

Abstract
The desire to rid the blood supply of pathogens of all types has led to the development of many technologies aimed at the same goal—eradication of the pathogen(s) without harming the blood cells or generating toxic chemical agents. This is a very ambitious goal, and one that has yet to be achieved. One approach is to shun the ‘one size fits all’ concept and to target pathogen-reduction agents at the Individual component types. This permits the development of technologies that might be compatible with, for example, plasma products but that would be cytocidal and thus incompatible with platelet concentrates or red blood cell units. The technologies to be discussed include solvent detergent and methylene blue treatments—designed to inactivate plasma components and derivatives; psoralens (S-59—amotosalen) designed to pathogen-reduce units of platelets; and two products aimed at red blood cells, S-303 (a Frale—frangible anchor-linker effector compound) and Inactine (a binary ethyleneimine). A final pathogen-reduction material that might actually allow one material to inactivate all three blood components—riboflavin (vitamin B2)—is also under development.
The sites of action of the amotosalen (S-59), the S-303 Frale, Inactine, and riboflavin are all localized in the nucleic acid part of the pathogen. Solvent detergent materials act by dissolving the plasma envelope, thus compromising the integrity of the pathogen membrane and rendering it non-infectious. By disrupting the pathogen's ability to replicate or survive, its infectivity is removed. The degree to which bacteria and viruses are affected by a particular pathogen-reducing technology relates to its Gram-positive or Gram-negative status, to the sporulation characteristics for bacteria, and the presence of lipid or protein envelopes for viruses.
Concerns related to photoproducts and other breakdown products of these technologies remain, and the toxicology of pathogen-reduction treatments is a major ongoing area of investigation. Clearly, regulatory agencies have a major role to play in the evaluation of these new technologies. This chapter will cover the several types of pathogen-reduction systems, mechanisms of action, the inactivation efficacy for specific types of pathogens, toxicology of the various systems and the published research and clinical trial data supporting their potential usefulness. Due to the nature of the field, pathogen reduction is a work in progress and this review should be considered as a snapshot in time rather than a clear picture of what the future will bring.
Key words: amotosalen, binary ethyleneimine, Frale, INTERCEPT blood system, methylene blue, pathogen reduction, PEN110, psoralen, riboflavin, S-303, S-59, solvent detergent, transfusion-associated graft versus host disease, transfusion-transmitted disease, Triton X-100, Uniplas®, vitamin B2
The problem of blood-borne disease transmission has been a concern since the earliest days of transfusion. Bacterial contamination was the impetus for the development of the closed system collection method by Walter and Murphy in 1952; and eventually led to established storage practices. In 1943, Paul Beeson described post-transfusion hepatitis; this was later recognized as being due to transmission of viral hepatitis A, B (HAV, HBV), and later non-A non-B (hepatitis C; HCV). In 1944, Cohn and co-workers pasteurized 25% albumin to remove viral hepatitis. As early as 1963, the connection between paid donors and disease transmission became evident and, 10 years later, National Blood Policy promulgated that blood donors be derived from a pool of voluntary donors.
Despite the risk of hepatitis, the number of transfusions per year continued to rise. The recognition of human immunodeficiency virus (HIV) as transmissible by transfusion in the 1980s resulted in a plateau of the number of transfusions per year, and this persisted through the mid-1990s.
The specter of acquired immunodeficiency syndrome (AIDS) induced widespread fear and led to many changes. Some of these changes included more judicious blood use, the use of autologous units when possible, a more rigorous donor interviewing process, improved arm preparation techniques, removal of the first aliquot (first aliquot diversion), improved serologic and nucleic acid testing of blood components, and—in some countries—the use of pathogen-reduction or eradication techniques.1, 2 Even though the blood supply in industrialized countries is now considered to be very safe, concerns regarding transmission of existing, as well as emerging, transfusion-related pathogens still exist. A number of less threatening, although still troublesome, pathogens are also known to be transmissible by blood but are currently not tested, mainly because of prevalence and—sometimes—cost.3 These include pathogens Babesia, Erlichia, parvovirus, cytomegalovirus (CMV), and enteroviruses.4 Some of these pathogens do not cause disease, or cause disease only in immune-compromised patients. The recent identification and outbreaks of severe acute respiratory syndrome (SARS) and West Nile Virus (WNV) have reinforced this fear and the need to develop new ways of removing or inactivating blood-borne pathogens.
Currently, the risks for viral infection from transfusion is now reported to be3, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16:
-
•
1:2 135 000 for HIV 1 and HIV 2
-
•
1:138 700 to 233 000 for HBV
-
•
1:1 935 000 for HCV
-
•
1:2 500 000 for human T lymphotropic virus 1 (HTLV 1) and HTLV 2 (after serologic and molecular testing are performed)
-
•
1:1 000 000 HAV
-
•
1:300 to 1:10 000 for parvovirus B19
-
•
1:50 for hepatitis G virus (single-strand RNA-enveloped virus)
-
•
1:250 for CMV in unfiltered blood
-
•
Prior to nucleic acid testing (NAT), the risk for WNV was 1:833; this risk is expected to decrease with NAT testing.
Other viruses known to be transmitted by transfusion, but with less or unknown clinical significance include: transfusion-transmitted virus (TTV) (a non-enveloped single-strand DNA virus) and SEN-V (a non-enveloped single-strand DNA virus involved in 83% of non-A—E hepatitis). Wide prevalence of these viruses has been demonstrated in donor populations.17 The risk for bacterial infection is 1:30 000 red blood cell (RBC) units or 1:1500–3000 for random-donor whole blood or apheresis platelet units. Two technologies are Food and Drug Administration (FDA) approved for bacterial screening of platelet units, although not all bacterial contamination is thought to be eliminated by such screening. Seroprevalence of the parasite Trypanosoma cruzi is estimated at 1:7500–33 000, and Babesia microti is estimated at levels at 1:100. Transmission levels vary depending on the donor population. Risks for leishmaniasis and toxoplasma are unknown.17 Although the overall risk is low, significant risk of transfusion-transmitted infection remains. Emerging pathogens and as yet unknown future pathogens for which no screening tests exist might contaminate blood components. However, a new tier of protection—pathogen-reduction (PR) technology—is being developed to further decrease risks.4 Thus, the development of pathogen-reduction and inactivation technologies have been avidly pursued over the last 10–15 years.4
Ideal qualities of pathogen-reduction agents
Pathogen reduction (PR) or pathogen inactivation (PI) is the use of a process that inactivates or removes a virus, bacteria, fungus, or protozoan pathogen from the product. Some criteria for these methods have been delineated by the FDA. The methods used should remove or inactivate pathogens, including emerging pathogens, without damaging the function or longevity of the blood product. In addition, any chemicals used and the resulting complexes must be demonstrated as non-toxic and non-immunogenic. Any risks of the altered products must be less than the risk of acquiring disease from unaltered products. An ideal PR agent does not currently exist.2, 18
Pathogen-reduction chemicals have multiple sites of actions. Cross-linking of lipids in cellular membranes and organelles are one type. PR techniques interacting with proteins can cause cellular signal transduction mistakes, respiratory pathway deficiencies, or structural abnormalities. Chemical binding to nucleic acids or intercalating nucleic acid chains can lead to errors in transcription, translation, or replication.2
Shortcomings of pathogen-reduction technologies
The PR process or technique could cause damage to the components, resulting in a decreased in vivo lifespan of red cells or platelets or a decrease in coagulation proteins in fresh frozen plasma (FFP). Toxicities remain a concern. Even after passing through phase III trials, toxicity might not be revealed until large-scale exposure is seen.2 Accordingly, phase IV post-marketing studies will be required when PR technology is FDA licensed.
Methods of evaluation
Evaluations of PR methods need to be performed early in product and process development. Models for in vitro testing of viruses, bacteria, and parasites also need to develop in a timely fashion. Ideally, at least a 6–10 log10 reduction in pathogen load is suggested to be an acceptable level of efficacy for the methodology. Peak viral load in the window period, however can be as high as 108–1014 units per milliliter. These high viral loads might prevent reduction of pathogen load to a level that does not transmit infection.2
Phase III trials demonstrating clinical efficiency can be clinically difficult. Many factors decrease phase III trial assessment feasibility: First, too large a study population might be necessary to see a statistical or clinical difference. Since the initiation of NAT assays, even larger cohorts are required to find additional benefits of PR above those achieved with testing alone because the risks of contamination with the current methods of testing are so small. Second, PR techniques are not applied to all blood products (i.e. cryoprecipitate) given to patients. Third, and epidemiologically, multiple transmission routes/vectors are present by which patients acquire infections making it difficult to prove the infection came from blood; therefore, reliance on phase IV or surveillance of use is required to evaluate for subtle risks and marginal benefits.2
Elimination of compounds that clearly damage labile blood components is necessary when evaluating PR procedures. Gross hemolysis (>1%), platelet dysfunction, and a >30% reduction in coagulation factor activity are problems to be avoided. Subtle damage is more difficult to assess. Radiolabeling cells for autologous transfusions and monitoring survival or recovery can be used to evaluate clinical efficacy. Clinically, bleeding time is inadequate to assess platelet function, and delivery of oxygen to tissues is inadequate to assess red cell function after treatments with this technique.2 The FDA currently favors a clinical assessment of bleeding using the WHO criteria.
The challenge of PR technologies is to continue or increase benefit as threat levels decrease. As techniques in donor selection, collection processes, screening test methodologies, and detection limits are improved, additional benefit from the use of PR techniques decreases. The risks to the recipients decrease as pathogen reduction increases, with development of less toxic and more efficient techniques. More benefit is apparent when new transfusion-transmissible pathogens emerge (SARS, Ebola virus). As PR technologies are developed, however, the likelihood of replacing pathogen-screening tests is not expected nor anticipated. Rather, such technologies should complement existing protocols.2
Numerous methods have been cultivated and are in various stages of development and use. Solvent detergent pathogen reduction of plasma is the most widely studied methodology and is currently in use across Europe. Other methods include photosensitizing chemicals that interact with pathogen DNA and/or RNA and cross-link following exposure to UVA or visible light, thus inactivating the nucleic acids. Alkylating agents and ethyleneimines are PR technologies that also cross-link nucleic acids but do not require light for activation.19 An interesting side benefit of these technologies is that white cells are also inactivated, thus potentially eliminating the risk of transfusion-associated graft-versus-host disease (TA-GVHD). This chapter will discuss the more recent developments and address those technologies either in use or likely to be used in the near future.
The solvent detergent process
Mechanism of action
Solvent detergent (SD) treatment disrupts the membranes of enveloped virus, bacteria, and eukaryotes. Infectivity is lost via this action. Many combinations of tri-(n-butyl)-phosphate (TNBP) or ethyl ether (organic solvents) with Tween 80, sodium cholate, and Triton X-100 (non-ionic detergents) have been used in the past. The SD combination most often used now is TNBP with Triton X-100. Because the chemical reaction is non-selective, the agents must be removed before the final product is transfused.20, 21
Molecular biology
The molecular structure of TNBP is shown in Figure 1 . This compound acts as an organic solvent and removes lipids during SD processing by extracting and sequestering them into a separate micellar (colloidal) phase.22
Figure 1.
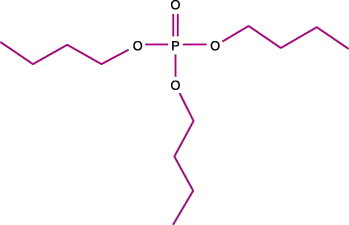
Molecular structure of tri-(n-butyl)-phosphate (TNBP). From Delipidation treatments for large scale protein purification process (from ref. 22).
The molecular structure of polyoxyethelene-p-t-octylphenol (Triton X-100) is shown in Figure 2 . This non-ionic detergent disrupts lipid bilayers for easier extraction and stabilizes TNBP.22
Figure 2.
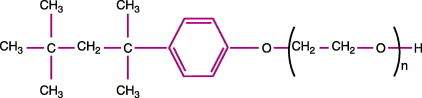
Partial molecular structure of Triton X-100 (from ref. 22).
History
SD viral inactivation technologies were first licensed for treatment of clotting factor concentrates in 1985.23, 24 Before 1984, a combination of ethyl ether (20%) and Tween 80 (1%) were found effective in inactivating HBV and NANBHV in factor VIII and IX concentrates with minimal loss in activity.25 In 1986, TNBP and 0.2% sodium cholate were shown to be effective at inactivating HBV, NANBHV, and HIV.
SD technology was first developed in 1987–1988. Use of SD treatment was shown to effect a 6 log10 viral load reduction in spiked samples. The SD-treated plasma coagulation factor profile was similar to untreated FFP. Removal of additives occurred at a 99.97% efficiency. During the period from 1991 to 1995, approximately 2×106 units of solvent detergent fresh frozen plasma (SD-FFP) were transfused without significant adverse sequelae. SD-FFP has been used in Germany, Switzerland, Austria, Belgium, France, the Netherlands, and Norway since the early 1990s. Initial tests from 1990 to 1992 used TNBP (1% organic solvent) and Triton X-100 (1% non-ionic detergent) to treat blood products. These additives were incubated with plasma for 4 hours at 30 °C. The pathogen reduction achieved was a >6 log10 reduction of HBV, >5 log10 reduction of HCV, and >6 log10 reduction of HIV. In France, 1% TNBP and 1% octoxynol 9 were evaluated as PR agents and were found to have similar coagulation factor activities compared to untreated FFP. In some countries in Europe, SD-FFP completely replaced conventional FFP. No systemic mutagenic or reproductive abnormalities have been noted.24, 25
The uses of SD-treated products from 1985 to 1997 have been wide and varied. Product use of this technology includes commercial factor VIII concentrates, factor IX concentrates, prothrombin complex, commercial factor VII, fibrinogen, protein C, factor XI, antithrombin III, fibrin glue, IM-Ig, IV-Ig, anti-D IgG, HBV-Ig, CMV-Ig, RSV-Ig, anti-tetanus, monoclonal antibody used in the treatment of malignancies, prothrombin complex, and plasma.24 ‘Double virus elimination’ procedures have been developed to decrease the risk of transmission of HAV. These procedures involve the use of monoclonal antibody affinity, chromatography, nano-filtration, heat, or ultraviolet light in combination with solvent detergent.24, 25
SD procedures, however, were discovered to be ineffective against non-enveloped viruses. In the US, development costs of SD plasma were never fully recovered because only 15% of the hospital market was using the product at peak usage. The reported occurrence of thrombosis and/or hemorrhage with use of SD-FFP in liver transplant patients markedly decreased usage in the US.26 Cost, risks, and administrative problems caused further declines in use of SD-FFP in the US. Fewer risks were noted in Europe, and use there continues. Indeed, SD technologies are the most widely used virucidal methods in the world. Before 1998, 35×106 doses of SD-treated products had been given. Even though SD plasma continues to be a licensed product in the US, the last lot was made in 2001 and is available only in Europe.24, 25, 26, 27, 28 Extensive studies for neonates, infants, and pregnant women are yet to be reported.
Treatment of labile blood products using SD technology
Many steps are required for the manufacture of SD-FFP. Up to approximately 300–1250 L (380–5000 donors) are processed simultaneously; the technique is not designed for single donor processing. The FFP is quick-thawed and passed through a 1-μm filter. This filtration, and an additional one, serves to remove cells, cell fragments, and membrane-associated viruses, assuring aggregate-free solutions. The plasma is then treated with 1% TNBP and 1% Triton X-100/polysorbate 80/octaxynol for 4 hours at 30 °C. A castor-oil extraction and phase separation are performed to remove TNBP, this is followed by a clear filtration. The Triton X-100 is removed by hydrophobic interaction chromatography. An additional sterile filtration at 0.2 μm is performed. The treated FFP is aseptically filled into 200 mL aliquots bags, sealed, fast frozen to ≤−60 °C, and stored at −30 °C. After quality control, the batch is released for transfusion. No impairment of viral inactivation is seen with protein concentrations to 90 mg/mL or lipid levels of 1064 mg/dL for triglycerides and 243 mg/dL cholesterol.23, 29, 30
Plasma protein derivatives
In preparation of plasma-derived products, two additional steps are performed. The first is prior to SD treatment, when a Cohn–Oncley fractionation procedure is utilized on the recovered plasma batch. Fractionation is not effective by itself as an adequate pathogen-reduction technique. Transmission of viral infections occurred after batched products were infused post-processing. Recipients of IV-Ig products in the 1990s had frequent incidences of infections following infusions. A minimal degree of protection is given by fractionation, although a decrease in viral particle titers does occur by about 2 log10. However, a second step with a pH 4 treatment at 21 °C is required before the final filtration. This step decreases aggregation and inactivates both enveloped and non-enveloped viruses by 2 log10.28, 31, 32, 33
Residual additives
Post-residual concentrations of TNBP and Triton X-100 present in the final product of several commercial manufacturers are as follows:
-
•
TNBP was undetectable in 73% of 130 batches; 27% of batches contained 0.5–1.7 μg/mL TNBP
-
•
Triton X-100 was undetectable in 91% of 130 batches; 9% of batches contained 1.0–1.6 μg/mL of the Octapharma product Octaplas®.
A study with the Vitex product VIPLAS-SD® revealed less than 2 ppm TNBP quantities, with one lot of 34 containing 2.1 ppm. Triton X-100 residual was ≤1 ppm with, two lots of 34 containing 1–2 ppm.24, 29
Efficacy of pathogen reduction
For the initial licensure of VIPLAS-SD®, viral inactivation rates were reported as vesicular stomatitis virus (VSV) >5.7 log10, Sindbis (SIN) virus ≥5.8 log10, HIV ≥6.0 log10, bovine viral diarrheal virus (BVDV) >6.0 log10, HBV ≥6.0 log10, and HCV >5.0 log10 reduction in active viruses. Non-enveloped viruses are less easily inactivated and show much lower log reductions. HAV had only a ≅1.22 log10 reduction in viral particles, leading to an outbreak in the 1990s with a factor VIII product. Seroconversions have also been noted to parvovirus B19 (PVB19).29, 34, 35
A benefit of the broad-spectrum activity of SD treatment is that enveloped pathogens not picked up on routine screening assays will be inactivated by this technique. Examples of these include HIV type O and variants of HBV.24, 29
Antibodies to non-enveloped viruses in the final products might protect recipients from these pathogens. Protection from acquiring HAV infection can occur as the result of the presence of antibody in the final SD product. Concentrations of anti-HAV antibodies in pooled SD-FFP are 30 times the prophylactic dose. These concentrations of antibody should protect recipients from HAV infection. SD-FFP has antibody concentrations of anti-PVB19 that are seen in IV-Ig used to treat chronic infections. These antibody concentrations should similarly protect recipients from acquiring parvovirus B19. Thus anti-HAV titers of 0.8 IU/mL and PVB19 titers of 8 IU/mL are thought to provide protection against their respective organisms. Screening with NAT is another way to decrease transfusion transmission because copy numbers as low as 10 copies/mL of pathogens can be detected.24, 29
Purity of product
The potency goal of PR-treated plasma as stated by the FDA is to have 70% activity of coagulation factors and 1.7 mg/mL of fibrinogen. SD-FFP meets this goal. In 34 lots tested, coagulation activity ranged from 0.83 to 1.08 U/mL for factors V, VII, X, XI, and XII (83–103%) with 2.67 mg/mL of fibrinogen.34
Effects of SD technology on blood proteins
Even though SD-FFP has activity >0.7 U/mL of most clotting factors, approximately 15–20% is lost for individual factors compared with untreated FFP. Some feel that this loss requires an increased number of transfusions of SD-FFP to achieve the same clinical efficacy as standard FFP. This could increase the risk of non-infectious secondary to increased transfusion requirements.27, 36
Protein S is decreased by 35–50% in SD-FFP, plasmin inhibitors are decreased by 76%, and alpha2-antiplasmin is decreased by 50%. These decreases in anticoagulation factors can lead to increased clotting risks in patients deficient in these factors.29, 37 Alpha2-antitrypsin is absent in SD-FFP. The clinical significance of the lack of this enzyme on acutely ill patients is unknown. Other therapies are available for patients with congenital deficiencies of this enzyme.27, 36
In the year 2000, in the US, six patients undergoing orthotopic liver transplants for various underlying causes of end-stage liver disease died of thrombotic or hemorrhagic events. It is not known if this occurrence was causative or coincidental. Shortly after this episode, the use of SD-FFP (PLAS+SD) fell into disfavor in the US. The last lots were made in 2001. Studies in the UK in 1999 showed equal efficacy of SD-FFP and FFP. In Germany (2001), open-heart surgeries using SD-FFP and FFP were compared and demonstrated equivalent improvements as relates to hemostasis and fibrinolysis.16, 26, 28, 38, 39, 40
Effects on viral pathogens
Vaccinia has been shown to be relatively resistant to SD treatment.20 Even though this is an enveloped virus, intracellular non-envelope forms exist and are infectious. The active intracellular particles can potentially lead to infection. This study implies that normally enveloped viruses might still be infectious and resistant to SD treatment while in an intracellular environment.20
SD techniques do not inactivate non-enveloped viruses. Each year between 1991 and 1998, outbreaks of HAV have occurred worldwide with factors VIII and IX concentrates.25, 38, 41 PVB19 is more difficult to evaluate secondary to increased incidence in the general population. Antibodies in pools of processed plasma might protect recipients from transmission. Nanofiltration can be used to remove non-enveloped viruses but a reduction in factor VII and in von Willebrand factor (vWF) is also seen. If higher titers of antibodies to these viruses are present in the final blood product, passive immunity might be acquired. SD technology cannot be used on cellular products because the plasma membranes will disintegrate.25, 31, 39, 41
Toxicity associated with SD technology
When compounds interact with nucleic acids, mutagenicity becomes a concern. If present, mutagenic compounds can lead to genotoxicity, carcinogenicity, or toxicity to the reproductive system. Compounds interacting with membrane lipids or protein can adversely affect cellular function. If such reactions are occurring, the processing must ensure the compounds are not present in the final product administered to the patient.2
The risk associated with SD treatment also includes the toxicity of the chemical additives per se. When evaluating the toxicity of Triton X-100, the lethal dose at which 50% of animals are killed (LD50) was used. Triton X-100 in animal studies has an LD50 of 1.2–1.8 g/kg via the oral route and LD50 of 108–150 mg/kg for the intraperitoneal or intravenous route. The lowest toxicity was found to be 33.7 mg/kg in mice and 15.7 mg/kg in rats, with a LD50 of 605–660 mg/kg in mice and 610–615 mg/kg in rats. No mutagenicity potential, embryo toxic, or teratogenic potential was demonstrated. A TTP patient could theoretically receive 63 L of PLAS/SD or Triton X-100 of 2.7 mg/kg. This is well below the toxicity levels. Because the chemical reaction is non-selective, the agents must be removed before the final product is transfused.20, 21 With present processing techniques, there often is no detectable TNBP or Triton X-100 in the final blood product.34, 42, 43
Benefits
Many of the benefits of SD treatment lie in the fact that in addition to pathogen reduction the process is unlikely to degrade the plasma proteins. Coagulation factor loss is not clinically significant; a loss of 5–20% activity is seen in multiple studies. Thirty percent activity is required to maintain normal hemostasis. Enveloped viruses are inactivated by this process. No transmission of such viruses was seen after the transfusion of 17 000 000 units between 1980 and 1993. In a 1995 study in the US, 3% of patients had hives, abdominal pain, nausea, vomiting, chills, headache, wheezing, fever, and hypertension, with an average recovery of 90% of clotting factors in this study population.25, 27, 28
Present research
Future studies are necessary to evaluate safety for use of these products in neonates, infants, and pregnant women. A study to evaluate the statistical/clinical relevance in the decrease of infection transmission risk would require hundreds of thousands in a cohort group. To be undertaken, this would require a multicenter trial over several years.2 SD-FFP is still FDA licensed. However, at this time, SD-FFP is not used in the US.25, 44
Costs
Producing and evaluating SD-FFP involves filtering, tracking, extracting, and monitoring residual pathogen or chemical agents. The additional steps and additives increase the cost of production to three to four times that of untreated FFP. Previous cost-effective analyses have suggested that SD-FFP costs go up to US$9 743 000 per quality of life year (QALY) gained. Some have argued that, taking into account the decreased incidence of transfusion-related acute lung injury (TRALI) with SD-FFP, the cost would be decreased to US$40 855–139 465 per QALY saved. The incidence of TRALI with SD-FFP is negligent given the dilutional affect of pooled plasma.45 In other aspects of medicine, an accepted procedure typically costs less than $30 000 per QALY gained, i.e. cholesterol reduction ($13 300) or coronary artery bypass ($26 117). In other aspects of transfusion medicine, however, high-cost safety measures have been adopted to reduce the risk of transfusion-transmitted diseases. Costs for these programs range from $235,000 to $2.3 million per QALY. Such procedures include autologous donations, as well as p24 antigen assay for HIV. The costs should, therefore, not be the only determining factor considered in rejecting or accepting SD technology.23, 30, 39, 45, 46, 47
Uniplas®
Another product currently available is Uniplas®, a solvent detergent-treated universal plasma. Anti-A and anti-B iso-agglutinin antibodies have been removed from this product. Immune complexes of anti-A and anti-B antibodies with soluble A and B antigens are hydrophobic and removed by SD treatment. Total protein and coagulation factors in Uniplas® are similar to SD-FFP. In a published study, no patients seroconverted to HIV, HTLV, HBC, CMV, HAV, or parvovirus B19 seropositivity.48 Two studies showed equivalent efficacy of Uniplas® with Octiplas® in open-heart surgeries48, 49, although Uniplas® has the additional benefit of being compatible with all blood groups. Further studies are recommended to determine if Uniplas® is otherwise equivalent to standard SD.48, 49
Phenothiazinium dyes: methylene blue
Methylene blue (MB) was first synthesized by Caro in 1876; in 1883 Bernsthen synthesized the parent ring system. Currently, this compound has been administered orally as an antiseptic, disinfectant, and an antidote for nitrate poisoning. MB has been used in the treatment of methemoglobinemia, in locating surgical sites, and for validation of properly following medication prescriptions. Long-term use in humans has not been associated with adverse effects in treating methemoglobin toxicities. In 1891, MB was instrumental in the cure of malaria in two patients. In 1928, it was demonstrated that MB could inactivate HSV vaccinia when exposed to light. Nucleic acids were found to be the target of MB in 1956, and oxygen was found to be necessary to this reaction in photodynamic activity reaction. Since 1962, MB has been known to photochemically degrade DNA.50, 51, 52, 53, 54 The use of MB for viral inactivation of plasma was first described in 1991.23
Ideally, a photochemical dye first needs to penetrate membranes (whether pathogen or plasma membrane) to inactivate intra- as well as extracellular pathogens. Second, by adhering to predominately or exclusively nucleic acid compounds, there should be little damage to red blood cells. Third, since hemoglobin absorbs light at wavelengths of less than 600 nm, the ideal photocompound should absorb light at other wavelengths. Last, the unbound dye should not cause excessive damage to RBC. The compound needs to be active only in the presence of light and to be inactive while blood components are in storage.55
Phenothiazine dyes are more favorable because unwanted cell damage can be limited by ensuring that no light exposure occurs after the initial treatment is complete. By contrast, for SD techniques the reaction ceases only when the PR compounds are removed. In the last 100 years, both bacterial and tropical diseases (i.e. malaria) have been treated with MB. In the past 50 years, as specific antibiotics to these pathogens were developed, use of MB has decreased. A renaissance of interest in pathogen reduction has developed as Plasmodium falciparum has demonstrated an increasing incidence of chloroquine resistance, drug-resistant bacteria (e.g. MRSA, VRE, multi-drug resistant tuberculosis) have become more common, and opportunistic organisms in HIV patients are more widespread.50, 55
Mechanism of action
MB binds DNA in two ways, depending on the ionic strength and concentration of Mg2+: MB can bind the outside of the DNA helix or intercalate between the rungs in the helix. Its precise mode of action is guanine-specific cleavage. In solutions lacking an oxygen environment, a direct electron transfer is probably responsible for strand breakage via direct cleavage of phosphodiester bonds; in an oxygen-rich solution, reactive oxygen species (iO2, OH, superoxides) are generated. MB has a peak absorption at 620–670 nm. Exposure to light of this wavelength produces a type I (redox) reaction or a type II (photodynamic, photo-oxidative) interaction. MB also binds to viral core proteins and has been shown to cause damage to bacteria (Proteus mirabilis) and RBC membrane proteins. Bacteria and hemoglobin can reduce MB and convert it to a leukomethylene form, which is neither a photosensitive nor an intercalater (maximum absorbance 340 nm). MB technology has been shown to inactivate enveloped and some non-enveloped viruses. However, given its hydrophobic nature at concentrations of 5 mM, this compound cannot penetrate plasma membranes and/or inactivate intracellular viruses.26, 39, 51, 52, 53, 54, 56, 57, 58, 59
Molecular biology
The formal chemical name for MB is3, 7 bis(dimethylamino)-phenothiazine-5-ium chloride; it is classed as a thiazine dye.60, 61 The structural formula for MB is shown in Figure 3 .50, 53, 62 The leukobase inactive form is shown in Figure 4 .53 MB is not very lipophilic. The polarity/charge makes the compound more hydrophilic. For this reason, MB does not easily penetrate membranes.53
Figure 3.
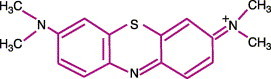
Molecular structure methylene blue (MB) (from refs 53, 56, 66).
Figure 4.
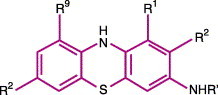
Generic molecular structure of the leukobase form of phenothiazinium dyes (from ref. 56).
A study of MB binding to organic compounds showed it attaching preferentially to DNA and negatively-charged lipids. Albumin and electrically neutral lipids had <3% binding with DNA, and negatively-charged lipids had 65 and 20% binding, respectively. Poly-guanine in the same solution accounted for the remaining 15% binding in that particular study.63
Present use in transfusion
After initial research in 1955 at Walter Reed Medical Center, MB-treated plasma became routine for use in 1992. A few countries in Europe still use this decontamination method. Between 1991 and 2000, 2 million units were transfused with no untoward events. MB has been used both as a marker in surgery and to reduce methemoglobin. As much as 50 mg t.i.d., for ifosfamide-induced encephalopathy, and 2 mg/kg, in septic shock, has been given to patients. Although MB has been used in much higher concentrations, the Paul Ehrlich Institute has refused to license MB-treated plasma because of toxicity concerns.64 Adverse reactions associated with MB include a burning sensation in the mouth, nausea, vomiting, diarrhea, and gastritis. Large doses can cause abdominal and chest pain, headache, profuse sweating, mental confusion, painful urination, and methemoglobinemia (see the Material Safety Data Sheets, refs. 60, 61). In life-threatening clinical situations, however, these adverse events do not outweigh the life-saving benefit of MB. Two different proprietary methods for processing FFP with MB—Pathinact MB® (Baxter) and Macotronic® (Macopharma)—are currently used in Europe.50, 53, 54, 55, 57, 65
Procedure
In general, the MB treatment of plasma utilizes a 1 μm concentration of MB followed by exposure to red light (600–700 nm) with a fluence rate of 10 mW/cm2 for 600 s. Freezing and thawing of the unit can liberate intracellular organisms. One study processed three units at a time by passing the contents of each unit through a 0.65-μm membrane filtration system.55 After filtration, a dry tab of MB (80 μg) was placed in the line; the MB dissolved during filtration. Approximately, 180 J/cm2 in less than 20 minutes was applied with 590 nm peak λ of light. The concentration of 1 μm MB in the unit was computer controlled.55 Other procedures process MB with 5 μm concentration and a fluence of 11.3 J/cm2. The product is then stored at −30 °C. When needed, each unit is then thawed and transfused in the same manner as untreated FFP.66, 67
Efficacy
The efficacy of MB reduction is increased in the presence of oxygen. This process has demonstrated an ability to inactivate enveloped viruses with DNA and RNA genomes (HIV 6.32 log10, VSV 6 log10). Some non-enveloped viruses with large capsid pores are inactivated (i.e. WNV 5.7 log10 reduction). However, non-encapsulated viruses with tightly interdigitated capsid proteins (e.g. encephalomyocarditis virus) are not inactivated. Little bacterial inactivation is observed under virucidal phototherapeutic conditions. MB is more effective against Gram-positive than Gram-negative organisms; Candida albicans and Trypanosoma brucei are also inactivated.
MB has difficulty in penetrating plasma membranes and, as such, intracellular organisms are not inactivated. The viability of red cells when MB is used with an RBC product is decreased. After 42 days of storage in MB, 0.8% of the red blood cells lyse. In addition, MB-treated RBC membranes show dramatically increased ion leakage. MB binds to IgG and albumin (serum proteins); this might further alter RBC membranes by attaching to these membrane proteins. Despite concern over adverse events, MB has a long history of use with minimal toxicity.11, 25, 52, 53, 56
Problems
Secondary to binding to proteins, 10–30% of coagulation factors and 20–24% of fibrinogen are inactivated after MB treatment of plasma. This loss of activity is thought to be secondary to the oxidation of histidine residues and other amino acids. MB also binds to the alpha subunits of fibrinogen resulting in lower platelet-receptor binding results.11, 25, 53, 54, 55, 57, 65, 66, 68, 69
MB does not inactivate intracellular pathogens or white blood cells. MB treatment of blood products cannot reduce the incidence TA-GVHD. In RBCs, up to one-half of the MB added is bound to membrane proteins or located within the RBCs. The compound within the RBCs is reduced to the leukobase form and can neither intercalate nor photosensitize. Binding of MB to RBC surface proteins also increases ion permeability of the membrane. MB also inactivates glutathione reductase and decreases red blood cells ability to handle oxidation toxicity. These effects decrease the life expectancy of red cells.11, 55, 66
Toxicity in animal studies reveals the LD50 oral route as being 1180 mg/kg and the intraperitoneal dose 180 mg/kg for rats. In mice studies, the oral LD50 was shown to be 3500 mg/kg; this indicates a low toxicity for this compound. MB is not reported to be carcinogenic, however, its toxocologic properties have not been fully investigated. At the concentrations used in clinical practice, toxicity appears to be minimal. This may relate to the final concentration in the bag at the micromolar level and not on the molar level. More research on the toxological properties of MB is needed.53, 60, 61
Amotosalen
Compounds known as photosensitizers were first used in around 1550bc in ancient Egypt and India to treat depigmented lesions. The chemicals were obtained from parsley, parsnip, and St John's wort, applied to the skin and exposed to sunlight, which caused skin damage, re-epithelialization, and re-pigmentation of the skin. In 1897, Oscar Raab observed that paramecia placed in acrydine dye died on exposure to light but were unaffected if no light exposure occurred. Subsequently, these compounds were identified as furocoumarins, a group that includes psoralens. In the modern era, furocoumarins have been applied to biologic systems to treat various skin tumors and conditions, and to deactivate microorganisms, as well as being employed as a research tool to increase our knowledge of nucleic acids.70
Amotosalen is a synthetic psoralen specifically engineered to inactivate pathogens in blood products. Psoralens are naturally occurring heterocyclic compounds and are common in numerous plants, such as those mentioned above. With the addition of amine side chains, the compound becomes water soluble, which increases it's affinity for nucleic acids. Specific characteristics necessary for the psoralens to be useful in inactivating microorganisms in blood products include: the chemical purity, activation only upon light exposure, preferentially retained in the target organism(s), and rapidly excretion or elimation with minimal toxicity. In addition, the chemical has to have a high quantum yield for the photochemical event.71, 72
Of the hundreds of psoralens evaluated, amotosalen was chosen because of its excellent activity against pathogens and minimal impact on the blood products, and because it essentially fulfilled the above criteria.
Mechanism of action
Amotosalen, also known as S-59, can be used to inactivate pathogens in plasma and platelets on exposure to UVA light at wavelengths of 320–400 nm. It cannot be used in red cell concentrates because hemoglobin absorbs the UVA light. Amotosalen works via a three-step chemical reaction: The chemical initially intercalates between strands of DNA or RNA. On exposure to UVA light, the molecule binds the olefinic moieties of the pyrimidine bases thymidine, cytidine, and uridine and forms a monoadduct. The monoadduction causes the helix to unwind slightly and, with continued exposure to UVA light, allows a second adduction to form, which results in cross-linkage. The nucleic acid then becomes non-functional because the cross-linkage is permanent and replication can no longer occur. The same process occurs in single-stranded nucleic acid structures that form hairpin turns and loops that allow for close proximity of pyrimidine bases, thus making them available for cross-linkage.72, 73 Thus, pathogens and white blood cells contained in the blood products are inactivated (Figure 5 and Table 1 ).
Figure 5.
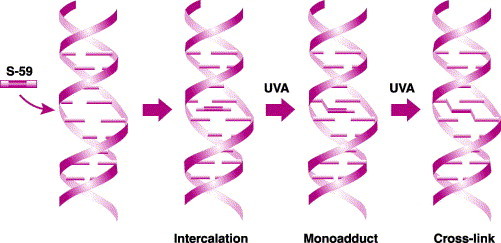
Mechanism of action of pathogen inactivation (PI). Amotosalen (S-59) is a synthetic psoralen that inhibits nucleic acid replication through UVA-light-mediated covalent addition to nucleic acids. Left: the molecule penetrates cells, viruses, bacteria, or other pathogens and seeks out DNA or RNA. 2nd left: amotosalen intercalates between the base pairs. 2nd right: once illuminated by UVA light, amotosalen forms monoadducts between pyrimidine bases. Right: another photon of light enables the molecule to form cross-links (diadducts) between DNA or RNA strands (from ref. 73).
Table 1.
Reported inactivation of pathogens in platelet concentrates after photochemical treatment with amotosalen with UVA light.
| Pathogen log-reduction in organisms | |
|---|---|
| Non-enveloped viruses | |
| Bluetongue virus | 6.1–6.4 |
| Parvovirus B19a | 4.0–4.9 |
| Enveloped viruses | |
| HIV (cell-free) | 6.2 |
| HIV (cell-associated) | 6.1 |
| CMV | 5.9 |
| Hepatitis B virus | 5.5 |
| Hepatitis C virus | 4.5 |
| Duck hepatitis B virus | 6.2 |
| Bovine viral diarrhea virus | 6.0 |
| Human T-cell leukemia virus type I/II | 4.7/5.1 |
| West Nile virus | 6.0 |
| Gram-negative bacteria | |
| Escherichia coli | 6.4 |
| Serratia marcescens | 6.7 |
| Klebsiella pneumoniae | 5.6 |
| Pseudomonas aeruginosa | 4.5 |
| Salmonella choleraesuis | 6.2 |
| Yersinia enterocolitica | 5.9 |
| Enterobacter cloacae | 5.9 |
| Gram-positive anerobic bacteria | |
| Lactobacillus species | 6.9 |
| Non-enveloped viruses | |
| Propionibacterium acnes | 6.7 |
| Clostridium perfringens | 7.0 |
| Bifidobacterium adolescentis | 6.5 |
| Protozoa | |
| Trypanosoma cruzi | 5.3 |
| Plasmodium falciparum | 7.0 |
| Leishmania mexicana | 5.2 |
Preliminary data: inactivation was performed in 35% B19-infected plasma and 65% PAS III (platelet additive solution III) in the absence of platelets. Studies included a 15- or 30-minute rest between addition of amotosalen and UVA treatment. Adapted from Ref. 79.
The Cerus Corporation that developed S-59 calls this Helinx technology. Combining Helinx technology with a compound adsorption device (CAD) results in a system known as the INTERCEPT Blood System (Figure 6 ). The CAD is composed of activated charcoal in a diethyl benzene matrix placed in a porous container. The device is located in the blood product storage bag where it can scavenge amotosalen and its photoproducts. An additional step is necessary for platelets, as the system works best when the plasma concentration in the platelet bag is lowered to 35% by adding platelet additive solution (PAS III).73 Amotosalen is added to plasma or platelets at a concentration of 150 μM/mL (50 μg/mL) and, while agitating, the bag is illuminated with 3 J/cm3 of UVA light. The plasma and/or platelets are subsequently exposed to the CAD, which adsorbs the excess amotosalen and its photoproducts. The product is then ready for storage and transfusion.74
Figure 6.
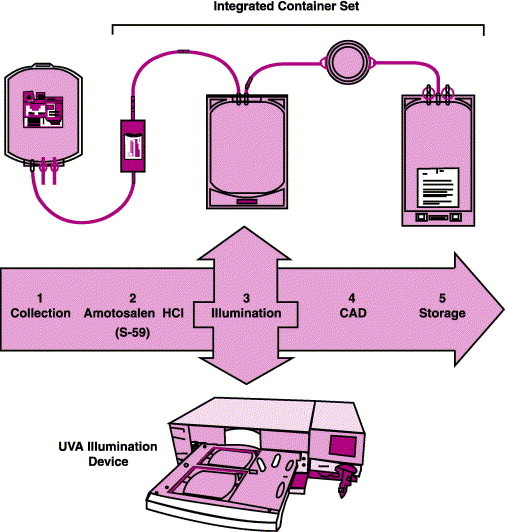
The INTERCEPT Blood System for plasma. Collected plasma is mixed with amotosalen and the mixture is placed in an ultraviolet A (UVA) illumination device. The contents are then passed through a Compound Adsorption Device (CAD) for reduction of amotosalen and free photoproducts. After CAD treatment, the plasma is placed in a final storage container until transfusion (from ref. 72).
Toxicology
Toxicology studies for amotosalen involved evaluation of the chemical itself and the photoproducts present in plasma after exposure to UVA light. These studies, reported by Ciaravino et al72, 73, included evaluations for reproductive, geno-, carcino-, and photo-toxicity, as well as acute and repeated dosing toxicity.
Photochemically treated plasma did not result in any reproductive changes. Genotoxicity was not identified in the in vitro assays (the bacterial mutagenicity assay, chromosome aberration assay, and mouse lymphoma assay) or the following in vivo assays: mouse micronucleus assay and unscheduled DNA synthesis assay, when using photochemically treated plasma without the CAD. However, when photochemically processing a single unit 25 times without a CAD, two of six assays were positive for genotoxicity. The authors attributed this to the abundant excess of amotosalen present in high concentrations, which was much higher than in any expected clinical dose. That part of the study evaluating carcinogenesis used heterozygous p53 transgenic mice.72 Unlike the homozygous p53 transgenic mice, 50% of which develop tumors by 4–5 months of age, 50% of the heterozygous mice do not develop tumors until 18 months. The heterozygous mice were exposed to high doses of amotosalen and its photoproducts intravenously over 6 months. There was no increase in tumors in the heterozygous p53 transgenic mice.72 Phototoxicity was not demonstrated, despite residual amotosalen levels of (10 μg/kg). In addition, studies to assess vein irritation and neoantigenicity were negative.
Amotosalen alone produced CNS effects at concentrations 6250-fold greater than the estimated clinical exposure of 4 μg/kg. Electrocardiogram changes were identified at doses 10 000-fold higher, and phototoxicity occurred at doses 250-fold higher than the estimated clinical dose. Genotoxicity occurred in three of the six assays, as expected in in vitro assays, due to the intercalating behavior of the amotosalen.73, 74 Based on these studies, amotosalen appears to be safe when used in blood components in the manner and dosages intended.
Effect on platelets
In addition to toxicology studies, determination of any adverse effects of amotosalen on the blood products, both in vitro and in vivo, is required. Numerous studies have been performed on platelet concentrates using various endpoints to determine the viability of the photochemically treated platelets.
In vitro studies have found that after processing through the INTERCEPT Blood System, there is a not insignificant drop—in the range of 8–10%—in the number of platelets present in the bag. This was due both to the increased amount of platelet sampling necessary to meet the demands of the studies and to the requirements of processing the platelets through the photochemical treatment, which involves manipulation of platelets through three different bags, and agitation needed to mix the amotosalen with platelets and to expose amotosalen and photoproducts to the compound absorption device. In the Moog et al study, single-donor platelet concentrates contained mean platelet counts of 3.06±0.27×1011.74 On completion of the photochemical treatment (PCT), the mean platelet count was 2.62±0.27×1011, representing a loss of 9.7%.74 Also in the Moog study, significant decreases in pH were identified after PCT, with a mean drop to 6.98±0.08. Compared to the control, however, this pH level was well above the required 6.8 limit required in Europe, and the pH remained above 6.8 until the end of the study, at 5 days of storage.74
Glucose decreased from 354±34 to 119±32 mg/dL after separation from plasma and following the addition of Intersol (PAS III) and remained essentially the same post-PCT at 122±10 mg/dL. As expected during storage, glucose continued to decrease and lactate dehydrogenase (LDH) to rise. A significant increase in LDH was evident after PCT, indicative of platelet lysis and activation. Subsequently, however, the LDH level remained stable throughout storage. The HSR decreased both after preparation and after PCT; however, the platelets appeared to achieve normal hemostasis upon transfusion.74
Parameters for apoptosis, function, and activation of platelets examined by Jansen et al75 included P-selectin, aggregation studies, hypotonic shock response, annexin V, and caspase 3 activity. PCT did not cause elevation of P-selectin, although the levels did rise over time during storage similar to control. Changes in HSR were not significant in this study compared with controls and caspase 3 activity and annexin V expression were unchanged, suggesting that apoptosis was not stimulated.75 Decreases in platelet aggregation were not significant after PCT. However, after storage, compared to controls, aggregation studies using collagen and thrombin were decreased, but not in those using ADP.75 These changes vary in extent from study to study and, to some extent, by technique. The results of a French validation study are consistent with the findings above.76 This study also investigated longer storage times, finding that the in vitro platelet parameters remained within satisfactory ranges for up to 8 days. The authors suggest that it might be possible to extend the expiration date of PCT platelets to 7 days.76
The changes that are induced by the PCT process might not always be significantly different from controls. In vitro platelet parameters remain within the guidelines set by various regulatory agencies and appear to be adequate for clinical use.
One in vivo study looked at PCT platelet survival following indium-111 radiolabeling of the treated autologous platelets. The normal span of platelet survival is broad, ranging from 5 to 7.3 days with 33–66% recovery. Recovery in PCT–CAD platelets was 43%±8.7 and lifespan was 4.8±1.3 days. Although within the lower range of normal and significantly lower than controls, the findings are consistent with those of the other studies. In addition, the authors found that, in in vitro studies, the label dissociation was higher in PCT platelets than in control platelets.77
Other in vivo studies have yielded similar data, with results showing small changes that might or might not be significant compared to controls, although the performance was still within regulatory guidelines and medically acceptable ranges. Endpoints in these studies included post-platelet counts at various time periods, number of transfusions required over a given time period, number of adverse reactions, platelet refractoriness, and bleeding episodes. Most studies also looked for possible antibodies to amotosalen neoantigens. Two large in vivo studies from the euroSPRITE (S-59 Platelet Recovery In Thrombocytopenia in Europe) and SPRINT (S-59 Platelet Recovery In Thrombocytopenia in the USA) trials have reported similar results.78, 79
The SPRINT trial was a nationwide study that enrolled 645 thrombocytopenic patients who were randomized to PCT–CAD-treated platelets or standard platelets. The platelets were single donor units collected by apheresis. The euroSPRITE trial was designed similarly. Both studies found that CCIs (corrected count increments) were lower in patients treated with PCT platelets. This finding in both studies is explained by the lower platelet number in the PCT units. In the SPRINT trial, however, after correcting for the lower dose per unit infused, investigators found no significant difference between PCT–CAD platelet and standard platelet units. For the same reasons, patients treated with PCT platelet units required more transfusions over time. Bleeding and refractoriness were essentially the same for controls and patients treated with PCT units in both trials. In the SPRINT trial, transfusion reactions were fewer in patients treated with PCT–CAD platelets (3 versus 4.4%, p=0.02). No antibodies to any amotosalen neoantigens were identified. Therefore, the PCT–CAD treated platelets were considered to be functionally as safe and effective as control platelets for patients.79, 80
In summary, the platelet studies as a whole demonstrated that the PCT–CAD platelets appear to be an acceptable alternative to untreated platelets given the added safety measure from PR process. Due to the loss of a fraction of the platelets during processing, the number of platelet transfusions required for each patient may be slightly higher.
Fresh frozen plasma
The function of FFP after PCT–CAD has also been validated in phase III trials in Norway and Germany.80 To support large-scale processing, FFP was pooled to a maximum quantity of 650 mL, allowing up to three units of FFP to be produced per PCT–CAD process. Assays of factors II, V, VII, VIII, IX, X, XI, XIII, proteins C and S, anti-thrombin III, alpha2-antiplasmin, and fibrinogen were assayed in the manufacturers own laboratory. Fibrinogen and factor VIII levels were maintained at 78–79% of pre-PCT–CAD levels. Factors II, V, VII, IX, X, XI, XIII, proteins C and S, anti-thrombin III, and alpha2 antiplasmin were maintained at 85–95% of pre-PCT–CAD levels. Thus, the quantity of anticoagulant and procoagulant factors declined minimally. This recalls the decline seen in PCT platelet numbers. However, clinical trials have demonstrated that PCT–CAD-treated plasma factors function adequately in vivo in patients with acquired and congenital coagulopathies and TTP, as well as those requiring warfarin reversal.81 Hambleton et al82 studied healthy volunteers who donated apheresis plasma. Half of the group was treated with PCT–CAD and the other half was processed in the usual fashion. Subsequently, subjects took warfarin for 4 days until factor VII levels were lowered to a mean concentration of 0.33 IU/mL. They were then infused with 1 L of either PCT-treated or untreated plasma. Two weeks later, the process was repeated and the subject received the other type of FFP. Mean peak increments of 0.10–0.12 IU/mL of factor VII were identified for both PCT–CAD-treated and standard FFP. The results reveal that, despite the mild decreased level of factors in PCT-treated FFP, the factor VII level increased to the same level in patients with either PCT-treated or untreated plasma. In addition, the study measured the PT to determine coagulation factor function. The change in PT levels closely reflected the drop in factor VII levels in the first 8 hours and decreased similarly in both control and test patients. At 24 hours after FFP infusions, however, the PT rebounded to above pre-infusion levels, more so in the PCT–CAD-treated FFP patients than in the patients treated with standard FFP.82
Another study reported in an abstract reviewed the effect of PCT–CAD-treated FFP on patients with an acquired coagulopathy due to liver disease requiring treatment with FFP. Several factor levels were evaluated but only the PT and PTT were reported. No adverse events were described and the PT and PTT decreased similarly both in controls and in treated patients. Active bleeding stopped in one patient given PCT–CAD-treated FFP.83 Von Willebrand factor was studied separately in another study, which again was reported in an abstract. Similarly, there was no statistical difference between PCT–CAD-treated plasma and conventional FFP.84 A more recent abstract reported by Mintz et al examined the efficacy of PCT–CAD plasma in TTP patients.85 The mean time to remission, remission rates, mean number of exchanges, and relapse rates were not significantly different than controls, with p values ranging from 0.58 to 1.0. No patients developed new antibodies, although one patient had a serious adverse event after 12 days of therapeutic plasma exchanges, which was not further described in the abstract. The patient was removed from the study and recovered without sequelae. Despite this event, the authors concluded that the PCT–CAD-treated FFP performed similarly to the control plasma in terms of efficacy and safety. Therefore, it is likely that the adverse event could have occurred with untreated plasma as well.85 Thus, numerous clinical trials have demonstrated that FFP treated with PCT–CAD appears to be as effective as standard FFP.
Pathogen inactivation
The ability of amotosalen to reduce pathogens contained in a unit of blood platelets or plasma is in little doubt. Numerous studies have demonstrated a minimum of a greater than 5 log reduction in most viruses, bacteria, and protozoan tested, whether extra- or intracellular. Experiments reported by Lin et al86 are typical. These researchers added high levels of pathogens to single-donor platelet units. In the case of HIV, they evaluated cell-free and cell-associated virus, as well as pro-viral HIV. Duck hepatitis B (DHBV) and bovine viral diarrhea virus (BVDV) were used as surrogates for human strains of hepatitis B and hepatitis C. A representative of Gram-positive Staphylococcus epidermidis and Gram-negative Klebsiella pnemoniae bacteria were studied by inoculating 104 colony-forming units (CFU)/mL into the platelet units. Before treatment, the bags were sampled in order to determine the viral or bacterial titer. Different levels of UVA light were used to determine the appropriate level of pathogen inactivation. Based on these studies, the UVA frequency is now set at 3 J/cm2. Starting with small doses allowed the authors to differentiate the speed and effectiveness of the process. HIV was readily eliminated and proviral HIV was even more sensitive to the process than the cell-free or cell-associated HIV. High levels of BVDV and DHBV were also readily inactivated at 3 J/cm2. Klebsiella bacteria, however, were more resistant, as two of four units were still infective after PCT with 3 J/cm2. When the experiment was repeated, all four units were negative. Gram-negative bacteria are known to be more resistant to PCT due to the lipopolysaccharide outer membrane, which serves to inhibit small organic molecules such as amotosalen. However, the authors concluded that the ability of the process to reduce the bacteria by a factor of greater than 105 was considered a success as clinically the presence of such high titers in blood products would be highly unlikely. S. epidermidis was readily inactivated by the process with as few as 0.5 J/cm2. Van Voorhis et al87 conducted similar experiments with T. cruzi and demonstrated the effectiveness of PCT–CAD in inactivating this parasite, which is likely to become a more of a concern in the future. Lin et al88 and Jordan et al89 demonstrated effective pathogen reduction of latent and free CMV in mice. Another experiment by Lin et al90, looked at several types of bacteria, including Gram-negative, Gram-positive, anaerobic, aerobic, and spirochetes (specifically Treponema pallidum and Borellia burgdorferi). The spirochetes were very sensitive to the PCT–CAD process and, again, the various other bacteria examined were effectively inactivated by the process.
Summary
As described above, the ability of the psoralen amotosalen (S-59) to reduce pathogens is excellent. However, further studies are needed to more fully explore the upper limit of the capacity of PCT–CAD (150 μg dose of amotosalen plus 3 J/cm2) to reduce pathogen activity. This might be most important for those pathogens more resistant to the process, such as Klebsiella pneumoniae and parvovirus B19. The safety of the amotosalen-based PCT–CAD process has been well demonstrated in the short-term in several phase III clinical trials. Long-term studies will be necessary to determine the risk, particularly of carcinogenesis. The amotosalen-based process for platelets is currently under review in the US by the FDA. In Europe, the process for platelets has received the CE mark and has been validated and implemented in some countries, including France and Norway. Cost-benefit analyses have begun and initial findings have been reported by the Netherlands and Germany, which conclude that the cost of INTERCEPT Blood System for platelets is comparable to that of other blood safety interventions, such as nucleic acid testing.91, 92 Currently, the INTERCEPT Blood System and its programs are in various stages of the development and regulatory approval process.
Frale
Numerous obstacles have beset the development of photochemical systems for red cells, including problems caused by the absorption of UVA light by hemoglobin. Further, the photochemically treated red cells often sustain damage that results in hemolysis and potassium leakage during storage. New methods are in the process of development. One of these methods, developed by the same manufacturer as amotosalen, is based on a compound known as S-303.93
Mechanism of action
S-303 is a part of class of compounds known as Frales (Frangible Anchor Linker Effectors). These tripartite molecules are composed of a nucleic acid anchor, an effector moiety, and a frangible linker. The effector moiety binds nucleic acids covalently, and the frangible linker breaks down forming an inactive negatively charged species (S-300). This prevents further binding to the nucleic acid, thus rendering the DNA or RNA non-functional.73 The completely light-independent reaction proceeds due to a shift in pH, which occurs as the S-303 is added to red blood cell concentrates. The process as developed requires the addition of S-303 to red cells at a concentration of 150 μg/mL, which is then incubated for 12 hours at room temperature. The red cells are incubated with a compound absorption device (CAD) for an additional 8 hours after transfer to a storage container to remove any residual S-303 and S-300. The CAD remains in the red cells throughout storage.73
Red blood cells
Most information published about S-303 pathogen reduction is in abstract form. The earliest abstract by Cook et al reported no biologically significant differences in S-303-treated red cells stored for 42 days, although the degree of hemolysis appeared to vary significantly among treated units.94 The same study evaluated S-303-treated RBC recovery in dogs and mice using a biotin recovery technique. The results were comparable with controls. A year later, Cook et al reported the results of another S-303-treated red cell study where recovery in dogs was not significantly different than controls. In addition, they conducted two further experiments. One involved replacing 80% of a dog's blood volume with S-303-treated cells and the other involved dogs receiving twelve 10-mL/kg transfusions over a month. The concentration of S-303 in each experiment reached 500 μg/mL, which was thought to be five times what a human would receive; no toxicity was identified. Flow cytometry was used to evaluate immunogenicity, which did not identify any antibodies specific to S-303.95
In early human studies, the findings have been similar. In 1999, Hambleton et al found greater than 75% RBC recovery in humans transfused with S-303-treated autologous blood. The study performed one clinical trial then re-enrolled the same subjects in a second trial. The subjects thus received more than one consecutive transfusion and evaluated the DAT antibody screen and cross-match data, all of which remained the same, as did in vivo recovery.96 Hambleton et al subsequently performed a cross-over study in 2002 and, again, found no statistical differences from controls in 24-hour red cell recovery after treatment with S-303.97
Phase III clinical trials were initiated based on these preliminary data. Despite promising results regarding the efficacy of S-303-treated red cells in patients98, 99, these trials have been halted due to antibody formation to S-303-treated cells. Patients requiring chronic transfusion for thalassemia or sickle-cell anemia were entered into a randomized, double-blinded, cross-over study. Each patient was to receive a total of six transfusions. The number of units per transfusion, as well as the frequency of transfusion, was determined by the treating physician. Two asymptomatic pediatric patients developed antibodies to S-303-treated red cells after transfusion; pre-transfusion IAT cross-matches were negative. Despite the antibody, there was no evidence of reduced red cell survival and a monocyte monolayer RBC phagocytic assay was negative, suggesting the antibody was not likely to have been clinically significant. The DAT in both patients was negative.99 In hapten inhibition assays, the S-303, but not glutathione, exhibited inhibition. The latter is used as the buffer during processing.99 Alterations in the pathogen-reduction process were made in an attempt to eliminate the antibody problem. In the original protocol, 200 μM of S-303 was added to 2 mM unbuffered GSH. Stassinopoulos et al100 altered the process by adding 20 mM GSH, which significantly reduced the presence of S-303 on the red cells. Sera from the patients who developed the antibodies was added to the modified treated S-303 red cells, and the IAT was negative. Pathogen reduction using the modified process remained effective.101 The modification essentially dilutes, rather than eliminates, the neo-antigen presence on the S-303-treated red cells. Further research will be required to determine if dilution alone will be adequate to prevent alloimmunization in red cells processed with S-303-treated red cells.
Pathogen inactivation
With regard to pathogen reduction, the S-303 system appears effective. In 1997, Cook et al reported that log inactivation depended linearly on the dose of S-303 added. In this study, they evaluated pathogen reduction of cell-free HIV, cell-associated HIV, duck hepatitis B virus, bovine viral diarrhea virus, herpes simplex virus-1, vesicular stomatitis virus, and Yersinia enterocolitica. Log10 inactivation ranged from >4.7 to >6.1. Whether the level of inactivation was related to the level of detection or not was not reported.94 In a second study, Cook et al again looked at cell-free HIV, duck hepatitis B virus, bovine viral diarrheal virus, and Y. enterocolitica and found even higher levels of inactivation ranging from 4.2±0.5 for the Yersinia to >7.3 for bovine viral diarrheal virus. The viruses were reduced to the level of detection of the assay.
Summary
Overall, the literature with regard to Frale technology is minimal due to the early phases of technological development. It appears to be effective as a pathogen-reduction agent, and the red cells function within the necessary clinical parameters both in vitro and in vivo. The phase III trials are now on hold due to the immune reactions of two patients as reported above. Even if the problem of antibody formation against S-303 is diminished, the level of risk for antibody production must be vanishingly small. The side effects of any pathogen-reduction process must be less than the risk of contracting the infectious diseases that it eradicates.
PEN110
PEN110 is another PR/PI compound that also disrupts nucleic acids. Like S-303, light is not required for the reaction and it can therefore be used to treat red cells. PEN110 is a compound chemically related to binary ethyleneimine and is known as an ethyleneimine oligomer. The altered molecule is a cation, selective for nucleic acids, and highly water soluble. As the molecule is very small, it readily diffuses through cell membranes, which, the manufacturer claims, makes it more effective against non-lipid enveloped viruses, such as parvoviruses, which are more resistant to heterocyclic compounds (see amotosalen, above). PEN110 forms ionic bonds with nucleic acids, activating the molecule by protonation of the aziridino nitrogen. The active form can then alkylate a proximal nucleophilic center such as the N7 position of guanine in DNA. This results in the opening of the imidazole ring structure of guanine, which creates a break in the strand and creates a stop message. The nucleic acid is thus inactivated and becomes useless as a template. Binary ethyleneimines, like psoralens, have been used in nucleic acid research since the 1980s. The development and use of PEN110 for pathogen reduction of blood products, however, has occurred only over the past 15 years. In the process known as INACTINE treatment, PEN110 is added to create a 0.1% (vol/vol) concentration and incubated at room temperature for 6 hours. PEN110 is then removed by washing with unbuffered saline to levels below the limit of detection when using HPLC (30 ng/mL). The units are then ready for storage.101, 104
Toxicology
Two main studies by Chapman, Butterwork, and Moore101, 102 describe the toxicology of PEN110. The PEN110 compound reacts with nucleic acids, and the Ames and mouse lymphoma assays are both positive, indicative of genotoxicity.102 PEN110 also induced unscheduled DNA synthesis (UDS) in rat hepatocyte cultures. However, chromosomal assays and whole animal toxicity assays were negative. PEN110 given to rats intravenously at high blood concentrations did not induce UDS in their hepatocytes suggestive of an effective clearance pathway.102 The compound appeared to be excreted quickly in the urine.102 The second study103, focusing on reproductive effects, was conducted on rats and rabbits. Male and female rats were regularly dosed with various quantities of PEN110 before and after mating. The animals were then killed after gestational day 14 or 16 and examined. Before euthanasia of the males, sperm analysis was conducted. As rabbits are known to be a more sensitive model for reproductive toxicology than rats, a second study arm was initiated using mated female rabbits dosed with PEN110 and killed on gestational day 29.102 The ability of PEN110-treated rats to become pregnant was not impaired. Further, the number of implantation sites, number of fetuses, and number of fetal losses were not significantly different from those found in controls. Fetal abnormalities were not identified. Numerous sperm characteristics were evaluated in males, none of which was significantly different than controls. Only female rats and rabbits receiving the highest doses of PEN110 demonstrated toxicity characterized by significantly decreased food consumption and decreased weight gain. The fetuses of rabbit mothers receiving high-dose PEN110 had significantly increased skeletal variations; however, these variations were not clinically significant and did not affect the viability to the fetuses. These effects were identified at dosing regimens of 0.1, 0.3, and 1.0 mg of PEN110 per kilogram of weight in rabbits and 0.05, 0.25, and 0.5 mg/kg in the rats. After washing, PEN110-treated blood (350 mL) contained 0.0175 mg of PEN110, which is equivalent to 0.00025 mg/kg of body weight for a 70-kg person. This level is 2000 times lower than the 0.5 mg/kg dose given the rats and, based on cumulative mg per kg figures, the dose is 48 000 times less. Therefore, even given that an individual human might receive multiple units at once or over time, the dose is believed to be far less than that required to induce reproductive or genotoxicity.102
Red blood cells
The majority of research thus far has been on the use of PEN110 for the inactivation of pathogens in units of red blood cells. In a preclinical trial, Purmal et al studied the effect of PEN110 pathogen inactivation, using 6- and 24-hour incubations, on a number of red cell parameters including hemolysis, RBC ATP, intracellular potassium, and red cell surface antigens.104 RBC ATP was significantly reduced in the PEN110-treated red cells compared to controls whether 6- or 24-hour incubations were used. However, the level of ATP was still within the clinically acceptable range. Hemolysis out to day 42 was not significantly increased compared to controls in the cells incubated for 24 hours with PEN110. The inactive PI/PR process had no effect on intracellular potassium.104 Snyder et al105 had similar results with lowered ATP in treated cells but no clinically significant difference in hemolysis for PEN110-treated versus control red cells. This group also studied 2,3-diphosphoglycerate (2,3-DPG), P50, sodium, methemoglobin, complete blood count (CBC), lactate, and glucose. Again, there was no significant difference between controls and PEN110-treated cells.105 ATP levels were also lower in a study by AuBuchon et al. The authors also noted lower levels of lactate and decreased glucose consumption in the PEN110-treated cells. These changes were interpreted as a ‘metabolic slowing’, although no further explanation was provided. Again, the changes were within acceptable ranges and clinically were not significantly different when compared with the control results.106
All three of these studies included in vivo study arms evaluated after 28 or 42 days of storage. Purmal et al104 removed red blood cells from baboons, double labeled them with 51Cr/125I, and re-infused them into the autologous animals. Red cell survival after 28 days of storage ranged from 66 to 76% in controls and from 72 to 80% in PEN110-treated cells. After 42 days of storage, red cell survival ranged from 39 to 61% in controls and 16 to 77% in PEN110-treated cells. The differences in survival time were not statistically significant. The Snyder et al study105 was a phase II clinical trial using healthy human volunteers who were re-infused with 51Cr-radiolabeled blood following treatment with PEN110 and 42 or 35 days of storage. Control units were processed in a similar manner. In vivo 24-hour survival in PEN110-treated cells stored for 42 days was 82.9±5.7% and for 35 days of storage was 84.7±4.8%. The 24-hour survival in controls was statistically similar; after 42 days storage it was 86.3±8.7%, and after 35 days storage, 85.7±3.5%. The mean rise in hemoglobin at 24 hours and 7 days after infusion of the remaining unlabeled autologous units of red blood cells was also evaluated and the PEN110-treated results were not statistically different from control, with 24-hour values of 0.8 versus 0.5 g/dL and 7-day values of 0.8 versus 0.9 g/dL. The in vivo portion of the AuBuchon et al study106 evaluated the survival of red cells in humans after 28 days of storage and determined T50 values. Again, there was no statistical difference between PEN110-treated cells in 24-hour survival. The T50 values also were similar to controls.
None of these studies found evidence of PEN110 induced neoantigenicity in any of their subjects (baboons or humans) who were monitored for 21 days104, 28 days105, and 56 days.106 Based on these results, PEN110 appears to be safe in animals and people, at least in the short term, and the PEN110-induced changes in red cell parameters are within acceptable ranges.
Pathogen inactivation
Extensive research has been conducted with regard to the pathogen inactivation capacity of PEN110. These studies are very specific in demonstrating the exact nature of the action of PEN110 on the virus and the kinetics of inactivation, as well as demonstrating activity in the presence of high and low titers of viruses.
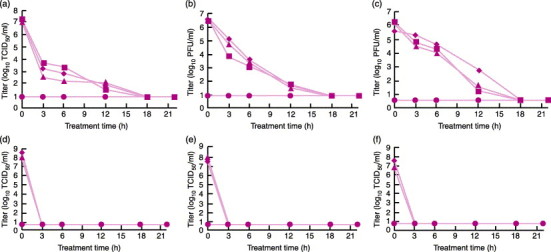
One of the initial studies evaluated the effect of PEN110 on four enveloped viruses, six non-enveloped viruses, and cell-associated HIV.107 The viruses were chosen to include a wide array of physiological characteristics such as size, structure, biochemical composition, and whether the virus was cell associated or cell free. Of the enveloped types, BVDV and pseudorabies virus (PRV) are recognized models for HCV and herpes virus. Other enveloped viruses included in the study were sinbis (SV) virus and vesicular stomatitis Indiana virus (VSIV). The non-enveloped viruses included porcine parvovirus (PPV), which is a model for B19 virus, and vesicular exanthema of swine virus (VESV), which is a model for hepatitis E and Norwalk viruses. The remaining viruses included were used to demonstrate the wide spectrum of activity of PEN110. In these studies, the inactivation process was altered by using 1 M sodium thiosulphate (STS) and 1 M 3-morpholinopropanesulphonic acid (MOPS) to chemically stop the PEN110 reaction (rather than washing) to ensure that the decreased viral cytotoxic effect was due to PEN110 alone and not washing. In addition, red cell units were collected in three different additive solution storage media CPD/AS-1, CP2D/AS-3, and CPD/AS-5. Virus titers were determined by tissue culture infectious dose 50% (TCID50) assay and plaque-forming units assay. The kinetics of viral reduction were determined by collecting samples at 3, 6, 12, 18, and 22 hours during incubation with PEN110. The results from these studies are seen in Figure 7, Figure 8 .
Figure 7.
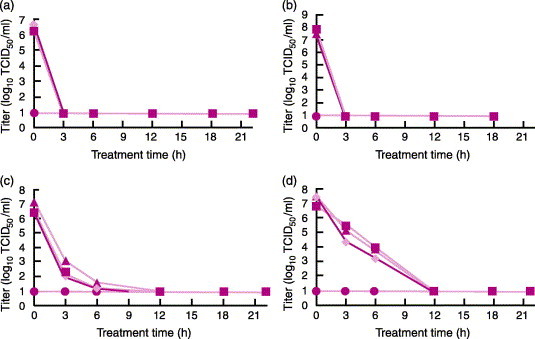
PEN110 kinetics of viral reduction of four enveloped viruses in CPD/AS-1, CP2D/AS-3 or CPD/AS-5 red blood cell concentrate (RBCC) units at 22±2 °C for up to 22±2 h. ♦, CPD/AS-1; ▪, CP2D/AS-3; ▴, CPD/AS-5; ●, limit of detection. (a) Inactivation kinetics of sindbis (SIN) virus; (b) inactivation kinetics of vesicular stomatitis Indiana virus (VSIV); (c) inactivation kinetics of bovine viral diarrhoea virus (BVDV); (d) inactivation kinetics of pseudorabies virus (PRV) (from ref. 107).
Figure 8.
PEN110 kinetics of viral reduction of six non-enveloped viruses in CPD/AS-1, CP2D/AS-3, or CPD/AS-5 red blood cell concentrate (RBCC) units at 22±2 °C for up to 22±2 hours. ♦, CPD/AS-1; ▪, CP2D/AS-3; ▴, CPD/AS-5; ●, limit of detection. (a) Inactivation kinetics of porcine parvovirus (PPV); (b) inactivation kinetics of reovirus 3 (Reo-3); (c) inactivation kinetics of human adenovirus 2 (Adeno-2); (d) inactivation kinetics of vesicular exanthema of swine virus (VESV); (e) inactivation kinetics of foot and mouth disease virus (FMDV); (f) inactivation kinetics of bluetongue virus (BTV) (from ref. 107).
Three of the non-enveloped (VESV, foot and mouth disease virus [FMDV], and bluetongue virus [BTV]) and two of the enveloped viruses (SV and VSIV) were reduced to the level of detection of the assay within 3 hours of incubation. These viruses were reduced by factors ranging from 5.4 to 7.5 log10 TCID50 per milliliter.107 BVDV and PRV took longer—12 hours—to be reduced to the level of detection. Three of the non-enveloped viruses—PPV, Reo-3, and Adeno-2—took 18 hours to be reduced to the level of detection. The reduction factors for these viruses were >6.2 log10 TCID50 per milliliter, >5.3 and >5.6 log10 PFU per milliliter, respectively. For cellular HIV, the reduction to the level of detection took 6 hours.
The same group of researchers conducted a series of experiments investigating PEN110 inactivation of HIV.108 They evaluated the effect of PEN110 on the structure of the virus by EM, the ability of the virus to enter cells after PEN110 treatment, and the reverse transcriptase activity after PEN110 treatment, in addition to the usual demonstration of the PEN110 treatment effect via the TCID50 assay. The structure of the virus did not change and the ability of the virus to infect cells remained intact after PEN110 treatment. However, the viral reverse transcriptase did not function, consistent with the alteration of nucleic acid by PEN110. The HIV reduction took place primarily in the first 6 hours and decreased to the level of detection after 12 hours. Large-volume infection required 18 hours to decrease to the level of detection. The reduction factors varied from 5.35 to 5.96 log TCID50 after 18 hours. Another study demonstrated that PEN110 was effective against HBV.109
In a study of West Nile virus, Mather et al110 explored the survival of the virus in PEN110-treated blood stored under the usual blood bank conditions and found that although the viral population decrease over time, the units remain ‘infectious’ throughout the 42 days of storage when spiked with high-titer viral concentrates; low-titer spiked units were non-infectious by 42 days. The doses chosen for high and low titers were based on the limited data available from donors with natural WNV infection. Infectivity was determined by PFU assays. Although, the study demonstrated viral survival, whether the dose present would be sufficient to cause infection in humans is not known. In addition, the same study demonstrated that the WNV was distributed in all blood components—red blood cells, plasma, and platelets—and that the virus can infect monocytes and may be harbored in these cells in vivo. PEN110 treatment reduced WNV to the level of detection of the assay within 3 hours and, after 24 hours, reduced the virus by a factor of 5.0 log TCID50 per milliliter. In another study111, the group compared the effect of PEN110 to gamma radiation on CMV-infected THP-1 cells, which are used to model latently infected white cells. In addition to demonstrating the efficiency of PEN110 against CMV-infected cells, the experiments demonstrated that gamma irradiation, despite inactivating white cells, has the capacity to actually increase CMV replication.
PEN110 inactivation of bacteria has been successful, although limitations are evident. Bacterial contamination of blood generally occurs due to organisms on the skin of donors at the time of collection. The most common organisms are therefore skin contaminants, such as Staphylococcus epidemidis and Propionibacterium acnes. Other organisms ubiquitous in the environment are also common, such as Clostridium perfringens, Pseudomonas fluorescens, Pseudomonas putida, and Acinetobacter species. Yersinia entercolitica is also associated with transfusion-related sepsis and is thought to come from bacteremia in a donor with Yersinia gastroenteritis. In studies using various titers of these organisms to infect red blood cell units, PEN110 successfully inactivated the majority of the organisms.112, 113, 114 However, some studies revealed that high titers of organisms could not be inactivated. Acinetobacter iwoffii and A. johnsonii were inactivated when the titer was less than 104 CFU/mL, but the bacteria persisted when units were infected with higher titers.112 Y. enterocolitica and P. fluorescens have demonstrated similar resistance to inactivation at higher titers in other studies.112 However, in an earlier study by the same group115, the titers (10–100 CFU) were chosen to simulate a worse-case scenario based on reported clinical levels detected in donated blood for Yersinia of 1 CFU/mL or lower.115 If this level of bacteremia is the norm clinically, the lack of pathogen reduction at levels above 104 CFU/mL is not relevant. However, the question of an upper limit of effectiveness for this pathogen-reduction technology remains a concern. Importantly, the concentration level of pathogens in the blood may be dependent on the organism involved. PEN110 was able to inactivate Mycoplasma pneumonia and M. arthritidis by more than 107 CFU/mL.113 Even if all of the organisms appear to be removed, however, they are only ‘removed’ to the level at which they can be detected. In protozoa studies, B. microti was eliminated below the level of detection by PEN110 and hamsters given the blood did not develop identifiable infection even by PCR testing. However, the hamsters did mount an IgG antibody response. This is not too surprising given that the inactivation occurs only on the nucleic acid level, leaving organism antigens in place. Other protozoa tested included P. falciparum and T. cruzi, which were effectively reduced by PEN110.114
Summary
PEN110 demonstrates robust viral and bacterial inactivation capacity. Using more sensitive PCR methods and high- and low-titer infections, investigators have revealed the higher and lower limits of this pathogen-reduction process. Given the cost of implementing any pathogen-reduction process, the hope would be that the process would reduce all pathogens to a non-infectious level despite the initial titer. Additional studies are needed to evaluate the pathogen-reduction process in clinical settings and under conditions involving mixtures of pathogens at clinically relevant levels in combination with the white cells present at different concentrations to determine the true capacity of these pathogen-reduction processes. The two phase III PEN110 studies involving patients requiring acute transfusion therapy for surgical support and chronic transfusion involving sickle-cell and thalassemia patients has been halted due to development of an immune response to PEN110-treated RBC and associated clinical findings.112 Immunogenicity is thus a problem for the pathogen-reduction chemicals. Even if the number of events is very small, because the risk of immunogenicity must be smaller than the risk of infection by untreated blood which is very small, it will take more research over time to determine how safe and effective PR/PI technologies really are.
Riboflavin
Riboflavin, also known as vitamin B2, has been widely investigated from a number of different perspectives for the past 70 years.116 Because of the extensive knowledge of this compound, much of the background work required for the development of riboflavin as a pathogen-reducing agent for blood has already been reported.
Toxicology and mechanism of action
Numerous nutritional studies have already been performed. In 1942, Unna et al investigated the toxicity of riboflavin in rats and found that excessive amounts of riboflavin taken by mouth were not toxic due to the inability of the gastrointestinal system to absorb sufficient quantities of the vitamin.117 Toxicity was observed in rats after intraperitoneal injections of 300 mg/kg, which resulted in renal failure due to yellow concretions that accumulated in the kidneys. Intravenous toxicity could not be determined due to the low solubility of riboflavin.
In 1996, Zempleni et al defined the pharmacokinetics of oral and intravenous riboflavin in humans using oral doses of 20, 40, and 60 mg and intravenous doses of 11.6 mg.118 Based on these and the numerous other studies performed over the years, riboflavin is now classified as a GRAS, or ‘generally regarded as safe’ compound. Also known for many years is the response of riboflavin to light, and the damage this reaction has on nucleic acids.
In 1965, Tsugita et al determined that the light reaction with riboflavin-inactivated s-RNA required the presence of oxygen to inactivate tobacco mosaic virus119 and they demonstrated that the inactivation was localized to guanine and adenine nucleotides. In addition, they compared riboflavin to lumichrome and MB and found riboflavin to be the most efficient sensitizer.119
The mechanism of action of the phototoxicity of riboflavin was described by Joshi in 1983. At that time, the photodynamic reaction of riboflavin and light was known to generate superoxide anion radicals and HO2, which damaged nucleic acids. Joshi demonstrated that singlet oxygen is also generated by these reactions, leading to photo-oxidation of the nucleotide guanine thus damaging nucleic acids.120 Two types of photoreactions can occur. Type I is a photoreaction between an electronically excited photosensitizer and a biological substrate. This reaction is oxygen independent and the substrate of concern is nucleic acids, although other biologic molecules can be affected, as well. Type II is a reaction of an activated photosensitizer molecule with oxygen resulting in the formation of oxygen radicals.120 The singlet oxygen radical is responsible for the majority of the damage to nucleic acids and results in strand breaks and fragmentation.
Kumar et al demonstrated the effectiveness of this process over UV light alone on bacteria, viruses, and white cells in 2004.121 In addition to the generation of oxygen radicals, the photoreaction of riboflavin also generates breakdown photoproducts of lumichrome, lumiflavin, 2′ketoflavin, 4′ketoflavin, flavin mononucleotide, and formylmethylflavin. Lumichrome and lumiflavin are the most abundant of these products; the others are present in small amounts best detected by HPLC.118
Lumichrome toxicity and genotoxicity were evaluated in in vitro and in vivo experiments by Piper et al, in 2002.122 In vitro genotoxicity was evaluated using the Ames assay72, 122 and was negative. Mouse fibroblasts were incubated with various concentrations of lumichrome, the highest of which was 90 μM (the limit of solubility). Cytotoxicity was not identified. The in vivo portion of the study involved injecting mice with 900 μg/kg of lumichrome IV. No acute toxicity was identified.122 The other major photo-product of riboflavin—lumiflavin—was evaluated by the Ames Salmonella assay, umu test, and SOS chromotest. The Ames and umu tests are bacterial genotoxicity test systems123, 124 and the SOS chromotest is a bacterial colorimetric assay for genotoxins.124 In this study, both riboflavin and lumiflavin were negative for genotoxicity. However, when lumiflavin was exposed to metabolic enzymes of the colon and liver, significant mutagenicity was identified by all three assays. The study was performed in order to ascertain the danger posed by riboflavin in food products.125 Investigation of mutagenicity of lumiflavin has not yet been investigated in blood products. The remaining photoproducts are present in small concentrations and often react to reform riboflavin and thus they are less likely to have an effect.
Platelets
Riboflavin and light (which can be visible light or light within the UV spectrum) can be used to pathogen-reduce all blood products; however, the majority of the studies reported thus far involve platelets. Platelet units subjected to the riboflavin light process have a decreased concentration due to dilution with the 500-μM riboflavin solution. The total platelet count is unchanged and is similar to controls, remaining stable throughout storage. In one study, some in vitro performance characteristics in treated units were significantly different after 5 days of storage.126 This difference appears to be due to metabolic changes, as the only parameter that changed significantly after photochemical treatment was a marked reduction in pO2 and a mild increase in P-selectin expression. The treatment also appears to increase glucose consumption. Despite these differences, the parameters were still within the clinically relevant limits for use. In vivo studies will be necessary to determine the significance of the in vitro findings.126
A recent study looked solely at the effect of the riboflavin and light process on platelets produced by the buffy coat method used commonly in Europe and platelets acquired via apheresis. Numerous platelet parameters including platelet integrity, morphology, and biological activity were examined. The results were similar to those of previous studies.127 Again, there was a marked decrease in pO2 after photochemical treatment, as expected given the mechanism of action of riboflavin and light. Glucose was decreased and P-selectin expression and lactate were increased after storage. pH was decreased over the storage period but remained above 7.0. Overall, there were changes in parameters compared to controls, but the numbers were still within defined clinical limits and suggested the platelets would be functional.127
Pathogen inactivation
Investigations of the capacity of riboflavin and light to reduce pathogens without harming the product are in the early stages and have primarily involved use of the technology in platelet concentrates. One of the initial studies placed 10 mM riboflavin into the storage media and exposed them to 419 nm of visible light at 40 and 80 J/cm2 energy dose. Bovine viral diarrheal virus (BVDV), a model for HCV, was used to infect the platelets. A 3.1 log reduction to the limit of detection was accomplished when the infection dose was 4.4 log (using 40 J/cm2 energy dose), and a 6.6 log reduction was identified when the initial infection dose was 6.8 log (using 180 J/cm2 energy dose). The platelet parameters in this study were minimally affected compared to controls.128
Goodrich et al looked at bacterial pathogen reduction in platelets in 2002.129 Riboflavin (50 μM) was added, and the units were spiked to 5–6 log/mL titers with S. epidermidis, Escherichia coli, S. aureus, B. cereus, and K. pneumoniae. Illumination this time was with 6–7 J/cm2 of UV light. A low-titer experiment paralleled the above, spiking the units to 2 log/mL titers and using 7 J/cm2 of UV light. Significant reduction occurred ranging from 3.1 to 5.5 log/mL reduction. Even the low titers were 100–1000 times higher than levels expected in platelets at the time of donation. Thus, the photoactivation of riboflavin appears to be adequate for pathogen reduction in blood products.129 At the same time, an experiment investigated riboflavin and light in red blood cell units. Using a 500-μM riboflavin solution and 447 nm of visible light for illumination, the red cells spiked with HIV, BVDV, and PSR were treated. Again, significant pathogen reduction occurred. The only red cell parameter examined, hemolysis, was not observed in this experiment.130
More extensive studies have been performed with a riboflavin level of 500 μm, which reaches 50 μM on dilution in the platelet concentrate. The light parameters chosen were UV light at 6.2 J/cm2 with an output ranging from 265 to 370 nm. Porcine parvovirus (PPV) was added to achieve a titer of 6.68±0.20 log TCID50. Cell-associated and cell-free HIV were added to a titer of 7.93±0.27 log TCID50 per milliliter, and intracellular HIV was added to a titer of 6.64±0.27 TCID50 per milliliter. WNV was added to achieve an initial titer of 7.06±0.26 TCID50 per milliliter. Interference and cytotoxicity were determined and factored into the results. Gram-negative bacteria represented by E. coli and Gram-positive bacteria represented by S. epidermidis were used to spike units to an initial titer of 5–6 CFUs/mL for the high-titer arm and 0.5–2 CFUs/mL for the low-titer arm. In vitro platelet parameters were measured and included pH, lactate, pO2, pCO2, platelet count, HSR, swirl scores, and P-selectin expression. The viral reduction levels observed exceeded the levels required to close the window period for HIV and reduced the titers of intracellular and free- and cell-associated HIV to below the levels of detection. The reduction in WNV occurred to an extent believed to close the total viremic period for WNV, due to inactivation of 5.11±0.5 log TCID50 per milliliter. Parvovirus was also reduced to the level of detection with a reduction factor of >5.03 log TCID50. This amount of reduction could close the chronic phase transmission window for some human parvovirus B19 infections. This last virus is difficult to eradicate, as titers in infected donors can exceed 1013 genome equivalents per milliliter. However, levels of B19 below 3.5 log/mL are thought not to be infectious. Both bacterial strains were reduced to below the level of detection in the high titer arm of the study. In the low titer arm, the concentrates remained culture negative after 5 days of storage.126
Summary
Riboflavin is photochemically activated by light and produces marked and robust pathogen reduction in platelets and red blood cells. The compound riboflavin is generally considered safe. Questions remain, however, regarding the photoproducts produced by this process; studies of the photoproducts are limited. Lumichrome appears to be safe in studies thus far, but more research is required. Lumiflavin is questionable and additional research will also be necessary. The early studies indicate that the effect of the process on platelets and red blood cells are clinically acceptable. More research on pathogen-reduction technology using riboflavin is needed.
Transfusion-associated graft-versus-host disease
Leukocyte inactivation
An unintended, but beneficial, result of nucleic-acid-altering pathogen-reduction technologies is that the processes also inactivate white blood cells, which can cause transfusion-associated graft-versus-host disease (TA-GVHD). Several studies using mice have demonstrated the efficiency of pathogen-reduction technologies in preventing GVHD. Grass et al131 compared amotosalen PCT and gamma irradiation in an experiment using strain A and B6AF1 mice. Strain A mice are homozygous at the H-2 locus whereas B6AF1 mice are heterozygous. Spleens from the donor mice were collected, processed to produce a splenocyte suspension, and treated in one of three ways. One-third of the suspension was treated with gamma irradiation, one-third was treated with amotosalen PCT, and one-third was untreated. The recipient mice (B6AF1 strain) each received transfusions of one of three suspensions or a suspension of splenocytes from another B6AF1 mouse, which were the controls. Several features of GVHD were followed over several weeks. Those that received untreated cells developed GVHD characterized by splenomegaly, decreased body weight, decreased skin integrity, and pancytopenia. After the animals were killed, the organs were examined histologically for evidence of GVHD. In addition, donor T cell engraftment was demonstrated by flow cytometry. None of the mice that received products treated by gamma irradiation or amotosalen PCT developed GVHD. All six mice that received the untreated product developed GVHD.131 Additional mouse-based studies132, 133, 134 demonstrated the efficiency of PEN110 in preventing GVHD. Riboflavin was demonstrated to be effective using a cell culture of Jurkat cells.135 Thus, based on existing data, pathogen-reduction technologies that disrupt nucleic acids also appear to prevent TA-GVHD.
Conclusion
Pathogen-reduction technologies show great promise for making the blood supply safer. Indeed, some PR-treated products are in use clinically. However, no PR technology is risk free. As we learn more about the pathogens that infect the blood supply, we realize that not all pathogens will be fully inactivated. Accordingly, it is unlikely that current practices testing for pathogens can be eliminated. The full clinical role of PR technologies in the field of transfusion medicine remains to be determined.
Footnotes
The views expressed in this article are those of the author and do not reflect the official policy or position of the United States Air Force, Department of Defense, or the US Government.
References
- 1.Teitel J.M. Viral safety of haemophilia treatment products. Annals Medicine. 2000;32(7):785. doi: 10.3109/07853890009002024. [DOI] [PubMed] [Google Scholar]
- 2.Epstein J.S., Vostal J.G. FDA approach to evaluation of pathogen reduction technology. Transfusion. 2003;43:1347. doi: 10.1046/j.1537-2995.2003.00584.x. [DOI] [PubMed] [Google Scholar]
- 3.Katz L. Blood bags for diversion of the initial collection: a statement of the American Association of Blood Banks before the Blood Products Advisory Committee; March 15, 2001.
- 4.Rossi E.C., Simon T.L. Transfusion in the new millennium. In: Simon T.L., editor. In: Rossi's Principles of Transfusion Medicine. third edn. Lippincott Williams and Wilkins; Philadelphia, PA: 2002. [Google Scholar]
- 5.Technical Manual. 14th edn. American Association of Blood Banks; Bethesda, MD: 2002. [Google Scholar]
- 6.Update on WNV-related activities and recommendations, American Association of Blood Banks, Bulletin 03–11; September 12 2003.
- 7.Biggeerstaff B.J., Petersen L.R. Estimated risk of transmission of the West Nile Virus through blood transfusions in the US, 2002. Transfusion. 2003;43(8):1007. doi: 10.1046/j.1537-2995.2003.00480.x. [DOI] [PubMed] [Google Scholar]
- 8.Yoto Y. Incidence of human parvovirus B19 DNA detection in blood donors. British Journal of Haematology. 1995;91:1017. doi: 10.1111/j.1365-2141.1995.tb05427.x. [DOI] [PubMed] [Google Scholar]
- 9.Wu Y.Y., Snyder E.L. Safety of the blood supply: role of pathogen reduction. Blood Reveiws. 2003;17:111. doi: 10.1016/s0268-960x(02)00063-2. [DOI] [PubMed] [Google Scholar]
- 10.Hirayama J., Wagner S.J., Abe H. Involvement of reactive oxygen species in hemoglobin oxidation and virus inactivation by 1,9-dimethylmethylene blue photo-treatment. Biological Pharmacy Bulletin. 2001;24(4):418. doi: 10.1248/bpb.24.418. [DOI] [PubMed] [Google Scholar]
- 11.Wagner S.J., Skripchenko A., Thompson-Montgomery D. Quinacrine enhances vesicular stomatitis virus inactivation and diminishes hemolysis of dimethylene blue-photo-treated red cells. Photochemistry and Photobiology. 2002;76(5):514. doi: 10.1562/0031-8655(2002)076<0514:qevsvi>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 12.van der Poel C.L. Hepatitis C virus and blood transfusion: past and present risks. Journal of Hepatology. 1999;31(S1):101. doi: 10.1016/s0168-8278(99)80384-5. [DOI] [PubMed] [Google Scholar]
- 13.Rollag H., Solheim B.G., Svennevig J.L. Viral safety of blood derivatives by immune neutralization. Vox Sanguinis. 1998;74(S1):213. doi: 10.1111/j.1423-0410.1998.tb05475.x. [DOI] [PubMed] [Google Scholar]
- 14.Horowitz B., Ben-Hur E. Efforts in minimizing risk of viral transmission through viral inactivation. Annals of Medicine. 2000;32:475. doi: 10.3109/07853890009002023. [DOI] [PubMed] [Google Scholar]
- 15.Dodd R.Y. Current viral risks of blood and blood products. Annals of Medicine. 2000;32:469. doi: 10.3109/07853890009002022. [DOI] [PubMed] [Google Scholar]
- 16.Haubelt H., Blome M., Kiessling A.H. Effects of solvent/detergent-treated plasma and fresh-frozen plasma on haemostasis and fibrinolysis in complex coagulopathy following open-heart surgery. Vox Sanguinis. 2002;82:9. doi: 10.1046/j.1423-0410.2002.00129.x. [DOI] [PubMed] [Google Scholar]
- 17.Snyder E.L., Dodd R.Y. Reducing the risk of blood transfusion. In: Schecuter G.P., Broudy V.C., Williams M.E., editors. Hematology, 2001. 2001. p. 433. (American Society of Hematology Education Program Book). [DOI] [PubMed] [Google Scholar]
- 18.FDA. Workshop on safety and efficacy of methods for reducing pathogens in cellular products used in transfusion, Bethesda, MD; 7 August 2002. Available at: http://www.fda.gov/cber/minutes/pathtrans080702.pdf
- 19.Cazenave J.P., Aleil B, Wiesel M.L. In vitro evaluation of pooled buffy coat platelets treated with photochemical pathogen inactivation using amotosalen. Vox Sanguinis. 2004;86:201–202. doi: 10.1111/j.0042-9007.2004.00417.x. [DOI] [PubMed] [Google Scholar]
- 20.Roberts P. Resistance of Vaccinia virus to inactivation by solvent/detergent treatment of blood products. Biologicals. 2000;28:29. doi: 10.1006/biol.1999.0236. [DOI] [PubMed] [Google Scholar]
- 21.Mohr H. Virus inactivation of fresh plasma. Vox Sanguinis. 1998;74(S2):171. doi: 10.1111/j.1423-0410.1998.tb05417.x. [DOI] [PubMed] [Google Scholar]
- 22.Delipidation treatments for large scale protein purification process. From http://scholar.lib.vt.edu/theses/public/etd-1454132679612381/etd-title.html
- 23.Pamphilon D. Viral inactivation of fresh frozen plasma. British Journal of Haematology. 2000;109:680. doi: 10.1046/j.1365-2141.2000.02019.x. [DOI] [PubMed] [Google Scholar]
- 24.Horowitz B., Lazo A., Grossberg H. Virus inactivation by solvent/detergent treatment and the manufacture of SD-plasma. Vox Sanguinis. 1998;74(S1):203. doi: 10.1111/j.1423-0410.1998.tb05473.x. [DOI] [PubMed] [Google Scholar]
- 25.Pehta J.C. Clinical studies with solvent detergent-treated products. Transfusion Medicine Reviews. 1996;10(4):303. doi: 10.1016/s0887-7963(96)80006-x. [DOI] [PubMed] [Google Scholar]
- 26.Letter to providers from V.I. Technologies Inc. (VITEX). Important prescribing information October 20, 2000.
- 27.Horowitz B, Klein HG, Cable R. Preparing for pathogen reduction. AABB Annual Meeting News; 2003, 7.
- 28.Williamson L.M., Llewelyn C.A., Fisher N.C. A randomized trial of solvent/detergent-treated and standard fresh-frozen plasma in the coagulopathy of liver disease and liver transplantation. Transfusion. 1999;39:1227. doi: 10.1046/j.1537-2995.1999.39111227.x. [DOI] [PubMed] [Google Scholar]
- 29.Beisert L., Suhartono H. Solvent/detergent treatment of human plasma—a very robust method for virus inactivation. Validation virus safety of OCTAPLAS®. Vox Sanguinis. 1998;74(S1):207. doi: 10.1111/j.1423-0410.1998.tb05474.x. [DOI] [PubMed] [Google Scholar]
- 30.AuBuchon J.P., Birkmeyer J.D. Safety and cost-effectiveness of solvent-detergent-treated plasma: in search of a zero risk blood supply. JAMA. 1994;272(15):1210. [PubMed] [Google Scholar]
- 31.Fricke W. FDA perspective on and response to risk of hepatitis A from blood products. Vox Sanguinis. 1994;67(S4):16. [PubMed] [Google Scholar]
- 32.Beisert L. Virus validation studies of immunoglobulin preparations. Clinical and Experimental Rheumatology. 1996;14(S15):S47. [PubMed] [Google Scholar]
- 33.Wagner S.J., Skripchenko A., Robinette D. The use of dimethylene blue for virus photo-inactivation of red cell suspensions. Development in Biological Standardization. 1999;102:125. [PubMed] [Google Scholar]
- 34.Summary of basis for approval: 93-0253 and 97-0141, plasma, solvent/detergent treated. VIPLAS/SDTM V.I. Technologies (VITEX). From www.fda.gov/cber/sba/sdplvit050698s.pdf
- 35.Chandra S., Groener A., Feldman F. Effectiveness of alternative treatments for reducing potential viral contaminants from plasma derived products. Thrombosis Research. 2002;105:391. doi: 10.1016/s0049-3848(02)00044-0. [DOI] [PubMed] [Google Scholar]
- 36.Horowitz M.S., Pehta J.C. SD plasma in TTP and coagulation factor deficiencies for which no concentrates are available. Vox Sanguinis. 1998;74(S1):231. doi: 10.1111/j.1423-0410.1998.tb05478.x. [DOI] [PubMed] [Google Scholar]
- 37.Breek H., Hellstern P. In vitro characterization of solvent/detergent-treated human plasma and of quarantine fresh frozen plasma. Vox Sanguinis. 1998;74(S1):219. doi: 10.1111/j.1423-0410.1998.tb05476.x. [DOI] [PubMed] [Google Scholar]
- 38.Freeman J.W., Williamson L.M., Llewelyn C. A randomized trial of solvent/detergent and standard fresh frozen plasma in the treatment of coagulopathy seen during orthotopic liver transplantation. Vox Sanguinis. 1998;74(S1):225. doi: 10.1111/j.1423-0410.1998.tb05477.x. [DOI] [PubMed] [Google Scholar]
- 39.Williamson L.M., Allain J.P. Virally inactivated fresh frozen plasma. Vox Sanguinis. 1995;69(3):159. doi: 10.1111/j.1423-0410.1995.tb02590.x. [DOI] [PubMed] [Google Scholar]
- 40.Sarode R., Yomtovian R. Efficacy of SD-treated plasma during liver transplantation. Transfusion. 2000;40:886. doi: 10.1046/j.1537-2995.2000.40070886.x. [DOI] [PubMed] [Google Scholar]
- 41.Fischer G., Hoots W.K., Abrams C. Viral reduction techniques: types and purpose. Transfusion Medicine Reviews. 2001;15(2 supplement 1):27. doi: 10.1053/tm.2001.25376. [DOI] [PubMed] [Google Scholar]
- 42.Material safety datae sheet for Triton-X. From: http://www.genomicsolutions.com/files/80-0178%20Triton%20X-100.pdf
- 43.FDA approves alternative to fresh frozen plasma. May 6, 1998. From: http://www.fda.gov/bbs/topics/ANSWERS/ANS00868.html
- 44.Piquet Y., Janvier G., Selosse P. Virus inactivation of fresh frozen plasma by a solvent detergent procedure: biological results. Vox Sanguinis. 1992;63(4):251. doi: 10.1111/j.1423-0410.1992.tb01230.x. [DOI] [PubMed] [Google Scholar]
- 45.Riedler G.F., Haycox A.R., Duggan A.K., Dakin H.A. Impact of TRALI on the cost-effectiveness of solvent-detergent-treated fresh-frozen plasma (Octaplas®) British Journal of Haematology. 2003;121(supplement 1):81. [Google Scholar]
- 46.Pereira A. Cost-effectiveness of transfusing virus-inactivated plasma instead of standard plasma. Transfusion. 1999;39:479. doi: 10.1046/j.1537-2995.1999.39050479.x. [DOI] [PubMed] [Google Scholar]
- 47.Jackson B.R., AuBuchon J.P., Birkmeyer J.D. Update of cost-effectiveness analysis for solvent-detergent-treated plasma. JAMA. 1999;282(4):329. doi: 10.1001/jama.282.4.329. [DOI] [PubMed] [Google Scholar]
- 48.Tollofsrud S., Noddeland H., Svennevig J.L. Universal fresh frozen plasma (Uniplas®): a safe product in open heart surgery. Intensive Care Medicine. 2003;29:1736. doi: 10.1007/s00134-003-1952-3. [DOI] [PubMed] [Google Scholar]
- 49.Noddeland H., Tollofsrud S., Svennevig J.L. Universal solvent/detergent-treated fresh frozen plasma (Uniplas®)—rationale and clinical properties. Thrombosis Research. 2002;107:S33. doi: 10.1016/s0049-3848(02)00150-0. [DOI] [PubMed] [Google Scholar]
- 50.Wainwright M. The use of dyes in modern medicine. Biotechnic and Histochemistry. 2003;78(3-4):147. doi: 10.1080/10520290310001602404. [DOI] [PubMed] [Google Scholar]
- 51.OhUigin C., McConnell D.J., Kelly J.M., van der Putten W.J.M. Methylene blue photosensitized strand cleavage of DNA: effects of dye binding and oxygen. Nucleic Acid Research. 1987;15(18):7411. doi: 10.1093/nar/15.18.7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Floyd R.A., Schneider J.E., Dittmer D.P. Methylene blue photo-inactivation of RNA viruses. Antiviral Research. 2004;61:141. doi: 10.1016/j.antiviral.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 53.Wainwright M. Methylene blue derivatives-suitable photomicrobials for blood product disinfection? International Journal of Antimicrobial Agents. 2000;16:381. doi: 10.1016/s0924-8579(00)00207-7. [DOI] [PubMed] [Google Scholar]
- 54.Wainwright M. Pathogen inactivation in blood products. Current Medicinal Chemistry. 2002;9:127. doi: 10.2174/0929867023371355. [DOI] [PubMed] [Google Scholar]
- 55.Hornsey V.S., Drummond O., Young D. A potentially improved approach to methylene blue virus inactivation of plasma: the Maco Pharma Maco-Tronic system. Transfusion Medicine. 2001;11:31. doi: 10.1046/j.1365-3148.2001.00282.x. [DOI] [PubMed] [Google Scholar]
- 56.Wagner S.J., Skripchenko A. Investigation of photosensitizing dyes for pathogen reduction in red cell suspensions. Biotechnic and Histochemistry. 2003;78(3-4):171. doi: 10.1080/1052029032000140894. [DOI] [PubMed] [Google Scholar]
- 57.Kelner M.J., Alexander N.M. Methylene blue directly oxidizes glutathione without the intermediate formation of hydrogen peroxide. The Journal of Biological Chemistry. 1985;260(28):15168. [PubMed] [Google Scholar]
- 58.Mohr H., Bachmann B., Klein-Struckmeier A., Lanbrecht B. Virus inactivation of blood products by phenothiazine dyes and light. Photochemistry and Photobiology. 1997;65(3):441. doi: 10.1111/j.1751-1097.1997.tb08586.x. [DOI] [PubMed] [Google Scholar]
- 59.Karl-Freidrich Bopp K., Morell A., Parkkinen J. Pathogen inactivation of labile blood products. Transfusion Medicine. 2001;11(3):149. doi: 10.1046/j.1365-3148.2001.00310.x. [DOI] [PubMed] [Google Scholar]
- 60.Material safety data sheet for methylene blue from http://ptcl.chem.ox.ac.uk/MSDS/ME/methylene_blue.html
- 61.Material safety data sheet: methylene blue from http://www.jtbaker.com/msds/englishhtml/m4381.htm
- 62.Wollowitz S. Fundamentals of the Psoralen-based helinx technology for inactivation of infectious pathogens and leukocytes in platelets and plasma. Seminars in Hematology 2001; 38(supplement 11): 4–11. [DOI] [PubMed]
- 63.Dardare N., Platz M.S. Binding affinities of commonly employed sensitizers of viral inactivation. Photochemistry and Photobiology. 2002;75(6):561. doi: 10.1562/0031-8655(2002)075<0561:baoces>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 64.Allain J., Seghatchain J. Current strategies on pathogen removal/inactivation: an overview. Transfusion and Apheresis Science. 2001;25:195. doi: 10.1016/s1473-0502(01)00122-7. [DOI] [PubMed] [Google Scholar]
- 65.Detty M.R., Gibson S.L., Wagner S.J. Current clinical and pre-clinical photosensitizers for use in photodynamic therapy. Journal of Medicinal Chemistry. 2004;47(16):3897–3915. doi: 10.1021/jm040074b. [DOI] [PubMed] [Google Scholar]
- 66.Skripchenko A.A., Wagner S.J. Inactivation of WBCs in RBC suspensions by photoactive phenothiazine dyes: comparison of dimethylene blue and MB. Transfusion. 2000;40:968. doi: 10.1046/j.1537-2995.2000.40080968.x. [DOI] [PubMed] [Google Scholar]
- 67.Wieding J.U., Hellstern P., Kohler M. Inactivation of viruses in fresh frozen plasma. Annals of Hematology. 1993;67(6):259. doi: 10.1007/BF01696345. [DOI] [PubMed] [Google Scholar]
- 68.Pereira A. Methylene-blue photo-inactivated plasma and its contribution to blood safety. Transfusion. 2004;44:948. doi: 10.1111/j.0041-1132.2004.359_6.x. [DOI] [PubMed] [Google Scholar]
- 69.Aznar J.A., Bonanad S., Montoro J.M. Influence of Methylene blue photoinactivation treatment on coagulation factors from fresh frozen plasma, cryoprecipitates, and cryosupernatants. Vox Sanguinis. 2000;79:156. doi: 10.1159/000031234. [DOI] [PubMed] [Google Scholar]
- 70.Wollowitz S. Fundamentals of the Psoralen-based helinx technology for inactivation of infectious pathogens and leukocytes in platelets and plasma. Seminars in Hematology 38(supplement 11): 4–11. [DOI] [PubMed]
- 71.Wu Y.Y., Snyder E.L. Safety of the blood supply: role of pathogen reduction. Blood Reviews. 2003;(17):111–122. doi: 10.1016/s0268-960x(02)00063-2. [DOI] [PubMed] [Google Scholar]
- 72.Ciaravino V., McCullough T., Cimino G., Sullivan T. Preclinical safety profile of plasma prepared using the INTERCEPT Blood System. Vox Sanguinis. 2003;85:171–182. doi: 10.1046/j.1423-0410.2003.00351.x. [DOI] [PubMed] [Google Scholar]
- 73.Ciaravino V., McCullough T., Cimino G. The role of toxicology assessment in transfusion medicine. Transfusion. 2003;43:1481–1492. doi: 10.1046/j.1537-2995.2003.00544.x. [DOI] [PubMed] [Google Scholar]
- 74.Moog R., Frohlich A., Mayaudon V., Lin L. In vitro evaluation of COM.TEC apheresis platelet concentrates using a preparation set and pathogen inactivation over a storage period of five days. Journal of Clinical Apheresis. 2004;19:185–191. doi: 10.1002/jca.20025. [DOI] [PubMed] [Google Scholar]
- 75.Jansen G., VanVliet H., Vermeij H. Functional characteristics of photochemically treated platelets. Transfusion. 2004;44:313–319. doi: 10.1111/j.1537-2995.2003.00588.x. [DOI] [PubMed] [Google Scholar]
- 76.Cazenave J., Aleil B., Wiesel M. In vitro evaluation of pooled buffy-coat platelets treated with photochemical pathogen inactivation using amotosalen. Letters. Vox Sanguinis. 2004;86:201–202. doi: 10.1111/j.0042-9007.2004.00417.x. [DOI] [PubMed] [Google Scholar]
- 77.Snyder E. Recovery and life span of 111 indium-radiolabeled platelets treated with pathogen inactivation with amotosalen HCL (S-59) and ultraviolet A light. Transfusion. 2004;44:1732–1740. doi: 10.1111/j.0041-1132.2004.04145.x. [DOI] [PubMed] [Google Scholar]
- 78.van Rhenen V. Transfusion of pooled buffy coat platelet components prepared with photochemical pathogen inactivation treatment: the euroSPRITE trial. Blood. 2003;101(6):2426–2433. doi: 10.1182/blood-2002-03-0932. [DOI] [PubMed] [Google Scholar]
- 79.McCullough J. Therapeutic efficacy and safety of platelets treated with a photochemical process for pathogen inactivation: the SPRINT Trial. Blood. 2004;104:1534–1541. doi: 10.1182/blood-2003-12-4443. [DOI] [PubMed] [Google Scholar]
- 80.Pinkoski T, et al. The INTERCEPT Blood System for plasma: process validation studies of coagulation factor activity and yield in two European blood centers. Abstract presented at American Society of Hematology Annual Meeting, San Diego, CA; December 4–7, 2004.
- 81.Corash L. Helinx technology for inactivation of infectious pathogens and leukocytes in labile blood components: from theory to clinical application. Transfusion and Apheresis Science. 2001;25:179–181. doi: 10.1016/s1473-0502(01)00116-1. [DOI] [PubMed] [Google Scholar]
- 82.Hambleton J. Pharmacokinetic study of FFP photochemically treated with amotosalen (S-59) and UV light compared to FFP in healthy volunteers anticoagulated with warfarin. Transfusion. 2002;42:1302–1307. doi: 10.1046/j.1537-2995.2002.00220.x. [DOI] [PubMed] [Google Scholar]
- 83.Wages D. Treatment of acquired coagulopathy by transfusion of fresh frozen plasma (FFP) prepared using a novel, single unit photochemical pathogen inactivation (P.I.) process. Blood. 1999;Nov. 15(supplement 1):247a. [Google Scholar]
- 84.Hillyer KL, et al. von Willebrand factor-cleaving protease (vWF-CP) in S-59 treated donor plasma. Abstract Presented at the American Society of Hematology, 42nd Annual Meeting; December 2000.
- 85.Mintz P.D. Theraputic plasma exchange for thrombotic thrombocytopenic purpura using plasma prepared with photochemical treatment (INTERCEPT Plasma) Blood. 2004;104:239a. [Google Scholar]
- 86.Lin L. Photochemical inactivation of viruses and bacteria in platelet concentrates by use of a novel psoralen and long-wavelength ultraviolet light. Transfusion. 1997;7:423–435. doi: 10.1046/j.1537-2995.1997.37497265344.x. [DOI] [PubMed] [Google Scholar]
- 87.van Voorhis W.C. Trypanosoma cruzi inactivation in human platelet concentrates and plasma by a psoralen (amotosalen HCL) and long-wavelength UV. Antimicrobial Agents of Chemotherapy. 2003;47(2):475–479. doi: 10.1128/AAC.47.2.475-479.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lin L. Inactivation of cytomegalovirus in platelet concentrates using Helinx™ technology. Seminars in Hematology. 2001;38(4):27–33. doi: 10.1016/s0037-1963(01)90121-0. [DOI] [PubMed] [Google Scholar]
- 89.Jordan C.T. Photochemical treatment of platelet concentrates with amotosalen hydrochloride and ultraviolet A light inactivates free and latent cytomegalovirus in a murine transfusion model. Transfusion. 2004;44:1159–1165. doi: 10.1111/j.1537-2995.2004.03351.x. [DOI] [PubMed] [Google Scholar]
- 90.Lin L. Photochemical treatment of platelet concentrates with amotosalen and longwavelength ultraviolet light inactivates a broad spectrum of pathogenic bacteria. Transfusion. 2004;44:1496–1504. doi: 10.1111/j.1537-2995.2004.04125.x. [DOI] [PubMed] [Google Scholar]
- 91.Bell CF, et al. Cost effectiveness of pathogen inactivation in blood platelet transfusion-a German model. Abstract presented at 27th Annual Congress of the International Society of Blood Transfusion, Vancouver, BC; August 2002.
- 92.van Hulst M, et al. Cost effectiveness of pathogen inactivation for platelet transfusions in Dutch cardiac surgery. Abstract presented at 27th Annual Congress of the International Society of Blood Transfusion, Vancouver, BC; August 2002.
- 93.Corash L. Inactivation of viruses, bacteria, protozoa, and leukocytes in platelet and red cell concentrates. Vox Sanguinis. 2000;78(supplement 2):205–210. [PubMed] [Google Scholar]
- 94.Cook D., Stassinopoulos A. Inactivation of pathogens in packed red cell concentrates using S-303. Blood. 1997;90(supplement 1):409a. [Google Scholar]
- 95.Cook D. In vivo analysis of packed red blood cells treated with S-303 to inactivate pathogens. Blood. 1998;92(supplement 1):503a. [Google Scholar]
- 96.Hambleton J. Post transfusion recovery after multiple exposures to red cell concentrates treated with a novel pathogen inactivation process. Blood. 1999;94(supplement 1):376a. [Google Scholar]
- 97.Hambleton J, et al. RBCs treated with Helinx™ pathogen inactivation have recovery and half-life comparable to conventional RBCs in a randomized cross-over trial. Presented at the Seventh Annual Congress of the European Hematology Association, Florence, Italy; June 2002.
- 98.Benjamin R.J. Transfusion of S-303 treated RBCs to treat acute anemia during or following cardiac surgery: results of a phase III trial. Abstract No. 381. Blood. 2004;104:112a. [Google Scholar]
- 99.Conlan M.G. Antibody formation to S-303 treated RBCs in the setting of chronic RBC transfusion. Abstract No. 382. Blood. 2004;104:112a. [Google Scholar]
- 100.Stassinopoulos A. Elimination of immunoreactivity of red cells treated with a modified S-303 pathogen inactivation process. Abstract No. 2703. Blood. 2004;104:738a. [Google Scholar]
- 101.Chapman J. Progress in improving the pathogen safety of red cell concentrates. Vox Sanguinis. 2000;78(supplement 2):203–204. [PubMed] [Google Scholar]
- 102.Chapman J. Pathogen inactivation of RBCs: PEN110 reproductive toxicology studies. Transfusion. 2003;43:1386–1393. doi: 10.1046/j.1537-2995.2003.00497.x. [DOI] [PubMed] [Google Scholar]
- 103.Chapman J., Butterworth B.E. Genetic toxicology profile of Inactine PEN110. Toxicologist. 2000;66:73. [Google Scholar]
- 104.Purmal A. Process for the preparation of pathogen-inactivated RBC concentrates by using PEN110 chemistry: preclinical studies. Transfusion. 2002;42:139–145. doi: 10.1046/j.1537-2995.2002.00020.x. [DOI] [PubMed] [Google Scholar]
- 105.Snyder E, et al. Pathogen inactivated red blood cells using INACTINE™ technology demonstrate 24 hours post transfusion recovery equal to untreated red cells after 42 days of storage. Presented at the American Society of Hematology, 43rd Annual Meeting, Orlando, FL; December 2001.
- 106.AuBuchon J. Production of pathogen-inactivated RBC concentrates using PEN110 chemistry: a phase I clinical study. Transfusion. 2002;42:146–152. doi: 10.1046/j.1537-2995.2002.00021.x. [DOI] [PubMed] [Google Scholar]
- 107.Lazo A. Broad-spectrum virus reduction in red cell concentrates using INACTIME™ PEN110 chemistry. Vox Sanguinis. 2002;83:313–323. doi: 10.1046/j.1423-0410.2002.00234.x. [DOI] [PubMed] [Google Scholar]
- 108.Ohagen A. Inactivation of HIV in blood. Transfusion. 2002;42:1308–1317. doi: 10.1046/j.1537-2995.2002.00217.x. [DOI] [PubMed] [Google Scholar]
- 109.Aytay S. Development of a sensitive PCR inhibition method to demonstrate HBV nucleic acid inactivation. Transfusion. 2004;44:476–484. doi: 10.1111/j.1537-2995.2003.03306.x. [DOI] [PubMed] [Google Scholar]
- 110.Mather T. West Nile virus in blood: stability, distribution, and susceptibility to PEN110 inactivation. Transfusion. 2003;43:1029–1037. doi: 10.1046/j.1537-2995.2003.00464.x. [DOI] [PubMed] [Google Scholar]
- 111.Ohagen A. Induction of latent human cytomegalovirus by conventional gamma irradiation and prevention by treatment with INACTINE PEN110. Vox Sanguinis. 2004;87:1–9. doi: 10.1111/j.1423-0410.2004.00532.x. [DOI] [PubMed] [Google Scholar]
- 112.Zavizion B. Inactivation of Gram-negative and Gram-positive bacteria in red cell concentrates using INACTINE PEN110 chemistry. Vox Sanguinis. 2004;87:143–149. doi: 10.1111/j.1423-0410.2004.00556.x. [DOI] [PubMed] [Google Scholar]
- 113.Zavizion B. Inactivation of mycoplasma species in blood by Inactine PEN110 process. Transfusion. 2004;44:286–293. doi: 10.1111/j.1537-2995.2004.00647.x. [DOI] [PubMed] [Google Scholar]
- 114.Zavizion B. Inactivation of protozoan parasites in red blood cells using Inactine PEN110 chemistry. Transfusion. 2004;44:731–738. doi: 10.1111/j.1537-2995.2004.03207.x. [DOI] [PubMed] [Google Scholar]
- 115.Zavizion B. Prevention of Yersinia enterolitica, Pseudomonas fluorescens, and Pseudamonas putida out growth in deliberately inoculated blood by a novel pathogen-reduction process. Transfusion. 2003;43:135–142. doi: 10.1046/j.1537-2995.2003.00294.x. [DOI] [PubMed] [Google Scholar]
- 116.Hardwick C. Separation, identification, and quantification of riboflavin and its photoproducts using high-performance liquid chromatography with fluorescence detection: a method to support pathogen reduction technology. Photochemistry and Photobiology. 2004;80:609–615. doi: 10.1562/2004-04-14-TSN-139. [DOI] [PubMed] [Google Scholar]
- 117.Unna K., Greslin J. Studies on the toxicity and pharmacology of riboflavin. The Journal of Pharmacology and Experimental Therapeutics. 1942;75:75–80. [Google Scholar]
- 118.Zempleni J. Pharmacokinetics of orally and intravenously administered riboflavin in health humans. American Journal of Clinical Nutrition. 1996;63:54–66. doi: 10.1093/ajcn/63.1.54. [DOI] [PubMed] [Google Scholar]
- 119.Tsugita A., Okada Y., Uehara K. Photosensitized inactivation of ribonucleic acid in the presence of riboflavin. Biochemica et Biophysica Acta. 1965;103:360–363. doi: 10.1016/0005-2787(65)90182-6. [DOI] [PubMed] [Google Scholar]
- 120.Joshi P. Comparison of the DNA-damaging property of photosensitized riboflavin via singlet oxygen and superoxide radical mechanisms. Toxicology Letters. 1985;26:211–217. doi: 10.1016/0378-4274(85)90169-9. [DOI] [PubMed] [Google Scholar]
- 121.Kumar V. Riboflavin and UV-light based pathogen reduction: extent and consequence of DNA damage at the molecular level. Photochemistry and Photobiology. 2004;80:15–21. doi: 10.1562/2003-12-23-RA-036.1. [DOI] [PubMed] [Google Scholar]
- 122.Piper J. Assessment of the acute toxicity and genotoxicity risks associated with lumichrome, the primary photoproduct of riboflavin. Abstract No. P575. Vox Sanguinis. 2002;83:192. [Google Scholar]
- 123.Maron D.M., Ames B.N. Revised methods for the Salmonella mutagenicity test. Mutation Research. 1983;113:173–215. doi: 10.1016/0165-1161(83)90010-9. [DOI] [PubMed] [Google Scholar]
- 124.Quillardet P., Hofnung M. The SOS chromotest, a calorimetric bacterial assay for genotoxins: procedures. Mutation Research. 1985;147:65–78. doi: 10.1016/0165-1161(85)90020-2. [DOI] [PubMed] [Google Scholar]
- 125.Kale H. Assessment of the genotoxic potential of riboflavin and lumiflavin. A. Effect of metabolic enzymes. Mutation Research. 1992;298:9–16. doi: 10.1016/0165-1218(92)90023-s. [DOI] [PubMed] [Google Scholar]
- 126.Ruane P.H. Photochemical inactivation of selected viruses and bacteria in platelet concentrates using riboflavin and light. Transfusion. 2004;44:855–877. doi: 10.1111/j.1537-2995.2004.03355.x. [DOI] [PubMed] [Google Scholar]
- 127.Li J. Pathogen reduction of buffy coat platelet concentrates using riboflavin and light: comparisons with pathogen-reduction technology-treated apheresis platelet products. Vox Sanguinis. 2004;87:82–90. doi: 10.1111/j.1423-0410.2004.00548.x. [DOI] [PubMed] [Google Scholar]
- 128.McBurney L.L. The use of riboflavin for the viral inactivation of platelets. Abstract No. 136-0401. Transfusion. 2000;40S:37S. [Google Scholar]
- 129.Goodrich L. Riboflavin photoinactivation procedure inactivates significant levels of bacteria and produces a culture negative product. Abstract No. S58-030J. Transfusion. 2002;42:16S. [Google Scholar]
- 130.Reddy H. Reduction of virus in red blood cell suspensions with riboflavin and light. Abstract No. S57-030J. Transfusion. 2002;42:16S. [Google Scholar]
- 131.Grass J. Prevention of transfusion associated graft versus host disease by photochemical treatment. Blood. 1999;93:3140–3147. [PubMed] [Google Scholar]
- 132.Fast L. Inhibition of xenogeneic GVHD by PEN 110 treatment of donor human PBMNCs. Transfusion. 2004;44:282–285. doi: 10.1111/j.1537-2995.2004.00639.x. [DOI] [PubMed] [Google Scholar]
- 133.Fast L. Inhibition of murine GVHD by PEN 110 treatment. Transfusion. 2002;42:1326–1332. doi: 10.1046/j.1537-2995.2002.00212.x. [DOI] [PubMed] [Google Scholar]
- 134.Fast L. PEN 110 treatment functionally inactivates the PBMNCs present in RBC units: comparison to the effects of exposure to gamma irradiation. Transfusion. 2002;42:1318–1325. doi: 10.1046/j.1537-2995.2002.00213.x. [DOI] [PubMed] [Google Scholar]
- 135.Reddy H, et al. Inactivation of T-lymphocytes with riboflavin and visible light. Abstract presented at the 27th Congress of the International Society of Blood Transfusion, Vancouver, BC, Canada; August 24–28, 2002.