

Abstract
Molecular diagnostic methods have evolved and matured considerably over the last several decades and are constantly being evaluated and adopted by clinical laboratories for the identification of infectious pathogens. Advancement in other technologies such as fluorescence, electronics, instrumentation, automation, and sensors have made the overall diagnostic process more accurate, sensitive, and rapid. Nucleic acid based detection procedures, which rely on the fundamental principles of DNA replication have emerged as a popular and standard diagnostic method, and several commercial assays are currently available based on different nucleic acid amplification techniques. This review focuses on the major amplification chemistries that are used for developing commercial assays and discusses their application in the clinical virology laboratory.
Keywords: PCR, LAMP, HDA, Amplification, NEAR, NASBA
1. Introduction
Clinical microbiologists are constantly striving to deliver accurate test results more quickly to ensure that appropriate treatment and infection control measures are implemented on time. Conventional diagnostic methods, such as microscopy and culture are laborious, time-consuming and require considerable technical skill for accurate analysis of the pathogen [1,2]. The technology revolution that has occurred over the last few decades has considerably improved and expanded the diagnostic capabilities of modern clinical microbiology laboratories. In particular, the development of nucleic acid amplification chemistries that allows rapid amplification and characterization of the target nucleic acid has aided in the development of numerous commercial assays for several relevant viral pathogens (Table 1 ). The amplification chemistries are often modified to detect multiple targets or to improve the overall sensitivity and specificity of the method, by controlling specific reaction conditions or by the addition of specially designed oligonucleotides and enzymes. Most of these diagnostic methods have been automated or converted to a sample-to-answer format that can manipulate smaller volumes of sample and can deliver accurate, quantifiable test results in a short amount of time. Furthermore, automation eliminates the need for extensive training of laboratory personnel and reduces the possibility of contamination and human error. This review focuses on the major amplification chemistries, including target amplification, signal amplification, and probe amplification that are currently being used for developing commercial assays for the detection of viral pathogens, discusses the advantages and disadvantages of each technique, and their application in the clinical virology laboratory.
Table 1.
Commercially available nucleic acid amplification assays for viral pathogens a.
| Product | Company | Chemistry | Viral Pathogens |
|---|---|---|---|
| Abbott RealTime CMV | Abbott | Real-time PCR | CMV |
| Aptima HPV 16 18/45 Genotype Assay | Hologic | Real-time PCR | HPV |
| artus CMV RGQ MDx Kit | Qiagen | Real-time PCR | CMV |
| artus HSV-1/2 QS-RGQ MDx Kit | Qiagen | Real-time PCR | HSV 1, HSV 2 |
| BD Onclarity™ HPV Assay | Becton Dickinson | Real-time PCR | HPV |
| COBAS® AmpliPrep/COBAS® TaqMan® CMV Test | Roche Molecular Diagnostics | Real-time PCR | CMV |
| COBAS®AmpliPrep/COBAS® TaqMan® HBV Test, v2 | Roche Molecular Diagnostics | Real-time PCR | HBV |
| cobas® CMV Test | Roche Molecular Diagnostics | Real-time PCR | CMV |
| cobas® HBV test | Roche Molecular Diagnostics | Real-time PCR | HBV |
| cobas® HPV Test | Roche Molecular Diagnostics | Real-time PCR | HPV |
| Lyra Adenovirus Assay | Quidel | Real-time PCR | Adenovirus |
| Prodesse ProAdeno™+ Assay | Hologic | Real-time PCR | Adenovirus |
| Sentosa SA201 HSV-1/2 PCR Test | Vela Diagnostics | Real-time PCR | HSV 1, HSV 2 |
| Simplexa™ HSV 1 & 2 Direct assay | Focus Diagnostics | Real-time PCR | HSV 1, HSV 2 |
| Simplexa™ Influenza A H1N1 (2009) | Focus Diagnostics | Real-time PCR | Flu A and 2009 H1N1 |
| FilmArray® Respiratory Panel (RP) | BioFire Diagnostics | Nested multiplex RT-PCR | Adenovirus, Coronavirus (229E, HKU1, NL63, OC43), hMPV, RSV, Flu A [Subtype A H1, A H1-2009, A H3], Flu B, Parainfluenza 1,2,3,4, Rhinovirus/ Enterovirus |
| FilmArray® Respiratory Panel 2 (RP2) | BioFire Diagnostics | Nested multiplex RT-PCR | Adenovirus, Coronavirus (229E, HKU1, NL63, OC43), hMPV, RSV, Flu A [Subtype A H1, A H1-2009, A H3], Flu B, Parainfluenza 1,2,3,4, Rhinovirus/ Enterovirus |
| FilmArray® Respiratory Panel EZ (RP EZ) | BioFire Diagnostics | Nested multiplex RT-PCR | Adenovirus, hMPV, Coronavirus, Rhinovirus/Enterovirus, Flu A [Subtype A H1, A H1-2009, A H3], Flu B, Parainfluenza, RSV |
| FilmArray Gastrointestinal (GI) Panel | BioFire Diagnostics | Nested multiplex RT-PCR | Adenovirus F 40/41, Astrovirus, Norovirus GI/GII, Rotavirus A · Sapovirus (I, II, IV, and V) |
| Abbott RealTime HCV Genotype II | Abbott | RT-PCR | HCV |
| Abbott RealTime HBV Viral Load Assay | Abbott | RT-PCR | HBV |
| Abbott RealTime HCV Viral Load Assay | Abbott | RT-PCR | HCV |
| artus® Infl A/B RG RT-PCR Kit | Qiagen | RT-PCR | Flu A, Flu B |
| Abbott RealTime HIV-1 Viral Load Assay | Abbott | Real-time RT-PCR | HIV-1 |
| AMPLICOR HIV-1 MONITOR™ Test, v1.5 | Roche Molecular Diagnostics | Real-time RT-PCR | HIV-1 |
| Aptima HPV 16 18/45 Genotype Assay | Hologic | Real-time PCR | HPV |
| artus CMV RGQ MDx Kit | Qiagen | Real-time PCR | CMV |
| artus HSV-1/2 QS-RGQ MDx Kit | Qiagen | Real-time PCR | HSV 1, HSV 2 |
| BD Onclarity™ HPV Assay | Becton Dickinson | Real-time PCR | HPV |
| CDC DENV-1-4 Real-Time RT-PCR Assay | CDC | Real-time RT-PCR | Dengue virus |
| COBAS® AMPLICOR HIV-1 MONITOR™ Test | Roche Molecular Diagnostics | Real-time RT-PCR | HIV-1 |
| COBAS® AmpliPrep/COBAS® TaqMan® CMV Test | Roche Molecular Diagnostics | Real-time PCR | CMV |
| COBAS® AmpliPrep/COBAS® TaqMan HIV-1 Test | Roche Molecular Diagnostics | Real-time RT-PCR | HIV-1 |
| COBAS® AmpliPrep/COBAS® TaqMan® HBV Test, v2 | Roche Molecular Diagnostics | Real-time PCR | HBV |
| COBAS® AmpliPrep/COBAS® TaqMan® HCV Test | Roche Molecular Diagnostics | Real-time RT-PCR | HCV |
| cobas® CMV Test | Roche Molecular Diagnostics | Real-time PCR | CMV |
| cobas® HCV Test | Roche Molecular Diagnostics | Real-time RT-PCR | HCV |
| Prodesse Pro hMPV + Assay | Hologic | Real-time RT-PCR | hMPV |
| RIDA GENE Norovirus GI/GII | R-Biopharm AG | Real-time RT-PCR | Norovirus GI/GII |
| Simplexa™ Flu A/B & RSV | Focus Diagnostics | Real-time RT-PCR | Flu A/B & RSV |
| Simplexa™ Flu A/B & RSV Direct | Focus Diagnostics | Real-time RT-PCR | Flu A, Flu B, RSV |
| eSensor® Respiratory Viral Panel | GenMark Diagnostics, Inc. | Multiplex real-time PCR | Adenovirus, Rhinovirus, hMPV, RSV A, RSV B, Flu A [Subtype A H1, A H1-2009, A H3], Flu B, Parainfluenza 1,2,3 |
| Lyra Direct HSV 1 + 2/VZV Assay | Quidel | Multiplex real-time PCR | HSV 1, HSV 2, VZV |
| Lyra RSV + hMPV Assay | Quidel | Multiplex real-time PCR | RSV, hMPV |
| Lyra™ Parainfluenza Virus Assay | Quidel | Multiplex real-time PCR | Parainfluenza 1,2,3 |
| Panther Fusion AdV/hMPV/RV assay | Hologic | Multiplex real-time PCR | Adenovirus, hMPV, Rhinovirus |
| Panther Fusion Flu A/B/RSV assay | Hologic | Multiplex real-time PCR | Flu A, Flu B, RSV |
| Panther Fusion Paraflu assay | Hologic | Multiplex real-time PCR | Parainfluenza 1,2,3,4 |
| Prodesse ProFAST®+ Assay | Hologic | Multiplex real-time PCR | Flu A/H1, seasonal influenza A/H3 and 2009 H1N1 |
| Prodesse ProFlu® + assay | Hologic | Multiplex real-time PCR | Flu A, Flu B, RSV |
| Prodesse ProParaflu + assay | Hologic | Multiplex real-time PCR | Parainfluenza 1, 2, 3 |
| cobas® Liat Influenza A/B & RSV | Roche Molecular Diagnostics | Multiplex real-time RT-PCR | Flu A, Flu B, RSV |
| ePlex® Respiratory Pathogen Panel | GenMark Diagnostics, Inc. | Multiplex PCR and multiplex RT-PCR | Adenovirus,Coronavirus (229E, HKU1, NL63, OC43), hMPV, RSV A, RSV B, Flu A [Subtype A H1, A H1-2009, A H3], Flu B, Parainfluenza 1,2,3,4, Rhinovirus/ Enterovirus |
| Idylla Respiratory (IFV-RSV) Panel | Janssen Diagnostics | Multiplex real-time RT-PCR | Flu A, RSV |
| Lyra Influenza A + B Assay | Quidel | Multiplex real-time RT-PCR | Flu A, Flu B |
| NxTAG® Respiratory Pathogen Panel | Luminex Corporation | Multiplex RT-PCR | Adenovirus, Coronavirus (229E, HKU1, NL63, OC43), hMPV, RSV, Flu A [Subtype A H1, A H3], Flu B, Parainfluenza 1,2,3,4, Rhinovirus/ Enterovirus, Bocavirus |
| VERIGENE® Respiratory Pathogens Flex Nucleic Acid Test (RP Flex) | Luminex Corporation | Multiplex RT-PCR + microarray detection | Adenovirus, hMPV, Flu A [Subtype H1, H3], Flu B, RSV A, RSV B, Parainfluenza 1,2,3,4, Rhinovirus |
| Xpert® Norovirus | Cepheid | Multiplex real-time RT-PCR | Norovirus GI/GII |
| Xpert® Xpress Flu/RSV | Cepheid | Multiplex real-time RT-PCR | Flu A, Flu B, RSV |
| xTAG® Gastrointestinal Pathogen Panel (GPP) | Luminex Corporation | Multiplex RT-PCR | Norovirus GI/GII, Rotavirus A |
| xTAG® Respiratory Viral Panel FAST | Luminex Corporation | Multiplex end point RT-PCR | Adenovirus, Coronavirus (229E, HKU1, NL63, OC43), hMPV, RSV, Flu A [Subtype A H1, A H1-2009, A H3], Flu B, Parainfluenza 1,2,3,4, Rhinovirus/ Enterovirus, Bocavirus |
| ARIES® HSV1&2 Assay | Luminex Corporation | MultiCode® RTx | HSV 1, HSV 2 |
| ARIES® Flu A/B & RSV Assay | Luminex Corporation | Multicode® RTx | Flu A, Flu B, RSV |
| NUCLISENS® CMV PP67 | Organon Teknika Corp. | NASBA | CMV |
| NUCLISENS® EASYQ® HIV-1 v2.0 | bioMérieux | Real-time NASBA | HIV-1 |
| NUCLISENS® EASYQ® HPV | bioMérieux | Real-time NASBA | HPV |
| Aptima HSV 1 & 2 assay | Hologic | Real-time TMA | HSV 1, HSV 2 |
| Aptima HCV Quant Dx Assay | Hologic | Real-time TMA | HCV |
| Aptima HBV Quant Dx Assay | Hologic | Real-time TMA | HBV |
| illumigene® HSV 1&2 DNA Amplification Assay | Meridian Bioscience, Inc. | LAMP | HSV 1, HSV 2 |
| Alere™ i Influenza A & B Test | Abbott | NEAR | Flu A, Flu B |
| Alere™ i RSV | Abbott | NEAR | RSV |
| AmpliVue® HSV 1 + 2 Assay | Quidel | HDA | HSV 1, HSV 2 |
| Solana® HSV 1 + 2/VZV Assay | Quidel | HDA | HSV 1, HSV 2, VZV |
| Solana® Influenza A + B Assay | Quidel | Real-time RT-HDA | Flu A, Flu B |
| Solana® Respiratory Viral Panel | Quidel | Real-time RT-HDA | RSV A, RSV B, hMPV, Flu A, Flu B |
| VERSANT® HIV-1 RNA 3.0 Assay (bDNA) | Siemens Healthcare Diagnostics | bDNA | HIV-1 |
| Versant HCV 3.0 Assay (bDNA) | Siemens Healthcare Diagnostics | bDNA | HCV |
| digene HC2 HPV DNA Test | Qiagen | Hybrid Capture | HPV |
FDA 510(k) Premarket Notification. (2018). Retrieved from https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm. Accessed on November 14, 2018.
2. Target amplification techniques
Target amplification is the most popular method for DNA and RNA analysis and has been widely implemented for the detection of viral pathogens. In this method, nucleic acids of interest are enzymatically amplified and the amplified products can be detected by various methods such as electrophoresis, electrochemiluminescence, or labeled oligonucleotides that hybridize to the complementary target sequence. Target amplification techniques include several chemistries such as the polymerase chain reaction (PCR), nucleic acid sequence–based amplification (NASBA), transcription mediated amplification (TMA), loop-mediated isothermal amplification (LAMP), and strand displacement amplification (SDA), which are described in detail below.
2.1. Polymerase chain reaction (PCR)
The development of PCR by Kary Mullis in the 1980s revolutionized the detection of microbial pathogens in clinical specimens. PCR is a highly sensitive method that involves cyclical heating and cooling of the reaction mixture to facilitate amplification of the target DNA [3]. The method consists of three basic steps (denaturation, annealing, and extension) and the reaction mixture requires four primary components (template DNA, primers, nucleotides, and a DNA polymerase) (Fig. 1 a). Double-stranded target DNA (dsDNA) is thermally denatured to form a single-stranded DNA template (ssDNA). Oligonucleotide primers anneal to complementary target sequences on the nucleic acid template and are extended by the DNA polymerase, thus creating the resultant PCR product (amplicon). The reaction occurs in a programmable thermal cycler, and the steps are repeated for several cycles (n) to produce multiple copies (2n) of the target sequences [4]. The end product formed can be detected by various methods such as agarose gel electrophoresis, colorimetric detection, and chemiluminescence. Gel electrophoresis uses fluorescent dyes such as ethidium bromide, SYBR Green, SYBR Gold or SYBR Safe, that can intercalate between the stacked DNA bases and fluoresce under UV exposure [5]. In colorimetric detection, labeled oligonucleotides are incorporated within the target DNA, and the complex formed is detected using a chromogenic substrate [6]. In chemiluminescence detection, enzyme catalyzed chemical reactions involving the amplified nucleic acid emit energy in the form of light, which can be visualized by autoradiography or CCD imaging [7].
Fig. 1.

a. Polymerase Chain Reaction; Figure 1b. SYBR Green and TaqMan Detection Chemistry; Figure 1c. Nested PCR; Figure 1d. Reverse-Transcription PCR (RT-PCR).
Alternatively, in real-time PCR (also known as quantitative PCR or qPCR), amplification and detection occur simultaneously, which can eliminate PCR post-processing steps and may reduce the overall turnaround time, depending on the assay design. The accumulated amplification product is measured in real time as the reaction progresses and thus quantified after each cycle, although real-time PCR can be used for both qualitative and quantitative DNA analysis. This method employs dsDNA intercalating dyes (SYBR Green I and EvaGreen) or fluorophore-labeled oligonucleotides (Scorpions, Amplifluor, TaqMan, etc.) for detection and quantification of the amplified products [8]. The fluorescent-labeled oligonucleotides can be further subdivided into three categories based on the type of fluorescent molecule used – primer probes (Scorpions, Amplifluor, etc.), hydrolysis probes (TaqMan, etc.), and hybridization probes (FRET, Molecular Beacons, etc.). While the dsDNA intercalating dyes increase fluorescence after binding to the minor groove of the DNA, the fluorophore-labeled oligonucleotides either hybridize to the PCR product, or undergo hydrolysis, which then separates the fluorophore from a quenching dye and results in an increased signal (Fig. 1b). Quenching chemistries such as MultiCode®-RTx uses the synthetic DNA basepair 2ʹ-deoxy-5-methyl-isocytidine (iC):2ʹ-deoxyisoguanosine (iG) (isobases) for site-specific incorporation of a quencher molecule that quenches fluorescence as a measure of amplification in real-time [9]. Commercial real-time PCR assays have been developed for the detection and quantification of most relevant viruses – Epstein-Barr virus (EBV) [10], hepatitis C virus (HCV) [11], hepatitis B virus (HBV) [12], herpes simplex virus 1&2 (HSV 1&2) [13], and human immunodeficiency virus (HIV) [14].
Conventional PCR has been modified over time to increase sensitivity and specificity (nested-PCR), detect RNA targets (reverse-transcription or RT-PCR), and amplify multiple targets in the same reaction mixture (multiplex PCR). Nested PCR uses two sets of amplification primers and two successive PCR reactions [15]. In the first stage PCR, the first set of primers binds to the DNA template on either side of the target sequence so that there is some sequence flanking both ends of the target (Fig. 1c). The amplified product generated is used as a template for the second stage reaction where a second set of primers anneals to sequences internal to the first set. The sensitivity and specificity are increased by this method as the first amplicon provides more template for the second reaction and the second primer set can only anneal if the first amplicon has complementary sequences. The amplified product can be detected in the same way as conventional PCR [16]. Some disadvantages of nested PCR can be long turnaround time due to time-consuming post-PCR steps, difficulty to automate the procedure if the reactions are not performed in a single tube and require tube transfer, and increased risk of amplicon contamination because of high amplicon yield, particularly when the process employs separate tubes for each stage [17,18]. Commercial nested PCR and multiplex nested PCR assays have largely overcome these limitations and have been developed for the molecular diagnosis of respiratory viruses such as Flu A & B, coronavirus, enterovirus, respiratory syncytial virus (RSV), parainfluenza, rhinovirus, adenovirus, bocavirus, and human metapneumovirus (hMPV) [19], viruses causing meningitis and encephalitis such as cytomegalovirus (CMV), enterovirus, HSV 1 & 2, human herpesvirus type 6 (HHV-6), human parechovirus, and varicella -zoster virus (VZV) [20], and gastrointestinal viruses such as adenovirus F 40/41, astrovirus, norovirus GI/GII, rotavirus A, and sapovirus [21]. These assays have demonstrated 94–100% sensitivity and >94% specificity for the viral targets in the various clinical trial studies.
PCR can also amplify RNA targets when preceded by a reverse transcription (RT) reaction at 42–55 °C [22]. In RT-PCR, target RNA templates are converted to their complementary DNA (cDNA) sequences by reverse transcriptase (Fig. 1d). The newly synthesized cDNA is then amplified by conventional PCR. Commercial RT-PCR kits are available for the detection of HCV [23], severe acute respiratory syndrome-associated coronavirus (SARS-CoV) [24], dengue virus [25], and HIV-1 virus [26].
PCR amplification can be also multiplexed to detect several targets in a single reaction which is beneficial for infection control, timely treatment decisions, and is substantially less expensive than detecting individual pathogens by monoplex real-time PCR [27,28]. In multiplex PCR, multiple primer pairs complementary to specific targets of interest are used to simultaneously amplify their corresponding DNA fragments in a single multiplex reaction or in multiple parallel singleplex reactions. For developing sensitive and specific multiplex PCR panels, care should be taken during design to ensure compatibility of primers and PCR conditions [29]. Commercial multiplex syndromic panels are available for the detection of respiratory [[30], [31], [32]] and gastrointestinal viruses [33].
2.2. Isothermal target amplification
The necessity of stringent temperature control can make PCR an expensive and time-consuming process that would be difficult to implement in low resource and field settings [34]. Unlike PCR, isothermal amplification methods are carried out at a constant temperature that eliminates the need for sophisticated and expensive thermal cyclers. Isothermal methods are rapid, sensitive, require less energy than PCR, and are easier to implement in a point-of-care or low resource setting. Additionally, the risk of contamination is reduced as these methods are usually performed in enclosed microsystems or portable devices. The primary challenges for isothermal amplification include designing compatible primer pairs, requirement for specific assay optimization, and potential for generation of nonspecific amplified products [35].
2.2.1. Nucleic acid sequence based amplification (NASBA)
NASBA is a sensitive, isothermal amplification system that is primarily used for the identification of RNA targets. Three enzymes are used in this reaction – avian myeloblastosis virus reverse transcriptase, RNase H, and T7 RNA polymerase that ultimately produce ssRNA as the terminal amplification product [36]. A sequence-specific primer (P1) binds the target sequence to be amplified and reverse transcriptase extends to produce a RNA–DNA hybrid. RNase H degrades the RNA portion of the hybrid and the second primer (P2) binds to the remaining cDNA (Fig. 2 ). A second primer extension generates the complementary strand, forming a dsDNA molecule. The P1 primer includes a T7 RNA polymerase promoter site so that multiple RNA copies can be transcribed from the DNA template. The ssRNA end product can be detected by various methods such as gel electrophoresis, enzyme-linked gel assay, enzymatic bead based detection, and electrochemiluminescence [37]. The primary concern for NASBA is RNA integrity and controlling the reaction temperature, as the enzymes involved are thermolabile. Commercial NASBA assays are clinically used for the detection of RSV [38], HIV-1 [39], CMV [40] and human papillomavirus (HPV) [41]. A multiplex NASBA assay for HIV-1 and HCV has been developed and demonstrated an analytical sensitivity of 1000 copies/mL, 93.3% clinical sensitivity, and 100% specificity [42].
Fig. 2.

Nucleic Acid Sequence Based Amplification (NASBA). Adapted from Tröger et al., 2015 [67].
2.2.2. Transcription mediated amplification (TMA)
Similar to NASBA, TMA is an isothermal amplification method that utilizes reverse transcriptase to synthesize cDNA from the target RNA template, and then uses RNA polymerase to generate complementary RNA molecules from the cDNA, thus amplifying the original RNA target of interest [43]. Since RNA is more labile than DNA, the possibility of carry-over contamination is reduced in TMA assays. The rapid kinetics of TMA can produce 100–1000 copies of target RNA per reaction cycle that ultimately results in greater than 109-fold amplification in a short period of time. The amplicons generated by TMA can be detected by gel electrophoresis or by oligonucleotide probes; however, detection using specific methods, such as hybridization protection assay (HPA) with radioactive or non-radioactive probes, allows amplification and detection in a single tube thereby minimizing the risk of amplicon contamination [44]. In HPA, acridinium ester (AE) labeled sequence-specific oligonucleotide probes are hybridized with the TMA end product, and the chemiluminescent signal from the probe-amplicon hybrids is measured [45]. The performance of TMA is critically dependent on primer design, and like NASBA, precise control of the reaction temperature is important for denaturing the secondary structures. Commercial TMA assays are available for the detection and quantification of HIV-1, HCV, and HBV and have demonstrated equivalent performance when compared to commercial real-time PCR assays [46].
2.2.3. Strand displacement amplification (SDA)
SDA is an isothermal amplification reaction for DNA and RNA targets that can be performed in three simple steps – a) heat denaturation of the target DNA in the presence of primers and other reagents; b) addition of HincII restriction endonuclease and 3′ → 5′ exo minus Klenow fragment; and c) incubation of the sample at 37 °C [47]. In the first stage of the reaction, the target DNA sequence is duplicated using four primers in the presence of DNA polymerase and results in the addition of restriction endonuclease sites at each end of the amplified target (Fig. 3 ). In the second stage, multiple cycles involving nicking of the restriction endonuclease site, extending the nicks by DNA polymerase, and strand displacement occur, resulting in exponential amplification of the target. The amplified target can be detected by photometry, sequence-specific labeled probes, and chemiluminescence [48,49]. The limitations of SDA include its inefficiency in amplifying long target sequences and the probability of producing considerable background reactions due to unspecific primer binding [50,51]. A commercial SDA assay developed for the detection of HSV 1 & 2 has demonstrated a sensitivity of 96.7–100% and 98.4–100%, and specificity of 95.1–99.4% and 80.6–97% for HSV-1 and HSV-2, respectively [52].
Fig. 3.

Strand Displacement Amplification (SDA). Adapted from Tröger et al., 2015 [67].
2.2.4. Loop-mediated isothermal amplification (LAMP)
LAMP is an isothermal amplification that uses 4–6 sets of different primers which can individually recognize up to 6–8 distinct sequences on the target DNA [53]. The four core primers employed by the LAMP system are FIP (forward inner primer), BIP (backward inner primer), F3 (forward primer) and B3 (backward primer). The LAMP reaction starts when FIP binds to the F2c region of the target DNA and initiates complementary strand synthesis [54] (Fig. 4 ). Following strand synthesis, the F3 primer hybridizes to the target DNA and is extended by DNA polymerase. Primer extension displaces the FIP-linked complementary strand which then forms a self-hybridizing loop structure at the 5’ end. This strand serves as a template for the BIP, which initiates complementary strand synthesis and opens up the 5’ end loop. After the complementary strand is synthesized, hybridization, extension, and displacement reactions are repeated with the B3 primer which generates a double looped dumbbell-shaped DNA. Nucleotides are added to the 3’ open end and the dumbbell-shaped DNA is converted to a stem-loop structure. This stem-loop DNA structure serves as the initiator for the second stage, i.e., exponential LAMP cycling. In this stage, the FIP hybridizes to the loop, initiates strand synthesis, thereby displacing the F1 strand to form a new loop at the 3’ end. The nucleotide addition step is repeated, followed by DNA extension that displaces the FIP strand and again forms a dumbbell-shaped DNA. Subsequent self-primed strand displacement DNA synthesis yields one complementary structure of the original stem-loop DNA and one gap repaired stem-loop DNA. Both of these products are used as a template by the BIP. The annealing and displacement cycles are repeated and as the reaction proceeds, the product grows to form long concatamers. The amplified products can be detected by indirect detection methods like turbidity and non-specific dyes [53].
Fig. 4.

Loop-mediated Isothermal Amplification (LAMP). Adapted from PREMIER Biosoft Tech Note. Accessed on October 30, 2018 from http://www.premierbiosoft.com/tech_notes/Loop-Mediated-Isothermal-Amplification.html.
Compared to conventional PCR, LAMP assays are highly sensitive, rapid, cost-effective, and can be easily implemented in resource-limited settings [55]. However, complicated primer design and the possibility of carry-over contamination still remain a concern for LAMP-based assays [50,56]. A commercial LAMP assay demonstrating >94% sensitivity and >95% specificity is available for the detection of HSV 1 & 2 [57]. LAMP-based assays have also been developed for the detection of emerging viral pathogens such as SARS-CoV [58], chikungunya, and dengue virus [59]. Reverse transcriptase LAMP (RT-LAMP) assays have been developed for the detection of several viruses such as foot-and-mouth disease virus [60], zika virus [61], influenza virus [62], CMV [63], HPV [64], BK [65], VZV and HSV viruses [66].
2.2.5. Nicking enzyme amplification reaction (NEAR)
NEAR is an isothermal amplification method that relies on the rapid identification of small DNA or RNA fragments that are directly generated from the target nucleic acid [67]. In this method, a set of target-specific primers, a nicking endonuclease, and a strand displacing DNA polymerase are used for isothermal unwinding and subsequent generation and amplification of these small fragments (Fig. 5 ). Insertion of a nicking enzyme recognition site inside the target sequence facilitates the amplification procedure. The DNA polymerase initiates primer extension at the nick created by a nicking enzyme and releases small oligonucleotide fragments. Repeated nicking, polymerization, and strand displacement result in linear amplification of these small oligonucleotides, and the amplicons generated can be detected using a variety of probe formats such as molecular beacons and lateral flow sandwich. The primary limitation of the NEAR reaction includes generation of a large number of non-specific products that not only inhibit the progression of the forward reaction, but also limit the detection sensitivity, and thus may require more specific and accurate amplification analysis methods for avoiding detection errors [68].
Fig. 5.

Nicking Enzyme Amplification Reaction (NEAR). Adapted from Niemz et al., 2011 [120].
Commercial point-of-care (POC) NEAR assays are available for Flu A/B and RSV which can provide results within 5–7 minutes and have demonstrated 91.8–97.8% sensitivity and 85.6–96.3% specificity for Flu A/B as compared to viral culture [69,70]. However, when compared to multiplex real-time RT-PCR assays, the POC NEAR assay has demonstrated 71.3–93.8% sensitivity for Flu A, and 91.8–93.3% sensitivity for Flu B [71,72]. The poorer sensitivity has been attributed to low positive samples that were below the limit of detection for the NEAR assay. The specificity reported was 62.5–100% for Flu A, and 53.6–100% for Flu B. The assay yielded the incorrect type of Flu virus for 22 out of 38 discrepant samples [71]. In addition, 17 of those 22 samples were actually positive for both Flu A and Flu B.
2.2.6. Helicase-dependent amplification (HDA)
HDA is an isothermal process of DNA amplification that relies on the complementary strand displacing ability of a thermostable DNA helicase [73]. Using the free energy from ATP hydrolysis, helicase, in the presence of the accessory protein MutL, catalyzes the unwinding of the dsDNA (Fig. 6 ). The unwound ssDNA strands are then coated with ssDNA-binding proteins, and sequence-specific primers are hybridized at the 3’-end of each ssDNA template. DNA polymerase extends the annealed primers and converts the ssDNA to dsDNA, which again acts as the substrate for helicase, thus initiating the subsequent rounds of the exponential amplification of the target sequence. Although helicases can unwind dsDNA at a single temperature, it has been observed that an initial heat-induced denaturation and annealing step can increase the amplification efficiency and the yield by 40%–60% [73]. The sensitivity, speed, and robustness of HDA assays can be further improved by restriction endonuclease-mediated DNA helicase homing, macromolecular crowding agents, and optimization of reaction enzyme mix [74]. HDA assays are critically dependent on primer design, and are often vulnerable to primer-dimer amplicon contamination [75]. Amplification of the primer-dimers in place of the target nucleic acid leads to erroneous detection and remains one of the major concerns for HDA based assays. Currently, HDA-based commercially available assays have been developed for the detection of HSV 1 & 2, Flu A & B, RSV, and hMPV. The HSV HDA assay has demonstrated a positive percent agreement (PPA) of 98.2% and a negative percent agreement (NPA) of 90.9% when compared to shell vial virus culture and immunofluorescence typing [76]. Like other isothermal reactions, HDA assays are rapid, sensitive, inexpensive, can be performed without sophisticated instrumentation, and can be successfully implemented in low resource settings [77].
Fig. 6.

Helicase-Dependent Amplification (HDA). Adapted from Tröger et al., 2015 [67].
2.2.7. Rolling circle amplification (RCA)
RCA is an isothermal process that uses DNA or RNA polymerase to generate ssDNA or RNA strands utilizing the principles of rolling circle replication [78]. An initiating primer is annealed to the replication origin site of the circular template and the polymerase facilitates the continuous addition of nucleotides to the leading strand, resulting in long ssDNA or RNA with tandem repeats (Fig. 7 ). Depending on the number of annealed primers, RCA can be classified as linear RCA (produces up to 105 copies) or exponential RCA (produces up to 109 copies) [79]. In linear RCA, a single complementary primer generates a single amplified product that is linked to the initiating primer. Exponential RCA uses two primers, where the second primer binds to the ssDNA product of the first primer and initiates further amplification. Circulating oligonucleotide probes or padlock probes (PLPs) can further enhance the specificity and multiplexing ability of RCA [80]. The 5′ and 3′ ends of these probes are designed to recognize adjacent sequences on the target strand, and when properly hybridized, the ends can be joined using ligase to result in circularized probes. Following circularization, the probe is replicated by RCA and the products are labeled and detected using various signal readout techniques such as gel electrophoresis, fluorescence spectroscopy, and microscopy [78]. High specificity is ensured since the hybridization and circularization can only occur when the target sequence is correctly recognized by both ends of the probe.
Fig. 7.

Rolling Circle Amplification (RCA).
RCA-based assays have been developed for the full-length genome amplification of HBV from samples with viral loads as low as 10 copies/reaction [81]. Commercial RCA kits that aid in the preparation of circular DNA templates for cycle sequencing, cloning, and transformation have been used to detect plant viruses [82]; however, currently no commercial RCA assays are available for detection of viral pathogens that affect humans. Several challenges have limited the application of RCA for commercial diagnostic purposes, including technical concerns such as preparation of large quantities of high purity circular templates, enzyme efficiency on small DNA substrates, and extensive optimization requirements for RCA product length, sequence, and composition [78].
3. Signal amplification techniques
Signal amplification is an alternative to enzymatic amplification that amplifies and detects the signal generated by an external probe molecule that binds to the target nucleic acid. The signal produced is directly proportional to the amount of target present in the reaction mix, and therefore this method is also suitable for developing quantitative assays. The sensitivity and specificity are determined by the design of the probe molecule that recognizes the target sequences.
3.1. Branched DNA assays (bDNA)
The bDNA technology is a powerful tool for reliable quantification of targeted nucleic acids and is primarily used for monitoring patients on antiviral therapy for HIV, HCV, and HBV [83]. Unlike PCR that relies on in vitro amplification of the target sequences, bDNA is based on a series of specific hybridization reactions and chemiluminescent detection of hybridized probes [84]. The assay uses multiple synthetic capture oligonucleotides (label extenders and capture extenders) that are hybridized to complementary regions of the target nucleic acid to form a sandwich complex of probes and target sequences (Fig. 8 ). The capture extenders can bind efficiently to the targeted nucleic acid sequences as well as the capture probes that are attached on an immobilized surface, such as a microtiter plate. Once the capture extenders are hybridized to the capture probes, the sample containing the viral nucleic acid is added for hybridization. Following hybridization, the label extenders are added to the mix and bind to contiguous regions on the target and also to a preamplifier oligonucleotide. Hybridization of the label extenders to the preamplifier molecule initiates the signal amplification process. The preamplifier molecule also harbors regions that hybridize to multiple bDNA amplifier molecules forming a branched structure. This branched structure is finally bound to alkaline phosphatase (AP)-labeled oligonucleotides, which are complementary to bDNA amplifier sequences. The chemiluminescent product of the AP reaction generates the bDNA signal that is directly proportional to the amount of DNA or RNA target present in the original sample.
Fig. 8.
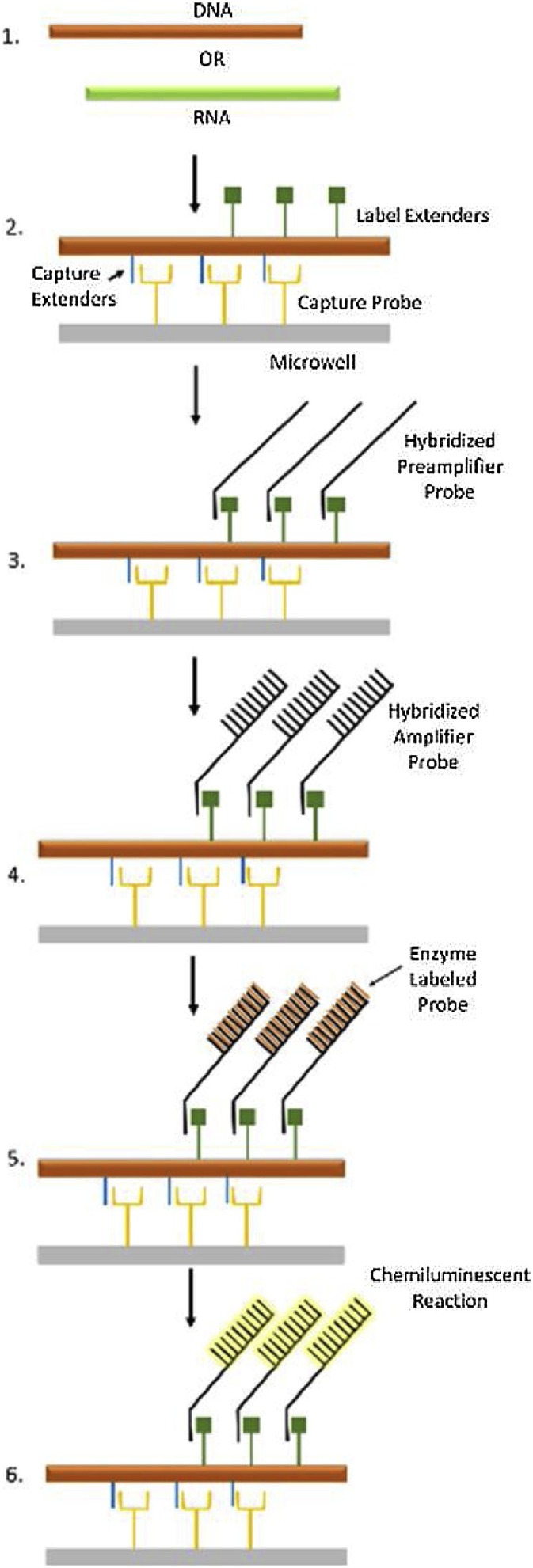
Branched DNA (bDNA). Adapted from Schutzbank, 2013 [85].
The complexity of the method in addition to the intellectual property surrounding bDNA technology has limited its application in the development of molecular infectious disease assays [85]. Currently, only two U.S. FDA approved commercial bDNA assays are available – Versant® HIV-1 RNA 3.0 assay for HIV and Versant® HCV 3.0 Assay for HCV. The dynamic range of the HIV and HCV assays are 7.5 × 101 – 5 × 105 copies/mL and 6.15 × 102 – 7.69 × 106 IU/mL, respectively [86,87]. However, when compared to commercial real-time PCR assays, the bDNA-based HBV assay has demonstrated lower sensitivity and less linear dynamic range (3.57 × 102 – 1.79 × 107 for bDNA vs. 5.6 × 101 – 5.00 × 108 for real-time PCR), thereby indicating that these assays might be insufficient for detecting very low levels of viral load [88]. In the last decade, bDNA technology has been used to develop modified fluorescent in situ hybridization (FISH) assays for the detection of HBV [89] and oncogenic FGFR3-TACC3 fusion genes [90]. bDNA has been also adapted to the Luminex xMAP® bead array technology for direct quantification of DNA, RNA, and protein targets [91].
3.2. Hybrid capture assays
Hybrid capture technology relies on the capture and detection of RNA and DNA hybrid complexes [85]. In this method, a target DNA is hybridized to synthetic complementary RNA probes, generating RNA-DNA hybrids (Fig. 9 ). The RNA-DNA hybrids are then captured onto a solid phase that is coated with universal capture antibodies specific for RNA-DNA hybrids. The antibody-hybrid conjugates are then detected by anti-RNA-DNA monoclonal antibody conjugated to several AP molecules. Multiple detection antibodies bind to the RNA-DNA hybrid, resulting in signal amplification. The amplified signal is then detected using a chemiluminescent substrate measured on a luminometer.
Fig. 9.

Hybrid Capture Assays.
Currently, the only commercial hybrid capture assay available is for the detection of HPV. The Hybrid Capture 1 (HC1) assay was the first commercially available assay for diagnosis of HPV [92]. Since then, an updated version of the HPV hybrid capture assay (HC2) has been released that has demonstrated comparable performance with other molecular diagnostic methods and is considered to be a reliable and robust assay for cervical cancer screening. [[93], [94], [95]]. However, the HC2 HPV assay has a relatively lower analytical sensitivity (˜5000 copies of HPV DNA) when compared to PCR [85]. But, for HPV screening, the clinical sensitivity and specificity are more important than analytical performance for predicting high-risk cases of cervical cancer over positive results which may be clinically irrelevant [96]. The primary limitation of the HC2 assay is its inability to distinguish between different strains of HPV which is essential for efficient risk analysis and to determine persistence of infection [97]. However, the future of hybrid capture assays seems questionable as the manufacturer appears to be more focused on traditional target amplification methodologies as well as other newer technologies [85].
4. Probe amplification techniques
Probe amplification techniques rely on the hybridization and amplification of synthetic oligonucleotide probes that are complementary to the target nucleic acid. The reaction generates multiple copies of the bound probe, which are then detected by various methods such as gel electrophoresis or fluorescent label detection. The method consists of four major steps – sample preparation and denaturation of the target DNA, hybridization of the labeled probe, removal of the unbound probe, and detection of the hybridized probe.
4.1. Ligase chain reaction (LCR)
LCR chemistry uses four synthetic oligonucleotides (each 20–35 nucleotides in length) – two adjacent oligonucleotide primers that uniquely hybridize to one strand of the target DNA and a complementary set of adjacent oligonucleotides that bind to the other strand of the target DNA [98] (Fig. 10 ). In a nick-closing reaction, thermostable DNA ligase joins the two adjacent oligonucleotide sets only when the nucleotides are perfectly basepaired at the oligonucleotide junction and are hybridized to the complementary sequence in a 3′ to 5′ orientation on the same strand of the target DNA [99]. The ligated products then serve as templates for the subsequent reaction cycles that lead to an exponential amplification, similar to PCR. The LCR products generated can be differentiated and detected by several methods such as polyacrylamide gel electrophoresis, fluorescent label detection, and autoradiography.
Fig. 10.
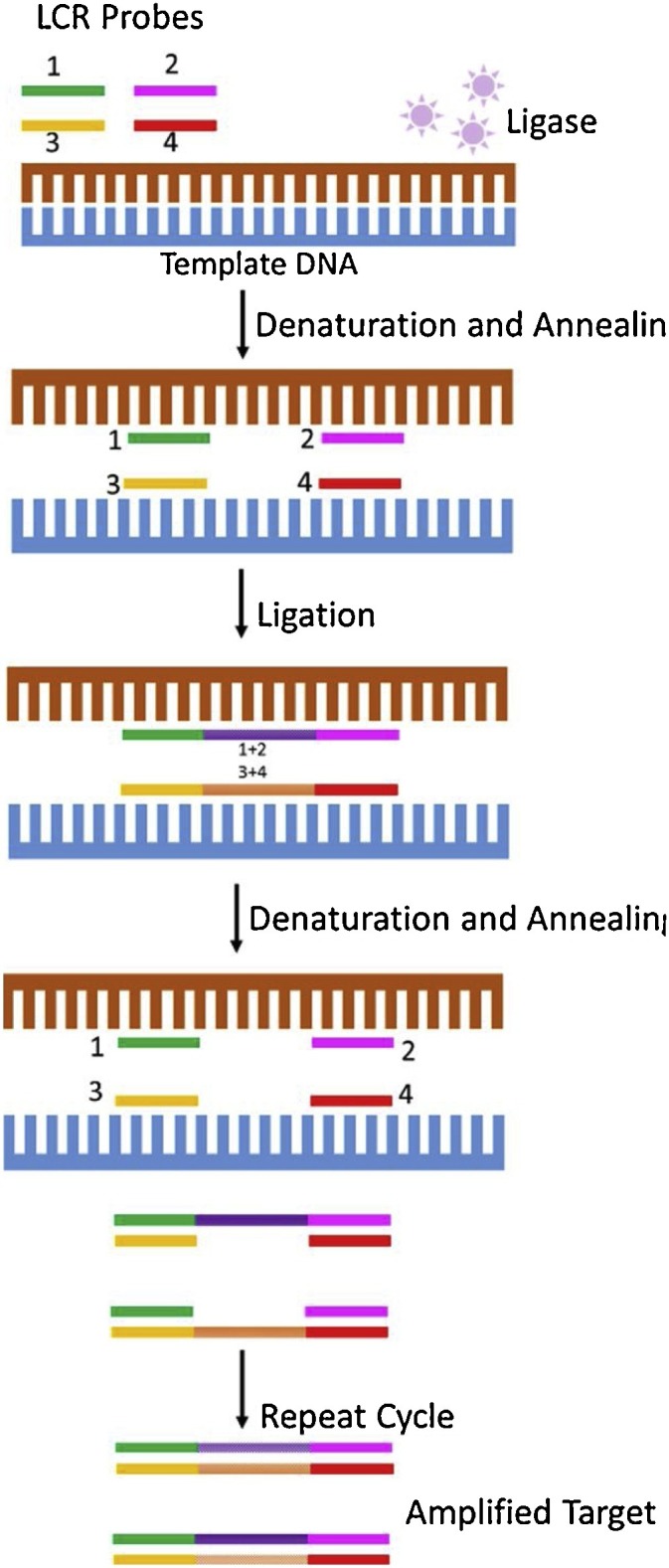
Ligase Chain Reaction (LCR). Adapted from Epicentre: An illumina company. Accessed on October 30, 2018 from http://www.epibio.com/enzymes/ligases-kinases-phosphatases/dna-ligases/ampligase-thermostable-dna-ligase?details.
LCR assays are sensitive, specific, easy to perform, and can be readily automated. Unlike PCR, no new DNA is synthesized and therefore misincorporated nucleotides or sequences not present in the target DNA are not replicated in the final product [99]. The potential drawbacks of LCR include increased background signal and false positive results when target independent ligation occurs in the absence of template DNA, difficulty in inactivating post-amplification products, and the inability to incorporate effective contamination control methods [100,101]. LCR assays have been successfully developed for the detection of several viruses such as HPV [102], HSV 1 & 2 [103], and HBV [104]; however, no commercial LCR-based viral assays are currently available in the U.S.
Gap-LCR or gLCR is a modified LCR that can reduce the background that is generated by target-independent, blunt-end ligation. In this reaction, the gap between the annealed probes is filled in by DNA polymerase which is then subsequently joined by DNA ligase [105]. Modified gap ligase chain reaction (gLCR) has been developed for detection of HCV which detected fewer than 50 copies of synthetic RNA transcript and demonstrated comparable results with nested RNA-PCR detection [106].
4.2. Cycling probe technology (CPT)
CPT is an isothermal amplification method that can detect and quantify low amounts of target DNA without amplification of the target DNA sequences. A sequence-specific chimeric probe (25–30 bases in length), typically DNA-RNA-DNA, anneals to the complementary single strand of the targeted DNA and acts as the substrate for a thermostable RNase H [107] (Fig. 11 ). RNase H specifically degrades the RNA portion of the chimeric probe and disassociates the shorter DNA probe fragments from the target DNA, making them available for hybridization with the next probe molecule. The accumulated probe fragments can be detected using fluorescence markers, gel electrophoresis, or can be amplified by a secondary amplification system [108].
Fig. 11.

Cycling Probe Technology (CPT).
Adapted from Suzuki et al., 2010 [111].
Commercial kits combining quantitative real-time PCR with CPT-based detection have been used for rapid detection and quantification of HHV-6 and can also discriminate between HHV-6A and HHV-6B [109]. CPT has also been employed for differentiating strains of VZV [110], rapid and specific detection of resistant influenza A viruses [111], and HCV [112].
4.3. Cleavase-invader technology
The cleavase-invader technology is an isothermal probe amplification method that can detect both DNA and RNA targets with high sensitivity and specificity [113]. The assay uses two oligonucleotide probes – the invader oligonucleotide and an allele-specific primary oligonucleotide. These two probes hybridize to a single-stranded target and form a unique partially overlapping three-dimensional invader or “flap” structure (Fig. 12 ). This three-dimensional structure is recognized by cleavase, a flap endonuclease (FEN) that cleaves the 5′ flap of the primary probe. The cleaved 5′ flap product subsequently anneals with a fluorescence resonance energy transfer (FRET) probe and initiates a second invasive cleavage reaction that releases a fluorescent dye detectable by a fluorometer. Measurable fluorescent signal is generated only when the primary probe matches the target sequence – the cleavase remains inactive if there is a mismatch.
Fig. 12.

Cleavase-Invader Technology.
Commercial invader assays have been developed for rapid differentiation of HCV genotypes and is compatible with various commercially available PCR-based HCV 5′ non-coding region amplification assays [114]. Invader assays have also been developed for classification of HBV [115], and commercial invader assays with improved specificity are available for detection of HPV [116]. Modified invader assays combining PCR with invader chemistry in a single, closed-tube, continuous-reaction format have been developed for simultaneous differentiation of HSV 1 & 2 with 100.0% clinical sensitivity and 98.6% specificity [117]. Additionally, invader assays have also been adapted for the detection of novel viruses, such as African swine fever virus which has demonstrated a dynamic range from 2.5 × 103 to 2.5 × 106 copies [118]. Invader assays have also been developed for SNP genotyping but have two major limitations [119]. First, these assays require either a large amount of sample DNA or an initial amplification of the sample DNA. Second, in its current format, these assays have demonstrated poor multiplexing ability and can only genotype one SNP per reaction.
5. Conclusion
Rapid and accurate diagnosis of infectious pathogens has been proven paramount for improving the overall clinical outcomes in patients and for infection control. In the last two decades, molecular testing methods have considerably improved the diagnosis of viral pathogens and are often being considered as the new “gold standard”. While most of these methods are implemented routinely in the clinical virology laboratory, some are still used only for reference testing. Commercial assays based on the fundamentals of nucleic acid amplification have been extensively developed for the detection, identification, and quantification of relevant viral pathogens. Most of these assays require minimal hands-on time, have short turnaround times, can be automated, and can provide results directly from the specimen, thus eliminating lengthy pre- and post-processing steps. The primary advantage of molecular assays is their high sensitivity and specificity when compared to conventional diagnostic methods. However, with higher rates of pathogen detection, testing should be performed and the results analyzed based on the clinical signs and symptoms before initiating a specific treatment. Primary concerns for molecular assays are their complexity, the level of technical expertise required, and potentially high cost per test. However, many of these chemistries have evolved to moderate or low complexity sample-to-answer systems and are even achieving CLIA-waived point-of-care (POC) status. Although diagnostic companies are developing strategies to help reduce cost, clinical laboratories should evaluate their patient population, their size and capacity, and perform a cost-analysis for their specific setting before implementing specific tests.
Authors statement
Both authors contributed equally to all phases of preparation of this review article.
Conflict of interest
S. Dunbar and S. Das are employees of Luminex Corporation.
Funding
None.
Ethical approval
Not applicable.
References
- 1.Zhang Y., Hung T., Song J., He J. Electron microscopy: essentials for viral structure, morphogenesis and rapid diagnosis. Sci. China Life Sci. 2013;56:5:421–430. doi: 10.1007/s11427-013-4476-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dilnessa T., Zeleke H. Cell culture, cytopathic effect and immunofluorescence diagnosis of viral infection. J. Microbiol. Modern Technol. 2017;2(1):102. [Google Scholar]
- 3.Garibyan L., Avashia N. Research techniques made simple: polymerase chain reaction (PCR) J. Invest. Dermatol. 2013;133(3):e6. doi: 10.1038/jid.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cobo F. Suppl 1: application of molecular diagnostic techniques for viral testing. Open Virol. J. 2012;6:104. doi: 10.2174/1874357901206010104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yılmaz M., Ozic C., Gok İ. Gel Electrophoresis-Principles and Basics. InTech; 2012. Principles of nucleic acid separation by agarose gel electrophoresis. [Google Scholar]
- 6.Kemp D.J., Smith D.B., Foote S.J., Samaras N., Peterson M.G. Colorimetric detection of specific DNA segments amplified by polymerase chain reactions. Proc. Natl. Acad. Sci. 1989;86(7):2423–2427. doi: 10.1073/pnas.86.7.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolcott M.J. Advances in nucleic acid-based detection methods. Clin. Microbiol. Rev. 1992;5(4):370–386. doi: 10.1128/cmr.5.4.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navarro E., Serrano-Heras G., Castaño M.J., Solera J. Real-time PCR detection chemistry. Clin. Chim. Acta. 2015;439:231–250. doi: 10.1016/j.cca.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 9.Piccirilli J.A., Benner S.A., Krauch T., Moroney S.E. Enzymatic incorporation of a new base pair into DNA and RNA extends the genetic alphabet. Nature. 1990;343(6253):33. doi: 10.1038/343033a0. [DOI] [PubMed] [Google Scholar]
- 10.Ruiz G., Pena P., de Ory F., Echevarría J.E. Comparison of commercial real-time PCR assays for quantification of Epstein-Barr virus DNA. J. Clin. Microbiol. 2005;43(5):2053–2057. doi: 10.1128/JCM.43.5.2053-2057.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halfon P., Bourlière M., Pénaranda G., Khiri H., Ouzan D. Real-time PCR assays for hepatitis C virus (HCV) RNA quantitation are adequate for clinical management of patients with chronic HCV infection. J. Clin. Microbiol. 2006;44(7):2507–2511. doi: 10.1128/JCM.00163-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim H., Hur M., Bae E., Lee K.A., Lee W.I. Performance evaluation of cobas HBV real-time PCR assay on Roche cobas 4800 System in comparison with COBAS AmpliPrep/COBAS TaqMan HBV Test. Clin. Chem. Lab. Med. (CCLM) 2018;56(7):1133–1139. doi: 10.1515/cclm-2017-1133. [DOI] [PubMed] [Google Scholar]
- 13.Kessler H.H., Mühlbauer G., Rinner B., Stelzl E., Berger A., Dörr H.W., et al. Detection of herpes simplex virus DNA by real-time PCR. J. Clin. Microbiol. 2000;38(7):2638–2642. doi: 10.1128/jcm.38.7.2638-2642.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Braun P., Ehret R., Wiesmann F., Zabbai F., Knickmann M., Kühn R., et al. Comparison of four commercial quantitative HIV-1 assays for viral load monitoring in clinical daily routine. Clin. Chem. Lab. Med. 2007;45(1):93–99. doi: 10.1515/CCLM.2007.008. [DOI] [PubMed] [Google Scholar]
- 15.Loeffelholz M., Deng H. Advanced Techniques in Diagnostic Microbiology. Springer; Boston, MA: 2006. PCR and its variations; pp. 166–183. [Google Scholar]
- 16.Carr J., Williams D.G., Hayden R.T. Molecular Diagnostics. 2010. Molecular detection of multiple respiratory viruses; pp. 289–300. [Google Scholar]
- 17.Wiedbrauk D.L. Molecular Diagnostics. 2010. Herpes simplex virus; pp. 453–460. [Google Scholar]
- 18.Khlif M., Mary C., Sellami H., Sellami A., Dumon H., Ayadi A., Ranque S. Evaluation of nested and real‐time PCR assays in the diagnosis of candidaemia. Clin. Microbiol. Infect. 2009;15(7):656–661. doi: 10.1111/j.1469-0691.2009.02762.x. [DOI] [PubMed] [Google Scholar]
- 19.Poritz M.A., Blaschke A.J., Byington C.L., Meyers L., Nilsson K., Jones D.E., et al. FilmArray, an automated nested multiplex PCR system for multi-pathogen detection: development and application to respiratory tract infection. PLoS One. 2011;6(10) doi: 10.1371/journal.pone.0026047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leber A.L., Everhart K., Balada-Llasat J.M., Cullison J., Daly J., Holt S., et al. Multicenter evaluation of the BioFire FilmArray meningitis encephalitis panel for the detection of bacteria, viruses and yeast in cerebrospinal fluid specimens. J. Clin. Microbiol. 2016 doi: 10.1128/JCM.00730-16. JCM-00730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buss S.N., Leber A., Chapin K., Fey P.D., Bankowski M.J., Jones M.K., et al. Multicenter evaluation of the BioFire FilmArray™ gastrointestinal panel for the etiologic diagnosis of infectious gastroenteritis. J. Clin. Microbiol. 2015 doi: 10.1128/JCM.02674-14. JCM-02674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mackay I.M. Real-time PCR in the microbiology laboratory. Clin. Microbiol. Infect. 2004;10(3):190–212. doi: 10.1111/j.1198-743x.2004.00722.x. [DOI] [PubMed] [Google Scholar]
- 23.Gerken G., Rothaar T., Rumi M.G., Soffredini R., Trippler M., Blunk M.J., et al. Performance of the COBAS AMPLICOR HCV MONITOR test, version 2.0, an automated reverse transcription-PCR quantitative system for hepatitis C virus load determination. J. Clin. Microbiol. 2000;38(6):2210–2214. doi: 10.1128/jcm.38.6.2210-2214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drosten C., Chiu L.L., Panning M., Leong H.N., Preiser W., Tam J.S., et al. Evaluation of advanced reverse transcription-PCR assays and an alternative PCR target region for detection of severe acute respiratory syndrome-associated coronavirus. J. Clin. Microbiol. 2004;42(5):2043–2047. doi: 10.1128/JCM.42.5.2043-2047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai H.P., Tsai Y.Y., Lin I.T., Kuo P.H., Chang K.C., Chen J.C., et al. Validation and application of a commercial quantitative real-time reverse transcriptase-PCR assay in investigation of a large dengue virus outbreak in southern Taiwan. PLoS Negl. Trop. Dis. 2016;10(10) doi: 10.1371/journal.pntd.0005036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giordano M., Kelleher T., Colonno R.J., Lazzarin A., Squires K. The effects of the Roche AMPLICOR HIV-1 MONITOR® UltraSensitive Test versions 1.0 and 1.5 viral load assays and plasma collection tube type on determination of response to antiretroviral therapy and the inappropriateness of cross-study comparisons. J. Clin. Virol. 2006;35(4):420–425. doi: 10.1016/j.jcv.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 27.Reijans M., Dingemans G., Klaassen C.H., Meis J.F., Keijdener J., Mulders B., et al. RespiFinder: a new multiparameter test to differentially identify fifteen respiratory viruses. J. Clin. Microbiol. 2008;46(4):1232–1240. doi: 10.1128/jcm.02294-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schreckenberger P.C., McAdam A.J. Point-counterpoint: large multiplex PCR panels should be first-line tests for detection of respiratory and intestinal pathogens. J. Clin. Microbiol. 2015;53(10):3110–3115. doi: 10.1128/jcm.00382-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elnifro E.M., Ashshi A.M., Cooper R.J., Klapper P.E. Multiplex PCR: optimization and application in diagnostic virology. Clin. Microbiol. Rev. 2000;13(4):559–570. doi: 10.1128/cmr.13.4.559-570.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brotons P., Henares D., Latorre I., Cepillo A., Launes C., Muñoz-Almagro C. Comparison of NxTAG respiratory pathogen panel and Anyplex II RV16 tests for multiple detection of respiratory pathogens in hospitalized children. J. Clin. Microbiol. 2016 doi: 10.1128/JCM.01243-16. JCM-01243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beckmann C., Hirsch H.H. Comparing Luminex NxTAG-respiratory pathogen panel and RespiFinder-22 for multiplex detection of respiratory pathogens. J. Med. Virol. 2016;88(8):1319–1324. doi: 10.1002/jmv.24492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruggiero P., McMillen T., Tang Y.W., Babady N.E. Evaluation of the BioFire FilmArray respiratory panel and the GenMark eSensor respiratory viral panel on lower respiratory tract specimens. J. Clin. Microbiol. 2014;52(1):288–290. doi: 10.1128/JCM.02787-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Navidad J.F., Griswold D.J., Gradus M.S., Bhattacharyya S. Evaluation of Luminex xTAG® gastrointestinal pathogen analyte specific reagents for high-throughput, simultaneous detection of bacteria, viruses, and parasites of clinical and public health importance. J. Clin. Microbiol. 2013 doi: 10.1128/JCM.00896-13. JCM-00896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zanoli L.M., Spoto G. Isothermal amplification methods for the detection of nucleic acids in microfluidic devices. Biosensors. 2012;3(1):18–43. doi: 10.3390/bios3010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gill P., Ghaemi A. Nucleic acid isothermal amplification technologies—A review. Nucleosides Nucleotides Nucleic Acids. 2008;27(3):224–243. doi: 10.1080/15257770701845204. [DOI] [PubMed] [Google Scholar]
- 36.Fakruddin M., Mazumdar R.M., Chowdhury A., Mannan K.S.B. Nucleic acid sequence based amplification (NASBA)-prospects and applications. Int. J. Life Sci. Pharma Res. 2012;2:106. [Google Scholar]
- 37.Sergentet-Thevenot D., Montet M.P., Vernozy-Rozand C. Challenges to developing nucleic acid sequence based amplification technology for the detection of microbial pathogens in food. Rev. Med. Vet. 2008;159:514–527. [Google Scholar]
- 38.Tillmann R.L., Simon A., Müller A., Schildgen O. Sensitive commercial NASBA assay for the detection of respiratory syncytial virus in clinical specimen. PLoS One. 2007;2(12):e1357. doi: 10.1371/journal.pone.0001357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoesley C.J., Allen S.A., Raper J.L., Musonda R., Niu Y., Gao F., et al. Comparative analysis of commercial assays for the detection and quantification of human immunodeficiency virus type 1 (HIV-1) RNA in plasma from patients infected with HIV-1 subtype C. Clin. Infect. Dis. 2002;35(3):323–325. doi: 10.1086/341490. [DOI] [PubMed] [Google Scholar]
- 40.Hebart H., Rudolph T., Loeffler J., Middeldorp J., Ljubicic T., Jahn G., Einsele H. Evaluation of the NucliSens CMV pp67 assay for detection and monitoring of human cytomegalovirus infection after allogeneic stem cell transplantation. Bone Marrow Transplant. 2002;30(3):181. doi: 10.1038/sj.bmt.1703604. [DOI] [PubMed] [Google Scholar]
- 41.Boulet G.A., Micalessi I.M., Horvath C.A., Benoy I.H., Depuydt C.E., Bogers J.J. Nucleic acid sequence-based amplification assay for human papillomavirus mRNA detection and typing: evidence for DNA amplification. J. Clin. Microbiol. 2010;48(7):2524–2529. doi: 10.1128/JCM.00173-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paryan M., Forouzandeh Moghadam M., Kia V., Mohammadi Yeganeh S., Raz A., Mirab Samiee S. A simple and rapid method for the detection of HIV-1/HCV in Co-infected patients. Iran. J. Biotechnol. 2013;11(2):74–79. [Google Scholar]
- 43.McDonough S.H., Bott M.A., Giachetti C. Nucleic Acid Amplification Technologies: Application to Disease Diagnosis. Eaton Publishing; Cambridge, Mass: 1997. Application of transcription-mediated amplification to detection of nucleic acids from clinically relevant organisms; pp. 113–123. [Google Scholar]
- 44.Brentano S.T., Mcdonough S.H. Nonradioactive Analysis of Biomolecules. Springer; Berlin, Heidelberg: 2000. Isothermal amplification of RNA by transcription-mediated amplification (TMA) pp. 374–380. [Google Scholar]
- 45.Langabeer S.E., Gale R.E., Harvey R.C., Cook R.W., Mackinnon S., Linch D.C. Transcription-mediated amplification and hybridisation protection assay to determine BCR-ABL transcript levels in patients with chronic myeloid leukaemia. Leukemia. 2002;16(3):393. doi: 10.1038/sj.leu.2402392. [DOI] [PubMed] [Google Scholar]
- 46.May S., Adamska E., Tang J.W. Evaluating the aptima HIV-1 quant dx, HCV quant dx and HBV quant assays against the Abbott HIV-1, HCV and HBV RealTime assays. J. Clin. Virol. 2018;106:7–10. doi: 10.1016/j.jcv.2018.06.015. [DOI] [PubMed] [Google Scholar]
- 47.Walker G.T., Fraiser M.S., Schram J.L., Little M.C., Nadeau J.G., Malinowski D.P. Strand displacement amplification—An isothermal, in vitro DNA amplification technique. Nucleic Acids Res. 1992;20(7):1691–1696. doi: 10.1093/nar/20.7.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walker G.T., Little M.C., Nadeau J.G., Shank D.D. Isothermal in vitro amplification of DNA by a restriction enzyme/DNA polymerase system. Proc. Natl. Acad. Sci. 1992;89(1):392–396. doi: 10.1073/pnas.89.1.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spargo C.A., Dean C.H., Nycz C.M., Walker G.T. Nonradioactive Analysis of Biomolecules. Springer; Berlin, Heidelberg: 2000. SDA target amplification; pp. 356–366. [Google Scholar]
- 50.Karami A., Gill P., Motamedi M.H.K., Saghafinia M. A review of the current isothermal amplification techniques: applications, advantages and disadvantages. J. Glob. Infect. Dis. 2011;3(3):293–302. [Google Scholar]
- 51.Walker G.T. Empirical aspects of strand displacement amplification. Genome Res. 1993;3(1):1–6. doi: 10.1101/gr.3.1.1. [DOI] [PubMed] [Google Scholar]
- 52.Van Der Pol B., Warren T., Taylor S.N., Martens M., Jerome K.R., Mena L., et al. Type specific identification of anogenital herpes simplex virus infections using a commercially available nucleic acid amplification test. J. Clin. Microbiol. 2012 doi: 10.1128/JCM.01685-12. JCM-01685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gadkar V.J., Goldfarb D.M., Gantt S., Tilley P.A. Real-time detection and monitoring of loop mediated amplification (LAMP) reaction using self-quenching and de-quenching fluorogenic probes. Sci. Rep. 2018;8(1):5548. doi: 10.1038/s41598-018-23930-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Notomi T., Okayama H., Masubuchi H., Yonekawa T., Watanabe K., Amino N., Hase T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28(12):e63. doi: 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rekha V., Rana R., Arun T.R., Aswathi P.B., Kalluvila J., John D.G., et al. Loop mediated isothermal amplification (LAMP) test–a novel nucleic acid based assay for disease diagnosis. Adv. Anim. Vet. Sci. 2014;2:344–350. [Google Scholar]
- 56.Dhama K., Karthik K., Chakraborty S., Tiwari R., Kapoor S., Kumar A., Thomas P. Loop-mediated isothermal amplification of DNA (LAMP): a new diagnostic tool lights the world of diagnosis of animal and human pathogens: a review. Pak. J. Biol. Sci. 2014;17(2):151–166. doi: 10.3923/pjbs.2014.151.166. [DOI] [PubMed] [Google Scholar]
- 57.Faron M.L., Ledeboer N.A., Patel A., Beqa S.H., Yen-Lieberman B., Kohn D., et al. Multicenter evaluation of the meridian bioscience molecular HSV 1&2 assay for the detection of herpes simplex virus 1 and 2 from clinical cutaneous and mucocutaneous specimens. J. Clin. Microbiol. 2016 doi: 10.1128/JCM.00483-16. JCM-00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Poon L.L., Leung C.S., Tashiro M., Chan K.H., Wong B.W., Yuen K.Y., et al. Rapid detection of the severe acute respiratory syndrome (SARS) coronavirus by a loop-mediated isothermal amplification assay. Clin. Chem. 2004;50(6):1050–1052. doi: 10.1373/clinchem.2004.032011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lu X., Li X., Mo Z., Jin F., Wang B., Zhao H., et al. Rapid identification of Chikungunya and Dengue virus by a real-time reverse transcription-loop-mediated isothermal amplification method. Am. J. Trop. Med. Hyg. 2012;87(5):947–953. doi: 10.4269/ajtmh.2012.11-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Farooq U., Latif A., Irshad H., Ullah A., Zahur A.B., Naeem K., et al. Loop-mediated isothermal amplification (RT-LAMP): a new approach for the detection of foot-and-mouth disease virus and its sero-types in Pakistan. Iran. J. Vet. Res. 2015;16(4):331. [PMC free article] [PubMed] [Google Scholar]
- 61.Sabalza M., Yasmin R., Barber C.A., Castro T., Malamud D., Kim B.J., et al. Detection of Zika virus using reverse-transcription LAMP coupled with reverse dot blot analysis in saliva. PLoS One. 2018;13(2) doi: 10.1371/journal.pone.0192398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharma V., Chaudhry D., Kaushik S. Evaluation of clinical applicability of reverse transcription-loop-mediated isothermal amplification assay for detection and subtyping of Influenza A viruses. J. Virol. Methods. 2018;253:18–25. doi: 10.1016/j.jviromet.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang X., Li X., Hu S., Qu H., Zhang Y., Ni H., Wang X. Rapid detection of active human cytomegalovirus infection in pregnancy using loop-mediated isothermal amplification. Mol. Med. Rep. 2015;12(2):2269–2274. doi: 10.3892/mmr.2015.3572. [DOI] [PubMed] [Google Scholar]
- 64.Rohatensky M.G., Livingstone D.M., Mintchev P., Barnes H.K., Nakoneshny S.C., Demetrick D.J., et al. Assessing the performance of a Loop Mediated Isothermal Amplification (LAMP) assay for the detection and subtyping of high-risk suptypes of Human Papilloma Virus (HPV) for Oropharyngeal Squamous Cell Carcinoma (OPSCC) without DNA purification. BMC Cancer. 2018;18(1):166. doi: 10.1186/s12885-018-4087-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bista B.R., Ishwad C., Wadowsky R.M., Manna P., Randhawa P.S., Gupta G., et al. Development of a loop-mediated isothermal amplification assay for rapid detection of BK virus. J. Clin. Microbiol. 2007;45(5):1581–1587. doi: 10.1128/JCM.01024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaneko H., Iida T., Aoki K., Ohno S., Suzutani T. Sensitive and rapid detection of herpes simplex virus and varicella-zoster virus DNA by loop-mediated isothermal amplification. J. Clin. Microbiol. 2005;43(7):3290–3296. doi: 10.1128/JCM.43.7.3290-3296.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tröger V., Niemann K., Gärtig C., Kuhlmeier D. Isothermal amplification and quantification of nucleic acids and its use in microsystems. J. Nanomed. Nanotechnol. 2015;6(3):1. [Google Scholar]
- 68.Qian C., Wang R., Wu H., Ji F., Wu J. Nicking enzyme-assisted amplification (NEAA) technology and its applications: a review. Anal. Chim. Acta. 2018 doi: 10.1016/j.aca.2018.10.054. [DOI] [PubMed] [Google Scholar]
- 69.Bell J., Bonner A., Cohen D.M., Birkhahn R., Yogev R., Triner W., et al. Multicenter clinical evaluation of the novel Alere™ i Influenza A&B isothermal nucleic acid amplification test. J. Clin. Virol. 2014;61(1):81–86. doi: 10.1016/j.jcv.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 70.Peters R.M., Schnee S.V., Tabatabai J., Schnitzler P., Pfeil J. Evaluation of Alere i RSV for rapid detection of respiratory syncytial virus in children hospitalized with acute respiratory tract infection. J. Clin. Microbiol. 2017;55(4):1032–1036. doi: 10.1128/JCM.02433-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chapin K.C., Flores-Cortez E.J. Performance of the molecular Alere i influenza A&B test compared to that of the Xpert Flu A/B assay. J. Clin. Microbiol. 2015;53(2):706–709. doi: 10.1128/JCM.02783-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nolte F.S., Gauld L., Barrett S.B. Direct comparison of Alere™ i and cobas® Liat Influenza A and B tests for rapid detection of influenza virus infection. J. Clin. Microbiol. 2016 doi: 10.1128/JCM.01586-16. JCM-01586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vincent M., Xu Y., Kong H. Helicase‐dependent isothermal DNA amplification. EMBO Rep. 2004;5(8):795–800. doi: 10.1038/sj.embor.7400200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tong Y., Lemieux B., Kong H. Multiple strategies to improve sensitivity, speed and robustness of isothermal nucleic acid amplification for rapid pathogen detection. BMC Biotechnol. 2011;11(1):50. doi: 10.1186/1472-6750-11-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen F., Zhang D., Zhang Q., Zuo X., Fan C., Zhao Y. Zero‐background helicase‐dependent amplification and its application to reliable assay of telomerase activity in cancer cell by eliminating primer–dimer artifacts. ChemBioChem. 2016;17(12):1171–1176. doi: 10.1002/cbic.201500605. [DOI] [PubMed] [Google Scholar]
- 76.Teo J.W., Chiang D., Jureen R., Lin R.T. Clinical evaluation of a helicase-dependant amplification (HDA)–based commercial assay for the simultaneous detection of HSV-1 and HSV-2. Diagn. Microbiol. Infect. Dis. 2015;83(3):261–262. doi: 10.1016/j.diagmicrobio.2015.07.018. [DOI] [PubMed] [Google Scholar]
- 77.Huang S., Do J., Mahalanabis M., Fan A., Zhao L., Jepeal L., et al. Low cost extraction and isothermal amplification of DNA for infectious diarrhea diagnosis. PLoS One. 2013;8(3) doi: 10.1371/journal.pone.0060059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ali M.M., Li F., Zhang Z., Zhang K., Kang D.K., Ankrum J.A., et al. Rolling circle amplification: a versatile tool for chemical biology, materials science and medicine. Chem. Soc. Rev. 2014;43(10):3324–3341. doi: 10.1039/c3cs60439j. [DOI] [PubMed] [Google Scholar]
- 79.Gusev Y., Sparkowski J., Raghunathan A., Ferguson H., Jr, Montano J., Bogdan N., et al. Rolling circle amplification: a new approach to increase sensitivity for immunohistochemistry and flow cytometry. Am. J. Pathol. 2001;159(1):63–69. doi: 10.1016/S0002-9440(10)61674-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Banér J., Nilsson M., Mendel-Hartvig M., Landegren U. Signal amplification of padlock probes by rolling circle replication. Nucleic Acids Res. 1998;26(22):5073–5078. doi: 10.1093/nar/26.22.5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Margeridon S., Carrouée-Durantel S., Chemin I., Barraud L., Zoulim F., Trépo C., Kay A. Rolling circle amplification, a powerful tool for genetic and functional studies of complete hepatitis B virus genomes from low-level infections and for directly probing covalently closed circular DNA. Antimicrob. Agents Chemother. 2008;52(9):3068–3073. doi: 10.1128/AAC.01318-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.James A.P., Geijskes R.J., Dale J.L., Harding R.M. Development of a novel rolling-circle amplification technique to detect Banana streak virus that also discriminates between integrated and episomal virus sequences. Plant Dis. 2011;95(1):57–62. doi: 10.1094/PDIS-07-10-0519. [DOI] [PubMed] [Google Scholar]
- 83.Collins M.L., Irvine B., Tyner D., Fine E., Zayati C., Chang C.A., et al. A branched DNA signal amplification assay for quantification of nucleic acid targets below 100 molecules/ml. Nucleic Acids Res. 1997;25(15):2979–2984. doi: 10.1093/nar/25.15.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tsongalis G.J. Branched DNA technology in molecular diagnostics. Am. J. Clin. Pathol. 2006;126(3):448–453. doi: 10.1309/90BU6KDXANFLN4RJ. [DOI] [PubMed] [Google Scholar]
- 85.Schutzbank T.E. Advanced Techniques in Diagnostic Microbiology. Springer; Boston, MA: 2013. Signal amplification technologies; pp. 327–344. [Google Scholar]
- 86.Gleaves C.A., Welle J., Campbell M., Elbeik T., Ng V., Taylor P.E., et al. Multicenter evaluation of the Bayer VERSANT™ HIV-1 RNA 3.0 assay: analytical and clinical performance. J. Clin. Virol. 2002;25(2):205–216. doi: 10.1016/s1386-6532(02)00011-2. [DOI] [PubMed] [Google Scholar]
- 87.Elbeik T., Surtihadi J., Destree M., Gorlin J., Holodniy M., Jortani S.A., et al. Multicenter evaluation of the performance characteristics of the Bayer VERSANT HCV RNA 3.0 assay (bDNA) J. Clin. Microbiol. 2004;42(2):563–569. doi: 10.1128/JCM.42.2.563-569.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Welzel T.M., Miley W.J., Parks T.L., Goedert J.J., Whitby D., Ortiz-Conde B.A. Real-time PCR assay for detection and quantification of hepatitis B virus genotypes A to G. J. Clin. Microbiol. 2006;44(9):3325–3333. doi: 10.1128/JCM.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang X., Yue L., Zhang Z., Yuan Z. Establishment of a fluorescent in situ hybridization assay for imaging hepatitis B virus nucleic acids in cell culture models. Emerg. Microbes Infect. 2017;6(11):e98. doi: 10.1038/emi.2017.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kurobe M., Kojima T., Nishimura K., Kandori S., Kawahara T., Yoshino T., et al. Development of RNA-FISH assay for detection of oncogenic FGFR3-TACC3 fusion genes in FFPE samples. PLoS One. 2016;11(12) doi: 10.1371/journal.pone.0165109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Grech G. Presented at Protein and Cell Analysis Education Series. 2017. The power of multiplexing and applications of the QuantiGene Plex Assay in oncology research and diagnostics [Webinar]https://www.labroots.com/ms/webinar/power-multiplexing-applications-quantigene-plex-assay-oncology-research-diagnostics (November 07) Retrieved from. [Google Scholar]
- 92.Schiffman M.H., Kiviat N.B., Burk R.D., Shah K.V., Daniel R.W., Lewis R., et al. Accuracy and interlaboratory reliability of human papillomavirus DNA testing by hybrid capture. J. Clin. Microbiol. 1995;33(3):545–550. doi: 10.1128/jcm.33.3.545-550.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sandri M.T., Lentati P., Benini E., Dell’Orto P., Zorzino L., Carozzi F.M., et al. Comparison of the Digene HC2 assay and the Roche AMPLICOR human papillomavirus (HPV) test for detection of high-risk HPV genotypes in cervical samples. J. Clin. Microbiol. 2006;44(6):2141–2146. doi: 10.1128/JCM.00049-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Poljak M., Oštrbenk A., Seme K., Učakar V., Hillemanns P., Bokal E.V., et al. Comparison of clinical and analytical performance of the Abbott RealTime high risk HPV test to the performance of hybrid capture 2 in population-based cervical cancer screening. J. Clin. Microbiol. 2011;49(5):1721–1729. doi: 10.1128/JCM.00012-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Levi A.W., Bernstein J.I., Hui P., Duch K., Schofield K., Chhieng D.C. A comparison of the Roche cobas HPV test with the Hybrid Capture 2 test for the detection of high-risk human papillomavirus genotypes. Arch. Pathol. Lab. Med. 2016;140(2):153–157. doi: 10.5858/arpa.2015-0027-OA. [DOI] [PubMed] [Google Scholar]
- 96.Arney A., Bennett K.M. Molecular diagnostics of human papillomavirus. Lab. Med. 2010;41(9):523–530. [Google Scholar]
- 97.Abreu A.L., Souza R.P., Gimenes F., Consolaro M.E. A review of methods for detect human Papillomavirus infection. Virol. J. 2012;9:262. doi: 10.1186/1743-422X-9-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Barany F. The ligase chain reaction in a PCR world. PCR Methods Appl. 1991;1(1):5–16. doi: 10.1101/gr.1.1.5. [DOI] [PubMed] [Google Scholar]
- 99.Benjamin W.H., Smith K.R., Waites K.B. PCR Protocols. Humana Press; 2003. Ligase chain reaction; pp. 135–149. [Google Scholar]
- 100.Li H., Tang Y.W. Advanced Techniques in Diagnostic Microbiology. Springer; Boston, MA: 2006. In vitro nucleic acid amplification: an introduction; pp. 158–165. [Google Scholar]
- 101.Gibriel A.A., Adel O. Advances in ligase chain reaction and ligation-based amplifications for genotyping assays: detection and applications. Mutat. Res. Mutat. Res. 2017;773:66–90. doi: 10.1016/j.mrrev.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Theelen W., Reijans M., Simons G., Ramaekers F.C., Speel E.J.M., Hopman A.H. A new multiparameter assay to assess HPV 16/18, viral load and physical status together with gain of telomerase genes in HPV‐related cancers. Int. J. Cancer. 2010;126(4):959–975. doi: 10.1002/ijc.24844. [DOI] [PubMed] [Google Scholar]
- 103.Rinehardt L., Hampl H., Laffler T.G. Ultrasensitive non-radioactive detection of herpes simplex virus by LCR, the ligase chain reaction. 20th Annual Meeting of the Keystone Symposia on Molecular and Cellular Biology. 1991:101. doi: 10.1128/JCM.00012-11. [DOI] [Google Scholar]
- 104.Trippler M., Hampl H., Goergen B., Spies U., Knolle P., Grimm B., et al. Ligase chain reaction (LCR®) assay for semi‐quantitative detection of HBV DNA in mononuclear leukocytes of patients with chronic hepatitis B. J. Viral Hepat. 1996;3(5):267–272. doi: 10.1111/j.1365-2893.1996.tb00054.x. [DOI] [PubMed] [Google Scholar]
- 105.Abravaya K., Carrino J.J., Muldoon S., Lee H.H. Detection of point mutations with a modified ligase chain reaction (Gap-LCR) Nucleic Acids Res. 1995;23(4):675–682. doi: 10.1093/nar/23.4.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Marshall R.L., Laffler T.G., Cerney M.B., Sustachek J.C., Kratochvil J.D., Morgan R.L. Detection of HCV RNA by the asymmetric gap ligase chain reaction. Genome Res. 1994;4(2):80–84. doi: 10.1101/gr.4.2.80. [DOI] [PubMed] [Google Scholar]
- 107.Bhatt R., Scott B., Whitney S., Bryan R.N., Cloney L., Lebedev A. Detection of nucleic acids by cycling probe technology on magnetic particles: high sensitivity and ease of separation. Nucleosides Nucleotides. 1999;18(6–7):1297–1299. doi: 10.1080/07328319908044696. [DOI] [PubMed] [Google Scholar]
- 108.Wolcott M.J. Advances in nucleic acid-based detection methods. Clin. Microbiol. Rev. 1992;5(4):370–386. doi: 10.1128/cmr.5.4.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ihira M., Yamaki A., Kato Y., Higashimoto Y., Kawamura Y., Yoshikawa T. Cycling probe‐based real‐time PCR for the detection of Human herpesvirus 6A and B. J. Med. Virol. 2016;88(9):1628–1635. doi: 10.1002/jmv.24513. [DOI] [PubMed] [Google Scholar]
- 110.Ihira M., Higashimoto Y., Kawamura Y., Sugata K., Ohashi M., Asano Y., Yoshikawa T. Cycling probe technology to quantify and discriminate between wild-type varicella-zoster virus and Oka vaccine strains. J. Virol. Methods. 2013;193(2):308–313. doi: 10.1016/j.jviromet.2013.06.031. [DOI] [PubMed] [Google Scholar]
- 111.Suzuki Y., Saito R., Zaraket H., Dapat C., Caperig-Dapat I., Suzuki H. Rapid and specific detection of amantadine-resistant influenza A viruses with a Ser31Asn mutation by the cycling probe method. J. Clin. Microbiol. 2010;48(1):57–63. doi: 10.1128/JCM.00698-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Uchida Y., Kouyama J.I., Naiki K., Sugawara K., Ando S., Nakao M., et al. Significance of variants associated with resistance to NS5A inhibitors in Japanese patients with genotype 1b hepatitis C virus infection as evaluated using cycling-probe real-time PCR combined with direct sequencing. J. Gastroenterol. 2016;51(3):260–270. doi: 10.1007/s00535-015-1106-8. [DOI] [PubMed] [Google Scholar]
- 113.Zhang D., Feng T., Ye F., Lee I., Wu J., Yin B. Advanced Techniques in Diagnostic Microbiology. Springer; Boston, MA: 2006. Recent advances in probe amplification technologies; pp. 210–227. [Google Scholar]
- 114.Germer J.J., Majewski D.W., Yung B., Mitchell P.S., Yao J.D. Evaluation of the invader assay for genotyping hepatitis C virus. J. Clin. Microbiol. 2006;44(2):318–323. doi: 10.1128/JCM.44.2.318-323.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tadokoro K., Kobayashi M., Yamaguchi T., Suzuki F., Miyauchi S., Egashira T., Kumada H. Classification of hepatitis B virus genotypes by the PCR-Invader method with genotype-specific probes. J. Virol. Methods. 2006;138(1–2):30–39. doi: 10.1016/j.jviromet.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 116.Ginocchio C.C., Barth D., Zhang F. Comparison of the Third Wave Invader human papillomavirus (HPV) assay and the digene HPV hybrid capture 2 assay for detection of high-risk HPV DNA. J. Clin. Microbiol. 2008;46(5):1641–1646. doi: 10.1128/JCM.01824-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Allawi H.T., Li H., Sander T., Aslanukov A., Lyamichev V.I., Blackman A., et al. Invader plus method detects herpes simplex virus in cerebrospinal fluid and simultaneously differentiates types 1 and 2. J. Clin. Microbiol. 2006;44(9):3443–3447. doi: 10.1128/JCM.01175-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hjertner B., Meehan B., McKillen J., McNeilly F., Belák S. Adaptation of an Invader® assay for the detection of African swine fever virus DNA. J. Virol. Methods. 2005;124(1–2):1–10. doi: 10.1016/j.jviromet.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 119.Olivier M. The invader assay for SNP genotyping. Mutat. Res. 2005;573(1-2):103–110. doi: 10.1016/j.mrfmmm.2004.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Niemz A., Ferguson T.M., Boyle D.S. Point-of-care nucleic acid testing for infectious diseases. Trends Biotechnol. 2011;29(5):240–250. doi: 10.1016/j.tibtech.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]