

Highlights
-
•
We identify the species and geographical origin of emergent avian virus, GVI_PMV1.
-
•
Europe was the epicenter for the rapid global dispersal of the emergent virus.
-
•
At least three independent introductions of GVI_PMV1 occurred into North America.
-
•
Introduced viruses are evolving rapidly and diversifying locally in North America.
-
•
Introduced viruses are recovered from an invasive European avian species.
Keywords: Evolution, Pigeon, Newcastle disease, Phylogeography, Cross-species transmission, Avian paramyxovirus
Abstract
The evolutionary history of avian paramyxovirus serotype 1 (PMV1), which includes the agents of Newcastle disease (ND), is characterized by a series of strain emergence events since viruses in this family were first recognized in the 1920s. Despite the importance of ND to the poultry industry, little is known about PMV1 strain emergence events and the subsequent dispersal and evolution of new strains. The genotype VI-PMV1 was first identified in the 1980s and has been named pigeon paramyxovirus-1 (PPMV1) because of unusual host specificity with Columbiformes (Collins et al., 1996); it has been responsible for panzootics in both chickens and pigeons during that time. Here, we used evolutionary analyses to characterize the emergence of this contemporary PMV1 lineage. We demonstrate that GVI-PMV1 arose through cross-species transmission events from Galliformes (i.e. chicken) to Columbiformes, and quickly established in pigeon populations. Our studies revealed a close association between the time of viral emergence and panzootic events of this virus. The virus appeared first in Southeastern Europe and quickly spread across the European continent, which became the epicenter for global virus dissemination. With new viral gene sequences, we show that GVI-PMV1 viruses currently circulating in North America resulted from multiple invasion events from Europe, one associated with an exotic European Columbiformes species, and that extant lineages have diversified locally. This study extends our understanding of successful viral emergence subsequent to cross-species transmission and dispersal patterns of newly emerged avian viruses, which may improve surveillance awareness and disease control of this and other important avian pathogens.
1. Introduction
The number of emergent or re-emergent infectious viral diseases that are linked to cross-species transmission from reservoirs to naive hosts is increasing in both animals and humans (Woolhouse, 2008). Notable examples include emergence of avian and swine influenza viruses, henipaviruses, and the severe acute respiratory syndrome coronavirus. The mechanisms in viral emergence are complex and include ecological, immunological, and genetic features of both virus and host (Lloyd-Smith et al., 2009, Parrish et al., 2008). Although the dispersal of viruses in humans following zoonotic transmission can be documented by integrating epidemiological data with virus sequence data, the events that precede productive viral infection in a new host are more difficult to untangle (Lloyd-Smith et al., 2009). Similarly, cross-species transmissions among animal species leading to successful strain emergence are poorly understood. With the increased availability of genetic data and associated temporal and geographic information from improved surveillance effort, it is now possible to obtain insights of inter-species transmission dynamics that give rise to new viral strains, which pose health risks in animals.
Avian paramyxovirus serotype 1 (PMV1) is a negative-sense single stranded RNA virus in the family Paramyxoviridae. It is best known for causing outbreaks of Newcastle disease (ND), a source of significant economic loss to the poultry industry worldwide. In the 1950s, 30 years after the first reported PMV1 outbreak, modified live vaccines derived from the dominant circulating strain were deployed to prevent and control the disease (Alexander, 1988a). Subsequently, a series of new strains have emerged from unknown sources that contribute to the ND global burden. There are now at least ten reported genotypes of PMV1 contributing to the extensive genetic diversity in avian populations worldwide (Aldous et al., 2004, Barbezange and Jestin, 2003, Barbezange and Jestin, 2005, Diel et al., 2012, Kim et al., 2008, Kommers et al., 2001, Liu et al., 2006, Mase et al., 2009, Meulemans et al., 2002, Panshin et al., 1999, Servan de Almeida et al., 2009, Sleeman, 2010, Tsai et al., 2004, Ujvari et al., 2003, Zhu et al., 2010, http://www.worldpoulty.net/). Understanding the mechanism of successful strain emergence in PMV1 can inform efforts to control ND and other viral diseases with similar evolutionary dynamics that threaten domestic and wild birds.
In the late 1970s, a distinct strain of PMV1 was sporadically isolated from pigeons in proximity to ND outbreaks in chickens (Alexander et al., 1984, Alexander et al., 1985a, Kaleta et al., 1985, Kaleta and Baldauf, 1988, Stewart, 1971). The new virus, called pigeon-associated PMV1 (PPMV1), was initially presumed to be of chicken origin and to infect pigeons only as a result of sporadic spill over events. However, in 1984, multiple ND outbreaks in Great Britain were initiated by PPMV1 and spread from pigeons to chickens (Alexander et al., 1985b). Subsequently, PPMV1 has been responsible for other chicken ND outbreaks worldwide (Aldous et al., 2004, Capua et al., 2002, Kommers et al., 2001, Liu et al., 2006, OIE, 2011a, OIE, 2011b, Toro et al., 2005, Werner et al., 1999) and has caused extensive panzootic infections in racing pigeons in European countries despite vaccination efforts (Alexander et al., 1997, Kommers et al., 2001, Meulemans et al., 2002, Werner et al., 1999, Zanetti et al., 2001). Genetic analysis of PPMV1 demonstrated that this strain, named genotype VI-PMV1 (GVI-PMV1) (Aldous et al., 2004), constituted a distinct monophyletic lineage with considerable genetic heterogeneity.
Although virulent PMV1 is considered to be exotic to US poultry, serological data evidenced GVI-PMV1 in ND outbreaks in pigeons during the 1980–1990s (Barton et al., 1992, Pearson et al., 1987, Tangredi, 1988). A recent molecular study (part of the West Nile Virus Surveillance program) (Kim et al., 2008) also confirmed that GVI-PMV1 is circulating in North American pigeon populations. The evolutionary dynamics of this pathogen in North America remains unclear because of limited sequence data and absence of coordinated national surveillance.
In this study, we aim to understand the events leading to the emergence and the molecular evolution and phylogeography of the new strain. We inferred the evolutionary timescale for the entire GVI-PMV1 lineage with statistical confidence. The chicken-to-pigeon transmission events leading to the establishment of the major GVI-PMV1 sub-lineage in Columbiformes populations was studied in details. Using advanced phylogenetic methods, we estimated the time and geographic origin of this important cross-species event. To better understand the viral spread in North America, we sequenced the F genes of 74 PMV1 isolates collected in northeastern United States (US) from 2001 to 2009. We demonstrate that there were at least three introductions of GVI-PMV1 into North America, including one associated with Eurasian collared doves. We also provide evidence for viral population expansion in one of the two circulating lineages.
2. Materials and methods
2.1. PMV1 field isolates
The Animal Diagnostic Laboratory at The Pennsylvania State University (PSU-ADL) conducted virus isolation tests on avian cases submitted from Pennsylvania and other states in the US. Bird tissue specimens and swab samples were processed for virus isolation in specific pathogen free (SPF) embryonated chicken eggs (ECE) and/or chicken embryo fibroblast (CEF) cell cultures following described procedures [(Alexander, 1988b) and OIE Terrestrial Manual, 2009 (Manual, 2009)]. Hemagglutination (HA) and hemagglutination-inhibition (HI) tests were conducted for PMV1 identification. PMV1 field strains isolated from various avian species at PSU-ADL between 1996 and 2009 were retrieved for this study.
2.2. Viral RNA extraction and sequencing
Viral RNA was extracted using the QIAamp Viral RNA extraction kit (Qiagen, Valencia, CA-USA) and was reverse transcribed using a specific PMV1 oligonucleotide targeting the 3′ untranslated region of the genome (5′-ACGGGTAGAAGGTGTGAATC-3′) with the AffinityScript™ Multiple Temperature Reverse Transcriptase Kit (Stratagene, Santa Clara, CA-USA). PCR amplification was performed targeting the entire coding region of the F gene using two pairs of specific primers (GVI-forward primer: 5′-TGCTCGGACCTTCTGTGCTTGTGA-3′; GVI-reverse primer: 5′-TGCGGACCTTGTTCTTGCTGCTAC-3′; GII-forward primer: 5′-TGACCGCCGACCACGAG-3′; GII-reverse primer: 5′-TCAGGAGAGGCCGATTCAAGTATT-3′) with the TaKaRa Ex Taq Hot Start PCR kit (Clontech, Mountain View, CA-USA). These primer sets also amplify the entire coding region of the HN gene. A total of 74 PMV1 isolates were amplified. PCR products were gel purified using the QIAquick Gel Extraction kit (Qiagen, Valencia, CA-USA) and sequenced. Sequences were assembled and edited using the Lasergene DNASTAR software package (DNASTAR Inc. WI, USA). Sequences have been deposited in GenBank (Accession numbers JX901304–JX901377; Table S1).
2.3. Sequence collection and alignment
A total of 3631 F gene sequences of PMV1 were downloaded from GenBank in April, 2011 and aligned using Muscle program (Edgar, 2004). Sequences with length shorter than 240 bp were removed. The 74 PMV1 sequences generated in this study were combined with GenBank sequences, and tested for recombination by RDP2 (Martin et al., 2005). Putative recombinants, which were confirmed by GARD method (Kosakovsky Pond et al., 2006), were removed. This resulted in a final data set of 3469 sequences, which included sequences ranging from 240 to 1662 bp. Of these 710 were full-length.
2.4. Phylogenetic analyses
A maximum likelihood (ML) panoramic phylogeny was built from the F gene sequence data set using FastTree v2.1 program (Price et al., 2010). The GVI-PMV1 lineage (n = 534, with known isolation dates; 512 were longer than 350nt and 108 were full-length) was identified and extracted from the panoramic phylogeny for further analyses (detailed taxon names are shown in Table S2). A refined ML phylogenetic tree of GVI-PMV1 was reconstructed by PhyML v3 program (Guindon et al., 2010), where the best tree was selected from subtree pruning and regrafting (SPR) and nearest-neighbor interchange (NNI) topological optimizations. The general time reversible with gamma distribution (GTR + Γ) nucleotide substitution model was used. Local topological supports were assessed by Shimodaira–Hasegawa (SH) tests (Guindon et al., 2010). Among the new PMV1 sequences generated in this study, 61 isolates were GVI and the remaining 13 isolates were grouped in genotype II (GII; the genotype to derive vaccine strain). These strains are indicated in Fig. 1 A.
Fig. 1.
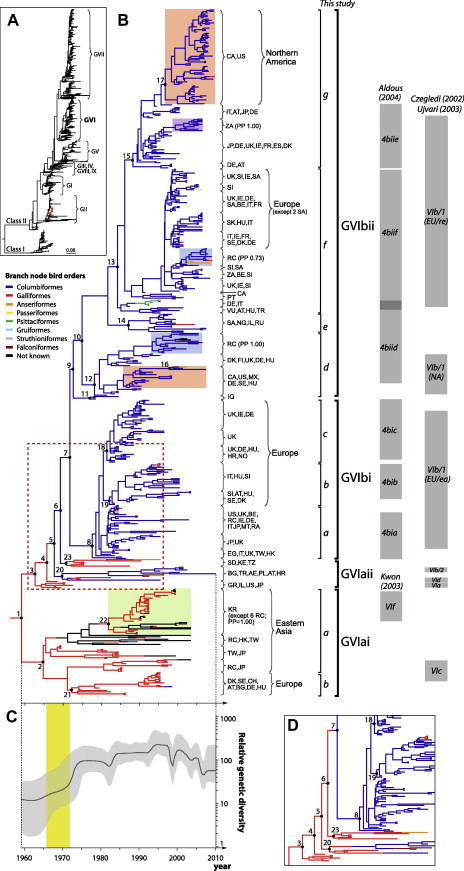
Phylogenetic trees of the F gene of PMV1. (A) Panoramic phylogeny of the F gene sequences of PMV1 (n = 3,469). Labels GI through GIX indicate different genotypes of the virus. Red dots indicate GII-PMV1 isolates sequenced in this study. (B) Time-scaled phylogeny of GVI-PMV1 inferred from the F gene. Colored-branch nodes indicate the bird orders (reconstructed based on ancestral bird order states estimated by parsimony method): blue indicates Columbiformes (i.e. pigeon and doves), red indicates Galliformes (mostly chicken; also includes pheasant, fowl, Hazel Grouse, and quail), orange indicates Anseriformes (i.e. duck), yellow indicates Passeriformes (i.e. migratory birds represented here by Japanese Blue Magpie and common blackbird), green indicates Psittaciformes (i.e. parrot), turquoise refers to Gruiformes (i.e. cranes and rails), grey refers to Struthioniformes (i.e. ostrich), brown refers to Falconiformes (i.e. falcon), and black colors indicates other unknown avian species. Places of sampling are shown as the two-letter ISO country codes with the right brace (code abbreviation in Table S5). Nomenclatures of GVI-PMV1 grouping used in this study are indicated with right brackets; while the phylogenetic groupings used in (Aldous et al., 2004, Czegledi et al., 2002, Ujvari et al., 2003) are indicated by grey vertical bars. Numbers adjacent to the selected nodes refer to topological supports summarized in Table 1, S4. Colored sub-clades indicate strong spatial clustering (to a particular country) with good topological supports – green, peach, blue and purple colors denote South Korean, North America, Republic of China and South African clusters, respectively. The red dashed box indicates one of the two parsimonious scenarios capturing the Galliformes-to-Columbiformes transmission event leading to the GVIb sublineage. (C) Bayesian skyline plot illustrates the changes of relative genetic diversity over time estimated from the F gene. The shaded yellow vertical bars indicate the time window of the first putative emergence of GVIb. (D) The alternative parsimonious scenario indicates the major events of Galliformes-to-Columbiformes transmissions. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2.5. Evolutionary timescale and population dynamics of GVI-PMV1
The time-scaled phylogeny of GVI-PMV1, along with various evolutionary parameters including substitution rate and population size, were estimated simultaneously by the Bayesian Markov Chain Monte Carlo (BMCMC) method implemented in BEAST v1.6.1 program (Drummond and Rambaut, 2007). Relaxed clock model with uncorrelated lognormal distributed rates was used. Hasegawa, Kishino, and Yano with gamma distribution (HKY + Γ) and GTR + Γ nucleotide substitution models were applied separately for comparison. Bayesian skyline demographic model was employed to estimate the effective virus population size over time (Drummond et al., 2005). Robustness of topology was assessed by the clade posterior probability (PP) in the maximum clade credibility (MCC) tree summarized from the BMCMC analysis. With sampling at every 103 steps and disposal of the first 10% of the steps, a total of 109 steps were run. Convergence was assessed in Tracer v1.5.1 program (http://beast.bio.ed.ac.uk/Tracer). Uncertainties in BMCMC estimates were indicated by the ranges of the 95% highest posterior density (HPD).
2.6. History of cross-species transmissions
The history of cross species transmissions was investigated using a comparative phylogenetic approach. Depending on the order of the avian species from which the virus sequence was obtained, one of nine discrete states including Columbiformes, Galliformes, Anseriformes, Passeriformes, Psittaciformes, Gruiformes, Struthioniformes, Falconiformes, and ‘unknown’ was assigned to each sequence. The species states of the sequences can be observed at the tree tips. The unobserved species states of the internal nodes of the MCC phylogeny were estimated using the Fitch parsimony method (Fitch, 1977) implemented in BEAST library (Drummond and Rambaut, 2007), where the parsimony rule – i.e. minimum state change – was complied in the algorithm. A change of species state between two nodes in the phylogeny was considered a putative cross-species transmission event. In addition, the ancestral host states in the internal nodes were also estimated with the Bayesian trait analysis (Lemey et al., 2009) implemented in BEAST software. In this full Bayesian probabilistic method, host states were treated as discrete traits, and their transitions in the tree were modeled in reversible continuous-time Markov chains. The GTR + Γ model was used for the sequence evolution and other BMCMC model specifications were similar to those described in Section 2.5.
2.7. Phylogeography of GVI-PMV1
The phylogeography of GVI-PMV1 was studied using a comparative phylogenetic approach similar to that described above. Briefly, unobserved ancestral geographic (discrete) states in the internal tree nodes were inferred based on the observed isolation localities (categorized as discrete states in a regional basis, i.e. either North America, Asia, Europe, South Africa or Middle East; see Table S3) of the sequences in the MCC phylogeny, using the Fitch parsimony method (Fitch, 1977). The history of geographic dispersion of the virus was reconstructed by tracing the changes of the estimated geographic states from the tree root to tips. These changes were considered as the dispersion events of the virus. Moreover, the ancestral geographic states in the internal nodes were estimated with the Bayesian phylogeographic analysis, treating localities as discrete traits (Lemey et al., 2009) using a similar approach to the host trait analysis described above.
To extrapolate finer resolution of ancestral geographic regions and migration trajectory of GVI-PMV1, geographic localities of the sequences were taken as continuous variables of the average latitudes and longitudes (according to CIA World Factbook; http://www.maxmind.com/app/country_latlon) of the countries where the viruses were isolated. Multivariate diffusion models (Lemey et al., 2010), employing uniform rates and variable rates of lognormal, gamma and Cauchy distributions of random walks were applied independently in the BMCMC analysis (with GTR + Γ) using the BEAST program, to reconstruct the ancestral geographic states (continuous variables, as latitude and longitude) in the phylogeny. Other BMCMC model specifications were set similarly as described above. The best-fit diffusion model was selected by the Bayes factor test.
The Slatkin–Maddison test (Slatkin and Maddison, 1989), which is a discrete parsimony approach, was used to test the hypothesis of random mixing (i.e. panmixis) for the dispersion of GVI-PMV. Under the assumption of panmixis, the expected number of migration (E) was the parsimony state change estimated from the phylogeny with random shuffling of the geographic locations among the taxa. A distribution of E was generated by random shuffling 10,000 times. If the observed number of migrations (O) were significantly less than those expected in panmixis (E), then we conclude that the virus movement was restricted.
The gene flow frequency of GVI-PMV1 between each pair of discrete geographic states was determined by estimating the scaled Slatkin–Maddison’s “s” as previously described (Lam et al., 2012b). Briefly, O was divided by E, which was generated by randomly shuffling the taxa locality in a pair-wise manner, to accommodate unequal sampling frequencies in different geographic states. In our case, only viruses isolated within one-year prior or after their isolation were shuffled to generate E values, to avoid generating excessive E that is unlikely to occur in panmixis due to the time–space difference between samples. To investigate the effect by excessive numbers of European sequences in our data set, random sub-samplings (n = 10) of European sequences were performed to create smaller data sets in which the number of European sequences at most equals the largest number of samples from other localities in each year. The analysis of gene flow frequency was applied similarly in these randomly sub-sampled data sets. Furthermore, a Bayes factor test was performed to identify statistically supported gene flows from a Bayesian stochastic search variable selection (BSSVS) procedure in the BMCMC phylogeographic analysis (Lemey et al., 2009).
3. Results
3.1. Emergence of GVI-PMV1 in Galliformes and Columbiformes
In the panoramic phylogeny of PMV1 (Fig. 1A), all genotypes except III, IV, VIII and IX formed distinct monophyletic lineages. GVI shares a most recent common ancestor (MRCA) with GVII. They have comparable intra-genotypic diversities (0.0423 [95% CI: 0.0132–0.1071] and 0.0538 [0.0108–0.0945] substitutions per site for GVII and GVI, respectively). There was a strong positive linear correlation between genetic divergence of GVI-PMV1 and their isolation dates (Fig. S1), suggesting that viruses in this lineage were evolving at a relatively constant substitution rate and that applying a molecular clock would yield a reliable time-scale of GVI-PMV1 evolutionary history.
In the estimated time-scaled phylogeny based on most available F gene sequences (n = 3,469) (Fig. 1A), the MRCA of all existent GVI-PMV1 (n = 534) was dated to 1959 (HPD = 1948–1965; Node 1 in Fig. 1B; Table 1 ). The GVI-PMVI lineage has two sub-lineages, denoted as GVIa and GVIb, which were primarily associated with chicken and pigeon hosts, respectively (Fig. 1B; detailed taxon names are shown in Fig. S2 and Table S2). The GVIa sub-lineage is comprised of a GVIai subgroup, which appeared around 1965 (HPD = 1960–1968; Node 2 in Fig 1B) in Eastern Asia, with local spread evidenced by a cluster of isolates from Korea (Green block; PP = 1.00). A European subgroup diverged from the Eastern Asian clusters around 1971 (HPD = 1968–1973; Node 21 in Fig. 1B; Table S4), and caused sporadic ND in chickens in Western Europe and Bulgaria in the mid-1990s [designated as ‘GVIc’ by Czegledi et al., 2002, Ujvari et al., 2003]. A number of sparsely distributed non-chicken isolates (color coded branches indicating different bird orders in Fig. 1B) is observed in the GVIai subgroup, suggestive of sporadic but unsustained cross-species transmissions from chickens. The phylogeny based on a mixture of full and partial F gene sequences (Fig. 1B) is consistent with that inferred from a smaller data set of exclusively full-length F gene sequences (n = 120), and of full-length HN gene sequences (n = 39) (Fig. S3).
Table 1.
Time estimates and topological confidence for nodes 1–7 shown in Fig 1B.
| Node | tMRCA (95% HPD) | Posterior clade probability | SH-like branch support | Host state estimate | Geographic estimate by discrete phylogeographic method |
|---|---|---|---|---|---|
| 1 | 1959 (1948–1965) | 1.00 | 1.00 | Galliformes (0.945) | Europe (0.621); Asia (0.251); Middle East (0.0263) |
| 2 | 1965 (1960–1968) | 0.91 | 0.96 | Galliformes (0.996) | Asia (0.941) |
| 3 | 1963 (1958–1966) | 0.38 | 0.96 | Galliformes (0.994) | Europe (0.789); Asia (0.206) |
| 4 | 1966 (1962–1968) | 0.93 | 0.84 | Galliformes (0.997) | Europe (0.993) |
| 5 | 1968 (1964–1970) | 0.66 | 0.75 | Galliformes (0.988) | Europe (0.997) |
| 6 | 1969 (1965–1972) | 1.00 | 0.93 | Galliformes (0.968) | Europe (0.963) |
| 7 | 1972 (1969–1974) | 1.00 | 0.97 | Columbiformes (0.980) | Europe (0.979) |
After the GVIai and GVIaii subgroups emerged in chicken populations at around 1965 and 1963 (Node 2 and 3 in Fig. 1B; Table 1), there was a successful chicken-to-pigeon transmission event that is associated with the divergence of a pigeon-associated GVIb sublineage from GVIaii (Fig. 1B; Nodes 4–7; species illustrated as colored branches in Fig. 1B and D). We applied the parsimony rule to determine the scenario with the least number of possible viral cross-species transmission steps to account for the successful viral introductions from Galliformes (i.e. chicken) to Columbiformes. Two equally plausible scenarios (red dashed box in Fig. 1B and in Fig. 1D) exist. In one scenario (Fig. 1B), the first successful transmission from Galliformes to Columbiformes is estimated to have occurred around 1967 (HPD = 1962–1970, at the branch from node 4 to node 5). This scenario presupposes backward transmission events from Columbiformes to Galliformes, the first around 1971–1973 (see node 20 in Fig. 1B; Table S4) is characterized by two isolates from Bulgaria in 1974–1982 and the second in Sudan, Kenya and Tanzania in 1969–1971 (between nodes 6 and 23 in Fig. 1B; Table S4); these strains persisted until the 1990s. An equally parsimonious scenario (Fig. 1D) suggests that the major event of Galliformes-to-Columbiformes transmission that gave rise to the current GVIb sublineage occurred around 1970 (HPD = 1965–1974; between node 6 and 7) and requires two additional transmissions from Galliformes to Columbiformes in Turkey in 1995 and in a geographically confined lineage of viruses found in Central/Eastern Europe from 1995–2010 (node 20 in Fig. 1D; Table S4). The result of the Bayesian discrete trait analysis supports this second parsimony scenario – i.e. the major cross-species event occurred between node 6 and 7 (Table 1).
After the successful introduction of PMV1 into Columbiformes, there were numerous sporadic pigeon-to-chicken transmissions (n = 14) and a smaller number of sporadic transmissions to other avian species including Anseriformes (n = 3), Psittaciformes (n = 6) and Passeriformes (n = 1). Many of these cross-species events were represented by only one or two isolates, suggesting that GVIb-PMV1 could not successfully transmit in non-Columbiformes or that these species are poorly represented in surveillance efforts.
We estimated the geographic origins of the major cross-species transmissions from chicken that resulted in successful adaptation of the GVIb lineage in pigeons. Nodes 4–7 in the phylogenetic tree (Fig. 1B) represent hypothetical virus ancestors in which such cross-species events might have occurred. Based on BMCMC estimation of geographic regions as discrete states, the posterior probabilities that Middle East, Europe, or South Africa were estimated in node 4 are 0.002, 0.993 and <0.001, respectively; 0.003, 0.997, and <0.001, respectively for node 5; <0.001, 0.963, and 0.017, respectively for node 6; and 0.02, 0.979, and 0.001, respectively for node 7. To increase the precision of geographical information used for analyses, we employed a recently developed Bayesian continuous diffusion model (Lemey et al., 2010) where the latitudes and longitudes of sequence isolations were used as inputs to estimate the location coordinates of ancestral nodes, by assuming variable spatial diffusion rates among branches. The relaxed random walk (RRW) model with gamma distributed rate variation was the best-fit model according to Bayes Factor tests (lnBF > 3), in which nodes 4–7 were estimated to Southeastern Europe (Table S5) with posterior probabilities of 0.994, 1, 0.99, and 0.864, respectively. This result is consistent with the detection of the earliest pigeon isolate of GVI-PMV1 in Iraq. Notably, the latitude and longitude estimates for these nodes were patchy at the country level. Therefore, the estimates were grouped into larger geographic regions (e.g. Southeastern Europe), to avoid potentially biased conclusions.
3.2. Global spread and evolution of GVIb lineage in Columbiformes
The adaptation of PMV1 to pigeons coincided with multiple worldwide outbreaks of PMV1 in Columbiformes at the end of the 1970s and extensively in the 1980s and resulted in rapid genetic diversification of this new lineage. The GVIb lineage split into two major extant subgroups (GVIbi and GVIbii; Fig. 1B) in 1978 (HPD = 1974–1980; Node 8) and 1975 (HPD = 1972–1978; Node 10) respectively. The GVIbi subgroup has diversified into three clades, termed ‘a’, ‘b’, and ‘c’, which is in agreement with the grouping proposed by Aldous et al. (2004). There is some geographic structure to this subgroup; GVIbi-a consists of viruses isolated from America and Eurasia, GVibi-b is dominated by Italian isolates with a substantial numbers of Central/Eastern European viruses, and GVIbi-c is mainly restricted to UK and Irish isolates.
GVIbii has diversified into four major clades, termed ‘d’, ‘e’, ‘f’, and ‘g’, with notable spatial structure due to sampling of small transmission clusters (color blocks; Fig. 1B). Ujvari et al. (2003) named this group ‘recent European viruses’ VIb/1(EU/re). European isolates are basal to the GVIbii strains derived from outbreaks in other international locations (Fig. 1B).
Based on F gene sequences of GVI-PMV1, we tested whether there was significant restriction to viral gene flow between different continents (i.e. random mixing), using the Slatkin–Maddison test. The mean number of observed geographical state change (O) values for all geographical state pairs (see Section 2.) was significantly lower than the null distribution of geographical state changes (E) value under panmixis (p < 0.0002). This suggests that geographical structure existed in the GVI-PMV1 population, and hence the viral gene flow between continents was restricted to a significant degree.
Using a modified version of Slatkin and Maddison’s “s” (Lam et al., 2012b) to quantify the level of gene flow, we found that viral gene flow between Europe and other regions was considerably higher (average: 0.287) than the flow among other regions (Fig. 2 ; Table S3), consistent with Europe being the epicenter of GVIb-PMV1 dispersal. The Bayesian phylogeographic analysis result also identifies gene flows from Europe as topologically significant by Bayes factor tests (lnBF > 3; Table S3). There was a weak signature of isolation-by-distance. For example, gene flow between North America and Europe and Asia was higher than gene flow between North America and Middle East and South Africa (Table S3). Furthermore, GVI-PMV1 gene flow between North America and other regions was less frequent than that among countries in the Eurasian-African continent (on average 0.039; Fig. 2; Table S3). Similar results were obtained if European samples were randomly down-sampled (see Section 2.) to obtain more geographically balanced data sets.
Fig. 2.
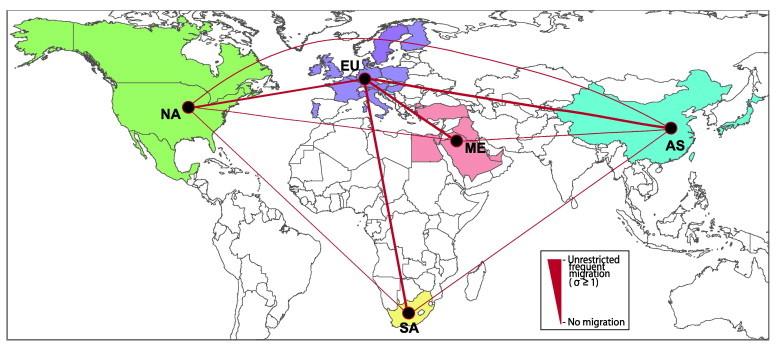
World map showing the average gene flows of GVI-PMV1 between continents. Thickness of red lines connecting two continents are scaled as shown in the box and indicates the extent of gene flow (scaled Slatkin–Maddison’s “s”) between the localities. Only countries with available sequences of Columbiforme-associated GVI are colored, and include Asia (AS), Europe (EU), Middle East (ME), North America (NA) and South Africa (SA).
3.3. Evolution of GVIb-PMV1 introduced into North American Columbiformes
The F gene of 74 PMV1 samples from several avian species submitted to PSU-ADL were sequenced, and analyzed with the limited North American PMV1-GVI sequences in GenBank (Table S1). The GVIbi-a clade was the first to disperse to North American Columbiformes. This small cluster consists of three pigeon isolates from New York and Maryland obtained in 1984, a Japanese chicken isolate, and other Columbiformes viruses detected during the first European panzootic of PMV1 in in the 1980s. There are no contemporary isolates that fall into this cluster, suggestive of lineage extinction.
GVIbii-d (Fig. 1B and 3A) was introduced to North America in the late 1980s; this lineage shares ancestry with isolates from China (blue block of GVIbii-d in Fig 1B; blue-colored strain names in Fig 3 A). European isolates form a basal group of GVIbii-d viruses, suggesting that they were introduced from the European continent to other countries, and their subsequent circulation was geographically restricted. The North American cluster consists of samples from Canada, Texas and Mexico collected during the late 1980s and 1990s (PP = 0.91 in Fig. 3A; yellow and pale blue dots plotted in Fig. 3C). Our newly obtained US sequences (collected between 2001 and 2007) indicate that these North American strains are still circulating but have diversified locally, leading to geographical clustering. Noteworthy, a long branch preceding this small cluster (node 16, orange block of Fig. 1B; Fig. 3A) indicates that the ancestral viruses were not sampled during the eight-year period that separates these groups.
Fig. 3.
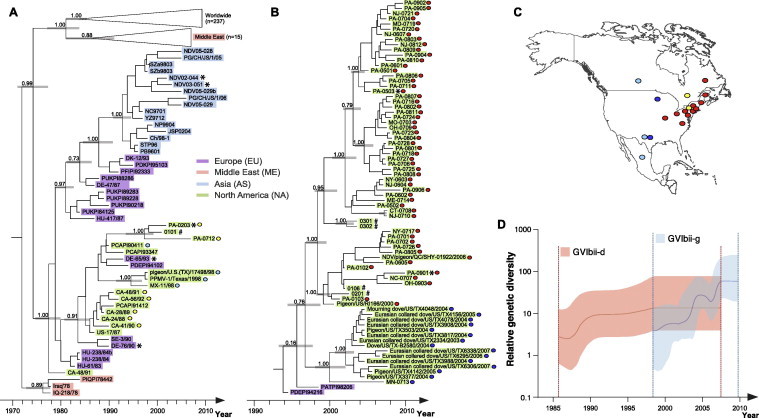
GVIbii-PMV1 in North America. Lineages were extracted from the complete time-scaled phylogeny of PMV1 in Fig. 1b. (A) Lineage GVIbii-d. The GVIbii-e, -f, -g lineages were collapsed as triangles for clarity. The colors indicate country where the sample was obtained. All isolates were derived from Columbiformes except those indicated by asterisks (∗) after the strain names in the phylogeny. Posterior probability values of 0.7 or greater are shown adjacent to the tree nodes. Isolates with unknown sampling locations within the North America continent, are indicated by # after the viral strain names in the phylogeny. (B) Lineage GVIbii-g. Support values for nodes and country color scheme are as in (A). (C) The red, yellow, pale blue and blue dots plotted on the map correspond to the known sampling localities of the viruses collected from North America. (D) Bayesian skyline plot illustrates the changes of relative genetic diversity over time estimated from the F gene for GVIbii North American clusters. Relative genetic diversities are expressed on a logarithmic scale (y-axis). The shaded red and blue bands give the 95% HPD intervals of the estimates for GVIbii-d and -g, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The majority of North American isolates are represented within the GVIbii-g clade (Fig. 1, Fig. 3B), which has an estimated time of introduction around 1996 (HPD = 1994–1999) (node 17, orange block of Fig. 1B) and includes a cluster of strains from Eurasian collared doves. Closely related strains have been recovered from a native US Columbiforme species (mourning dove) and a sick racing pigeon from Minnesota indicating that the US GVbii-g clade has successfully dispersed in several wild and domestic US species.
Within GVIbii-g, there is a clear geographical separation of isolates into Northeastern and Central North America (Fig. 3). This could be due to multiple introductions of closely related viruses. Alternatively, it is possible that the virus dispersed from a point source into multiple populations that are geographically or ecologically isolated. The Northeastern US monophyletic lineage is comprised primarily of our newly sequenced isolates and one sample each from Quebec and Rhode Island (Kim et al., 2008). The geographical proximity of the sampling locations (red dots plotted on the map in Fig. 3C) indicates that GVIbii-g effectively dispersed within the Northeastern US over a decade. The Central US cluster consists of Texas and Minnesota isolates (blue dots plotted on the map in Fig. 3C). Noteworthy, European isolates from Austria and Germany (purple strain name, Fig. 3B) are basal to the entire cluster of North American isolates within GVIbii-g clade, supporting our result that global dispersal of GVI virus originated in Europe.
Bayesian estimates of the evolutionary rate for the global dataset of GVI-PMV1 (n = 534) based on the F gene are between 1.48 and 1.81 × 10−3 substitutions/site/year. The Bayesian skyline plot (BSP), which infers the relative genetic diversity over time, demonstrates an abrupt expansion of the population in the early 1970s, coincident with the time of emergence of both GVIbi and ii subgroups (Fig. 1C). The population size remained constant or grew slowly for the following 15 years, with a number of small fluctuations. From the 1990s onwards, the global GVI-PMV1 population size has a declining trend. The Bayesian estimates of the evolutionary rates for both North American GVIbii-d and -g clusters are consistent with the global rate, at 1.14–2.34 × 10−3 and 1.30–2.14 × 10−3 substitutions/site/year, respectively and are comparable to those previously estimated from all PMV1 genotypes (Chong et al., 2010). In North America, GVIbii-g lineage has experienced exponential-like population growth (Fig. 3C, blue), and the GVIbii-d lineage has been constant over time (Fig. 3C, red).
4. Discussion
Our studies integrated large amounts of genetic data with associated temporal and geographic information to investigate the emergence and global dispersal of GVI-PMV1. We provide quantitative data that the successful establishment of Columbiformes GVIb lineage occurred in the 1980s, likely in Southeastern Europe, and that the ancestral host was Galliformes. The virus dispersed worldwide from Europe, including multiple introductions into North America.
The ultimate establishment of GVIb in Columbiformes was preceded by a number of sporadic unsuccessful transmissions from chickens to pigeons. Once GVIb became established in Columbiformes, spillovers of GVIb-PMV1 to non-Columbiformes bird species were detected along terminal branches; these included multiple transmissions to Galliformes and Psittaciformes and infrequent spillovers to Anseriformes and Passeriformes. This latter spill over is consistent with the presumed route by which pathogenic viruses including H5N1 influenza and other PMV1 genotypes that circulate in wild birds have been introduced to domestic chickens (Kim et al., 2009, Miller et al., 2010). However, our data demonstrate that viral strains circulating in wild birds can be derived from domestic poultry, evolve in the new host, and be reintroduced to poultry, providing an additional model to explain epizootic viral emergence in commercial flocks. These data suggest that detection of endemic poultry virus strains in wild and peri-domestic species could presage a virus emergence event.
Establishment of GVIb PMV1 in pigeons was more likely a result of adaptation to the new host than a host range expansion for several reasons. With range expansion, we would expect to observe similarly high associations of contemporary GVIb isolates with both pigeons and chickens. However, both epidemiological reports (Abolnik et al., 2008, Hassan et al., 2010, Pedersen et al., 2004) and our phylogenetic data support that such adaptation is unidirectional host switch and not a range expansion. The observed back transmission from pigeons to chicken may reflect the tight ecological interface between their populations, and possibly a relative lower species barrier. Experimental studies confirm that pigeon derived GVIb strains do not readily infect chickens but, significantly, their ability to infect and cause disease in chickens requires only a few serial passages in chickens (Alexander and Parsons, 1984, Collins et al., 1996, Kommers et al., 2003). Reverse genetics studies and complete sequence analysis further confirmed that three substitutions in the P and L proteins are sufficient to increase replication efficiency in chickens (Dortmans et al., 2011). Notably, the number of reports of GVIb-PMV1 in poultry farms is increasing recently (Abolnik et al., 2008, Hassan et al., 2010, Pedersen et al., 2004). Therefore, it is important to determine whether the natural evolutionary trajectory of GVIb isolates in different regions will result in increased risk of backward transmission into chicken populations.
The global dispersal of GVIb-PMV1 after emergence into Columbiformes was rapid. International and local transportation of birds may have played an important role in spreading the new virus (Alexander et al., 1985a, Kaleta et al., 1985, Stewart, 1971). Rapid early dispersal could account for the lineage radiation that took place within the first decade after emergence. The two founding Columbiformes lineages, GVIbi and GVIbii, that branched in the early 1980s, have diversified into several sub-lineages, some of which are still circulating. GVIbii viruses expanded into 28 countries involving Africa, Asia, Europe North America and Oceania continents, whereas GVIbi viruses have been recorded in 20 countries of the same continents except Oceania. The extensive diversity that has occurred in GVIb has confounded nomenclature, and we reconcile our data with that found in the literature (Fig. 1) (Aldous et al., 2004, Czegledi et al., 2002, Kwon et al., 2003, Ujvari et al., 2003).
Our phylogenetic analysis of migration patterns revealed a strong spatial structure in GVIb-PMV1 globally, which may reflect the geophysical barriers to gene flow between continents following the initial dispersal and establishment of the virus. Recent studies have also demonstrated strong spatial isolation of both low and high pathogenic avian influenza amongst wild and domestic birds (Lam et al., 2012a, Lam et al., 2012b, Wallace and Fitch, 2008), and suggested that long distance movement was less favored possibly due to migration patterns of wild birds or by trade restrictions imposed at international borders. Increased awareness of the global impact of avian diseases in response to high pathogenic avian influenza could also be impacting the contemporary dispersal patterns of PMV1 genotypes.
Our data indicate that GVIb-PMV1 was introduced to North America from Europe on multiple occasions and the virus is now maintained as an endemic infection in wild (Kim et al., 2008) and domestic birds. The first introduction event of GVIbi-a either failed to sustain in North America, or that the strain was missed by sampling efforts. The identification of GVIbii-d and g clades indicates that there were at least two additional introduction events. Given the extensive dispersal of PMV1 from Europe to other countries, GVIb could have been brought into North America through legal or illegal transport of infected birds. It is noteworthy that GVIbii-g strains have been isolated from the Eurasian collared dove. This invasive species was first identified in Florida in the mid-1980s, likely from escaped captive birds brought into the Bahamas for the pet bird trade in the 1970s (Smith, 1987). Our phylogenetic data and molecular dating analyses estimate that the North American GVI-PMV1bii-g was introduced in the mid-1990. We cannot confirm whether the Eurasian collared dove was the source of the introduced North American GVIbii-g lineage because there are no samples from the late 1980s from this species. However, the close affiliation of GVIbii-g strains isolated from domestic pigeons, Eurasian collared doves, and mourning doves indicates that collared doves are competent hosts and that their rapid range expansion, which now extends to southern Alaska, could facilitate dispersal of the virus throughout North America.
It is noteworthy that vaccination policy to prevent ND, which is applied in many countries, has not stopped the emergence of new PMV1 strains in domestic birds. The two genotypes responsible for most of the recent ND outbreaks in poultry and wild birds, GVI- and GVII-PMV1, diverged from a MRCA in the mid-1960s (Fig. 1A), approximately a decade after vaccination was implemented. The rapid global dispersal of new genotypes as they emerge implicate human involvement in establishing new viral genotypes, although we cannot rule out the bird migration could contribute in some areas. It is likely that the number of molecular changes that occurred to allow GVIaii viruses to successfully establish in pigeons is small given that only a few changes are needed for GVIb viruses to replicate in chickens. Our data demonstrate that PMV1 has a rapid evolutionary rate and that one of the extant sub-lineages introduced into North America is currently undergoing a population expansion. This suggests that the mixing of Columbiformes species with domestic chickens could facilitate the adaptation of potentially virulent viruses to chickens.
Acknowledgments
We acknowledge technical contributions from Daniel Elleder and Brian Huylebroeck and thank Gina Riggio for critical reading of the manuscript. We thank the personnel in Animal Diagnostic Laboratory from The Pennsylvania State University, Dr. Eva Nagy (University of Guelph, Canada) and Dr. Ruth Manvell (Animal Health and Veterinary Laboratories Agency, AHVLA, UK) for providing data on some PMV1 sequences used in this study, and to the Penn State Genomics Core Facility, University Park, PA for sequencing service. We also appreciate the advice given by Dr. Philippe Lemey on phylogenetic analyses and thank two anonymous reviewers for insightful comments. YLC is supported by the Malaysian Academic Training Scheme for Institutions of Higher Education Scholarship. TTYL is supported by Newton International Fellowship of Royal Society, UK. This work was partially supported by USDA grant 2009-35204-20082 and by the RAPIDD program of the Science and Technology Directorate, US Department of Homeland Security, and the Fogarty International Center, NIH.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.meegid.2013.04.025.
Appendix A. Supplementary data
The genetic distances of GVI-PMV1 isolates from the root of the phylogeny plotted against the years of their isolation. The solid straight line is the linear regression of the genetic distance and year. The 95% predicted confidence bands of regression are shown as dashed lines. The Pearson’s coefficient (R) of the regression is also shown.
Time-scaled phylogeny of GVI-PMV1 inferred from the F gene as shown in Fig. 1. Branch labels indicate the complete information of the virus strain name with GenBank accession numbers. Numbers adjacent to the internal nodes refer to topological supports based on posterior clade probability.
Phylogenetic trees of GVI-PMV1 inferred from the full length HN and F gene sequences. Branch labels indicate the complete information of the virus strain name with GenBank accession numbers. Numbers adjacent to the internal nodes refer to topological supports based on 1000 bootstrapping trees.
Supplementary Tables.
References
- Abolnik C., Gerders G.H., Kitching J., Swanepoel S., Romito M., Bisschop S.P.R. Characterization of pigeon paramyxoviruses (Newcastle disease virus) isolated in South Africa from 2001 to 2006. Onderstepoort J. Vet. Res. 2008;75:147–152. doi: 10.4102/ojvr.v75i2.13. [DOI] [PubMed] [Google Scholar]
- Aldous E.W., Fuller C.M., Mynn J.K., Alexander D.J. A molecular epidemiological investigation of isolates of the variant avian paramyxovirus type 1 virus (PPMV-1) responsible for the 1978 to present panzootic in pigeons. Avian Pathol. 2004;33:258–269. doi: 10.1080/0307945042000195768. [DOI] [PubMed] [Google Scholar]
- Alexander D.J. Kluwer Academic Publishers; Boston: 1988. Developments in Veterinary Virology: Newcastle Disease Virus. [Google Scholar]
- Alexander D.J. Newcastle disease diagnosis. In: Alexander D.J., editor. Developments in Veterinary Virology: Newcastle Disease. Kluwer Academic Publishers; Massachusetts: 1988. [Google Scholar]
- Alexander D.J., Manvell R.J., Lowings J.P., Frost K.M., Collins M.S., Russell P.H., Smith J.E. Antigenic diversity and similarities detected in avian paramyxovirus type 1 (Newcastle disease virus) isolates using monoclonal antibodies. Avian Pathol. 1997;26:399–418. doi: 10.1080/03079459708419222. [DOI] [PubMed] [Google Scholar]
- Alexander D.J., Parsons G. Avian paramyxovirus type 1 infections of racing pigeons: 2 pathogenicity experiments in pigeons and chickens. Vet. Rec. 1984;114:466–469. doi: 10.1136/vr.114.19.466. [DOI] [PubMed] [Google Scholar]
- Alexander D.J., Russell P.H., Collins M.S. Paramyxovirus type 1 infections of racing pigeons: 1 characterisation of isolated viruses. Vet. Rec. 1984;114:444–446. doi: 10.1136/vr.114.18.444. [DOI] [PubMed] [Google Scholar]
- Alexander D.J., Russell P.H., Parsons G., Elzein E.M., Ballouh A., Cernik K., Engstrom B., Fevereiro M., Fleury H.J., Guittet M., Kaleta E.F., Kihm U., Kosters J., Lomniczi B., Meister J., Meulemans G., Nerome K., Petek M., Pokomunski S., Polten B., Prip M., Richter R., Saghy E., Samberg Y., Spanoghe L., Tumova B. Antigenic and biological characterisation of avian paramyxovirus type I isolates from pigeons – an international collaborative study. Avian Pathol. 1985;14:365–376. doi: 10.1080/03079458508436238. [DOI] [PubMed] [Google Scholar]
- Alexander D.J., Wilson G.W., Russell P.H., Lister S.A., Parsons G. Newcastle disease outbreaks in fowl in Great Britain during 1984. Vet. Rec. 1985;117:429–434. doi: 10.1136/vr.117.17.429. [DOI] [PubMed] [Google Scholar]
- Barbezange C., Jestin V. Molecular characterisation of three avian paramyxovirus type 1 isolated from pigeons in France. Virus Genes. 2003;26:175–183. doi: 10.1023/a:1023439530750. [DOI] [PubMed] [Google Scholar]
- Barbezange C., Jestin V. Quasispecies nature of an unusual avian paramyxovirus type-1 isolated from pigeons. Virus Genes. 2005;30:363–370. doi: 10.1007/s11262-004-6780-1. [DOI] [PubMed] [Google Scholar]
- Barton J.T., Bickford A.A., Cooper G.L., Charlton B.R., Cardona C.J. Avian paramyxovirus type 1 infections in racing pigeons in California. I. clinical signs, pathology, and serology. Avian Dis. 1992;36:463–468. [PubMed] [Google Scholar]
- Capua I., Terregino C., Dalla Pozza M., Marangon S., Mutinelli F. Newcastle disease outbreaks in Italy during 2000. Vet. Rec. 2002;150:565–568. doi: 10.1136/vr.150.18.565. [DOI] [PubMed] [Google Scholar]
- Chong Y.L., Padhi A., Hudson P.J., Poss M. The effect of vaccination on the evolution and population dynamics of avian paramyxovirus-1. PLoS Pathog. 2010;6:e1000872. doi: 10.1371/journal.ppat.1000872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins M.S., Strong I., Alexander D.J. Pathogenicity and phylogenetic evaluation of the variant Newcastle disease viruses termed “pigeon PMV-1 viruses” based on the nucleotide sequence of the fusion protein gene. Arch. Virol. 1996;141:635–647. doi: 10.1007/BF01718322. [DOI] [PubMed] [Google Scholar]
- Czegledi A., Herczeg J., Hadjiev G., Doumanova L., Wehmann E., Lomniczi B. The occurrence of five major Newcastle disease virus genotypes (II, IV, V, VI and VIIb) in Bulgaria between 1959 and 1996. Epidemiol. Infect. 2002;129:679–688. doi: 10.1017/s0950268802007732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diel D.G., Miller P.J., Wolf P.C., Mickley R.M., Musante A.R., Emanueli D.C., Shively K.J., Pedersen K., Afonso C.L. Characterization of Newcastle disease viruses isolated from cormorant and gull species in the United States in 2010. Avian Dis. Digest. 2012;7:e20–e21. doi: 10.1637/9886-081111-Reg.1. [DOI] [PubMed] [Google Scholar]
- Dortmans J.C.F.M., Rottier P.J.M., Koch G., Peeters B.P.H. Passaging of a Newcastle disease virus pigeon variant in chickens results in selection of viruses with mutations in the polymerase complex enhancing virus replication and virulence. J. Gen. Virol. 2011;92:336–345. doi: 10.1099/vir.0.026344-0. [DOI] [PubMed] [Google Scholar]
- Drummond A.J., Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A.J., Rambaut A., Shapiro B., Pybus O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005;22:1185–1192. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch W.M. On the problem of discovering the most parsimonious tree. Am. Nat. 1977;111:223–257. [Google Scholar]
- Guindon S., Dufayard J.-F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Hassan W., Khair S.A., Mochotlhoane B., Abolnik C. Newcastle disease outbreaks in the Sudan from 2003 to 2006 were caused by viruses of genotype 5d. Virus Genes. 2010;40:106–110. doi: 10.1007/s11262-009-0424-4. [DOI] [PubMed] [Google Scholar]
- Kaleta E.F., Alexander D.J., Russell P.H. The first isolation of the avian pmv-1 virus responsible for the current panzootic in pigeons? Avian Pathol. 1985;14:553–557. doi: 10.1080/03079458508436258. [DOI] [PubMed] [Google Scholar]
- Kaleta E.F., Baldauf C. Newcastle disease in free living and pet birds. In: Alexander D.J., editor. Newcastle Disease. Kluwer Academic Publishers; Boston: 1988. pp. 197–246. [Google Scholar]
- Kim J.-K., Negovetich N., Forrest H., Webster R.G. Ducks: the “Trojan Horses” of H5N1 Influenza. Influenza Other Respi. Viruses. 2009;3:121–128. doi: 10.1111/j.1750-2659.2009.00084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim L.M., King D.J., Guzman H., Tesh R.B., Travassos da Rosa A.P., Bueno R., Jr., Dennett J.A., Afonso C.L. Biological and phylogenetic characterization of pigeon paramyxovirus serotype 1 circulating in wild North American pigeons and doves. J. Clin. Microbiol. 2008;46:3303–3310. doi: 10.1128/JCM.00644-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kommers G.D., King D.J., Seal B.S., Brown C.C. Virulence of pigeon-origin Newcastle disease virus isolates for domestic chickens. Avian Dis. 2001;45:906–921. [PubMed] [Google Scholar]
- Kommers G.D., King D.J., Seal B.S., Brown C.C. Virulence of six heterogeneous-origin Newcastle disease virus isolates before and after sequential passages in domestic chickens. Avian Pathol. 2003;32:81–93. doi: 10.1080/0307945021000070750. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond S.L., Posada D., Gravenor M.B., Woelk C.H., Frost S.D.W. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 2006;23:1891–1901. doi: 10.1093/molbev/msl051. [DOI] [PubMed] [Google Scholar]
- Kwon H.J., Cho S.H., Ahn Y.J., Seo S.H., Choi K.S., Kim S.J. Molecular epidemiology of Newcastle disease in Republic of Korea. Vet. Microbiol. 2003;95:39–48. doi: 10.1016/s0378-1135(03)00130-5. [DOI] [PubMed] [Google Scholar]
- Lam T.T.-Y., Hon C.-C., Lemey P., Pybus O.G., Shi M., Tun H.M., Li J.U.N., Jiang J., Holmes E.C., Leung F.C.-C. Phylodynamics of H5N1 avian influenza virus in Indonesia. Mol. Ecol. 2012;21:3062–3077. doi: 10.1111/j.1365-294X.2012.05577.x. [DOI] [PubMed] [Google Scholar]
- Lam T.T., Ip H.S., Ghedin E., Wentworth D.E., Halpin R.A., Stockwell T.B., Spiro D.J., Dusek R.J., Bortner J.B., Hoskins J., Bales B.D., Yparraguirre D.R., Holmes E.C. Migratory flyway and geographical distance are barriers to the gene flow of influenza virus among North American birds. Ecol. Lett. 2012;15:24–33. doi: 10.1111/j.1461-0248.2011.01703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P., Rambaut A., Drummond A.J., Suchard M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009;5:e1000520. doi: 10.1371/journal.pcbi.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P., Rambaut A., Welch J.J., Suchard M.A. Phylogeography takes a relaxed random walk in continuous space and time. Mol. Biol. Evol. 2010;27:1877–1885. doi: 10.1093/molbev/msq067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Wang Z., Son C., Wang Y., Yu B., Zheng D., Sun C., Wu Y. Characterization of pigeon-origin Newcastle disease virus isolated in China. Avian Dis. 2006;50:636–640. doi: 10.1637/7618-042606R1.1. [DOI] [PubMed] [Google Scholar]
- Lloyd-Smith J.O., George D., Pepin K.M., Pitzer V.E., Pulliam J.R., Dobson A.P., Hudson P.J., Grenfell B.T. Epidemic dynamics at the human-animal interface. Science. 2009;326:1362–1367. doi: 10.1126/science.1177345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manual, O.T., 2009. Manual of diagnostic tests and vaccines for terrestrial animals 2009 summary. In: Newcastle Disease, pp. 576–589 (Chapter 2.3.14).
- Martin D.P., Williamson C., Posada D. RDP2: recombination detection and analysis from sequence alignments. Bioinformatics. 2005;21:260–262. doi: 10.1093/bioinformatics/bth490. [DOI] [PubMed] [Google Scholar]
- Mase M., Inoue T., Imada T. Genotyping of Newcastle disease viruses isolated from 2001 to 2007 in Japan. J. Vet. Med. Sci. 2009;71:1101–1104. doi: 10.1292/jvms.71.1101. [DOI] [PubMed] [Google Scholar]
- Meulemans G., van den Berg T.P., Decaesstecker M., Boschmans M. Evolution of pigeon Newcastle disease virus strains. Avian Pathol. 2002;31:515–519. doi: 10.1080/0307945021000005897. [DOI] [PubMed] [Google Scholar]
- Miller P.J., Decanini E.L., Afonso C.L. Newcastle disease: evolution of genotypes and the related diagnostic challenges. Infect. Genet. Evol. 2010;10:26–35. doi: 10.1016/j.meegid.2009.09.012. [DOI] [PubMed] [Google Scholar]
- OIE, 2011a. WAHID Interface – Animal Health Information. Weekly Disease Information, 04/02/2011: Newcastle disease, Sweden (Immediate notification).
- OIE, 2011b. WAHID Interface – Animal Health Information. Weekly Disease Information, 04/01/2011: Newcastle disease, France (Immediate notification).
- Panshin A., Shihmanter E., Weisman Y., Orvell C., Lipkind M. Comparative characteristics of the Israeli Newcastle disease virus field strains by means of a wide panel of monoclonal antibodies against hemagglutinin-neuraminidase glycoprotein. Comp. Immunol. Microbiol. Infect. Dis. 1999;22:103–124. doi: 10.1016/s0147-9571(98)00025-3. [DOI] [PubMed] [Google Scholar]
- Parrish C.R., Holmes E.C., Morens D.M., Park E.-C., Burke D.S., Calisher C.H., Laughlin C.A., Saif L.J., Daszak P. Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol. Mol. Biol. Rev. 2008;72:457–470. doi: 10.1128/MMBR.00004-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson J.E., Senne D.A., Alexander D.J., Taylor W.D., Peterson L.A., Russell P.H. Characterization of Newcastle disease virus (Avian Paramyxovirus-1) isolated from pigeons. Avian Dis. 1987;31:105–111. [PubMed] [Google Scholar]
- Pedersen J.C., Senne D.A., Woolcock P.R., Kinde H., King D.J., Wise M.G., Panigrahy B., Seal B.S. Phylogenetic relationships among virulent Newcastle disease virus isolates from the 2002–2003 outbreak in California and other recent outbreaks in North America. J. Clin. Microbiol. 2004;42:2329–2334. doi: 10.1128/JCM.42.5.2329-2334.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M.N., Dehal P.S., Arkin A.P. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servan de Almeida R., Maminiaina O.F., Gil P., Hammoumi S., Molia S., Chevalier V., Koko M., Andriamanivo H.R., Traore A., Samake K., Diarra A., Grillet C., Martinez D., Albina E. Africa, a reservoir of new virulent strains of Newcastle disease virus? Vaccine. 2009;27:3127–3129. doi: 10.1016/j.vaccine.2009.03.076. [DOI] [PubMed] [Google Scholar]
- Slatkin M., Maddison W.P. A cladistic measure of gene flow inferred from the phylogenies of alleles. Genetics. 1989;123:603–613. doi: 10.1093/genetics/123.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleeman, J., 2010. Wildlife Health Bulletin 2010-07: Summary of 2010 Newcastle Disease virus outbreaks in wild birds in upper Midwest and the Northeast, Madison.
- Smith P.W. The Eurasian collared-dove arrives in the Americas. Am. Birds. 1987;41:1370–1379. [Google Scholar]
- Stewart G.H. Naturally occurring clinical Newcastle disease in the racing pigeon (Columba livia) Vet. Rec. 1971;89:225–226. doi: 10.1136/vr.89.8.225. [DOI] [PubMed] [Google Scholar]
- Tangredi B.P. Avian paramyxovirus type 1 infection in pigeons: recent changes in clinical observations. Avian Dis. 1988;32:839–841. [PubMed] [Google Scholar]
- Toro H., Hoerr F.J., Farmer K., Dykstra C.C., Roberts S.R., Perdue M. Pigeon paramyxovirus: association with common avian pathogens in chickens and serologic survey in wild birds. Avian Dis. 2005;49:92–98. doi: 10.1637/7268-083104R1. [DOI] [PubMed] [Google Scholar]
- Tsai H.J., Chang K.H., Tseng C.H., Frost K.M., Manvell R.J., Alexander D.J. Antigenic and genotypical characterization of Newcastle disease viruses isolated in Taiwan between 1969 and 1996. Vet. Microbiol. 2004;104:19–30. doi: 10.1016/j.vetmic.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Ujvari D., Wehmann E., Kaleta E.F., Werner O., Savic V., Nagy E., Czifra G., Lomniczi B. Phylogenetic analysis reveals extensive evolution of avian paramyxovirus type 1 strains of pigeons (Columba livia) and suggests multiple species transmission. Virus Res. 2003;96:63–73. doi: 10.1016/s0168-1702(03)00173-4. [DOI] [PubMed] [Google Scholar]
- Wallace R.G., Fitch W.M. Influenza A H5N1 immigration is filtered out at some international borders. PLoS One. 2008;3:e1697. doi: 10.1371/journal.pone.0001697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner O., Romer-Oberdorfer A., Kollner B., Manvell R.J., Alexander D.J. Characterization of avian paramyxovirus type 1 strains isolated in Germany during 1992 to 1996. Avian Pathol. 1999;28:79–88. doi: 10.1080/03079459995082. [DOI] [PubMed] [Google Scholar]
- Woolhouse M.E.J. Epidemiology: emerging diseases go global. Nature. 2008;451:898–899. doi: 10.1038/451898a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanetti F., Mattiello R., Garbino C., Kaloghlian A., Terrera M.V., Boviez J., Palma E., Carrillo E., Berinstein A. Biological and molecular characterization of a pigeon paramyxovirus type-1 isolate found in Argentina. Avian Dis. 2001;45:567–571. [PubMed] [Google Scholar]
- Zhu W., Dong J., Xie Z., Liu Q., Khan M.I. Phylogenetic and pathogenic analysis of Newcastle disease virus isolated from house sparrow (Passer domesticus) living around poultry farm in southern China. Virus Genes. 2010;40:231–235. doi: 10.1007/s11262-009-0436-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The genetic distances of GVI-PMV1 isolates from the root of the phylogeny plotted against the years of their isolation. The solid straight line is the linear regression of the genetic distance and year. The 95% predicted confidence bands of regression are shown as dashed lines. The Pearson’s coefficient (R) of the regression is also shown.
Time-scaled phylogeny of GVI-PMV1 inferred from the F gene as shown in Fig. 1. Branch labels indicate the complete information of the virus strain name with GenBank accession numbers. Numbers adjacent to the internal nodes refer to topological supports based on posterior clade probability.
Phylogenetic trees of GVI-PMV1 inferred from the full length HN and F gene sequences. Branch labels indicate the complete information of the virus strain name with GenBank accession numbers. Numbers adjacent to the internal nodes refer to topological supports based on 1000 bootstrapping trees.
Supplementary Tables.