

Summary
Ribosomes are specialized entities that participate in regulation of gene expression through their rRNAs carrying ribozyme activity. Ribosome biogenesis is overactivated in p53-inactivated cancer cells, although involvement of p53 on ribosome quality is unknown. Here, we show that p53 represses expression of the rRNA methyl-transferase fibrillarin (FBL) by binding directly to FBL. High levels of FBL are accompanied by modifications of the rRNA methylation pattern, impairment of translational fidelity, and an increase of internal ribosome entry site (IRES)-dependent translation initiation of key cancer genes. FBL overexpression contributes to tumorigenesis and is associated with poor survival in patients with breast cancer. Thus, p53 acts as a safeguard of protein synthesis by regulating FBL and the subsequent quality and intrinsic activity of ribosomes.
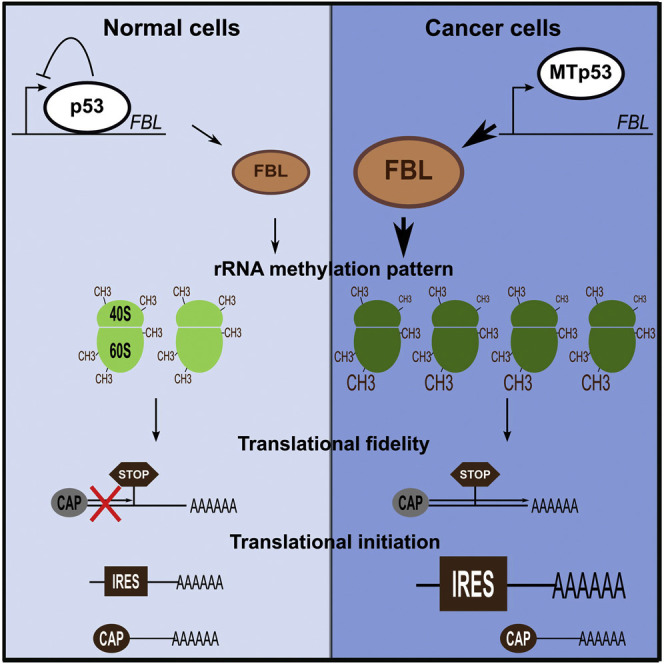
Graphical Abstract
Highlights
-
•
p53 directly represses transcription of the rRNA methyl-transferase fibrillarin
-
•
rRNA methylation pattern is altered in response to p53 inactivation
-
•
p53 inactivation promotes IRES-dependent translation in an FBL-dependent manner
-
•
High FBL mRNA levels are a marker of poor prognosis in breast cancer
Significance
Ribosome production is increased in cancer cells. This enhanced production of ribosomes plays a crucial role in tumor progression. Our results show that p53 regulates the pattern of ribosomal RNA posttranscriptional modification that impairs translational fidelity and increases the initiation of internal ribosome entry site (IRES)-dependent translation. By demonstrating that p53 regulates the biogenesis and intrinsic activity of ribosomes, our study demonstrates that p53 deficiency participates in the “translational reprogramming” in cancer cells and contributes to the uncontrolled expression of key oncogenic genes.
Introduction
Recent findings support the emerging notion that the dysregulation of ribosome biogenesis in cancer cells is not just a consequence of oncogenesis, but represents a key step in this complex process (Barna et al., 2008, Bywater et al., 2012, Yoon et al., 2006). Ribosome biogenesis is a multistage process that involves transcriptional and posttranscriptional regulation and a stringent quality control to produce functional ribosomes (Bashan and Yonath, 2008, Henras et al., 2008). Ribosome biogenesis is overactivated in cancer cells, notably by a loss of function of RNA polymerase I (Pol I) repressors such as p53 (Bywater et al., 2012, Zhai and Comai, 2000).
The posttranscriptional steps of ribosomal RNA (rRNA) processing determine the structure and function of the mature molecule (King et al., 2003, Baxter-Roshek et al., 2007, Puglisi, 2009). Indeed, rRNAs are ribozymes that support the decoding and proofreading steps and catalyze the formation of the peptide bond during translation (Cech, 2000, Demeshkina et al., 2012). rRNAs are subjected to intense and highly specific chemical modifications (methylations and pseudouridylations). The exact role of these modifications has not yet been fully elucidated, although it appears that they help optimize the complex ribosomal architecture required to produce an efficient ribosome (Baxter-Roshek et al., 2007).
Changes in the posttranscriptional modifications of rRNAs influence translational fidelity (i.e., nonsense suppression or amino acid misincorporation) and the mode of translation initiation (i.e., CAP versus internal ribosome entry site [IRES]) of key cancer genes (Ruggero, 2013, Basu et al., 2011, Baxter-Roshek et al., 2007). Moreover, a defect in rRNA methylation or pseudouridylation may cause specific clinical syndromes and is correlated with an increased incidence of cancer (Belin et al., 2009, Montanaro et al., 2008). This study aims to determine whether p53 controls rRNA methylation and subsequently affects translational regulation.
Results
FBL Expression Is Inversely Associated with p53 Activity in Cell Lines and Human Breast Cancer Samples
Fibrillarin (FBL) is an indispensable, highly conserved protein essential in the processing of pre-rRNAs (Newton et al., 2003, Tollervey et al., 1993). In eukaryotes, it is the only known methyltransferase that performs the specific 2′-O-ribose-methylation directed by a large family of small trans-acting guide RNA (box C/D antisense snoRNAs). Because abnormal rRNA methylation could influence translational control and because p53 regulates rRNA transcription, we explored whether FBL expression was associated with p53 activity. We measured FBL mRNA and protein levels in different cell lines in which p53 expression and/or activity was modulated by different strategies. In immortalized human mammary epithelial cells (HME), p53 activity was impaired either by reducing its expression using an shRNA approach (HME-shp53) or by inactivation using an SV40 T/t antigen trapping strategy (HMLE; Elenbaas et al., 2001). In response to p53 knockdown or inactivation, there was a significant increase in FBL expression: 1.5-fold for the mRNA and 2-fold for the protein (Figures 1 A–1D; Figures S1A and S1B available online). As expected, we observed a decrease in the expression of CDKN1A, which encodes p21, and MDM2, two p53 target genes, which validated the reduction in p53 activity in these cellular models (Figures 1B, 1D, S1A, and S1B). The inverse correlation between FBL and p53 expression was confirmed in a second series of immortalized HMEC lines (Figures S1C and S1D) and in an isogenic human HCT-116 colorectal cellular model: HCT-116-p53+/+ cells that express wild-type p53 protein and HCT-116-p53−/− cells that lack p53 protein expression (Bunz et al., 1998). FBL mRNA and protein levels were increased in HCT-116-p53−/− cells compared to those of HCT-116-p53+/+ cells (Figures 1E, 1F, and S1E), demonstrating that the increase in expression of FBL in response to p53 inactivation is not restricted to mammary cell lines.
Figure 1.

p53 Regulates FBL Expression at both the mRNA and Protein Levels
The expression of endogenous FBL in the indicated cell lines was analyzed at the mRNA level by RT-qPCR (A, C, and E) and at the protein level by western blot (B, D, and F). All graphs represent mean and SD of at least three experiments. ∗p < 0.05 and ∗∗p < 0.01 according to Student’s t test.
See also Figure S1.
To assess more directly the impact of p53 on FBL expression, HME cells were treated with a p53-specific siRNA instead of an shRNA to induce a transient knockdown of p53 expression (Figures 2A, 2B, S2A, and S2B). In this condition, increased FBL expression at the mRNA and protein levels correlated with the inhibition of p53 expression (Figures 2A and 2B). Taken together, these results exclude the possibility that the changes in FBL expression levels resulted from an off-target effect and firmly link FBL expression to p53 expression. Additionally, to investigate the impact of p53 activation on FBL expression, we treated HME cells with the topoisomerase inhibitors doxorubicin and camptothecin, which are prominent activators of p53. The results showed that the expected p53 induction in response to treatment was accompanied by a decrease in FBL mRNA and protein levels (Figures 2C and 2D). Quantification of the western blots demonstrated a significant association between the increase of p53 protein levels and the decrease of FBL protein levels (Figures S2C–S2E).
Figure 2.

Modulation of p53 Expression Alters FBL Expression
Endogenous FBL expression was analyzed in HME cells at the mRNA level by RT-qPCR (A and C) and at the protein level by western blot (B and D). The p53 expression is modulated by using an siRNA (A and B) or by treating or not (NT) with 2 μg/ml doxorubicin or 1 nM camptothecin (C and D). The p21 lanes in (D) were spliced together from discontinuous lanes of the same blot as indicated by dotted lines. All graphs represent mean and SD of at least three experiments. ∗p < 0.05 and ∗∗∗p < 0.001 according to Student’s t test.
See also Figure S2.
In human cancers, the TP53 tumor suppressor gene is frequently inactivated, mainly by mutations (Olivier et al., 2006). To determine whether FBL expression is associated with p53 mutation status independently of other genetic variations in cell lines, we analyzed the FBL expression levels in a panel of three wild-type p53 breast cell lines and five mutant p53 breast cancer cell lines. High levels of FBL mRNA and protein were significantly associated with the expression of mutant p53 (Figures 3A, 3B, and S3). This result prompted us to investigate the clinical correlation between the p53 mutation status and FBL expression. We analyzed the FBL mRNA expression levels by RT-qPCR in relation to the p53 mutation status in a cohort of 80 randomly selected primary breast tumors (Table S1). Consistent with the results obtained in cell lines, FBL mRNA levels were significantly higher in mutant p53 tumors compared to wild-type p53 tumors (Figure 3C). We also performed a retrospective statistical analysis of the gene expression array data described by Miller and colleagues (Miller et al., 2005). FBL mRNA levels were significantly higher in mutant p53 tumors (n = 58) than in wild-type p53 tumors (n = 193; p < 10−4, t test). Altogether, these results show a significant and reproducible inverse association between the p53 level and/or activity and the expression of FBL at both the mRNA and protein levels, suggesting that p53 can repress FBL expression in cellular models of breast and colon cancer as well as in human breast tumors.
Figure 3.

p53 Regulates FBL Expression in Human Breast Cell Lines and Tumors
(A) Quantification of FBL mRNA expression analyzed by RT-qPCR and normalized to RNA18S.
(B) Quantification of FBL protein expression analyzed by western blot. All graphs represent mean and SD of at least three experiments.
(C) Box-and-whisker plots of FBL mRNA expression quantification in wild-type p53 (n = 59) versus mutant p53 (n = 21) primary breast tumor samples. The bottom and the top of the boxes represent the 25th and 75th percentiles, respectively. The median values are visible as a line, the mean as a cross in the box and SD as error bars. The p value has been determined by a Mann-Whitney W test.
p53 Represses FBL Expression by Directly Binding to DNA
Using the MatInspector software and the p53FamTag database, two putative p53 responsive elements (p53RE-1 and p53RE-2) were identified within the FBL intron 1, suggesting a direct transcriptional regulation of FBL expression by p53 (Cartharius et al., 2005, Sbisà et al., 2007; Figures 4 A and S4A). Based on these predictions, we developed a luciferase reporter (pFBL-Luc) assay to assess whether p53 regulates FBL promoter activity. HCT-116-p53−/− cells were cotransfected with pFBL-Luc and a plasmid expressing either wild-type or mutant p53 protein at detectable protein levels (Figures 4B and S4B). The coexpression of wild-type p53 significantly decreased the luciferase activity by 80%, while no significant variation in luciferase activity was observed after co-expression of any p53 mutant. Similar results were observed in HME-shp53, the coexpression of wild-type, but not mutant, p53 protein reducing the luciferase activity (Figure 4C). These results suggest that p53 represses promoter activity through intron 1 of FBL in both breast and colon cellular models.
Figure 4.

p53 Represses FBL Promoter Activity by Directly Binding to DNA
(A) Alignment of the two putative p53 response elements (p53RE-1 and p53RE-2, black arrows) located within the intron 1 of FBL with the p53RE consensus (R, G/A; W, A/T; Y, C/T). n, spacer within p53RE consensus; dotted box, nucleotide region of the FBL gene cloned in the pFBL-Luc reporter vector; P1 and P2, primers pairs used in ChIP assays.
(B and C) Luciferase reporter assays were performed in the absence of p53 (−) and in the presence of the wild-type (WT) or the indicated mutant p53 protein in HCT-116-p53−/− (B) and in HME-shp53 cells (C). Firefly luciferase activity is normalized to the renilla luciferase activity. Basic, luciferase reporter vector with no FBL sequence.
(D and E) ChIP using an anti-p53 antibody and primer pairs P1 (D) or P2 (E) were performed in nontreated (NT) or 1 nM camptothecin-treated HME cells, or in HME-shp53 cells. All graphs represent mean and SD of at least three experiments. ∗p < 0.05 and ∗∗∗p < 0.001 according to Student’s t test.
See also Figure S4.
To determine whether p53 directly binds to FBL gene DNA, chromatin immunoprecipitation (ChIP) assays were performed in HME-derived cells (Figures 4D, 4E, S4C, and S4D). Compared to nontreated HME cells, camptothecin treatment increased p53 binding to both the CDKN1A promoter and the intron 1 of FBL at p53RE-1 and p53RE-2. In contrast, decrease in p53 expression in HME-shp53 cells was associated with a drastic reduction in p53 binding to both the CDKN1A promoter and intron 1 of FBL compared with nontreated HME cells (Figures 4D, 4E, S4C, and S4D). These data show that p53 binds the FBL intron 1 both in the basal condition and when p53 is activated. Altogether, these results demonstrate that FBL is a p53 target gene and that FBL expression is directly repressed by p53.
p53-Mediated Alteration of the rRNA Methylation Pattern
Because p53 directly represses FBL expression, we determined whether p53 inactivation alters rRNA methylation by using a previously described, site-specific semiquantitative RT-qPCR-based method (Belin et al., 2009, Maden, 1988). We analyzed the change of rRNA methylation at 18 sites distributed along the 5.8S, 18S, and 28S rRNAs that are known to be methylated. These sites include those localized within key functional domains of rRNAs, i.e., the decoding center (DC) in the 18S rRNA, and the peptidyl transferase center (PTC) and the helix 69 (H69) of 28S rRNA. In general, most of the sites were significantly more frequently methylated in HME-shp53 than in HME cells (Figures 5 A and S5A). This overall increase in the site-specific rRNA methylation pattern is consistent with our finding that FBL expression level is higher in HME-shp53 than in HME cells, and suggests that methylation could be regulated in a site-by-site manner.
Figure 5.

p53 Regulates the rRNA Methylation Pattern and the Translational Fidelity of Ribosomes
(A) The fold difference in rRNA methylation at 18 sites distributed throughout the 5.8S, 18S, and 28S rRNAs between HME-shp53 and HME cells were analyzed by RT-qPCR.
(B–J) Translational fidelity was analyzed by transfecting cells with the pGL3mut1 vector (premature stop mutant, B–D), the pGL3mut2 vector (amino acid substitution mutant, E–G), or the SARS-CoV −1 programmed ribosome frameshift vector (H–J) in the indicated cells. (G) Translational fidelity was analyzed in nontransfected cells (NT) and after transfection of siRNA control (sc) or siRNA targeting FBL (si-FBL). FBL expression levels were verified by western blot (G, lower panel). All graphs represent mean and SD of at least three experiments. ∗p < 0.05 and ∗∗∗p < 0.001 according to Student’s t test.
See also Figure S5.
As expected, metabolic labeling with [5,6-3H]-uridine and L-[methyl-3H]-methionine showed that p53 inactivation led to a significant increase in rRNA synthesis (Figure S5B; Zhai and Comai, 2000), with a faint increase in the global rRNA methylation rate (Figure S5C). This showed that the amount and/or activity of the rRNA methylation machinery was sufficient to sustain the global rRNA methylation rate following the increase in rRNA synthesis after p53 inactivation. In addition, because selection of site methylation is ensured by guide C/D-box snoRNAs that complex with FBL, we verified whether snoRNA expression levels were altered in p53-inactivated cells. Northern blot analyses revealed a modification of snoRNA levels according to p53 levels, suggesting that p53 could also be involved in the regulation of some C/D-box snoRNA as is the case for some H/ACA snoRNP (Figures S5D–S5F; Krastev et al., 2011). However, no correlation was found between the amount of snoRNA and the level of the corresponding rRNA methylation sites (Figures 5A and S5A). These results show that inactivation of p53 resulted in a site-specific modification of the rRNA methylation pattern that correlates with FBL expression.
p53 Alters the Translational Fidelity by Modulating FBL Expression
The chemical modifications of rRNA that have been conserved throughout evolution in all species are involved, at least in eukaryotes, in the control of translational fidelity and in the control of translation initiation modalities (i.e., CAP-dependent versus IRES-dependent; Ruggero, 2013, Baxter-Roshek et al., 2007, Chaudhuri et al., 2007, Ruggero et al., 2003). We first analyzed two different examples of translational fidelity, nonsense suppression and amino acid misincorporation (Belin et al., 2009). The bypass of a premature stop-codon (Figures 5B–5D) and the misincorporation of amino acids (Figures 5E–5G) were both significantly increased after p53 inhibition or inactivation in different cell lines. This suggested that the translational alteration could be due to p53-mediated increase of FBL expression level. To investigate this possibility, we analyzed the misincorporation of amino acids in response to knockdown of FBL expression. As shown in Figure 5G, reduction of FBL expression in HCT-116-p53−/− prevented the increase in amino acids misincorporation, demonstrating that the decrease in translational quality control in response to p53 inhibition is dependent on FBL overexpression. In contrast, the ability of ribosomes to induce a −1 frameshift from a severe acute respiratory syndrome-coronavirus (SARS-CoV)-1 programmed ribosome frameshift signal was similar in the three cell lines (Figures 5H–5J). These results suggest that p53 inactivation could decrease translational fidelity in an FBL-dependent manner.
p53 Increases IRES-Mediated Initiation of Translation by Modulating FBL Expression
To determine whether the CAP- or IRES-dependent mode of translation was modified, we used a bi-cistronic vector strategy that has been extensively used to identify a vast number of IRES-containing sequences (Belin et al., 2009, Komar and Hatzoglou, 2011). At this stage of the study, we focused our analysis on IGF1R due to its role in tumorigenesis and because IGF1R possesses the longest GC-rich 5′UTR that contains a well-identified IRES among all human transcripts yet characterized (Giraud et al., 2001, Pollak et al., 2004). Using a bi-cistronic luciferase assay, we observed a significant increase in the global luciferase activity in the p53-inactivated HME-shp53 and HCT-116-p53−/− cells compared with cells expressing wild-type p53 (Figure 6 A). This increase was due to a preference to initiate IRES-dependent translation. Indeed, the translation of the firefly luciferase driven by the IRES of IGF1R was significantly increased, while no significant variation was detected for the renilla luciferase driven by a CAP-dependent mechanism (Figures S6A and S6B). Similar results were obtained in other HME-derived cell lines (Figures 6B, white bars; Figures S6C and S6D, sc bars). Moreover, HME and HMLE cells expressed similar levels of firefly luciferase mRNA and renilla luciferase mRNA independently of variations in p53 protein levels, which supports the hypothesis of IRES-mediated translational regulation rather than transcriptional regulation of IGF1R expression (Figure S6E).
Figure 6.

p53 Regulates the IGF1R IRES-Dependent Translation
(A and B) The IGF1R IRES-dependent translation initiation was determined by using luciferase bi-cistonic vectors in the indicated cells (A) and in cells after the downregulation of FBL by siRNA approach (B).
(C) Analysis of the IGF1R IRES-dependent translation initiation in a panel of breast cell lines expressing either wild-type or mutant p53 proteins.
(D) Typical polysomal profiles after fractionation of postmitochondrial supernatants from HME and HME-shp53 cells in a 10%–40% sucrose gradient are shown (upper). The distribution of the IGF1R mRNA within polysomes was determined by RT-qPCR (lower).
(E and F) Endogenous IGF1R expression at mRNA (E, black bars) and protein levels (E, white bars; and F) was analyzed in HME and HMLE cells. All graphs represent mean and SD of at least three experiments. ∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001 according to Student’s t test.
See also Figure S6.
To evaluate whether a similar regulation exists in human cell lines, we explored the IGF1R IRES activity in breast cell lines that express wild-type or mutant p53 protein (Figure 6C). The activity level of the IGF1R IRES was significantly higher in the mutant p53 cells that expressed high levels of FBL compared with the wild-type p53 cells that expressed low levels of FBL. Thus, translation initiation mediated by the IGF1R IRES was higher in breast cell lines that had inactivated p53 instead of wild-type p53.
To confirm that the IRES activity was modulated by rRNA methylation, we analyzed the IGF1R IRES activity after using an FBL-siRNA knockdown approach that reduced the FBL protein level (Figures S6C, S6D, and S6F). As shown in Figure 6B, reduction of FBL expression significantly decreases the IRES-dependent translation initiation of IGF1R in HME, HME-shp53 and HMLE cells. This result demonstrates that the efficiency of IRES-dependent translational initiation is modulated by rRNA methylation through modulation of FBL expression.
To determine whether the p53-mediated translational control of the IGF1R mRNA observed with recombinant expression vectors also occurs with endogenous IGF1R mRNA, we compared the distribution of the IGF1R mRNA within polysomal fractions of HME and HME-shp53 cells. The polysomal fraction corresponds to mRNA bound to more than one ribosome and therefore contains actively translated mRNA. As shown in Figure 6D, a significant 12-fold polysomal enrichment in IGF1R mRNA was observed in HME-shp53 compared with HME cells. In addition, a significant increase in the IGF1R protein level was observed in p53-inactivated HMLE cells compared to HME cells independently of any variation in IGF1R mRNA levels (Figures 6E and 6F). These results demonstrate that p53 inactivation increases the translation of IGF1R mRNA.
To investigate whether this p53-mediated translational control is restricted to IGF1R mRNA, we analyzed translational control of several other known cellular and viral mRNAs containing IRESs, including MYC, FGF1, FGF2, and VEGFA that play key roles in oncogenesis (Dang, 2012, Turner and Grose, 2010, Carmeliet and Jain, 2011) and encephalomyocarditis virus. Bi-cistronic luciferase assays and mRNA polysomal profiling assays showed an increase of translational efficiency of these mRNA in p53 inactivated cells compared to p53 wild-type cells (Figures S6G–S6I). Altogether these results show that p53 inactivation impacts the translational control of several genes involved in tumorigenesis through deregulation of FBL expression.
FBL Overexpression Contributes to Tumorigenesis
Six independent stable clones derived from MCF7 cells overexpressing either a FBL-GFP protein or a GFP protein were developed (Figure S7A) to determine the effect of FBL overexpression on several characteristics of cancer cells. Cell proliferation of the stable clones was first monitored for 72 hr and the index of cell proliferation was calculated from the slope of the growth curve (Figure 7 A). Cells overexpressing FBL-GFP exhibit a significant 2-fold increase in proliferation rate compared with GFP control cells. To determine whether the FBL-induced cell proliferation is mediated by IGF1R, whose translation is increased in FBL-overexpressing cells, proliferation of two clones was monitored in response to Osi-906, an IGF1R tyrosine kinase activity inhibitor (Figure 7B; Mulvihill et al., 2009). Inhibition of IGF1R pathway activity abolished the difference in proliferation rate between GFP-G3 and FBL-GFP-F2 clones, suggesting that IGF1R pathway activity is required in these cells, for FBL-induced cell proliferation.
Figure 7.

Contribution of FBL Overexpression to Cancer Phenotype
(A) Cell proliferation of three each independent stable MCF7 clones expressing FBL-GFP (F1, F2, and F3) or GFP (G1, G2, and G3).
(B) Proliferation of the indicated cell clones not treated (NT) or treated with 1 μM Osi-906 for 72 hr.
(C and D) Anchorage-independent growth of MCF7 clones using soft agar assay. Representative images are shown in (C) and the numbers of colonies determined in three experiments are shown in (D).
(E) Impact of FBL overexpression on drug response was investigated by determining the IC50 of doxorubicin using MTS assays. Error bars represent SD. The p values have been determined by a Mann-Whitney W test.
(F and G) Kaplan-Meier analysis of relapse-free survival rates (event = relapse) (F) and of breast cancer-specific survival rates (event = death related to breast cancer disease) (G) according to FBL mRNA level in primary breast tumor samples. The data are dichotomized at the upper quartile value into high and low expression groups.
Anchorage-independent cell proliferation was then investigated using soft agar assays. Compared to GFP control clones, those overexpressing FBL-GFP formed significantly more colonies (Figures 7C and 7D). Finally, the effect of FBL overexpression on cell growth in response to doxorubicin treatment was investigated (Figure 7E). A significantly higher concentration of doxorubicin is required to reach 50% of inhibitory effect in FBL-GFP cells compared to GFP control cells (mean half-maximal inhibitory concentration [IC50]: 24.6 μM versus 53.1 μM, respectively). Altogether, these data show that FBL overexpression promotes cell proliferation in both an anchorage-dependent and -independent manner and protects the MCF7 breast cancer cells from doxorubicin. Moreover, the FBL-induced cell proliferation required IGF1R pathway, supporting the notion that FBL overexpression can directly contribute to tumorigenesis by altering translational control of key cancer genes.
High Levels of FBL Are an Independent Marker of a Poor Outcome in Breast Cancer
To investigate whether the level of FBL mRNA in tumors is associated with prognosis, we analyzed the relapse-free survival and the breast cancer-specific survival of patients with breast cancer in regard to FBL expression. High expression of FBL mRNA was significantly associated with a poor relapse-free survival rate and poor breast cancer-specific survival rate (Figures 7F and 7G). We also performed retrospective statistical analyses of published gene expression array data (Györffy et al., 2010, Sabatier et al., 2011, Weigelt et al., 2005). This investigation confirmed that the high levels of FBL mRNA are associated with poor breast cancer-specific survival and relapse-free survival rates (Figures S7B–S7F).
However, because the high levels of FBL expression are associated with p53 mutations and because p53 mutations are known to be associated with poor disease-free and overall survival rates (Olivier et al., 2006), a multivariate analysis was conducted to adjust for possible confounding variables. The analysis included the number of invaded lymph nodes, histological grade, estrogen and progesterone receptors status, ErbB2 status, p53 mutation status, and FBL mRNA levels. The best model associated with poor survival contained two independent markers: progesterone receptor-negative status and high FBL expression (Table S2). These analyses showed that FBL expression is associated with poor survival independent of other commonly used clinical markers.
Discussion
It is now clearly established that ribosome synthesis is increased in cancers due to the overexpression of oncogenes or the inactivation of tumor suppressor genes leading to a sustained increase in RNA Pol I activity (Bywater et al., 2012). Moreover, studies performed in different animal and cellular models of various eukaryotic organisms have shown that heterogeneity in ribosome composition, due to regulated posttranscriptional modifications of ribosomal proteins and rRNA, is likely to be the more common mechanism (Xue and Barna, 2012). Xue and Barna have made detailed contributions to extend the concept of “specialized ribosomes” to eukaryotes and highlighted the adaptive capabilities of the ribosomes in the control of cell fate through selective protein synthesis. Moreover, it is now well demonstrated that within the ribosomes, the rRNAs catalyze and control protein synthesis through their ribozyme activity that could be finely optimized by their rRNAs methylations and pseudouridylations (Baxter-Roshek et al., 2007, Belin et al., 2009). In this study, we show a p53-mediated alteration of ribosome biogenesis and translational control of cancer cells that could contribute in gene expression dysregulation and cancer development (Ruggero, 2013).
We demonstrate a role of p53 in the control of rRNA methylation patterning by directly regulating FBL expression levels that leads to the synthesis of modified “cancer ribosomes.” The notion that FBL is a p53-target gene is supported by genome-wide analyses, such as ChIP-seq assays showing that p53 binds the FBL gene (ENCODE database, Nikulenkov et al., 2012) and by transcriptomic analysis showing an inverse expression of FBL in response to p53 inactivation by siRNA or activation by doxorubicin in several cell lines (Troester et al., 2006). p53 response elements have also been identified in the first intron of approximately 25% of p53 target genes, some of them being associated with gene repression, including genes involved in ribosome biogenesis such as NOLC1, a snoRNP chaperone gene (Menendez et al., 2009, Krastev et al., 2011).
We found that the methylation pattern of rRNAs varies between sites, which is consistent with published data (Baxter-Roshek et al., 2007, Belin et al., 2009, Basu et al., 2011) and suggests that the rRNA modification pattern is modulated in a site-by-site manner. Our data showed that p53 inactivation is sufficient to alter rRNA methylation patterning. In addition to regulating FBL expression, the role of p53 in optimizing the rRNA functional quality is reinforced by our observations that the level of some C/D-box snoRNAs is modulated according to the p53 status and that p53 is involved in the assembly process of the other major family of snoRNP (H/ACA box; Krastev et al., 2011). Deciphering the mechanisms by which p53 inactivation alters the site-specific rRNA methylation pattern through FBL induction will require biochemical and structural studies dedicated to the understanding of formation, dynamics, and activities of the rRNA methylation complex. However, we can hypothesize that the improper induction of FBL expression observed in cancer cells leads to an alteration of the coordination between pre-rRNA production and the rRNA methylation enzymatic machineries.
Today, several pieces of data, including ours, indicate that modulation of methylation at only some rRNA sites is sufficient to affect the translational regulation process and that it could alter cellular behavior without inducing a lethal phenotype (King et al., 2003, Baudin-Baillieu et al., 2009, Higa-Nakamine et al., 2012). Indeed, the depletion of methylation of several rRNA methylation sites in yeast and human cells has been associated with a decrease in translation fidelity (such as an increase in nonsense suppression, frameshifts, and amino acid misincorporation; Baxter-Roshek et al., 2007, Baudin-Baillieu et al., 2009). Moreover, modulating rRNA methylation by RPL13a depletion in HeLa cells modified the control of the translation initiation by IRES (Basu et al., 2011). Modification in the rRNA methylation pattern in breast cancer cells exhibiting an induced aggressive phenotype was also associated with the alteration of IRES activity of key factors such as vascular endothelial growth factor (VEGF) and p53 (Belin et al., 2009). In the present study, we demonstrate that the repression of FBL expression by p53 is accompanied by an increase of IRES-dependent translation initiation, affecting cellular as well as a viral IRES-containing mRNAs. These data are consistent with previous reports showing that the FGF2 mRNA translation is inhibited by p53, whereas the FGF2 mRNA IRES is aberrantly activated in transformed cells when p53 is inactivated (Galy et al., 2001). It remains to systematically explore the effect of methylation sites, individually and as a pattern, in the intrinsic activity of the ribosome.
Modulation of intrinsic activity of the ribosome by altering rRNA methylation may involve structural changes of ribosomes. The inhibition of rRNA methylation altered the IRES translation initiation by impairing the association of the 40S and 60S subunits (Basu et al., 2011). Moreover, structural and biochemical studies showed that ribose methylation modifies the conformational state of the RNA backbone, stabilizes the RNA loops, and influences the overall structure of the modified RNA regions. Ribose methylation helps maintain the tertiary structure of rRNAs and potentially the rRNA-mRNA, rRNA-tRNA or rRNA-protein interactions (Blanchard and Puglisi, 2001, Liang et al., 2009). Consistently, several 18S rRNA regions promote structural modifications when a viral IRES is bound to the 40S subunit (Spahn et al., 2004) and the efficient translation of IGF1R mRNA results from its IRES directly contacting an 18S rRNA domain through a Shine-Dalgarno-like interaction (Meng et al., 2010). These data support the notion that rRNA methylation could participate in translational control by regulating IRES translation initiation.
Our clinical analysis shows that a high level of FBL in primary breast tumors is associated with poor survival independent of other biological markers. Elevated expression levels of FBL were previously reported in primary and metastatic prostate cancers compared with normal prostate epithelium and in squamous cell cervical carcinoma compared with normal cervix samples (Choi et al., 2007, Koh et al., 2011, Su et al., 2013). Furthermore, we have shown the direct contribution of FBL overexpression in tumorigenesis. The maintenance and progression of cancer phenotype induced by FBL-mediated enhanced translation may involve several key cancer proteins whose synthesis is dependent upon IRES-containing mRNA. As shown here, these proteins may include IGF1R, which is involved in tumor progression, cell survival, and response to chemotherapy (Pollak et al., 2004), c-Myc, which exhibits pleiotropic pro-oncogenic functions (Dang, 2012), FGF1/2, which are involved in epithelial-mesenchymal transition (Sakuma et al., 2012), and VEGFA, which is involved in tumoral angiogenesis (Carmeliet and Jain, 2011). Thus, high levels of FBL observed in human samples could have a role in tumor progression and could affect the clinical outcome of patients through alteration of translational regulation.
Our results allow us to propose a model in which p53 regulates not only the ribosome production rate, but also their structure, function, and intrinsic activity (Figure 8 ). In this model, p53 alteration in pathological cells results in the production of ribosomes with decreased translational fidelity and increased translation of the IRES-containing mRNAs selectively. Thus, the p53-mediated ribosome alterations could be in part responsible for the “translational instability” of cancer cells and contribute to the expression of the continuously growing class of translationally regulated cancer-promoting genes (Ruggero, 2013).
Figure 8.

Model of the Implication of p53 in the Control of Ribosomes Quantity and Ribosomes Quality, and Their Consequences on Translation
In cells expressing functional p53 (top), p53 negatively regulates RNA Pol I activity to control ribosome quantity and FBL expression levels to control ribosome quality. This regulation would aim to coordinate the methylation of ribosomes and the rate of ribosome production according to the cell needs. These quality-controlled ribosomes allow a high translational fidelity together with a correct control of the balance between CAP- and IRES-dependent initiation of translation. In cells expressing a mutant or nonfunctional p53 (bottom), loss of the repression of RNA Pol I activity leads to an increase in rRNA synthesis. In parallel, p53 inactivation leads to an increase in FBL expression levels, resulting in a modification of the rRNA methylation patterns. Ribosomes with modified rRNA methylated translate mRNA with a lower fidelity (bypass of stop codon, amino acid misincorporation) and are more likely to initiate translation through IRES of mRNA coding for pro-oncogenic, anti-apoptotic, and survival proteins.
Finally, the detailed description of rRNA chemical modification patterning in cancer cells, occurring in part through the p53-mediated regulation of ribosome biogenesis enzymatic machineries, and the increasing knowledge of the ribosome structure at the atomic level (Anger et al., 2013), opens up the possibility to target these “cancer ribosomes” to develop anticancer molecules using strategies similar to those used for the development of antibiotics specifically targeting prokaryotic ribosomes (Yonath, 2009).
Experimental Procedures
Cell Culture, Transfection, and Luciferase Assay
Cells were maintained in culture following ATCC recommendations. siRNA and plasmids (Belin et al., 2009) were transfected using lipofectamine 2000 (Invitrogen). Cells were treated with 2 μg/ml doxorubicin or 1 nM camptothecin (Sigma). Luciferase activity was measured using the Dual Luciferase Reporter Assay kit (Promega). Anchorage-dependent cell proliferation was analyzed using a real-time monitoring cell proliferation assay based on variation of electric impedance using the xCELLigence RTCA system (ACEA Biosciences) for 72 hr in nontreated cells or in presence of 1 μM Osi-906 (Selleckchen). Anchorage-independent cell proliferation was analyzed by soft agar assays. The IC50 values for doxorubicin were determined by MTS assays (Promega).
Western Blot and Chromatin Immunoprecipitation
Protein extraction and western blot were performed as described (Belin et al., 2010) using the following antibodies: anti-FBL (38F3, Abcam); anti-p53 (DO-1, Santa Cruz); anti-β-actin (AC-15, Sigma); anti-Mdm2 (4B2, Bethyl Laboratories); anti-p21 (F-5, Santa Cruz); and anti-IGF1Rα (N20, Santa Cruz). For ChIP assays, chromatin was prepared from 1% formaldehyde fixed cells by sonication. Immunoprecipitation was performed on 200-1000 bp DNA fragments using the DO-1 anti-p53 antibody and immunoprecipitated DNA was quantified by qPCR using Sybr Green technology.
Total mRNA, Polysomal mRNA, and rRNA Methylation Quantification
Total RNA and RNA issued from cytosolic and polysomal fractions was extracted and purified using either Trizol reagent (Invitrogen) or TriPure Isolation reagent (Roche). Cytosolic ribosomes were obtained from postmitochondrial fractions, and polysomal ribosomes by separation of postmitochondrial fractions on a 10%–40% sucrose gradient by ultracentrifigation. Total, cytosolic, and polysomal mRNA levels were quantified by RT-qPCR using M-MLV and Sybr Green technologies as described (Ghayad et al., 2009). Site-specific rRNA methylation was quantified using a RT-qPCR based method, which relies on the inhibition of reverse transcription reaction by ribose methylation at low dNTP concentration and on the detection of total rRNA as an internal reference, by reverse transcription at high dNTP concentration (Belin et al., 2009). RT products were then quantified by qPCR.
Breast Tumor Samples
FBL mRNA expression was analyzed in a cohort of 80 primary breast tumors collected at Ninewells Hospital from white women who received no neoadjuvant treatment prior surgery (Tayside Tissue Bank, Dundee; Table S1). Informed consent was obtained from all patients and ethical approval was received from the Tayside Tissue Bank (REC Reference 07/S1402/90) under delegated authority from the Local Research Ethics Committee. Relapse-free survival was calculated among breast cancer patients from the date of diagnosis to the date of relapse (event = relapse). Breast cancer-specific survival was calculated from the date of diagnosis to the date of breast cancer specific death (event = death related to breast cancer disease). The characterization of the classical molecular markers (histological grade, invaded lymph node, p53 mutation as well as estrogen, progesterone, and errB2 status) was previously determined and reported (Bourdon et al., 2011).
Statistical Analyses
Statistical analyses were performed using the Statgraphics 3 plus software (Statgraphics Centurion). The log-rank test (univariate analysis), Kaplan-Meier plots, and Cox proportional hazards model (multivariate analysis) were performed using SPSS Software. A p value < 0.05 was considered to be statistically significant. All graphs present the mean and standard variations of at least three independent experiments and Student’s t test has been performed for experimental data.
Experimental procedures are detailed in the Supplemental Experimental Procedures.
Acknowledgments
This work was supported by Centre Léon Bérard, CNRS, INSERM, Université Claude Bernard Lyon 1, Ecole Normale Supérieure de Lyon, Cancéropôles GSO and CLARA, ARC, FRM, ANR (Oncoscreen), and Ligue Contre le Cancer. It was funded by grants from Région Rhône-Alpes (thématiques prioritaires and Cluster 10), Ligue Contre le Cancer (Comité du Rhône), Institut National contre le Cancer (RIBOCAN). V.M. is a recipient of a postdoctoral fellowship from the Centre Léon Bérard. S.B. and G.T. are recipients of doctoral fellowships from the Ligue Contre le Cancer and from ARC for S.B. We thank Carine Jolyon for her technical assistance.
Published: September 9, 2013
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.ccr.2013.08.013.
Supplemental Information
References
- Anger A.M., Armache J.P., Berninghausen O., Habeck M., Subklewe M., Wilson D.N., Beckmann R. Structures of the human and Drosophila 80S ribosome. Nature. 2013;497:80–85. doi: 10.1038/nature12104. [DOI] [PubMed] [Google Scholar]
- Barna M., Pusic A., Zollo O., Costa M., Kondrashov N., Rego E., Rao P.H., Ruggero D. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature. 2008;456:971–975. doi: 10.1038/nature07449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashan A., Yonath A. Correlating ribosome function with high-resolution structures. Trends Microbiol. 2008;16:326–335. doi: 10.1016/j.tim.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Basu A., Das P., Chaudhuri S., Bevilacqua E., Andrews J., Barik S., Hatzoglou M., Komar A.A., Mazumder B. Requirement of rRNA methylation for 80S ribosome assembly on a cohort of cellular internal ribosome entry sites. Mol. Cell. Biol. 2011;31:4482–4499. doi: 10.1128/MCB.05804-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin-Baillieu A., Fabret C., Liang X.-H., Piekna-Przybylska D., Fournier M.J., Rousset J.-P. Nucleotide modifications in three functionally important regions of the Saccharomyces cerevisiae ribosome affect translation accuracy. Nucleic Acids Res. 2009;37:7665–7677. doi: 10.1093/nar/gkp816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter-Roshek J.L., Petrov A.N., Dinman J.D. Optimization of ribosome structure and function by rRNA base modification. PLoS ONE. 2007;2:e174. doi: 10.1371/journal.pone.0000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin S., Beghin A., Solano-Gonzàlez E., Bezin L., Brunet-Manquat S., Textoris J., Prats A.-C., Mertani H.C., Dumontet C., Diaz J.-J. Dysregulation of ribosome biogenesis and translational capacity is associated with tumor progression of human breast cancer cells. PLoS ONE. 2009;4:e7147. doi: 10.1371/journal.pone.0007147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin S., Kindbeiter K., Hacot S., Albaret M.A., Roca-Martinez J.X., Thérizols G., Grosso O., Diaz J.-J. Uncoupling ribosome biogenesis regulation from RNA polymerase I activity during herpes simplex virus type 1 infection. RNA. 2010;16:131–140. doi: 10.1261/rna.1935610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard S.C., Puglisi J.D. Solution structure of the A loop of 23S ribosomal RNA. Proc. Natl. Acad. Sci. USA. 2001;98:3720–3725. doi: 10.1073/pnas.051608498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdon J.-C., Khoury M.P., Diot A., Baker L., Fernandes K., Aoubala M., Quinlan P., Purdie C.A., Jordan L.B., Prats A.-C. p53 mutant breast cancer patients expressing p53γ have as good a prognosis as wild-type p53 breast cancer patients. Breast Cancer Res. 2011;13:R7. doi: 10.1186/bcr2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz F., Dutriaux A., Lengauer C., Waldman T., Zhou S., Brown J.P., Sedivy J.M., Kinzler K.W., Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Bywater M.J., Poortinga G., Sanij E., Hein N., Peck A., Cullinane C., Wall M., Cluse L., Drygin D., Anderes K. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell. 2012;22:51–65. doi: 10.1016/j.ccr.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P., Jain R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartharius K., Frech K., Grote K., Klocke B., Haltmeier M., Klingenhoff A., Frisch M., Bayerlein M., Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- Cech T.R. Structural biology. The ribosome is a ribozyme. Science. 2000;289:878–879. doi: 10.1126/science.289.5481.878. [DOI] [PubMed] [Google Scholar]
- Chaudhuri S., Vyas K., Kapasi P., Komar A.A., Dinman J.D., Barik S., Mazumder B. Human ribosomal protein L13a is dispensable for canonical ribosome function but indispensable for efficient rRNA methylation. RNA. 2007;13:2224–2237. doi: 10.1261/rna.694007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y.W., Kim Y.W., Bae S.M., Kwak S.Y., Chun H.J., Tong S.Y., Lee H.N., Shin J.C., Kim K.T., Kim Y.J., Ahn W.S. Identification of differentially expressed genes using annealing control primer-based GeneFishing in human squamous cell cervical carcinoma. Clin. Oncol. (R Coll Radiol) 2007;19:308–318. doi: 10.1016/j.clon.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Dang C.V. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeshkina N., Jenner L., Westhof E., Yusupov M., Yusupova G. A new understanding of the decoding principle on the ribosome. Nature. 2012;484:256–259. doi: 10.1038/nature10913. [DOI] [PubMed] [Google Scholar]
- Elenbaas B., Spirio L., Koerner F., Fleming M.D., Zimonjic D.B., Donaher J.L., Popescu N.C., Hahn W.C., Weinberg R.A. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galy B., Créancier L., Zanibellato C., Prats A.-C., Prats H. Tumour suppressor p53 inhibits human fibroblast growth factor 2 expression by a post-transcriptional mechanism. Oncogene. 2001;20:1669–1677. doi: 10.1038/sj.onc.1204271. [DOI] [PubMed] [Google Scholar]
- Ghayad S.E., Vendrell J.A., Bieche I., Spyratos F., Dumontet C., Treilleux I., Lidereau R., Cohen P.A. Identification of TACC1, NOV, and PTTG1 as new candidate genes associated with endocrine therapy resistance in breast cancer. J. Mol. Endocrinol. 2009;42:87–103. doi: 10.1677/JME-08-0076. [DOI] [PubMed] [Google Scholar]
- Giraud S., Greco A., Brink M., Diaz J.-J., Delafontaine P. Translation initiation of the insulin-like growth factor I receptor mRNA is mediated by an internal ribosome entry site. J. Biol. Chem. 2001;276:5668–5675. doi: 10.1074/jbc.M005928200. [DOI] [PubMed] [Google Scholar]
- Györffy B., Lanczky A., Eklund A.C., Denkert C., Budczies J., Li Q., Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010;123:725–731. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- Henras A.K., Soudet J., Gérus M., Lebaron S., Caizergues-Ferrer M., Mougin A., Henry Y. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell. Mol. Life Sci. 2008;65:2334–2359. doi: 10.1007/s00018-008-8027-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higa-Nakamine S., Suzuki T., Uechi T., Chakraborty A., Nakajima Y., Nakamura M., Hirano N., Suzuki T., Kenmochi N. Loss of ribosomal RNA modification causes developmental defects in zebrafish. Nucleic Acids Res. 2012;40:391–398. doi: 10.1093/nar/gkr700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King T.H., Liu B., McCully R.R., Fournier M.J. Ribosome structure and activity are altered in cells lacking snoRNPs that form pseudouridines in the peptidyl transferase center. Mol. Cell. 2003;11:425–435. doi: 10.1016/s1097-2765(03)00040-6. [DOI] [PubMed] [Google Scholar]
- Koh C.M., Gurel B., Sutcliffe S., Aryee M.J., Schultz D., Iwata T., Uemura M., Zeller K.I., Anele U., Zheng Q. Alterations in nucleolar structure and gene expression programs in prostatic neoplasia are driven by the MYC oncogene. Am. J. Pathol. 2011;178:1824–1834. doi: 10.1016/j.ajpath.2010.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komar A.A., Hatzoglou M. Cellular IRES-mediated translation: the war of ITAFs in pathophysiological states. Cell Cycle. 2011;10:229–240. doi: 10.4161/cc.10.2.14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krastev D.B., Slabicki M., Paszkowski-Rogacz M., Hubner N.C., Junqueira M., Shevchenko A., Mann M., Neugebauer K.M., Buchholz F. A systematic RNAi synthetic interaction screen reveals a link between p53 and snoRNP assembly. Nat. Cell Biol. 2011;13:809–818. doi: 10.1038/ncb2264. [DOI] [PubMed] [Google Scholar]
- Liang X.H., Liu Q., Fournier M.J. Loss of rRNA modifications in the decoding center of the ribosome impairs translation and strongly delays pre-rRNA processing. RNA. 2009;15:1716–1728. doi: 10.1261/rna.1724409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maden B.E. Locations of methyl groups in 28 S rRNA of Xenopus laevis and man. Clustering in the conserved core of molecule. J. Mol. Biol. 1988;201:289–314. doi: 10.1016/0022-2836(88)90139-8. [DOI] [PubMed] [Google Scholar]
- Menendez D., Inga A., Resnick M.A. The expanding universe of p53 targets. Nat. Rev. Cancer. 2009;9:724–737. doi: 10.1038/nrc2730. [DOI] [PubMed] [Google Scholar]
- Meng Z., Jackson N.L., Shcherbakov O.D., Choi H., Blume S.W. The human IGF1R IRES likely operates through a Shine-Dalgarno-like interaction with the G961 loop (E-site) of the 18S rRNA and is kinetically modulated by a naturally polymorphic polyU loop. J. Cell. Biochem. 2010;110:531–544. doi: 10.1002/jcb.22569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller L.D., Smeds J., George J., Vega V.B., Vergara L., Ploner A., Pawitan Y., Hall P., Klaar S., Liu E.T., Bergh J. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc. Natl. Acad. Sci. USA. 2005;102:13550–13555. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montanaro L., Calienni M., Ceccarelli C., Santini D., Taffurelli M., Pileri S., Treré D., Derenzini M. Relationship between dyskerin expression and telomerase activity in human breast cancer. Cell. Oncol. 2008;30:483–490. doi: 10.3233/CLO-2008-0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvihill M.J., Cooke A., Rosenfeld-Franklin M., Buck E., Foreman K., Landfair D., O’Connor M., Pirritt C., Sun Y., Yao Y. Discovery of OSI-906: a selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med Chem. 2009;1:1153–1171. doi: 10.4155/fmc.09.89. [DOI] [PubMed] [Google Scholar]
- Newton K., Petfalski E., Tollervey D., Cáceres J.F. Fibrillarin is essential for early development and required for accumulation of an intron-encoded small nucleolar RNA in the mouse. Mol. Cell. Biol. 2003;23:8519–8527. doi: 10.1128/MCB.23.23.8519-8527.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikulenkov F., Spinnler C., Li H., Tonelli C., Shi Y., Turunen M., Kivioja T., Ignatiev I., Kel A., Taipale J., Selivanova G. Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell Death Differ. 2012;19:1992–2002. doi: 10.1038/cdd.2012.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier M., Langerød A., Carrieri P., Bergh J., Klaar S., Eyfjord J., Theillet C., Rodriguez C., Lidereau R., Bièche I. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin. Cancer Res. 2006;12:1157–1167. doi: 10.1158/1078-0432.CCR-05-1029. [DOI] [PubMed] [Google Scholar]
- Pollak M.N., Schernhammer E.S., Hankinson S.E. Insulin-like growth factors and neoplasia. Nat. Rev. Cancer. 2004;4:505–518. doi: 10.1038/nrc1387. [DOI] [PubMed] [Google Scholar]
- Puglisi J.D. Resolving the elegant architecture of the ribosome. Mol. Cell. 2009;36:720–723. doi: 10.1016/j.molcel.2009.11.031. [DOI] [PubMed] [Google Scholar]
- Ruggero D. Translational control in cancer etiology. Cold Spring Harb. Perspect. Biol. 2013;5 doi: 10.1101/cshperspect.a012336. a012336–a012336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggero D., Grisendi S., Piazza F., Rego E., Mari F., Rao P.H., Cordon-Cardo C., Pandolfi P.P. Dyskeratosis congenita and cancer in mice deficient in ribosomal RNA modification. Science. 2003;299:259–262. doi: 10.1126/science.1079447. [DOI] [PubMed] [Google Scholar]
- Sabatier R., Finetti P., Cervera N., Lambaudie E., Esterni B., Mamessier E., Tallet A., Chabannon C., Extra J.M., Jacquemier J. A gene expression signature identifies two prognostic subgroups of basal breast cancer. Breast Cancer Res. Treat. 2011;126:407–420. doi: 10.1007/s10549-010-0897-9. [DOI] [PubMed] [Google Scholar]
- Sakuma K., Aoki M., Kannagi R. Transcription factors c-Myc and CDX2 mediate E-selectin ligand expression in colon cancer cells undergoing EGF/bFGF-induced epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA. 2012;109:7776–7781. doi: 10.1073/pnas.1111135109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbisà E., Catalano D., Grillo G., Licciulli F., Turi A., Liuni S., Pesole G., De Grassi A., Caratozzolo M.F., D’Erchia A.M. p53FamTaG: a database resource of human p53, p63 and p73 direct target genes combining in silico prediction and microarray data. BMC Bioinformatics. 2007;8(Suppl 1):S20. doi: 10.1186/1471-2105-8-S1-S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn C.M., Jan E., Mulder A., Grassucci R.A., Sarnow P., Frank J. Cryo-EM visualization of a viral internal ribosome entry site bound to human ribosomes: the IRES functions as an RNA-based translation factor. Cell. 2004;118:465–475. doi: 10.1016/j.cell.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Su H., Xu T., Ganapathy S., Shadfan M., Long M., Huang T.H., Thompson I., Yuan Z.M. Elevated snoRNA biogenesis is essential in breast cancer. Oncogene. 2013 doi: 10.1038/onc.2013.89. Published online April 1, 2013. [DOI] [PubMed] [Google Scholar]
- Tollervey D., Lehtonen H., Jansen R., Kern H., Hurt E.C. Temperature-sensitive mutations demonstrate roles for yeast fibrillarin in pre-rRNA processing, pre-rRNA methylation, and ribosome assembly. Cell. 1993;72:443–457. doi: 10.1016/0092-8674(93)90120-f. [DOI] [PubMed] [Google Scholar]
- Troester M.A., Herschkowitz J.I., Oh D.S., He X., Hoadley K.A., Barbier C.S., Perou C.M. Gene expression patterns associated with p53 status in breast cancer. BMC Cancer. 2006;6:276. doi: 10.1186/1471-2407-6-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N., Grose R. Fibroblast growth factor signalling: from development to cancer. Nat. Rev. Cancer. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- Weigelt B., Hu Z., He X., Livasy C., Carey L.A., Ewend M.G., Glas A.M., Perou C.M., Van’t Veer L.J. Molecular portraits and 70-gene prognosis signature are preserved throughout the metastatic process of breast cancer. Cancer Res. 2005;65:9155–9158. doi: 10.1158/0008-5472.CAN-05-2553. [DOI] [PubMed] [Google Scholar]
- Xue S., Barna M. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat. Rev. Mol. Cell Biol. 2012;13:355–369. doi: 10.1038/nrm3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonath A. Large facilities and the evolving ribosome, the cellular machine for genetic-code translation. J. R. Soc. Interface. 2009;6(Suppl 5):S575–S585. doi: 10.1098/rsif.2009.0167.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon A., Peng G., Brandenburger Y., Zollo O., Xu W., Rego E., Ruggero D. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science. 2006;312:902–906. doi: 10.1126/science.1123835. [DOI] [PubMed] [Google Scholar]
- Zhai W., Comai L. Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol. Cell. Biol. 2000;20:5930–5938. doi: 10.1128/mcb.20.16.5930-5938.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.