

Abstract
Severe Acute Respiratory Syndrome (SARS) is a respiratory disease caused by a zoonotic coronavirus (CoV) named SARS-CoV (SCoV), which rapidly swept the globe after its emergence in rural China during late 2002. The origins of SCoV have been mysterious and controversial, until the recent discovery of SARS-like CoV (SLCoV) in bats and the proposal of bats as the natural reservior of the Coronaviridae family. In this article, we focused on discussing how phylogenetics contributed to our understanding towards the emergence and transmission of SCoV. We first reviewed the epidemiology of SCoV from a phylogenetic perspective and discussed the controversies over its phylogenetic origins. Then, we summarized the phylogenetic findings in relation to its zoonotic origins and the proposed inter-species viral transmission events. Finally, we also discussed how the discoveries of SCoV and SLCoV expanded our knowledge on the evolution of the Coronaviridae family as well as its implications on the possible future re-emergence of SCoV.
Keywords: SARS, SARS-like coronaviruses, Inter-species transmission, Evolution
1. Introduction
1.1. The disease and its epidemiology
Severe acute respiratory syndrome (SARS) is a highly contagious respiratory disease caused by a previously unknown coronavirus (CoV) named as SARS-CoV (SCoV) (Ksiazek et al., 2003, Peiris et al., 2003). The major outbreak started from November 2002 as a rapid wave of epidemic from Guangdong Province of China and spread through 29 regions around the world (WHO, 2004a). The epidemic was effectively controlled under vigorous quarantine measures and no new case was reported after July 2003. Six months after its disappearance, SCoV re-emerged in December 2003 as four sporadic cases in Guangdong Province, causing no fatality or secondary transmission (WHO, 2004b). SCoV is probably one of the very few examples of zoonotic viral emergence ‘caught-in-the-act’ with adequate sequences sampled from different phases of the epidemic (CSMEC, 2004, Wang et al., 2005a), as well as highly relevant samples from its zoonotic origins (Guan et al., 2003, Song et al., 2005, Ren et al., 2006). Taking advantage of the wealth of these sequence data, the evolutionary behaviors of SCoV during the epidemic have been investigated extensively, e.g. chains of transmission, theoretic time of epidemic onset, rate of substitutions and mode of natural selection acting on the viral genome, etc. In the first half of this article, applications of phylogenetics for investigating the emergence and transmission of SCoV were reviewed.
1.2. The phylogenetic and zoonotic origins
Prior to the discovery of SCoV, members of the Coronaviridae family can be unambiguously classified into three phylogenetic groups. Although initial phylogenetic analyses did not confidently classify SCoV as a member of any of the three existing groups of CoV, futher analyses suggested SCoV might share an ancient and distant ancestry with Group 2 CoVs (Snijder et al., 2003, Gorbalenya et al., 2004). SCoVs were also isolated from small mammals such as civets in wet markets, suggesting these mammals may have been the direct zoonotic origin of SCoV (Guan et al., 2003). Excitingly, wider animal surveys revealed unanticipated high levels of genetic diversity of CoV in bats (Cui et al., 2007). A group of CoVs named as SARS-like CoVs (SLCoVs) which shares 88–92% nucleotide identity with SCoV, was also identified from bats (Lau et al., 2005, Li et al., 2005b). These findings lead to the hypothesis that bats are the natural reservoir of SCoV and other members of the Coronaviridae family (Tang et al., 2006, Vijaykrishna et al., 2007). The biological and evolutionary aspects related to the conjectured inter-species transmission of SLCoV from its natural reservoir (i.e. bats) to the intermediate host (i.e. civets), and finally to human, have been the center of discussion (Shi and Hu, 2008). In the second half of this article, the phylogenetic origins of SCoV, as well as the diversity of CoVs in its zoonotic sources and the phylogenetic aspects of the speculated inter-species transmission events, were reviewed.
2. Dissecting the epidemiology of SARS outbreaks from a phylogenetic perspective
2.1. Super-spreading ‘caught-in-the-act’
Epidemiology of the SARS epidemic has been well-documented and one of its most intriguing characteristics is the concept of “super-spreading events” (SSE) (Lloyd-Smith et al., 2005), which contributed significantly to the rapid spread of the disease locally and globally (Li et al., 2004, Shen et al., 2004, Chen et al., 2006, Zhong et al., 2003). In mid-November 2002, the epidemic started as a series of seemingly independent cases and followed by the first documented SSE in Hospital HSZ-2 in Guangdong at the end of January 2003 (Zhong et al., 2003). By then, an infected nephrologist traveled from Guangdong to Hong Kong and initiated another SSE in Hotel M at the end of February 2003, leading to the worldwide transmission of SARS and subsequent outbreaks in Hong Kong (Ruan et al., 2003, Guan et al., 2004). The epidemic has been divided into three phases (Fig. 1 ) based on the occurrence of the above two mentioned SSEs (CSMEC, 2004). The early phase refers to the period prior to the SSE in Hospital HSZ-2 while the late phase refers to the period after to the SSE in Hotel M. The middle phase refers to the period between these two SSEs.
Fig. 1.
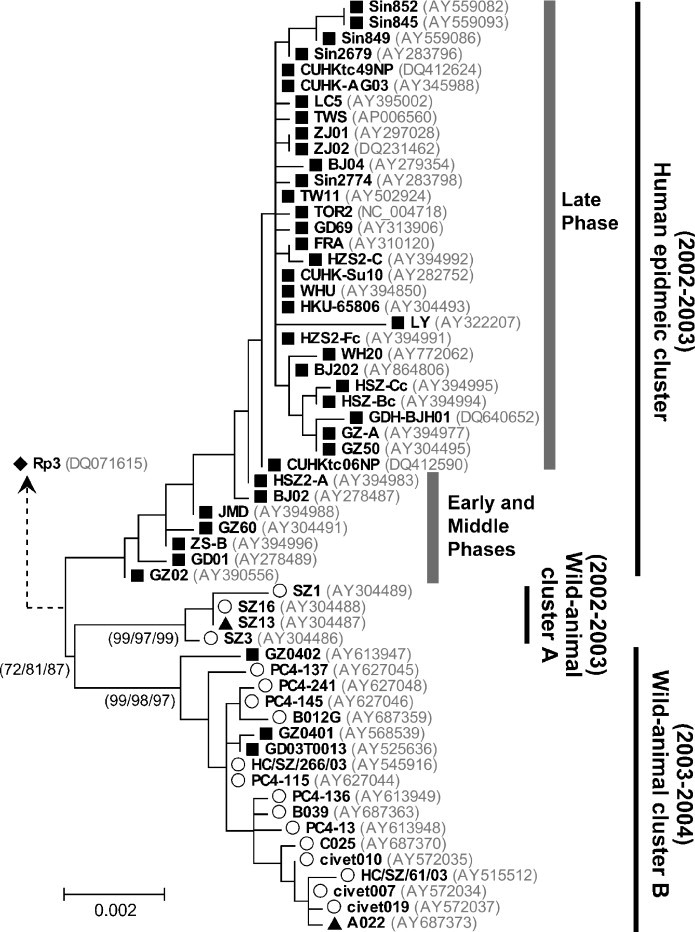
A phylogeny of spike gene nucleotide sequences from SCoV isolated from humans, civets and raccoon dogs. This phylogeny was modified and adopted from Lam et al. (2008a). Sequences from humans, civets, raccoon dogs and bats were indicated with symbols ■, ○, ▴ and ♦, respectively. The tree was constructed using ML method, with confidences of topology summarized from 5000 trees sampled from ML and NJ bootstrap replicates and BMCMC samples. Only confidence values of major clusters were shown (ML/NJ/BMCMC, in the parenthesis). The human epidemic cluster (2002–2003) was divided into late, early and middle phases according to a previous study (CSMEC, 2004). Accession numbers of the sequences are shown within round brackets after their strain names (in bold). The distance unit was substitutions/site. Rp3 isolated from bats (♦) was used as an out-group to root the tree, and the genetic distance of its branch is not shown.
Phylogenetic analyses enabled researchers to better understand the transmission chains and trace the sources of viral epidemics, which provide important information for making public health policy. Broadly speaking, phylogenies of SCoV sequences generally agreed with the documented contact histories, and additionally provided evidence to support some uncorroborated epidemiological speculations (Zhao, 2007). Phylogenies reconstructed by Neighbor Joining (NJ) (CSMEC, 2004, Guan et al., 2004), Maximum Likelihood (ML) (Zhang et al., 2006, Tang et al., 2007) and Bayesian methods (Tang et al., 2009) consistently distinguished the late phase strains as a monophyletic cluster, which included the index patient of the SSE in Hotel M and the primary cases in Vietnam, Singapore and Canada (Guan et al., 2004), supporting the viewpoint that the SARS outbreaks later in Hong Kong, as well as those in other parts of the world, were largely, if not completely, originated from a common source. In fact, analyses of the S gene sequences suggested multiple strains might have been independently introduced into Hong Kong from Guangdong before the SSE in Hotel M, although none of these strains contributed substantially to the subsequent outbreaks (Tsui et al., 2003, Guan et al., 2004). On the other hand, the strains from early and middle phases are relatively more diverse and appeared as multiple distinct clusters on the phylogenies (CSMEC, 2004, Song et al., 2005, Wang et al., 2005b), agreeing with the epidemiological investigations that SCoVs have been circulating in Guangdong and caused seemingly independent outbreaks prior to the SSE in Hotel M (Zhong et al., 2003). This observation fits the model put forward by Antia et al. (2003) and implies the possible occurrence of multiple zoonotic transmissions of phylogenetically distinct, but yet similar, SCoVs to human in the early phase of the epidemic.
2.2. Tracing the source of local outbreaks in the late phase epidemic
The epidemiology of some local outbreaks in the late phase was also investigated using phylogenetics. Firstly, two major subsequent outbreaks in Hong Kong, the Amoy Gardens outbreak (Ng, 2003) and the Hospital P outbreak (Tomlinson and Cockram, 2003), were phylogenetically demonstrated to be directly linked to the SSE in Hotel M (Chim et al., 2003, Guan et al., 2004). Secondly, phylogenies suggested the Taiwan outbreaks were likely to be derived from multiple sources (Lan et al., 2005b, Shih et al., 2005), including the Amoy Gardens outbreak (Chiu et al., 2003, Lan et al., 2005a) and the SSE in Hotel M (Yeh et al., 2004). Thirdly, the Singapore outbreak was traced back to two separate initial introductions, but both were directly linked to the SSE in Hotel M (Ruan et al., 2003, Vega et al., 2004), which later led to several other subsequent outbreaks within Singapore (Chen et al., 2006) and a case in Germany (Liu et al., 2005a). Lastly, in Beijing, although the first case was reported approximately a week after the SSE in Hotel M (Bi et al., 2003), phylogenetic analyses suggested not all the cases in Beijing were related to the SSE in Hotel M and some of the cases were likely originated from the Guangdong prior SSE (Liu et al., 2005c, Zhao, 2007).
2.3. Estimating the theoretical onset of the epidemic
Robust estimations on the theoretical onset of a viral epidemic provide evidence to testify the hypotheses on viral origins, e.g. AIDS pandemic (Korber et al., 2000, Lemey et al., 2003, Lemey et al., 2004, Worobey et al., 2004). Under the assumption of adequate sampling, the theoretical onset of a viral epidemic can be inferred as the time of the most recent common ancestor (tMRCA) of all sampled sequences. tMRCA can be estimated using a range of phylogenetic methods (Drummond et al., 2003), but all methods are primarily based on the assumption of molecular clocks of different extents (Pybus, 2006). It is noted that this tMRCA should theoretically be older than the earliest documented case, given the fact that there must be a window period between the emergence and recognition of the epidemic and the length of this window period is often a significant epidemiological interest (Stumpf and Pybus, 2002, Worobey et al., 2004).
In the case of SCoV, linear regression methods which correlate divergence with sampling dates were commonly used to estimate the tMRCA in the early studies. Based on simple linear regression, Zeng et al. (2003a) estimated the tMRCA of all human SCoVs at around December 2002, with the 95% credible interval (CI) from September 2002 to January 2003, which is comparable to a later similar study, with the same estimate at around November 2002 (95% CI from June 2002 to December 2002) (CSMEC, 2004). Later, Zhao et al. (2004) adopted three strategies to minimize possible errors and inferred the oldest bound of this tMRCA as early as spring 2002. A least square method, evaluated using Monte Carlo simulations, has also been applied to estimate the onset of the epidemic as August to September 2002 (Lu et al., 2004). While all the above studies relied on the assumption of a constant evolutionary rate over all lineages, i.e. a strict molecular clock (Pybus, 2006), enforcing a molecular clock to non-clocklike datasets could lead to biased estimations (Yoder and Yang, 2000). More recently, Salemi et al. (2004) applied a ML method to testify the clocklike behavior of a dataset of ten taxa and placed this tMRCA between September and November of 2002.
Despite the differences in methods and datasets, the above studies generally inferred the oldest bound of the onset of the epidemic at around mid-late 2002. The earliest documented case of SARS was identified on 16th November 2002 (Zhong et al., 2003), which is remarkably close to the oldest bounds of its theoretical onset when compared with other well-known viral epidemics (Stumpf and Pybus, 2002, Worobey et al., 2004). This finding suggests the SCoV has been quickly recognized after its emergence in human, partially reflecting the explosive mode of transmission in early phase of the epidemic.
After the discovery of SCoV in civets and sporadic re-emergence of SCoV in December 2003, these SCoV strains have been incorporated into datasets for estimation of their tMCRA (Song et al., 2005, Vijaykrishna et al., 2007, Hon et al., 2008). This tMRCA represents the upper limit of the time (i.e. oldest bound) of the inter-species transmission of SCoV from civets to human, which is phylogenetically different from the tMRCA of all human SCoVs discussed above and should theoretically be older. The estimated time of this inter-species transmission event will be discussed later in Section 4.
2.4. Sporadic re-emergence in December 2003
In December 2003, four seemingly independent cases of SCoV infection were reported (Liang et al., 2004, WHO, 2004b). All the patients have direct or indirect contact history with wild animals (Song et al., 2005, Wang et al., 2005a). Phylogenetic analyses of human SCoVs from these sporadic cases and civet SCoVs collected from the same period formed a monophyletic group distinct from the SCoVs of the 2002–2003 epidemic (Fig. 1), suggesting these sporadic cases were likely caused by inter-species transmissions that were independent from the previous outbreak (Song et al., 2005, Lam et al., 2008a). These epidemiological and phylogenetic findings provided convincing evidence for direct transmission of SCoV from civets to human (discussed later in Section 4).
In summary, the above findings demonstrated the vital roles of phylogenetics in understanding the emergence and transmission of SCoV in such a short-lasting but sweeping epidemic. The adequacy of early phase sampling and the relatively short duration of the SARS epidemic make it an excellent textbook example on how phylogenetic analyses can be used as an auxiliary tool in combination with classical epidemiological investigations to study the emergence of a viral epidemic.
3. Controversies over the phylogenetic origins of SCoV
3.1. The ancient history of a novel CoV
Which of the existing groups of CoV is phylogenetically closest to SCoV? This question has been under the spotlight of most discussions because the answer might help to trace the zoonotic origin of SCoV that is fundamentally important to public health. Prior to the availability of its complete genome sequences, initial phylogenetic analyses of a fragment of ORF1 suggested that SCoV may represent a novel group that is independent to the other three existing groups (Drosten et al., 2003, Ksiazek et al., 2003). Similar conclusions were reached based on phylogenetic analyses on multiple viral proteins after the complete genome sequences were available (Marra et al., 2003, Rota et al., 2003, Zeng et al., 2003b). Based on the observation that SCoV appears to be genetically equidistant to other known CoV groups, Holmes and Enjuanes (2003) concluded that SCoV is neither a recent host-range mutant of a known CoV nor a recent recombinant between known CoVs, but it probably evolved separately from an ancestor of the known CoVs in an unidentified host for a remarkably long period of time before its emergence in human.
Although the above argument was generally well-received, two follow-up questions were then raised. The first question is: If SCoV represents a lineage that anciently diverged from an ancestor of the existing CoVs, would this ancestor belong to one of the existing CoV groups? It later led to the concept of “early split-off from Group 2 CoV” for describing the phylogenetic origin of SCoV (Snijder et al., 2003). The second question is: Is SCoV an “ancient-recombinant” (if not a “recent-recombinant”) between the ancestors of any existing CoV groups? Due to relatively high divergence between SCoV and other CoVs (Rota et al., 2003) as well as the lack of a robust out-group (Holmes and Rambaut, 2004), the conclusions derived from various recombination analyses were yet intricate and inconsistent (Gorbalenya et al., 2004). A number of reasons were then proposed to explain the observed phylogenetic incongruence might not truly reflect the recombinant origin of SCoV (Holmes and Rambaut, 2004). The following sessions summarized the findings from numerous studies regarding the controversial phylogenetic origins of SCoV.
3.2. SCoV as an early-split off from Group 2 CoV: arguments and contentions
Following the initial analyses (Marra et al., 2003, Rota et al., 2003, Zeng et al., 2003b), the phylogenetic position of SCoV have been vigorously reevaluated with various approaches that were mainly different in their choices of genome regions (Eickmann et al., 2003, Zhu and Chen, 2004, Kim et al., 2006) and rooting strategies (Snijder et al., 2003, Gibbs et al., 2004, Lio and Goldman, 2004). Inspired by the positional conservation of cysteine residues between the S1 region of SCoV and Group 2 CoVs, Snijder et al. (2003) constructed an unrooted NJ phylogeny based on the amino acid sequences of S1 region and concluded SCoV is “closely related” to Group 2 CoVs (Eickmann et al., 2003). In two later studies, concatenated amino acid alignments of multiple viral proteins were used to construct unrooted phylogenies using various methods and the results consistently demonstrated the monophyletic relationship between SCoV and Group 2 CoVs (Zhu and Chen, 2004, Kim et al., 2006). In addition to these unrooted phylogenies, rooted phylogenies based on ORF1 alignments reached similar conclusions that SCoV and Group 2 CoV shared the last common ancestor (Snijder et al., 2003, Gibbs et al., 2004, Lio and Goldman, 2004). However, depends on the choices of out-groups, the rooting positions in these studies were inconsistent, which might be related to the possibly compromised accuracy of alignment if the out-groups are too diverged to be aligned (Holmes and Rambaut, 2004).
Nonetheless, the consensus of above findings is that SCoV could be classified as a “distant” member of Group 2 CoVs (Gorbalenya et al., 2004). This conclusion was reached primarily based on the robustness of the monophyletic relationship between SCoV and Group 2 CoVs. However, considering the actual genetic distance between SCoV and Group 2 CoVs, which is comparable to the inter-group distance among the three CoV groups, the biological significance of classifying SCoV as a distant member of Group 2 CoV is relatively low at best. We believe classification of a novel CoV should not be merely based on the branching orders of phylogenies and quantitative measurement of genetic distance should also be taken into account.
On the other hand, reconstructing the evolutionary relationships between highly divergent taxa has been proven as a difficult phylogenetic task (Philippe and Laurent, 1998). In particular, the observed branching order of the deep nodes in the highly divergent CoV phylogenies may not reflect their true evolutionary history, due to the possible influences of the long-branch attraction and rate variation among lineages (Gribaldo and Philippe, 2002, Holmes and Rambaut, 2004). In fact, Holmes and Rambaut reevaluated the tree topologies depicting the three possible phylogenetic positions of SCoV using Kishino-Hasegawa test, which is not biased by rate variations, and that demonstrated the topology with the best likelihood is not necessarily significantly better than the other two topologies, even the deep nodes were well-supported by quartet puzzling support values (Holmes and Rambaut, 2004). This finding indicated careful interpretations are needed before reaching a firm conclusion from the branching order of deep phylogenies.
Until recently, the addition of several newly discovered CoV lineages to the phylogeny broke up some of those long branches (Holmes and Rambaut, 2004), which tends to distribute the convergent and parallel mutations more evenly across the tree and hence reduce the problem of long-branch attraction (Hillis, 1996). In Fig. 2 , the closer phylogenetic relationship between SCoV and Group 2 CoVs suggested from the previous studies is also supported in this relatively well-sampled phylogeny. To this end, the question of whether it is appropriate to classify SCoV as a distant member of Group 2 CoVs seems to be a taxonomic problem more than a virological one, since SCoV and Group 2 CoVs are so divergent that they are likely to possess a substantial number of unique biological characteristics, e.g. host ranges and receptor usages (Haijema et al., 2003, Li et al., 2003). With the recent discoveries of novel CoV lineages, the classification of coronaviruses has to be revised systematically in both biological and phylogenetic context.
Fig. 2.

A phylogeny of all known CoVs (n = 40). The phylogeny was constructed based on the amino acid sequences of the RNA-dependent RNA Polymerase region (length = 163 a.a.). The phylogeny was constructed using BEAST (Drummond and Rambaut, 2007) under a uncorrelated lognormally relaxed clock model (Drummond et al., 2006). The number at the nodes indicates the Bayesian posterior probability support (as percentages) summarized from trees sampled at every 1000th step of a BMCMC chain of 10,000,000 steps where values lower than 80% were not shown. The mean substitution rate was fixed at 1.0 and the branch length was expressed in units of substitutions per site. FIPV, Feline infectious peritonitis virus (AY994055); CFBGDDM9503, Chinese ferret badger CoV 2003 (EU769560); PRCV, Porcine respiratory CoV (DQ811787); TGEV, Transmissible gastroenteritis virus (NC_002306); PEDV, Porcine epidemic diahrrea virus (NC_003436); BtCoV5122005, Bat CoV 512-2005 (NC_009657); BatCoVHKU6, Bat CoV HKU6 (DQ249224); BtCoV1A, Bat CoV 1A (NC_010437); BtCoV1B, Bat CoV 1B (NC_010436); BatCoVHKU8, Bat CoV HKU8 (NC_010438); HCoV229E, Human CoV 229E (NC_002645); HCoVNL63, Human CoV NL63 (NC_005831); BatCoVHKU2, Bat CoV HKU2 (EF203064); HCoVOC43, Human CoV OC43 (NC_005147); BCoV, Bovine CoV (NC_003045); AntelopeCoV, Sable antelope CoV (EF424621); GiCoV, Giraffe CoV (EF424622); PHEV, Porcine hemagglutinating encephalomyelitis virus (NC_007732); SDAV, Rat sialodacryoadenitis CoV (AF124990); MHV, Mouse hepatitis virus (NC_006852); HCoVHKU1, Human CoV HKU1 (NC_006577); BatCoVHKU9, Bat CoV HKU9 (NC_009021); SCoV (NC_004718); Rp3, Bat SLCoV (NC_009693); BatSCoVHKU3, Bat SLCoV HKU3 (NC_009694); Rm1, Bat SLCoV (NC_009696); Rf1, Bat SLCoV (NC_009695); BtCoV1332005, Bat CoV-133-2005 (NC_008315); BatCoVHKU4, Bat CoV HKU4 (NC_009019); BatCoVHKU5, Bat CoV HKU5 (NC_009020); IBV, Infectious bronchitis virus (NC_001451); IBVpeafowl, peafowl CoV (AY641576); IBVpartridge, partridge CoV (AY646283); TCoV, Turkey CoV (NC_010800); SW1, Beluga whale CoV (NC_010646); BuCoVHKU11, Bulbul CoV (NC_011548); ThCoVHKU12, Thrush CoV (NC_011549); MuCoVHKU13, Munia CoV (NC_011550); ALCGXF23006, Asian leopard cat CoV 2006 (EF584908); CFBGXF24706, Chinese ferret badger CoV 2006 (EF584911).
3.3. Phylogenetic incongruence and ancient recombination: facts and cautions
Phylogenetic incongruence, which is often justified by incompatible tree topologies for different genome regions (Holmes et al., 1999), has been widely used as an indicator for homologous recombination among viral genomes (Posada et al., 2002). Several studies demonstrated phylogenetic incongruence within the genome of SCoV based on a wide range of methods (Rest and Mindell, 2003, Stanhope et al., 2004, Stavrinides and Guttman, 2004, Zhang et al., 2005b). Despite the statistical significance of the phylogenetic incongruence observed in these studies, we could not conclude a consensus pattern of recombination since their findings were generally inconsistent. As an example, the potential recombination events within S gene proposed by Zhang et al. (2005b) were different from those proposed by Stavrinides and Guttman (2004), and were undetectable from the study of Rest and Mindell (2003). In fact, the considerable divergence between SCoV and the existing CoV groups has already excluded the possibility that SCoV is a recent recombinant from the existing CoV groups. Alternatively speaking, if recombination events had occurred, they have to be the ancient ones (Bosch, 2004), i.e. both the parents and daughter could have been evolved considerably after the recombination events. A simulation study demonstrated the accuracy for detecting these ancient recombination events can be substantially diminished if they were significantly obscured by the subsequent post-recombination substitutions (Chan et al., 2006). Therefore, the intricacy of the findings on the phylogenetic incongruence within the genome of SCoV may be a reflection of the varying sensitivity of the detection methods on the ancient and obscured recombination events, if any.
To further investigate the reported phylogenetic incongruence within the genome of SCoV, Rambaut and Holmes re-evaluated the study of Stavrinides and Guttman (2004) and suggested the patterns cited as evidence for recombination are more probably caused by a variation in substitution rate among lineages (Holmes and Rambaut, 2004). In addition, the author also stated the effect of the long-branch attraction on the branching order of deep phylogenies may also be a source of artifact, although unproven. Moreover, even if recombination did not occurred, given the stochastic nature of evolution, the authors would expect to observe phylogenetic incongruence among small genome fragments of a set of divergent taxa like SCoV and other CoVs. Therefore, the observed phylogenetic incongruence among the highly divergent genomes of CoV and its possible indication on ancient recombination events should be interpreted with extra cautions. Up to this point, the current phylogenetic evidence supporting the recombinant history of SCoV is weak at best (Holmes and Rambaut, 2004).
Putting aside the intricate phylogenetic evidence, the presence of stem-loop II motif (S2m) in the genome of SCoV have also been taken as an indication for recombination (Marra et al., 2003). S2m is a conserved RNA motif present in the genomes of several members of Astroviridae, Coronaviridae, and Picornaviridae family (Jonassen et al., 1998) while SCoV and Group 3 CoVs are the only members of the Coronaviridae that posse the motif. Assuming the motif in SCoV and Group 3 CoV were not acquired independently, the explanation of the co-presence of S2m should be either, (1) SCoV and Group 3 CoVs share the same ancestry, or (2) SCoV have acquired the motif from Group 3 CoVs through recombination (or vice versa). Currently, the phylogenetic data does not provide a better support for any of the above scenarios but a wider survey for the presence of S2m in other unknown CoV lineages will certainly provide insights to the ancient evolutionary history of the Coronaviridae family.
4. Zoonotic origins of SCoV
4.1. Civets as the immediate source of SARS epidemics
Since the early SARS cases in Guangdong seems to be related to restaurant workers handling wild mammals, Guan et al. (2003) surveyed the wild animals in a local market and isolated CoVs from Himalayan palm civets, which share 99.8% genome sequence identity to the SCoVs in human. Initial phylogenetic analyses suggested SCoVs from human and civets formed two distinct clades (Guan et al., 2003), but the SCoVs from civets are phylogenetically closer to the SCoVs of the early-phase epidemic than to those in the late-phase epidemic (Kan et al., 2005). These findings strongly suggest civets were the immediate sources of the SCoVs leading to the earliest SARS cases in Guangdong. The role of civets as the immediate zoonotic source of the SARS epidemic was further revealed during the sporadic re-emergence of SCoV in December 2003 (Wang et al., 2005a), based on the observation that the SCoVs isolated from civets and those patients of the same period formed a monophyletic cluster (Song et al., 2005, Lam et al., 2008a). These findings suggested that the emergence of SCoV in human is likely to be resulted from direct transmissions of SCoVs from civets. In addition, phylogenetic analyses demonstrated that the SCoVs from the 2002–2003 epidemic and the 2003–2004 re-emergence are phylogenetically distinct, suggesting the inter-species transmission events from civets to human in the two outbreaks might be independent (Song et al., 2005, Wang et al., 2005a).
However, a large-scale survey of SCoVs of civets in market in China suggested the lack of widespread infections in civets (Kan et al., 2005). In addition, the genetic diversity of civet SCoVs was relatively limited and was comparable to that of human SCoVs (Song et al., 2005, Lam et al., 2008a). These findings suggested civets might not be the natural reservoir of SCoV and it only acquired SCoVs shortly before the emergence of SCoV in human. Based on phylogenetic analyses using dynamic homology, Janies et al. (2008) speculated that civets might also acquired SCoV from other species, possibly human, even after the emergence of SCoV in 2002. An experimental evidence suggesting civets may not be the natural reservoir of SCoV is the observed symptoms in civets experimentally infected with SCoV (Wu et al., 2005), based on the fact that the natural reservoir hosts usually do not display severe signs of infection (Hudson et al., 2002). The above observations lead to the speculation of another natural reservoir host, which harbors a diverse group of SCoV-related CoVs and transmitted SCoV to civets prior to the emergence of SCoV in human.
4.2. Horseshoe bats as the natural reservoir of SCoV and SLCoV
As a result of extensive searches for the natural reservoir of SCoV, two groups of researchers independently identified a diverse group of CoVs from various species of horseshoe bats (Rhinolophus spp.). These CoVs shared 87–92% genome nucleotide identity with SCoVs and formed a distinct monophyletic cluster with SCoVs, therefore they were named SARS-like CoV (Lau et al., 2005, Li et al., 2005b). The close evolutionary relationship between SCoV and SLCoV is also supported by the presence of the S2m motif in their 3′UTR (Tang et al., 2006, Shi and Hu, 2008). Based on the phylogenetic analyses of the four characterized full genomes of SLCoV in horseshoe bats, the remarkably high genetic diversity of SLCoVs strongly suggested horseshoe bats are the natural reservoir of SCoV and SLCoV (Ren et al., 2006). This concept is further supported by the relatively high prevalence of SLCoV in R. sinicus (Lau et al., 2005) and the geographically widespread infections of SLCoVs in bats from distinct locations in China (Li et al., 2005b). The current hypothesis is that civets might have acquired SLCoVs from horseshoe bats and transferred to human, which is consistent with the observation that the 29-nt deletion in ORF8 are retained in bat SLCoVs, civet SCoVs and early phase human SCoV (Lau et al., 2005).
4.3. The missing link between SCoVs in civets and currently sampled SLCoVs in bats
Although the hypothesis of horseshoe bats as the natural reservoir for SLCoV and SCoV is relatively sensible, how bats SLCoV were transmitted to civets is still unexplained. If civets acquired SLCoV from bats shortly prior to its emergence in human as proposed, this bat SLCoV strain should be genetically very similar to the SCoVs sampled from civets. However, based on the relatively distant phylogenetic relationship between SCoVs and SLCoVs, none of the currently sampled SLCoVs in bats is the descendant of the direct ancestor of SCoVs in human and civet (Hon et al., 2008). In particular, Li and coworkers pointed out that substantial genetic changes in the S protein of the currently sampled SLCoV are likely to be necessary for the virus to infect civets or human (Li et al., 2006). Therefore, the direct ancestor of SCoVs in human and civets remains elusive.
More recently, Hon et al. (2008) demonstrated significant phylogenetic discordances among different genome regions of SLCoV strain Rp3 and speculated its potentially recombinant origin. Phylogenetic analysis of the parental regions of Rp3 genome suggested the presence of an uncharacterized bat SLCoV lineage (i.e. HB-SLCoV in Fig. 3 ) that is phylogenetically closer to SCoVs than any of the currently sampled bat SLCoVs. Based on the relatively high genetic diversity among the currently sampled SLCoVs in bats, the existence of a phylogenetically distinct lineage of SLCoV not yet sampled is highly possible. Thus, the authors speculated that the direct ancestor of SCoVs was as a descendant derived from this not yet sampled lineage, which crossed from a horseshoe bat species to civets (Hon et al., 2008).
Fig. 3.
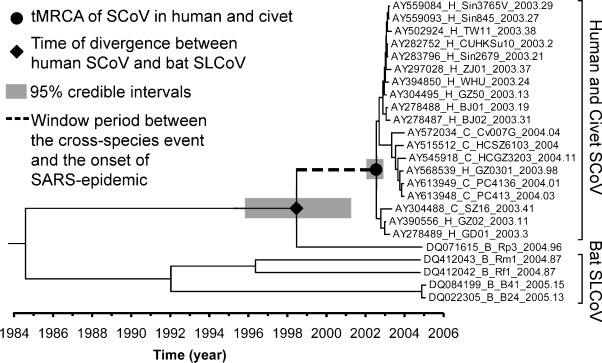
A time-scaled phylogeny of SCoV and SLCoV. This phylogeny was modified and adopted from Hon et al. (2008). The phylogeny was summarized from all MCMC phylogenies of the Orf1 data set analyzed under a Bayesian relaxed clock model. Height of the nodes was represented by the median of its estimates. The window period between the cross-species event and the onset of SARS epidemic was indicated as a dotted line. In the taxa labels, H, C and B represent host of human, civets and bats, respectively.
4.4. Estimated time of the inter-species transmission events and their implications
Determining the time of inter-species transmission events might help us to comprehend the viral zoonosis of the virus from an evolutionary standpoint. The oldest bound of the inter-species transmission of SCoV from civets to human is theoretically correspondent to the tMRCA of all human and civet SCoVs. Firstly, Song et al. (2005) estimated the synonymous substitution rate of SCoVs using linear regression and placed this tMRCA to be around early December 2002 without providing CI. In a later study, Vijaykrishna et al. (2007) investigated this tMRCA by applying a Bayesian relaxed clock model (Drummond et al., 2006) to a phylogeny of all representatives from the CoV family and estimated it at around 1999 with 95% posterior bounds of 13 years (i.e. 1990–2003). More recently, Hon et al. (2008) reconsidered this tMRCA using various clock models and estimated it at around September 2002 with 95% CIs between January and December of 2002. This tMRCA is very close to the observed first case of SARS (16th November 2002) as well as the estimated onset of the epidemic (as discussed earlier), suggesting SCoVs might have crossed from civets to human just months before the outbreak, supporting the view that civets are the immediate zoonotic source of SCoVs in human. It also implies the civet SCoVs might have adapted quickly, or alternatively speaking, a ‘by-pass’ host with only minimal adaptation is needed, to establish a sustainable chain of transmission in human.
On the other hand, the time of the speculated inter-species transmission of SLCoV from bats to civets has also been investigated. In the study described earlier (Vijaykrishna et al., 2007), the oldest bound of the time of this event was first estimated at a mean of 1986 with a 38-year credible interval (i.e. 1964–2002). Later, Hon et al. (2008) employed the concept of estimating the period between time of divergence (tDIV) and tMCRA proposed by Lam et al. (2008b), and speculated this inter-species transmission event might have happened with a median of 4.08 years before onset of the epidemic (credible intervals of 1.45–8.84 years) (Fig. 3). The above two estimates are not contradictory since the later estimate falls within the credible intervals of the former estimate but with an improved precision. Based on this relatively short window period between the inter-species transmission event and onset of the epidemic, Hon et al. (2008) speculated that civets might have acquired the ancestor of SCoV from the host species of SLCoV strain Rp3 directly and the involvement of other intermediate species may be unlikely. Therefore, authors suggested more focused surveillance on the host species of SLCoV strain Rp3 may shed light on the zoonotic origin of the direct ancestor of SCoV in civets.
4.5. Adaptive mutations potentially relevant to inter-species transmission
The host specificity of CoVs is mainly determined by the binding between the spike (S) protein and its cellular receptors (Haijema et al., 2003). Angiotensin I-converting enzyme 2 (ACE2) is a functional receptor for SCoVs in both human (Li et al., 2003) and civets (Li et al., 2005c), and was demonstrated to interact with the receptor binding domain (RBD) in the S1 subunit of the S protein (Wong et al., 2004). Based on the observed mutations within the RBD in strains from different epidemic phases and hosts, Li et al. (2005c) demonstrated that binding of S1 subunit to human and civet ACE2 can be significantly altered by mutating only two residues on the RBD (residue 479 and 487), suggesting these two residues may contribute to the adaptation of SCoV from civet to human. Later, this viewpoint was further supported by the location of these two residues at the binding interface between RBD and ACE2 in crystal structures (Li et al., 2005a, Li, 2008).
As observed in other examples of viral host shifts (Parrish and Kawaoka, 2005), the molecular determinants for host specificity, e.g. RBD residues related to adaptation to human in the case of SCoV, are likely to be subjected to an elevated level of selection pressure during the acquisition of a new host. The ratio of non-synonymous (dN) to synonymous substitution rate (dS), i.e. ω, which is widely used as a measure for selection pressure (Yang and Nielsen, 2002), has been employed in several studies to detect positive selection on viral genes of SCoV and SLCoV. Firstly, the ω values of structural genes were found to be higher than that of the non-structural region, suggesting the structural proteins might have been under a stronger selection pressure (Zhao et al., 2004, Song et al., 2005). Moreover, the ω value of S gene in the strains from early phase is significantly larger than that of those from middle and late phases (CSMEC, 2004). The above findings provided preliminary phylogenetic evidence for the potential role of S protein in adaptation of SCoV from civets to human.
Additionally, lineage-specific ω in the S gene phylogeny (n = 11) was estimated using a codon-based genetic algorithm (Kosakovsky Pond et al., 2006), and a significantly higher ω value was observed along the lineage leading to the human cluster (Vijaykrishna et al., 2007). More recently, Tang et al. (2009) comprehensively analyzed a larger dataset (n = 59) using a ML branch-site codon model (Zhang et al., 2005a), aimed at comparing the ω values of lineages of various epidemic phases or hosts (i.e. foreground) versus the rest of the phylogeny (i.e. background). The authors concluded that the 2002 early and middle phase human lineages and the 2003 sporadic-re-emergence human-civet lineages are under positive selection. Despite the differences in methodologies and datasets, both studies demonstrated phylogenetic evidence for positive selection along lineages relevant to the civets-to-human inter-species transmission on the S gene phylogeny.
On the other hand, positively selected residues on S protein have been identified in a number of similar studies, primarily by applying ML codon models to similar datasets but with different epidemic groupings (Zhang et al., 2005a, Zhang et al., 2006, Shi et al., 2006). Although the residues identified from these studies are not completely overlapping, probably due to the differences in taxa groupings, these sites are mainly located in the S1 subunit and residue 479 is consistently detected to be under positive selection, supporting its speculated roles in adaptation for new hosts (Song et al., 2005). It should be noted that positively selected residues were not identified in the S genes from neither the late epidemic phase (Zhang et al., 2006) nor the SLCoV in bats (Zhang et al., 2005a). The mode of selection relevant to the inter-species transmission from bats to civets has not been investigated systematically, primarily due to the lack of bat SLCoV S gene sequences that are phylogenetically close enough for robust analysis.
5. The continuing story of CoV diversity
5.1. The expanding diversity and widening host range
As a result of the extensive efforts in searching for the zoonotic origins of SCoV, our knowledge on the host range and diversity of CoVs has been expanded rapidly. For example, in the last couple of years, the number of avian species detected to harbor Group 3 CoVs has been doubled (Cavanagh, 2005). Furthermore, in addition to the two newly discovered human CoVs in the existing Group 1 and 2, i.e. NL63 (Fouchier et al., 2004) and HKU1 (Woo et al., 2005a), respectively, a number of divergent CoVs have been identified from Asian Leopard Cats and Chinese Ferret Badgers (Dong et al., 2007), wild birds (Woo et al., 2009) and a beluga whale (Mihindukulasuriya et al., 2008). These findings largely expanded the known diversity of CoVs and the long-established classification of the Coronaviridae family as three distinct groups needs to be revised systematically. On the other hand, a number of diverse CoVs belonging to Group 1 and 2 have been identified from a range of bat species (Woo et al., 2006). The surprisingly high diversity of bat CoVs rationally leads to the hypothesis that bats are the natural reservoir of all CoVs (Tang et al., 2006). According to a time-scaled phylogeny of representative CoVs from all groups, Vijaykrishna et al. (2007) concluded that bats are likely to be the host of the ancestor for all presently known CoV lineages. Furthermore, based on the results of their Bayesian coalescent analyses, the authors speculated a diverse group of CoVs may be endemic in various bat species, with repeated introduction to other animals and occasional establishment of new lineages in other species (Vijaykrishna et al., 2007). In fact, other than the speculated inter-species transmission of CoVs from bats to other animal species, hosts shifts of bat CoVs between different Rhinolophus spp. were also proposed based on the incongruence between the phylogenies of CoVs and their host Rhinolophus spp. (Cui et al., 2007).
Additionally, inter-species transmissions of CoVs between other non-bat species were also proposed. The first animal-human zoonotic pair of CoVs being detailedly analyzed was Bovine CoV and Human CoV-OC43 (Vijgen et al., 2005). Based on a combination of molecular clock analyses, the authors estimated the tMRCA of these two CoVs at around 1890 and speculated an inter-species transmission event from Bovine CoV to human might have occurred around this period (Vijgen et al., 2005). Moreover, a number of CoVs are documented to infect multiple closely related host species, e.g. bovine CoVs has been isolated from captive wild ruminants (Alekseev et al., 2008); closely related Group 3 CoVs have been isolated from various avian species (Cavanagh et al., 2002, Jonassen et al., 2005, Liu et al., 2005b). These findings implied the relatively promiscuous nature of the host specificity of CoVs, which was best demonstrated by the evidence of inter-species transmission of highly similar if not identical SCoVs between two distantly related species, i.e. human and civets (Wang et al., 2005a).
5.2. How much did recombination contribute to the diversity of CoV?
Last but not least, the relatively high rate of homologous recombination has been speculated to facilitate the inter-species transmissions of CoVs (Baric et al., 1995, Baric et al., 1997). Despite the lack of relevant examples to support this hypothesis, naturally occurring recombination between CoV strains of the same species (Jia et al., 1995), as well as between strains of different CoV species, have been documented (Herrewegh et al., 1998, Hon et al., 2008, Decaro et al., 2009), suggesting the generation of CoV diversity through recombination has been happening in the field. In fact, the two recently discovered human CoVs were proposed to have a recombinant history based on the observed phylogenetic incongruence between different genome regions (Woo et al., 2005b, Pyrc et al., 2006). As discussed earlier, due to the relatively high divergence between different groups of CoV, cautions have to be taken to interpret these phylogenetic incongruences as evidence for ancient recombination events (Holmes and Rambaut, 2004, Chan et al., 2006). In summary, the current data only suggests the occurrence of recombination between closely related CoV strains, but provides no direct evidence to support a role of recombination in the emergence of novel CoVs in novel host species, e.g. emergence of SCoV in human.
6. The lesson learnt and an alarm for next zoonosis
The SARS epidemic in 2003 offers a solid lesson on the application of phylogenetics in understanding the epidemiology of a newly established epidemic, as well as the evolutionary basis of a zoonotic viral emergence. Phylogenetics has played an indispensable role in the prompt identification of the transmission chains and its zoonotic origins, which provided important clues for the policy-making in public health, e.g. customs and border control, quarantine measures, culling of civets and the continuous search for the viral natural reservoir. These valuable experiences should help the community to better prepare for the next zoonotic viral epidemic.
More importantly, while most of the attentions have been focused on preparing for the known potential pandemics like avian influenza (Fauci, 2006), the unanticipated strike of SARS epidemic alarmed public health officials and researchers for the neglected possibility of deadly viral emergence from an unexpected origin. For many years, CoVs have long been regarded as relatively “mild” viruses with broad but yet restricted host ranges. However, according to our current understanding to the diversity and evolution of CoVs (Vijaykrishna et al., 2007), at least five divergent species of CoV are known to have zoonotic transmission into the human population and this cross species event will be likely to continue, and the zoonosis of SCoV was just the consequence of one of these inter-species transmission events. Although the prevention of zoonosis from an unexpected origin seems to be impractical, sufficiently flexible and stringent surveillance strategies provide us an opportunity to anticipate the disease in a population and prevent its further spreading by implementing appropriate control measures. The sporadic re-emergence of SCoV in the early 2004 illustrated the importance of stringent surveillance in preventing the further spread of a zoonotic virus in the early phase. One step further, the surveillance strategies must be adopted in a way that we could learn more about the diversity of potentially zoonotic viruses in their reservoirs. With CoV as an example, given the relatively promiscuous nature of its host specificity as discussed above, extensive and regular surveillance of known CoVs in pets and agricultural animals, which are in close contact with human, should be set up. Well-established knowledge on the diversity of potentially zoonotic viruses certainly accelerates the identification of its animal origin once it emerges in human. Last but not least, although the immediate zoonotic source of SCoV seems to be eliminated by culling of civets in Mainland China, the uncertainties about the diversity of SLCoV in bats still pose threat on the recurrence SCoV (Hon et al., 2008). Therefore, we emphasize the importance of continuous surveillance on the genetic diversity of SLCoV in bats.
Acknowledgements
This work is partially supported by Research Fund for the Control of Infectious Diseases (reference number 06060672) from Hong Kong SAR government and the Strategic Research Theme of Infection and Immunology, The University of Hong Kong.
References
- Alekseev K.P., Vlasova A.N., Jung K., Hasoksuz M., Zhang X., Halpin R., Wang S., Ghedin E., Spiro D., Saif L.J. Bovine-like coronaviruses isolated from four species of captive wild ruminants are homologous to bovine coronaviruses, based on complete genomic sequences. J. Virol. 2008;82(24):12422–12431. doi: 10.1128/JVI.01586-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antia R., Regoes R.R., Koella J.C., Bergstrom C.T. The role of evolution in the emergence of infectious diseases. Nature. 2003;426(6967):658–661. doi: 10.1038/nature02104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baric R.S., Fu K., Chen W., Yount B. High recombination and mutation rates in mouse hepatitis virus suggest that coronaviruses may be potentially important emerging viruses. Adv. Exp. Med. Biol. 1995;380:571–576. doi: 10.1007/978-1-4615-1899-0_91. [DOI] [PubMed] [Google Scholar]
- Baric R.S., Yount B., Hensley L., Peel S.A., Chen W. Episodic evolution mediates interspecies transfer of a murine coronavirus. J. Virol. 1997;71(3):1946–1955. doi: 10.1128/jvi.71.3.1946-1955.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi S., Qin E., Xu Z., Li W., Wang J., Hu Y., Liu Y., Duan S., Hu J., Han Y., Xu J., Li Y., Yi Y., Zhou Y., Lin W., Xu H., Li R., Zhang Z., Sun H., Zhu J., Yu M., Fan B., Wu Q., Tang L., Yang B., Li G., Peng W., Jiang T., Deng Y., Liu B., Shi J., Wei W., Liu H., Tong Z., Zhang F., Zhang Y., Wang C., Ye J., Gan Y., Ji J., Li X., Tian X., Lu F., Tan G., Yang R., Liu S., Li S., Cao W., Yu J., Dong X., Yang H. Complete genome sequences of the SARS-CoV: the BJ Group (Isolates BJ01-BJ04) Genomics Proteomics Bioinform. 2003;1(3):180–192. doi: 10.1016/S1672-0229(03)01023-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch X. SARS: an amalgam of avian and mammalian viruses? Lancet Infect. Dis. 2004;4(2):65. doi: 10.1016/S1473-3099(04)00913-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D. Coronaviruses in poultry and other birds. Avian Pathol. 2005;34(6):439–448. doi: 10.1080/03079450500367682. [DOI] [PubMed] [Google Scholar]
- Cavanagh D., Mawditt K., Welchman Dde B., Britton P., Gough R.E. Coronaviruses from pheasants (Phasianus colchicus) are genetically closely related to coronaviruses of domestic fowl (infectious bronchitis virus) and turkeys. Avian Pathol. 2002;31(1):81–93. doi: 10.1080/03079450120106651. [DOI] [PubMed] [Google Scholar]
- Chan C.X., Beiko R.G., Ragan M.A. Detecting recombination in evolving nucleotide sequences. BMC Bioinform. 2006;7:412. doi: 10.1186/1471-2105-7-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.I., Loon S.C., Leong H.N., Leo Y.S. Understanding the super-spreading events of SARS in Singapore. Ann. Acad. Med. Singapore. 2006;35(6):390–394. [PubMed] [Google Scholar]
- Chim S.S., Tsui S.K., Chan K.C., Au T.C., Hung E.C., Tong Y.K., Chiu R.W., Ng E.K., Chan P.K., Chu C.M., Sung J.J., Tam J.S., Fung K.P., Waye M.M., Lee C.Y., Yuen K.Y., Lo Y.M. Genomic characterisation of the severe acute respiratory syndrome coronavirus of Amoy Gardens outbreak in Hong Kong. Lancet. 2003;362(9398):1807–1808. doi: 10.1016/S0140-6736(03)14901-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu R.W., Chim S.S., Lo Y.M. Molecular epidemiology of SARS—from Amoy Gardens to Taiwan. N. Engl. J. Med. 2003;349(19):1875–1876. doi: 10.1056/NEJM200311063491923. [DOI] [PubMed] [Google Scholar]
- CSMEC, 2004. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science 303(5664), 1666–1669. [DOI] [PubMed]
- Cui J., Han N., Streicker D., Li G., Tang X., Shi Z., Hu Z., Zhao G., Fontanet A., Guan Y., Wang L., Jones G., Field H.E., Daszak P., Zhang S. Evolutionary relationships between bat coronaviruses and their hosts. Emerg. Infect. Dis. 2007;13(10):1526–1532. doi: 10.3201/eid1310.070448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro N., Mari V., Campolo M., Lorusso A., Camero M., Elia G., Martella V., Cordioli P., Enjuanes L., Buonavoglia C. Recombinant canine coronaviruses related to transmissible gastroenteritis virus of Swine are circulating in dogs. J. Virol. 2009;83(3):1532–1537. doi: 10.1128/JVI.01937-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong B.Q., Liu W., Fan X.H., Vijaykrishna D., Tang X.C., Gao F., Li L.F., Li G.J., Zhang J.X., Yang L.Q., Poon L.L., Zhang S.Y., Peiris J.S., Smith G.J., Chen H., Guan Y. Detection of a novel and highly divergent coronavirus from asian leopard cats and Chinese ferret badgers in Southern China. J. Virol. 2007;81(13):6920–6926. doi: 10.1128/JVI.00299-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348(20):1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- Drummond A., Pybus O.G., Rambaut A. Inference of viral evolutionary rates from molecular sequences. Adv. Parasitol. 2003;54:331–358. doi: 10.1016/s0065-308x(03)54008-8. [DOI] [PubMed] [Google Scholar]
- Drummond A.J., Ho S.Y., Phillips M.J., Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4(5):e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A.J., Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickmann M., Becker S., Klenk H.D., Doerr H.W., Stadler K., Censini S., Guidotti S., Masignani V., Scarselli M., Mora M., Donati C., Han J.H., Song H.C., Abrignani S., Covacci A., Rappuoli R. Phylogeny of the SARS coronavirus. Science. 2003;302(5650):1504–1505. doi: 10.1126/science.302.5650.1504b. [DOI] [PubMed] [Google Scholar]
- Fauci A.S. Pandemic influenza threat and preparedness. Emerg. Infect. Dis. 2006;12(1):73–77. doi: 10.3201/eid1201.050983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouchier R.A., Hartwig N.G., Bestebroer T.M., Niemeyer B., de Jong J.C., Simon J.H., Osterhaus A.D. A previously undescribed coronavirus associated with respiratory disease in humans. Proc. Natl. Acad. Sci. U.S.A. 2004;101(16):6212–6216. doi: 10.1073/pnas.0400762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs A.J., Gibbs M.J., Armstrong J.S. The phylogeny of SARS coronavirus. Arch. Virol. 2004;149(3):621–624. doi: 10.1007/s00705-003-0244-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Snijder E.J., Spaan W.J. Severe acute respiratory syndrome coronavirus phylogeny: toward consensus. J. Virol. 2004;78(15):7863–7866. doi: 10.1128/JVI.78.15.7863-7866.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribaldo S., Philippe H. Ancient phylogenetic relationships. Theor. Popul. Biol. 2002;61(4):391–408. doi: 10.1006/tpbi.2002.1593. [DOI] [PubMed] [Google Scholar]
- Guan Y., Peiris J.S., Zheng B., Poon L.L., Chan K.H., Zeng F.Y., Chan C.W., Chan M.N., Chen J.D., Chow K.Y., Hon C.C., Hui K.H., Li J., Li V.Y., Wang Y., Leung S.W., Yuen K.Y., Leung F.C. Molecular epidemiology of the novel coronavirus that causes severe acute respiratory syndrome. Lancet. 2004;363(9403):99–104. doi: 10.1016/S0140-6736(03)15259-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y., Zheng B.J., He Y.Q., Liu X.L., Zhuang Z.X., Cheung C.L., Luo S.W., Li P.H., Zhang L.J., Guan Y.J., Butt K.M., Wong K.L., Chan K.W., Lim W., Shortridge K.F., Yuen K.Y., Peiris J.S., Poon L.L. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302(5643):276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- Haijema B.J., Volders H., Rottier P.J. Switching species tropism: an effective way to manipulate the feline coronavirus genome. J. Virol. 2003;77(8):4528–4538. doi: 10.1128/JVI.77.8.4528-4538.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrewegh A.A., Smeenk I., Horzinek M.C., Rottier P.J., de Groot R.J. Feline coronavirus type II strains 79-1683 and 79-1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J. Virol. 1998;72(5):4508–4514. doi: 10.1128/jvi.72.5.4508-4514.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillis D.M. Inferring complex phylogenies. Nature. 1996;383(6596):130–131. doi: 10.1038/383130a0. [DOI] [PubMed] [Google Scholar]
- Holmes E.C., Rambaut A. Viral evolution and the emergence of SARS coronavirus. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2004;359(1447):1059–1065. doi: 10.1098/rstb.2004.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E.C., Worobey M., Rambaut A. Phylogenetic evidence for recombination in dengue virus. Mol. Biol. Evol. 1999;16(3):405–409. doi: 10.1093/oxfordjournals.molbev.a026121. [DOI] [PubMed] [Google Scholar]
- Holmes K.V., Enjuanes L. Virology. The SARS coronavirus: a postgenomic era. Science. 2003;300(5624):1377–1378. doi: 10.1126/science.1086418. [DOI] [PubMed] [Google Scholar]
- Hon C.C., Lam T.Y., Shi Z.L., Drummond A.J., Yip C.W., Zeng F., Lam P.Y., Leung F.C. Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus. J. Virol. 2008;82(4):1819–1826. doi: 10.1128/JVI.01926-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson P.J., Rizzoli A., Grenfell B.T., Heesterbeek H., Dobson A.P. Oxford University Press; Oxford (UK): 2002. The Ecology of Wildlife Disease. [Google Scholar]
- Janies D., Habib F., Alexandrov B., Hill A., Pol D. Evolution of genomes, host shifts and the geographic spread of SARS-CoV and related coronaviruses. Cladistics. 2008;24:111–130. doi: 10.1111/j.1096-0031.2008.00199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia W., Karaca K., Parrish C.R., Naqi S.A. A novel variant of avian infectious bronchitis virus resulting from recombination among three different strains. Arch. Virol. 1995;140(2):259–271. doi: 10.1007/BF01309861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonassen C.M., Jonassen T.O., Grinde B. A common RNA motif in the 3’ end of the genomes of astroviruses, avian infectious bronchitis virus and an equine rhinovirus. J. Gen. Virol. 1998;79(Pt 4):715–718. doi: 10.1099/0022-1317-79-4-715. [DOI] [PubMed] [Google Scholar]
- Jonassen C.M., Kofstad T., Larsen I.L., Lovland A., Handeland K., Follestad A., Lillehaug A. Molecular identification and characterization of novel coronaviruses infecting graylag geese (Anser anser), feral pigeons (Columbia livia) and mallards (Anas platyrhynchos) J. Gen. Virol. 2005;86(Pt 6):1597–1607. doi: 10.1099/vir.0.80927-0. [DOI] [PubMed] [Google Scholar]
- Kan B., Wang M., Jing H., Xu H., Jiang X., Yan M., Liang W., Zheng H., Wan K., Liu Q., Cui B., Xu Y., Zhang E., Wang H., Ye J., Li G., Li M., Cui Z., Qi X., Chen K., Du L., Gao K., Zhao Y.T., Zou X.Z., Feng Y.J., Gao Y.F., Hai R., Yu D., Guan Y., Xu J. Molecular evolution analysis and geographic investigation of severe acute respiratory syndrome coronavirus-like virus in palm civets at an animal market and on farms. J. Virol. 2005;79(18):11892–11900. doi: 10.1128/JVI.79.18.11892-11900.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim O.J., Lee D.H., Lee C.H. Close relationship between SARS-coronavirus and group 2 coronavirus. J. Microbiol. 2006;44(1):83–91. [PubMed] [Google Scholar]
- Korber B., Muldoon M., Theiler J., Gao F., Gupta R., Lapedes A., Hahn B.H., Wolinsky S., Bhattacharya T. Timing the ancestor of the HIV-1 pandemic strains. Science. 2000;288(5472):1789–1796. doi: 10.1126/science.288.5472.1789. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond S.L., Posada D., Gravenor M.B., Woelk C.H., Frost S.D. GARD: a genetic algorithm for recombination detection. Bioinformatics. 2006;22(24):3096–3098. doi: 10.1093/bioinformatics/btl474. [DOI] [PubMed] [Google Scholar]
- Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348(20):1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Lam T.T., Hon C.C., Lam P.Y., Yip C.W., Zeng F., Leung F.C. Comments to the predecessor of human SARS coronavirus in 2003-2004 epidemic. Vet. Microbiol. 2008;126(4):390–393. doi: 10.1016/j.vetmic.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam T.Y., Hon C.C., Wang Z., Hui R.K., Zeng F., Leung F.C. Evolutionary analyses of European H1N2 swine influenza A virus by placing timestamps on the multiple reassortment events. Virus Res. 2008;131(2):271–278. doi: 10.1016/j.virusres.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Lan Y.C., Liu H.F., Shih Y.P., Yang J.Y., Chen H.Y., Chen Y.M. Phylogenetic analysis and sequence comparisons of structural and non-structural SARS coronavirus proteins in Taiwan. Infect. Genet. Evol. 2005;5(3):261–269. doi: 10.1016/j.meegid.2004.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y.C., Liu T.T., Yang J.Y., Lee C.M., Chen Y.J., Chan Y.J., Lu J.J., Liu H.F., Hsiung C.A., Ho M.S., Hsiao K.J., Chen H.Y., Chen Y.M. Molecular epidemiology of severe acute respiratory syndrome-associated coronavirus infections in Taiwan. J. Infect. Dis. 2005;191(9):1478–1489. doi: 10.1086/428591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S.K., Woo P.C., Li K.S., Huang Y., Tsoi H.W., Wong B.H., Wong S.S., Leung S.Y., Chan K.H., Yuen K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. U.S.A. 2005;102(39):14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P., Pybus O.G., Rambaut A., Drummond A.J., Robertson D.L., Roques P., Worobey M., Vandamme A.M. The molecular population genetics of HIV-1 group O. Genetics. 2004;167(3):1059–1068. doi: 10.1534/genetics.104.026666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P., Pybus O.G., Wang B., Saksena N.K., Salemi M., Vandamme A.M. Tracing the origin and history of the HIV-2 epidemic. Proc. Natl. Acad. Sci. U.S.A. 2003;100(11):6588–6592. doi: 10.1073/pnas.0936469100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. Structural analysis of major species barriers between humans and palm civets for severe acute respiratory syndrome coronavirus infections. J. Virol. 2008;82(14):6984–6991. doi: 10.1128/JVI.00442-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309(5742):1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Shi Z., Yu M., Ren W., Smith C., Epstein J.H., Wang H., Crameri G., Hu Z., Zhang H., Zhang J., McEachern J., Field H., Daszak P., Eaton B.T., Zhang S., Wang L.F. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310(5748):676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- Li W., Wong S.K., Li F., Kuhn J.H., Huang I.C., Choe H., Farzan M. Animal origins of the severe acute respiratory syndrome coronavirus: insight from ACE2-S-protein interactions. J. Virol. 2006;80(9):4211–4219. doi: 10.1128/JVI.80.9.4211-4219.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Zhang C., Sui J., Kuhn J.H., Moore M.J., Luo S., Wong S.K., Huang I.C., Xu K., Vasilieva N., Murakami A., He Y., Marasco W.A., Guan Y., Choe H., Farzan M. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24(8):1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Yu I.T., Xu P., Lee J.H., Wong T.W., Ooi P.L., Sleigh A.C. Predicting super spreading events during the 2003 severe acute respiratory syndrome epidemics in Hong Kong and Singapore. Am. J. Epidemiol. 2004;160(8):719–728. doi: 10.1093/aje/kwh273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G., Chen Q., Xu J., Liu Y., Lim W., Peiris J.S., Anderson L.J., Ruan L., Li H., Kan B., Di B., Cheng P., Chan K.H., Erdman D.D., Gu S., Yan X., Liang W., Zhou D., Haynes L., Duan S., Zhang X., Zheng H., Gao Y., Tong S., Li D., Fang L., Qin P., Xu W. Laboratory diagnosis of four recent sporadic cases of community-acquired SARS, Guangdong Province China. Emerg. Infect. Dis. 2004;10(10):1774–1781. doi: 10.3201/eid1010.040445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lio P., Goldman N. Phylogenomics and bioinformatics of SARS-CoV. Trends Microbiol. 2004;12(3):106–111. doi: 10.1016/j.tim.2004.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Lim S.L., Ruan Y., Ling A.E., Ng L.F., Drosten C., Liu E.T., Stanton L.W., Hibberd M.L. SARS transmission pattern in Singapore reassessed by viral sequence variation analysis. PLoS Med. 2005;2(2):e43. doi: 10.1371/journal.pmed.0020043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Chen J., Kong X., Shao Y., Han Z., Feng L., Cai X., Gu S., Liu M. Isolation of avian infectious bronchitis coronavirus from domestic peafowl (Pavo cristatus) and teal (Anas) J. Gen. Virol. 2005;86(Pt 3):719–725. doi: 10.1099/vir.0.80546-0. [DOI] [PubMed] [Google Scholar]
- Liu W., Tang F., Fontanet A., Zhan L., Wang T.B., Zhang P.H., Luan Y.H., Cao C.Y., Zhao Q.M., Wu X.M., Xin Z.T., Zuo S.Q., Baril L., Vabret A., Shao Y.M., Yang H., Cao W.C. Molecular epidemiology of SARS-associated coronavirus, Beijing. Emerg. Infect. Dis. 2005;11(9):1420–1424. doi: 10.3201/eid1109.040773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Smith J.O., Schreiber S.J., Kopp P.E., Getz W.M. Superspreading and the effect of individual variation on disease emergence. Nature. 2005;438(7066):355–359. doi: 10.1038/nature04153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H., Zhao Y., Zhang J., Wang Y., Li W., Zhu X., Sun S., Xu J., Ling L., Cai L., Bu D., Chen R. Date of origin of the SARS coronavirus strains. BMC Infect. Dis. 2004;4:3. doi: 10.1186/1471-2334-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. The Genome sequence of the SARS-associated coronavirus. Science. 2003;300(5624):1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- Mihindukulasuriya K.A., Wu G., St Leger J., Nordhausen R.W., Wang D. Identification of a novel coronavirus from a beluga whale by using a panviral microarray. J. Virol. 2008;82(10):5084–5088. doi: 10.1128/JVI.02722-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng S.K. Possible role of an animal vector in the SARS outbreak at Amoy Gardens. Lancet. 2003;362(9383):570–572. doi: 10.1016/S0140-6736(03)14121-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish C.R., Kawaoka Y. The origins of new pandemic viruses: the acquisition of new host ranges by canine parvovirus and influenza A viruses. Annu. Rev. Microbiol. 2005;59:553–586. doi: 10.1146/annurev.micro.59.030804.121059. [DOI] [PubMed] [Google Scholar]
- Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., Cheng V.C., Chan K.H., Tsang D.N., Yung R.W., Ng T.K., Yuen K.Y. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361(9366):1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe H., Laurent J. How good are deep phylogenetic trees? Curr. Opin. Genet. Dev. 1998;8(6):616–623. doi: 10.1016/s0959-437x(98)80028-2. [DOI] [PubMed] [Google Scholar]
- Posada D., Crandall K.A., Holmes E.C. Recombination in evolutionary genomics. Annu. Rev. Genet. 2002;36:75–97. doi: 10.1146/annurev.genet.36.040202.111115. [DOI] [PubMed] [Google Scholar]
- Pybus O.G. Model selection and the molecular clock. PLoS Biol. 2006;4(5):e151. doi: 10.1371/journal.pbio.0040151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrc K., Dijkman R., Deng L., Jebbink M.F., Ross H.A., Berkhout B., van der Hoek L. Mosaic structure of human coronavirus NL63, one thousand years of evolution. J. Mol. Biol. 2006;364(5):964–973. doi: 10.1016/j.jmb.2006.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren W., Li W., Yu M., Hao P., Zhang Y., Zhou P., Zhang S., Zhao G., Zhong Y., Wang S., Wang L.F., Shi Z. Full-length genome sequences of two SARS-like coronaviruses in horseshoe bats and genetic variation analysis. J. Gen. Virol. 2006;87(Pt 11):3355–3359. doi: 10.1099/vir.0.82220-0. [DOI] [PubMed] [Google Scholar]
- Rest J.S., Mindell D.P. SARS associated coronavirus has a recombinant polymerase and coronaviruses have a history of host-shifting. Infect. Genet. Evol. 2003;3(3):219–225. doi: 10.1016/j.meegid.2003.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300(5624):1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Ruan Y.J., Wei C.L., Ee A.L., Vega V.B., Thoreau H., Su S.T., Chia J.M., Ng P., Chiu K.P., Lim L., Zhang T., Peng C.K., Lin E.O., Lee N.M., Yee S.L., Ng L.F., Chee R.E., Stanton L.W., Long P.M., Liu E.T. Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection. Lancet. 2003;361(9371):1779–1785. doi: 10.1016/S0140-6736(03)13414-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salemi M., Fitch W.M., Ciccozzi M., Ruiz-Alvarez M.J., Rezza G., Lewis M.J. Severe acute respiratory syndrome coronavirus sequence characteristics and evolutionary rate estimate from maximum likelihood analysis. J. Virol. 2004;78(3):1602–1603. doi: 10.1128/JVI.78.3.1602-1603.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z., Ning F., Zhou W., He X., Lin C., Chin D.P., Zhu Z., Schuchat A. Superspreading SARS events, Beijing, 2003. Emerg. Infect. Dis. 2004;10(2):256–260. doi: 10.3201/eid1002.030732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi P., Yu L., Fu Y.X., Huang J.F., Zhang K.Q., Zhang Y.P. Evolutionary implications of Avian Infectious Bronchitis Virus (AIBV) analysis. Cell Res. 2006;16(3):323–327. doi: 10.1038/sj.cr.7310041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z., Hu Z. A review of studies on animal reservoirs of the SARS coronavirus. Virus Res. 2008;133(1):74–87. doi: 10.1016/j.virusres.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih M.C., Peck K., Chan W.L., Chu Y.P., Chen J.C., Tsai C.H., Chang J.G. SARS-CoV infection was from at least two origins in the Taiwan area. Intervirology. 2005;48(2–3):124–132. doi: 10.1159/000081739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003;331(5):991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H.D., Tu C.C., Zhang G.W., Wang S.Y., Zheng K., Lei L.C., Chen Q.X., Gao Y.W., Zhou H.Q., Xiang H., Zheng H.J., Chern S.W., Cheng F., Pan C.M., Xuan H., Chen S.J., Luo H.M., Zhou D.H., Liu Y.F., He J.F., Qin P.Z., Li L.H., Ren Y.Q., Liang W.J., Yu Y.D., Anderson L., Wang M., Xu R.H., Wu X.W., Zheng H.Y., Chen J.D., Liang G., Gao Y., Liao M., Fang L., Jiang L.Y., Li H., Chen F., Di B., He L.J., Lin J.Y., Tong S., Kong X., Du L., Hao P., Tang H., Bernini A., Yu X.J., Spiga O., Guo Z.M., Pan H.Y., He W.Z., Manuguerra J.C., Fontanet A., Danchin A., Niccolai N., Li Y.X., Wu C.I., Zhao G.P. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl. Acad. Sci. U.S.A. 2005;102(7):2430–2435. doi: 10.1073/pnas.0409608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanhope M.J., Brown J.R., Amrine-Madsen H. Evidence from the evolutionary analysis of nucleotide sequences for a recombinant history of SARS-CoV. Infect. Genet. Evol. 2004;4(1):15–19. doi: 10.1016/j.meegid.2003.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavrinides J., Guttman D.S. Mosaic evolution of the severe acute respiratory syndrome coronavirus. J. Virol. 2004;78(1):76–82. doi: 10.1128/JVI.78.1.76-82.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf M.P., Pybus O.G. Genetic diversity and models of viral evolution for the hepatitis C virus. FEMS Microbiol. Lett. 2002;214(2):143–152. doi: 10.1111/j.1574-6968.2002.tb11338.x. [DOI] [PubMed] [Google Scholar]
- Tang J.W., Cheung J.L., Chu I.M., Ip M., Hui M., Peiris M., Chan P.K. Characterizing 56 complete SARS-CoV S-gene sequences from Hong Kong. J. Clin. Virol. 2007;38(1):19–26. doi: 10.1016/j.jcv.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X., Li G., Vasilakis N., Zhang Y., Shi Z., Zhong Y., Wang L.F., Zhang S. Differential stepwise evolution of SARS coronavirus functional proteins in different host species. BMC Evol. Biol. 2009;9(1):52. doi: 10.1186/1471-2148-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.C., Zhang J.X., Zhang S.Y., Wang P., Fan X.H., Li L.F., Li G., Dong B.Q., Liu W., Cheung C.L., Xu K.M., Song W.J., Vijaykrishna D., Poon L.L., Peiris J.S., Smith G.J., Chen H., Guan Y. Prevalence and genetic diversity of coronaviruses in bats from China. J. Virol. 2006;80(15):7481–7490. doi: 10.1128/JVI.00697-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson B., Cockram C. SARS: experience at Prince of Wales Hospital, Hong Kong. Lancet. 2003;361(9368):1486–1487. doi: 10.1016/S0140-6736(03)13218-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui S.K., Chim S.S., Lo Y.M. Coronavirus genomic-sequence variations and the epidemiology of the severe acute respiratory syndrome. N. Engl. J. Med. 2003;349(2):187–188. doi: 10.1056/NEJM200307103490216. [DOI] [PubMed] [Google Scholar]
- Vega V.B., Ruan Y., Liu J., Lee W.H., Wei C.L., Se-Thoe S.Y., Tang K.F., Zhang T., Kolatkar P.R., Ooi E.E., Ling A.E., Stanton L.W., Long P.M., Liu E.T. Mutational dynamics of the SARS coronavirus in cell culture and human populations isolated in 2003. BMC Infect. Dis. 2004;4:32. doi: 10.1186/1471-2334-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijaykrishna D., Smith G.J., Zhang J.X., Peiris J.S., Chen H., Guan Y. Evolutionary insights into the ecology of coronaviruses. J. Virol. 2007;81(8):4012–4020. doi: 10.1128/JVI.02605-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijgen L., Keyaerts E., Moes E., Thoelen I., Wollants E., Lemey P., Vandamme A.M., Van Ranst M. Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J. Virol. 2005;79(3):1595–1604. doi: 10.1128/JVI.79.3.1595-1604.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Yan M., Xu H., Liang W., Kan B., Zheng B., Chen H., Zheng H., Xu Y., Zhang E., Wang H., Ye J., Li G., Li M., Cui Z., Liu Y.F., Guo R.T., Liu X.N., Zhan L.H., Zhou D.H., Zhao A., Hai R., Yu D., Guan Y., Xu J. SARS-CoV infection in a restaurant from palm civet. Emerg. Infect. Dis. 2005;11(12):1860–1865. doi: 10.3201/eid1112.041293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.G., Zheng Z.H., Shang L., Li L.J., Cong L.M., Feng M.G., Luo Y., Cheng S.Y., Zhang Y.J., Ru M.G., Wang Z.X., Bao Q.Y. Molecular evolution and multilocus sequence typing of 145 strains of SARS-CoV. FEBS Lett. 2005;579(22):4928–4936. doi: 10.1016/j.febslet.2005.07.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO, 2004a. Summary of Probable SARS Cases With Onset of Illness from 1 November 2002 to 31 July 2003.
- WHO, 2004b. Update 4: Review of Probable and Laboratory-Confirmed SARS Cases in Southern China.
- Wong S.K., Li W., Moore M.J., Choe H., Farzan M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 2004;279(5):3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P.C., Lau S.K., Chu C.M., Chan K.H., Tsoi H.W., Huang Y., Wong B.H., Poon R.W., Cai J.J., Luk W.K., Poon L.L., Wong S.S., Guan Y., Peiris J.S., Yuen K.Y. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2005;79(2):884–895. doi: 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P.C., Lau S.K., Huang Y., Tsoi H.W., Chan K.H., Yuen K.Y. Phylogenetic and recombination analysis of coronavirus HKU1, a novel coronavirus from patients with pneumonia. Arch. Virol. 2005;150(11):2299–2311. doi: 10.1007/s00705-005-0573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P.C., Lau S.K., Lam C.S., Lai K.K., Huang Y., Lee P., Luk G.S., Dyrting K.C., Chan K.H., Yuen K.Y. Comparative analysis of complete genome sequences of three avian coronaviruses reveals a novel group 3c coronavirus. J. Virol. 2009;83(2):908–917. doi: 10.1128/JVI.01977-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P.C., Lau S.K., Li K.S., Poon R.W., Wong B.H., Tsoi H.W., Yip B.C., Huang Y., Chan K.H., Yuen K.Y. Molecular diversity of coronaviruses in bats. Virology. 2006;351(1):180–187. doi: 10.1016/j.virol.2006.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worobey M., Santiago M.L., Keele B.F., Ndjango J.B., Joy J.B., Labama B.L., Dhed A.B., Rambaut A., Sharp P.M., Shaw G.M., Hahn B.H. Origin of AIDS: contaminated polio vaccine theory refuted. Nature. 2004;428(6985):820. doi: 10.1038/428820a. [DOI] [PubMed] [Google Scholar]
- Wu D., Tu C., Xin C., Xuan H., Meng Q., Liu Y., Yu Y., Guan Y., Jiang Y., Yin X., Crameri G., Wang M., Li C., Liu S., Liao M., Feng L., Xiang H., Sun J., Chen J., Sun Y., Gu S., Liu N., Fu D., Eaton B.T., Wang L.F., Kong X. Civets are equally susceptible to experimental infection by two different severe acute respiratory syndrome coronavirus isolates. J. Virol. 2005;79(4):2620–2625. doi: 10.1128/JVI.79.4.2620-2625.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 2002;19(6):908–917. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]
- Yeh S.H., Wang H.Y., Tsai C.Y., Kao C.L., Yang J.Y., Liu H.W., Su I.J., Tsai S.F., Chen D.S., Chen P.J. Characterization of severe acute respiratory syndrome coronavirus genomes in Taiwan: molecular epidemiology and genome evolution. Proc. Natl. Acad. Sci. U.S.A. 2004;101(8):2542–2547. doi: 10.1073/pnas.0307904100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder A.D., Yang Z. Estimation of primate speciation dates using local molecular clocks. Mol. Biol. Evol. 2000;17(7):1081–1090. doi: 10.1093/oxfordjournals.molbev.a026389. [DOI] [PubMed] [Google Scholar]
- Zeng F., Chow K.Y., Leung F.C. Estimated timing of the last common ancestor of the SARS coronavirus. N. Engl. J. Med. 2003;349(25):2469–2470. doi: 10.1056/NEJM200312183492523. [DOI] [PubMed] [Google Scholar]
- Zeng F.Y., Chan C.W., Chan M.N., Chen J.D., Chow K.Y., Hon C.C., Hui K.H., Li J., Li V.Y., Wang C.Y., Wang P.Y., Guan Y., Zheng B., Poon L.L., Chan K.H., Yuen K.Y., Peiris J.S., Leung F.C. The complete genome sequence of severe acute respiratory syndrome coronavirus strain HKU-39849 (HK-39) Exp. Biol. Med. (Maywood) 2003;228(7):866–873. doi: 10.1177/15353702-0322807-13. [DOI] [PubMed] [Google Scholar]
- Zhang C.Y., Wei J.F., He S.H. Adaptive evolution of the spike gene of SARS coronavirus: changes in positively selected sites in different epidemic groups. BMC Microbiol. 2006;6:88. doi: 10.1186/1471-2180-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Nielsen R., Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005;22(12):2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- Zhang X.W., Yap Y.L., Danchin A. Testing the hypothesis of a recombinant origin of the SARS-associated coronavirus. Arch. Virol. 2005;150(1):1–20. doi: 10.1007/s00705-004-0413-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G.P. SARS molecular epidemiology: a Chinese fairy tale of controlling an emerging zoonotic disease in the genomics era. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2007;362(1482):1063–1081. doi: 10.1098/rstb.2007.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z., Li H., Wu X., Zhong Y., Zhang K., Zhang Y.P., Boerwinkle E., Fu Y.X. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol. Biol. 2004;4:21. doi: 10.1186/1471-2148-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong N.S., Zheng B.J., Li Y.M., Poon, Xie Z.H., Chan K.H., Li P.H., Tan S.Y., Chang Q., Xie J.P., Liu X.Q., Xu J., Li D.X., Yuen K.Y., Peiris, Guan Y. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People's Republic of China, in February, 2003. Lancet. 2003;362(9393):1353–1358. doi: 10.1016/S0140-6736(03)14630-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G., Chen H.W. Monophyletic relationship between severe acute respiratory syndrome coronavirus and group 2 coronaviruses. J. Infect. Dis. 2004;189(9):1676–1678. doi: 10.1086/382892. [DOI] [PMC free article] [PubMed] [Google Scholar]