discordance is observed between phylogenies and none of the method provide significant support to the nodes that link Betacoronavirus and Alphacoronavirus (posterior probability: 0.69 and bootstrap values: 0.21; 0.06; 0.24).
Interestingly, our study revealed that the Betacoronavirus detected in Hipposideridae in South-East Asia was related to those that currently circulate in Africa, according to the analysis of a RdRp fragment. Our analyses strengthen the diversity of Betacoronavirus-b clade and extend their broad distribution. Indeed, SARS-CoV emerged from this Betacoronavirus-b clade (Fig. 1A) which is present in Africa, Europe, Asia and even Australia.
Quan and collaborators pushed forward the possible occurrence of migration events from Africa to Asia to explain the basal phylogenetic position of an African Betacoronavirus (compared to Asian rhinolophidae SARSr-CoV) together with the apparent absence of SARSr-CoV in Asian Hipposideridae (Quan, 2010). With the detection of Betacoronavirus in hipposiderids in Asia, this hypothesis, while still possible, becomes less necessary to explain phylogenetic and geographic patterns of Hipposideridae and Rhinolophidae betacoronaviruses. The clustering of Betacoronavirus-b that correlates with the host phylogeny rather than with virus geolocalization suggests that the phylogenetic relationships between betacoronaviruses-b is mainly driven by the host phylogeny (Fig. 1 and S1). Accordingly, the phylogenetic relationships of the Hl BCoV detected here (from South-East Asia) with the SARSr-CoV (from Africa) and other betacoronaviruses support the following hypothesis. Betacoronaviruses-b ancestors, meaning SARSr-CoVs ancestors, could have been historically hosted by the common ancestor of the Rhinolophidae and Hipposideridae and could have later evolved independently in the lineages leading towards Rhinolophidae and Hipposideridae betacoronaviruses. This point of view is supported by the topology of Betacoronavirus clade that exhibits the monophyly of Rhinolophidae betacoronaviruses despite their broad geographic distribution (from Africa and central Europe to South-East Asia and Northern Australia – Fig. 1) and shows the close relationship of Hipposideridae betacoronaviruses. Nevertheless, it is unlikely that putative host switching between Rhinolophidae and Hipposideridae betacoronaviruses (ancestors), eventually associated with ancient migration events, would still be detectable. More investigations are necessary to address these points and test corresponding hypotheses. As well, new investigations would be necessary to confirm this suggested predominant influence of the host phylogeny on Betacoronavirus-b clustering and whether other lineage would exist in Hipposideridae, as suggested by the report of a Betacoronavirus from Australia (C. Smith pers. com. – Fig. 1).
The position of the Hipposideridae Betacoronavirus from Australia (basal to the Rhinolophidae betacoronaviruses), suggests the existence of an intermediary subgroup (Fig. 1 and S1) between Hipposideridae and Rhinolophidae betacoronaviruses that survived within a rare endemic Australian bat species. The phylogenetic status of this host, Rhinonicteris aurantia, is not well defined but this species is described as intermediary between Hipposideridae and Rhinolophidae, while considered an Hipposideridae (Hand and Kirsch, 1998, Simmons, 2007, Armstrong, 2006; Armstrong com. pers.). Albeit this putative intermediary betacoronavirus group count a unique member to date and its associated posterior probability is weak, its phylogenetic position correlates with that of its host. Therefore, we are looking forward new investigations that would address these elements and screen betacoronaviruses in other Hipposideridae and Rhinolophidae.
The ecological niche of these bats and their behaviour provide them with frequent opportunities to spread viral variants among other nocturnal mammals such as other Chiroptera or Carnivora Viverridae (civet family) (Song et al., 2005, Guan et al., 2003, Gonzalez et al., 2008). Many factors have been suggested to explain the occurrence of emerging viruses in bats such as diversity (in term of species and ecology), ability to fly, immunology, physiology, seasonal migration and roosting behaviour (Dobson, 2005, Kunz and Fenton, 2005, Calisher et al., 2006). Another stated that population genetic structure correlates with viral richness (Turmelle and Olival, 2009). In the case of Betacoronavirus, the role of: (i) the fragmented biotope observed today (the Thai forest biotope has drastically reduced over the past decades, leading to the formation of species refuge areas promoting species interactions (Harvey et al., 2006, Trisurat et al., 2010)) and, (ii) the segregation behaviour and the mobility patterns of bats (metapopulations, migrations), should be investigated for their impact on the mosaic pattern of Betacoronavirus.
6. Conclusion
This study suggests that Hipposideridae have been underestimated in the study of coronaviruses in the wild. Both an Alphacoronavirus and a Betacoronavirus (more precisely the sub-group b that includes SARS-CoV) were detected in Hipposideridae in South-East Asia (Thailand). In contrast to what is reported for Rhinolophidae, Hipposideridae are able to host Betacoronavirus close to SARS-CoV over long periods of time, suggesting that they could be a source of virus for other species under specific circumstances. Moreover, the phylogeny of the RdRp, when compared to the biogeography, suggests that these two bat families may share an ancient relationship with independent lineage of Betacoronavirus. Consequently, and by contrast to what have been generally reported for coronaviruses, we suggest that the host phylogeny globally drive the pathogen clustering in the particular relationship between Betacoronavirus and Rhinolophoidea. As for other emerging viruses, interactions between wildlife refuges (whether considered in spatial or organic dimensions) and human populations, likely enhanced by fragmentation, changes, cultural behaviour and secular traditions, contribute to drive the species barrier crossing of coronaviruses to humans.
7. Financial Disclosure
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. This work benefited from the financial support of Carrefour International Fondation, Episars 511063 E.U. funded project, IRD intramural research funds and Science Foundation Ireland’s, Research Frontiers Programme, GEN 0056.
Acknowledgments
The research was approved by the Faculty of Science of the Mahidol University, Thailand and the French National Museum of Natural History. This work would never have been possible without the hosting by the Center of Excellence for Vectors and Vector-Borne Diseases (CEVVD, F. S., M. U., Thailand), the participation of the French Institute for Research and Development (IRD – UR178, Thailand) and the support of the Carrefour International Fondation. We are also grateful to W. Uamcharoen, K. Armstrong and C. Smith for their kind collaboration.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.meegid.2011.06.021.
Contributor Information
Meriadeg Ar Gouilh, Email: merry@pasteur.fr.
Jean-Claude Manuguerra, Email: jmanugu@pasteur.fr.
Appendix A. Supplementary data
Supplementary Fig. 1.
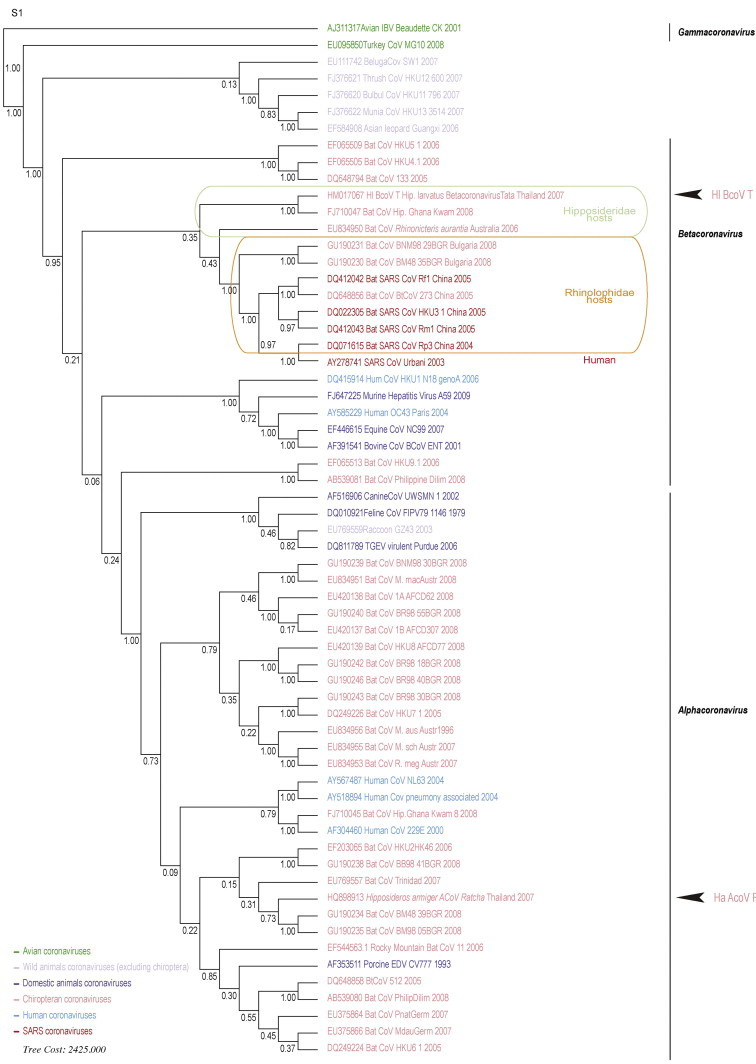
Phylogenetic reconstruction of main coronaviruses based on the analysis of 333 bases of the RNA-dependent polymerase protein nsp12 inferred by POY (same data as used with Bayesian method reconstruction in figure 1). Bootstrap values are specified for each node.
References
- Armstrong K.N. Phylogeographic structure in Rhinonicteris aurantia (Chiroptera: Hipposideridae): implications for conservation. Acta Chiropterologica. 2006;8(1):63–81. [Google Scholar]
- Benda P. et al., 2003. Bats (Mammalia: Chiroptera) of the Eastern Mediterranean. Part 3. Review of bat distribution in Bulgaria, 67, 245-357.
- Baric R.S. High recombination and mutation rates in mouse hepatitis virus suggest that coronaviruses may be potentially important emerging viruses. Advances in Experimental Medicine and Biology. 1995;380:571–576. doi: 10.1007/978-1-4615-1899-0_91. [DOI] [PubMed] [Google Scholar]
- Baric R.S. Episodic evolution mediates interspecies transfer of a murine coronavirus. Journal of Virology. 1997;71(3):1946–1955. doi: 10.1128/jvi.71.3.1946-1955.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baric R.S. Persistent infection promotes cross-species transmissibility of mouse hepatitis virus. Journal of Virology. 1999;73(1):638–649. doi: 10.1128/jvi.73.1.638-649.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Brand J.M.A. Pathology of experimental sars coronavirus infection in cats and ferrets. Veterinary Pathology Online. 2008;45(4):551–562. doi: 10.1354/vp.45-4-551. [DOI] [PubMed] [Google Scholar]
- Brandao P. A coronavirus detected in the vampire bat Desmodus rotundus. Brazilian Journal of Infectious Diseases. 2008;12:466–468. doi: 10.1590/s1413-86702008000600003. [DOI] [PubMed] [Google Scholar]
- Bruyndonckx N. Molecular cophylogenetic relationships between European bats and their ectoparasitic mites (Acari, Spinturnicidae) Molecular Phylogenetics and Evolution. 2009;51(2):227–237. doi: 10.1016/j.ympev.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Calisher C.H. Bats: important reservoir hosts of emerging viruses. Clinical Microbiology Review. 2006;19(3):531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington C.V.F. Detection and phylogenetic analysis of group 1 coronaviruses in south american bats. Emerging Infectious Diseases. 2008;14(12):1890–1893. doi: 10.3201/eid1412.080642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro C., Arnold J.J., Cameron C.E. Incorporation fidelity of the viral RNA-dependent RNA polymerase: a kinetic, thermodynamic and structural perspective. Virus Research. 2005;107(2):141–149. doi: 10.1016/j.virusres.2004.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomel B.B. New emerging zoonoses: a challenge and an opportunity for the veterinary profession. Comparative Immunology, Microbiology and Infectious Diseases. 1998;21(1):1–14. doi: 10.1016/s0147-9571(97)00018-0. [DOI] [PubMed] [Google Scholar]
- Chu D.K.W. Coronaviruses in bent-winged bats (Miniopterus spp.) Journal of General Virology. 2006;87(Pt 9):2461–2466. doi: 10.1099/vir.0.82203-0. [DOI] [PubMed] [Google Scholar]
- Cleaveland S., Laurenson M.K., Taylor L.H. Diseases of humans and their domestic mammals: pathogen characteristics, host range and the risk of emergence. Philosophical Transactions of the Royal Society of London. Series B. 2001;356(1411):991–999. doi: 10.1098/rstb.2001.0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J. Evolutionary relationships between bat coronaviruses and their hosts. Emerging Infectious Diseases. 2007;13(10):1526–1532. doi: 10.3201/eid1310.070448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daszak P. Bats, in black and white. Science. 2010;329(5992):634–635. doi: 10.1126/science.1194089. [DOI] [PubMed] [Google Scholar]
- Daszak P., Cunningham A.A., Hyatt A.D. Emerging Infectious diseases of wildlife – threats to biodiversity and human health. Science. 2000;287(5452):443–449. doi: 10.1126/science.287.5452.443. [DOI] [PubMed] [Google Scholar]
- Dobson A.P. What links bats to emerging infectious diseases? Science. 2005;310(5748):628–629. doi: 10.1126/science.1120872. [DOI] [PubMed] [Google Scholar]
- Domingo E., Holland J.J. RNA Virus mutations and fitness for survival. Annual Review of Microbiology. 1997;51(1):151–178. doi: 10.1146/annurev.micro.51.1.151. [DOI] [PubMed] [Google Scholar]
- Dominguez S.R. Detection of group 1 coronaviruses in bats in North America. Emerging Infectious Diseases. 2007;13(9):1295–1300. doi: 10.3201/eid1309.070491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler J.F. Genomic characterization of severe acute respiratory syndrome-related coronavirus in european bats and classification of coronaviruses based on partial rna-dependent rna polymerase gene sequences. Journal of Virology. 2010;84(21):11336–11349. doi: 10.1128/JVI.00650-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A.J., Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39(4):783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Gallagher, S.R. and Desjardins, P.R., 2006. Quantitation of DNA and RNA with Absorption and Fluorescence Spectroscopy. Available at: http://onlinelibrary.wiley.com/doi/10.1002/0471140864.psa04ks52/abstract. [DOI] [PubMed]
- Gannon W.L., Sikes R.S. Guidelines of the American society of mammalogists for the use of wild mammals in research. Journal of Mammalogy. 2007;88(3):809–823. doi: 10.1093/jmammal/gyw078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González J.M. A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae. Archives of Virology. 2003;148(11):2207–2235. doi: 10.1007/s00705-003-0162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez, J.P., Ar Gouilh, M., Reynes, J.M., Leroy, E., 2008. Bat-borne viral diseases. In: Pierce Colfer, C.J., Kleinau, E., (Eds.), Human Health and Forests. A Global Overview of Issues, Practice and Policy (Chapter 8).
- Gorbalenya Alexander E., Snijder E.J., Spaan W.J.M. Severe acute respiratory syndrome coronavirus phylogeny: toward consensus. Journal of Virology. 2004;78(15):7863–7866. doi: 10.1128/JVI.78.15.7863-7866.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M., Guindon S., Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Molecular Biology and Evolution. 2010;27(2):221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- Guan Y. Isolation and characterization of viruses related to the sars coronavirus from animals in Southern China. Science. 2003;302(5643):276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- Hand, S.J. & Kirsch, J.A., 1998. A southern origin for the Hipposideridae (Microchiroptera)? Evidence from the Australian fossil record. Bat biology and conservation. Washington, D.C: T.H. Kunz and P.A. Racey, 72–90.
- Harvey C.A. Patterns of animal diversity in different forms of tree cover in agricultural landscapes. Ecological Applications: A Publication of the Ecological Society of America. 2006;16(5):1986–1999. doi: 10.1890/1051-0761(2006)016[1986:poadid]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Herrewegh A.A. Feline coronavirus type ii strains 79–1683 and 79–1146 originate from a double recombination between feline coronavirus type i and canine coronavirus. Journal of Virology. 1998;72(5):4508–4514. doi: 10.1128/jvi.72.5.4508-4514.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutson T., 2003. Bats; ISBN: 089658500X, 9780896585003, 72p, Voyageur Press, 2003.
- Janies, D.A., Wheeler, W.C. 2002. Theory and practice of parallel direct optimization, Exs (92), 115–123. [DOI] [PubMed]
- Jia W. A novel variant of avian infectious bronchitis virus resulting from recombination among three different strains. Archives of Virology. 1995;140(2):259–271. doi: 10.1007/BF01309861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K.E. Global trends in emerging infectious diseases. Nature. 2008;451(7181):990–993. doi: 10.1038/nature06536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck J.G. Multiple recombination sites at the 5′-end of murine coronavirus RNA. Virology. 1987;156(2):331–341. doi: 10.1016/0042-6822(87)90413-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keesing F., Belden L.K. Impacts of biodiversity on the emergence and transmission of infectious diseases. Nature. 2010;468(7324):647–652. doi: 10.1038/nature09575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiken T. Emerging viral infections in a rapidly changing world. Current Opinion in Biotechnology. 2003;14(6):641–646. doi: 10.1016/j.copbio.2003.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz, T.H. and Fenton, M.B., 2005. Bats, emerging virusinfections, and the rabies paradigm. Dans Bat Ecology. Chicago: Thomas H. Kunz & M. Brock Fenton, 622–679.
- Lai M.M. Recombination between nonsegmented RNA genomes of murine coronaviruses. Journal of Virology. 1985;56(2):449–456. doi: 10.1128/jvi.56.2.449-456.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S.K.P. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proceedings of the National Academy of Science of the United States of America. 2005;102(39):14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S.K.P. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related rhinolophus bat coronavirus in china reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. Journal of Virology. 2010;84(6):2808–2819. doi: 10.1128/JVI.02219-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.-W., Jackwood M.W. Evidence of genetic diversity generated by recombination among avian coronavirus IBV. Archives of Virology. 2000;145(10):2135–2148. doi: 10.1007/s007050070044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. Bats are natural reservoirs of SARS-like coronaviruses. Science (New York, NY) 2005;310(5748):676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- Li W. Animal origins of the severe acute respiratory syndrome coronavirus: insight from ACE2-S-protein interactions. Journal of Virology. 2006;80(9):4211–4219. doi: 10.1128/JVI.80.9.4211-4219.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur, R.H. & Wilson, E.O., 1967. The theory of island biogeography, Princeton University Press.
- Makino S. High-frequency RNA recombination of murine coronaviruses. Journal of Virology. 1986;57(3):729–737. doi: 10.1128/jvi.57.3.729-737.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D.P. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26(19):2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina, Byron E E. et al., 2003. Virology: SARS virus infection of cats and ferrets. Nature, 425(6961), 915. [DOI] [PMC free article] [PubMed]
- Mueller, O., Lightfoot, S. and Schroeder, A., 2004. RNA Integrity Number (RIN) –Standardization of RNA Quality Control. Available at: http://www.genomics.agilent.com/.
- Müller M.A. Coronavirus antibodies in African bat species. Emerging Infectious Diseases. 2007;13(9):1367–1370. doi: 10.3201/eid1309.070342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon K.C. The parsimony ratchet, a new method for rapid parsimony analysis. Cladistics. 1999;15(4):407–414. doi: 10.1111/j.1096-0031.1999.tb00277.x. [DOI] [PubMed] [Google Scholar]
- Nunn C.L. Parasites and the evolutionary diversification of primate clades. The American Naturalist. 2004;164(S5):S90–S103. doi: 10.1086/424608. [DOI] [PubMed] [Google Scholar]
- Pfefferle S. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229e in bats, ghana. Emerging Infectious Diseases. 2009;15(9):1377–1384. doi: 10.3201/eid1509.090224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D. JModelTest: phylogenetic model averaging. Molecular Biology and Evolution. 2008;25(7):1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- Poon L.L.M. Identification of a novel coronavirus in bats. Journal of Virology. 2005;79(4):2001–2009. doi: 10.1128/JVI.79.4.2001-2009.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qinfen Z. The life cycle of SARS coronavirus in Vero E6 cells. Journal of Medical Virology. 2004;73(3):332–337. doi: 10.1002/jmv.20095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan, P.-L. et al., 2010. Identification of a severe acute respiratory syndrome coronavirus-like virus in a leaf-nosed bat in Nigeria. mBio, 1(4). [DOI] [PMC free article] [PubMed]
- Reperant, L.A., 2010. Applying the theory of island biogeography to emerging pathogens: toward predicting the sources of future emerging zoonotic and vector-borne diseases. Vector Borne and Zoonotic Diseases (Larchmont, N.Y.), 10(2), 105–110. [DOI] [PubMed]
- Schrag S.J., Wiener P. Emerging infectious disease: what are the relative roles of ecology and evolution? Trends in Ecology & Evolution. 1995;10(8):319–324. doi: 10.1016/s0169-5347(00)89118-1. [DOI] [PubMed] [Google Scholar]
- Simmons, N.B., 2007. Order Chiroptera. Mammalian Species of the world: A Taxonomic and Geographic Reference. D. E. Wilson, D. M. Reeders.
- Snijder E.J. Comparison of the genome organization of toro- and coronaviruses: evidence for two nonhomologous RNA recombination events during Berne virus evolution. Virology. 1991;180(1):448–452. doi: 10.1016/0042-6822(91)90056-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. Journal of Molecular Biology. 2003;331(5):991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H.-D. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(7):2430–2435. doi: 10.1073/pnas.0409608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler K. SARS – beginning to understand a new virus. Nature Reviews Microbiology. 2003;1(3):209–218. doi: 10.1038/nrmicro775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streicker D.G. Host phylogeny constrains cross-species emergence and establishment of rabies virus in bats. Science. 2010;329(5992):676–679. doi: 10.1126/science.1188836. [DOI] [PubMed] [Google Scholar]
- Tang X.C. Prevalence and genetic diversity of coronaviruses in bats from China. Journal of Virology. 2006;80(15):7481–7490. doi: 10.1128/JVI.00697-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeling E.C. A molecular phylogeny for bats illuminates biogeography and the fossil record. Science. 2005;307(5709):580–584. doi: 10.1126/science.1105113. [DOI] [PubMed] [Google Scholar]
- Thackray L.B., Holmes K.V. Amino acid substitutions and an insertion in the spike glycoprotein extend the host range of the murine coronavirus MHV-A59. Virology. 2004;324(2):510–524. doi: 10.1016/j.virol.2004.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong S. detection of novel sars-like and other coronaviruses in bats from Kenya. Emerging Infectious Diseases. 2009;15(3):482–485. doi: 10.3201/eid1503.081013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trisurat Y., Alkemade R., Verburg P.H. Projecting land-use change and its consequences for biodiversity in northern Thailand. Environmental Management. 2010;45(3):626–639. doi: 10.1007/s00267-010-9438-x. [DOI] [PubMed] [Google Scholar]
- Troyer J.L. FIV cross-species transmission: an evolutionary prospective. Veterinary Immunology and Immunopathology. 2008;123(1–2):159–166. doi: 10.1016/j.vetimm.2008.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turmelle A.S., Olival K.J. Correlates of viral richness in bats (order Chiroptera) EcoHealth. 2009;6(4):522–539. doi: 10.1007/s10393-009-0263-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VandeWoude S., Troyer J., Poss M. Restrictions to cross-species transmission of lentiviral infection gleaned from studies of FIV. Veterinary Immunology and Immunopathology. 2010;134(1–2):25–32. doi: 10.1016/j.vetimm.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varón A., Vinh L.S., Wheeler W.C. POY version 4: phylogenetic analysis using dynamic homologies. Cladistics. 2010;26(1):72–85. doi: 10.1111/j.1096-0031.2009.00282.x. [DOI] [PubMed] [Google Scholar]
- Wheeler W. Optimization alignment: the end of multiple sequence alignment in phylogenetics? Cladistics. 1996;12(1):1–9. [Google Scholar]
- WHO, 2003. WHO Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003. Available at: <http://www.who.int/csr/sars/country/table 2004_04_21/en/index.html>.
- Woo P.C.Y. Molecular diversity of coronaviruses in bats. Virology. 2006;351(1):180–187. doi: 10.1016/j.virol.2006.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolhouse M.E.J., Gowtage-Sequeria S. Host range and emerging and reemerging pathogens. Emerging Infectious Diseases. 2005;11(12):1842–1847. doi: 10.3201/eid1112.050997. [DOI] [PMC free article] [PubMed] [Google Scholar]