

Abstract
After decoding the genome of SARS-coronavirus (SARS-CoV), next challenge is to understand how this virus causes the illness at molecular bases. Of the viral structural proteins, the N protein plays a pivot role in assembly process of viral particles as well as viral replication and transcription. The SARS-CoV N proteins expressed in the eukaryotes, such as yeast and HEK293 cells, appeared in the multiple spots on two-dimensional electrophoresis (2DE), whereas the proteins expressed in E. coli showed a single 2DE spot. These 2DE spots were further examined by Western blot and MALDI-TOF/TOF MS, and identified as the N proteins with differently apparent pI values and similar molecular mass of 50 kDa. In the light of the observations and other evidences, a hypothesis was postulated that the SARS-CoV N protein could be phosphorylated in eukaryotes. To locate the plausible regions of phosphorylation in the N protein, two truncated N proteins were generated in E. coli and treated with PKCα. The two truncated N proteins after incubation of PKCα exhibited the differently electrophoretic behaviors on 2DE, suggesting that the region of 1–256 aa in the N protein was the possible target for PKCα phosphorylation. Moreover, the SARS-CoV N protein expressed in yeast were partially digested with trypsin and carefully analyzed by MALDI-TOF/TOF MS. In contrast to the completely tryptic digestion, these partially digested fragments generated two new peptide mass signals with neutral loss, and MS/MS analysis revealed two phosphorylated peptides located at the “dense serine” island in the N protein with amino acid sequences, GFYAEGSRGGSQASSRSSSR and GNSGNSTPGSSRGNSPARMASGGGK. With the PKCα phosphorylation treatment and the partially tryptic digestion, the N protein expressed in E. coli released the same peptides as observed in yeast cells. Thus, this investigation provided the preliminary data to determine the phosphorylation sites in the SARS-CoV N protein, and partially clarified the argument regarding the phosphorylation possibility of the N protein during the infection process of SARS-CoV to human host.
Keywords: Severe Acute Respiratory Syndrome (SARS), N protein, Phosphorylation, Matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF/TOF) mass spectrometry, Two-dimensional electrophoresis (2DE)
1. Introduction
The infectious disease, Severe Acute Respiratory Syndrome (SARS) defined by the World Health Organization, was initially reported in Guangdong province, China, in 2003 [1]. SARS caused considerable morbidity and mortality with more than 50% infection rate to the people in close contact with SARS patients [2]. Although few of cases have been reported in last 4 years worldwide, it is necessitated to maintain high vigilance to SARS revival. Understanding of molecular mechanism of SARS-coronavirus (SARS-CoV), the essential factor of this infectious disease, is a primary step towards preventing and protecting SARS.
The genome size of SARS-CoV is approximately 29 kb long and has 11 open reading frames (ORFs), composed of a stable region encoding a RNA-dependent RNA polymerase with two ORFs, a variable region representing four coding sequences (CDSs) for viral structural genes, spike (S protein), envelope (E protein), membrane (M protein) and nucleocapsid (N protein), and five putative uncharacterized proteins (PUPs) [3], [4]. The N protein is a key component in the coronaviral core and has attracted research attention for a long period [5]. As compared with the genomes of coronaviruses discovered so far, the N protein in SARS-CoV exhibits highly variable in amino acid composition, but may perform similar physical properties. Up to date, most of the N proteins contain 380–460 amino acids, with a preponderance of basic residues (58–72 arginine and lysine) and 7–10% serine content [6]. Also the N proteins range isoelectric points of 10.3–10.7 but with markedly acidic at their carboxy termini (pI 4.5–5.3) [7]. The N protein of SARS-coronavirus sizes up 422 amino acids with 60 basic amino acids and 8% of serine; and it has a theoretical pI value of 10.3 for the overall sequence and an acidic C-terminus (30 amino acids within C-terminus) with pI of 4.5. Therefore, this new coronavirus N protein is deduced to perform the similar functions as other N proteins for the viral formation or infection in host cells.
It is generally accepted that the N proteins function as the stabilizers for the viral genomic RNAs due to the complexes of helical ribonucleocapsid (RNP) generated from the N protein and RNA [8]. The phosphorylation of viral proteins, particularly for the positive charged nucleic acid binding proteins, can profoundly modulate the interactions between host cells and viruses. In rabies virus, like SARS-CoV as a single-stranded sense RNA, the dephosphorylated N protein binds more strongly to leader RNA than its phosphorylated form, resulting in a dramatic decrease of RNA transcription and replication [9]. It was postulated to attribute the stronger capacity of the dephosphorylated N protein binding with RNA to their high positive charges [10]. Whether the N protein of SARS-CoV is phosphorylated in the host cells, however, has been argued among some investigators. With proteomic means, two groups did not detect a single mass signal of phosphorylated peptide from the N protein even though the sequence coverage of peptide detection over 90% [11], [12]. Through the measurement of the intact protein mass, Ying et al. claimed no phosphorylation in the SARS N protein [12]. No evidence of the phosphorylation of the N protein was given in Krokhin's with the approaches of the de novo analysis to the N protein [11]. Lal's group described the contrary results and claimed that the N proteins of SARS-CoV localized at the nucleus as well as cytoplasm were phosphorylated by CDK [13], [14]. This group further revealed that the phosphorylation status of the N protein in SARS-CoV could perform a significant impact to S phase progression in mammalian cells [14]. This issue, however, has not been solved completely. First of all, there has been lack of accurate identification to the phosphorylation sites located at the N protein of SARS-CoV, even though the multiple phosphorylation sites could be theoretically predicted. Secondly, the evidence for the phosphorylation of the N protein was achieved from the experiments in vitro and site-mutagenesis. As a matter of fact, the real status of phosphorylated N protein in the SARS-CoV infected cells has not been clearly clarified regardless of which approaches have been employed so far.
The present study was undertaken to systematically explore whether the N protein of SARS-CoV was phosphorylated in prokaryote and in eukaryote. Two-dimensional electrophoresis (2DE), Western blot, and mass spectrometry were first employed to separate and identify the modified N proteins. Furthermore, the phosphorylation regions amongst the N protein were analyzed using the approaches of molecular biology as well as phosphorylation of protein kinase. Finally the phosphorylated sites in the N protein were identified with the strategy of partial tryptic digestion plus MALDI-TOF/TOF MS. These experiments took the first step to define the phosphorylation sites of the N protein, and offered the solid evidence to support the hypothesis that the SARS-CoV N protein was indeed phosphorylated during virus infection to human host.
2. Materials and methods
2.1. Materials
All chemicals employed for electrophoresis were from Amersham Biosciences (Uppsala, Sweden). IPG strips were purchased from Bio-Rad Laboratories (Hercules, CA). All chemicals of analytical grade were from Sigma (St Louis, MO). Modified trypsin (sequence grade) was acquired from Promega (Madison, WI). All HPLC grade solvents were from J.T. Baker (Phillipsburg, NJ).
2.2. Plasmids construction and proteins expression in E. coli
The N gene was derived from SARS-coronavirus strain BJ01. The viral genomic RNA was prepared using TRIzol reagent (Invitrogen). First-strand cDNA synthesis was carried out as described in the company manual (Promega). The full length of the N gene was amplified by PCR using a pair primer, 3′-primer (ATAAGAATGCGGCCGCTTATGCCTGAGTTGAA) with Not I site and 5′-primer (CGGGATCCATGTCTGATAATGGACCCCA) with BamH I site. To generate the truncated fragments of ΔN256 and ΔN124, the pairs of primer were designed as, 3′-primer (ATAAGAATGCGGCCGCTTATGCCTGAGTTGAA) with Not I site and 5′-primer (CGGGATCCCCTCGCCAAAAACGTACT) with BamH I site for ΔN256, and 3′-primer (AAGAATGCGGCCGCTTATGCCTGAGTTGAA) with Not I site and 5′-primer (CGGGATCCGCTAACAAAGAAGGCATCGTA) with Bam H I site for ΔN124, respectively. After restriction digestion, these N fragments were ligated with a linearized pET32a vector. The expression vectors pET32-N, pET32-ΔN256 and pET32-ΔN124 were transformed into strain BL21 (DE3) of E. coli. The transformed bacteria were cultured at 37 °C in LB medium containing 50 μg/ml ampicillin. The expression of the N proteins was induced by addition of 1 mM isopropyl-β-thiogalactoside. The bacteria were lysed by sonication in a buffer consisting of 20 mM Tris–HCl, pH 7.9, 200 mM NaCl, and 5 mM imidazole. The cell debris was pelleted by centrifugation, and the supernatant was applied to a Ni-NTA Superflow column (Qiagen) mounted on AKATA FPLC system (Amersham) that was pre-equilibrated with the lysis buffer. The bound proteins were eluted with a linear gradient of imidazole from 50 to 300 mM.
2.3. The N gene expression in the eukaryote systems
In yeast expression system, full length of N gene was inserted into pEGH vector with two sites of restriction enzyme, Hind III and Xba I. The expression of the N protein was induced with 0.2% galactose. The GST-fusion protein was purified following the recommended protocol from the manufactory (Amersham).
In mammalian expression, full length of N gene was inserted into the vector pCDNA3.1 with two sites of restriction enzyme, BamH I and EcoR I. The HEK293 cells were cultured in DMEM containing 10% FBS at 37 °C and 5% CO2/95% air, and transfected when the confluence reached 80%. For each 10 cm2 tissue culture dish, 5 μg of transfection vector was used at a DNA/Lipofectamine ratio of 1:5. The transfected cells were harvested 48 h after transfection.
2.4. Generation of polyclonal antibody against the N protein of SARS-CoV
Approximately 500 μg recombinant proteins in complete Freund's adjuvant (1:1) was immunized New Zealand white rabbit, followed by three boosts with the same amount proteins in incomplete Freund's adjuvant (1:1) after the periods of the first immunization. The rabbit sera were collected and purified through protein A affinity chromatography (Amersham).
2.5. Two-dimensional electrophoresis and Western blot
The protein solutions were incubated with commercial IPG strips (non-linear, pH 3–10, Amersham) for rehydration overnight. The rehydrated strips were electrofocused for 20 kVh (7 cm strip) using IPGphor (BioRad) at 20 °C. Prior to the second dimension, the IPG strips were sequentially equilibrated with two buffers, reducing buffer containing 50 mM Tris–HCl, pH 8.8, 6 M urea, 30% glycerol, 2% SDS, trace of bromphenol blue and 1% DTT and alkylating buffer containing 50 mM Tris–HCl, pH 8.8, 6 M urea, 30% glycerol, 2% SDS, a trace of bromphenol blue and 2.5% iodoacetamide. The electrophoresed strips were loaded and run on 12% acrylamide gels using Bio-Rad MINI-PROTEAN II with a constant voltage at 120 V. To specifically monitor the electrophoretic behavior of the N protein, the proteins resolved in polyacryamide gels were further transferred to PVDF membrane. The polyclonal antibody against the SARS-CoV N protein was used as the primary antibody, and the HRP-conjugated goat anti-rabbit Ig G antibody was adopted as the secondary antibody. The protein signals immune-recognized were visualized using ECL method (Amersham).
2.6. Trypsin digestion and peptide identification by mass spectrometry
The 2DE spots were excised, successively destained and dehydrated with 50% acetonitrile. The gel particles were reduced with 10 mM DTT at 56 °C for 1 h and alkylated by 55 mM iodoacetamide in dark at room temperature for 45 m. Finally, the gel pieces were thoroughly washed with 25 mM ammonium bicarbonate in water/acetonitrile (50/50) and completely dried in a Speedvac. The in-gel digestion was conducted in 25 μl modified trypsin solution (10 ng/μl in 25 mM ammonium bicarbonate) with incubation overnight at 37 °C. For partial digestion, the incubation period was shortened to 3 h.
The digestions were applied onto AnchorChip™ target (Bruker) followed by adding matrix solution consisting of α-cyano-4-hydroxycinnamic acid (4 mg/ml) in 70% acetonitrile with 0.1% TFA. The loaded target was subjected into the mass spectrometer, UltroFlex MALDI-TOF/TOF MS (Bruker). Positively charged ions were analyzed in the reflector mode, using delayed extraction. Typically 100 shots were accumulated per spectrum in MS mode and 400 shots in MS/MS mode. The spectra were processed using the FlexAnalysis 2.2 and BioTools 2.2 software tools (Bruker).
Monoisotopic peptide masses obtained from MALDI-TOF/TOF MS were used to search the database of SARS-CoV genome using MasCot program (Matrix Science). The range of molecular weights for protein search was set between 1000 and 100,000 Da with fragment ion mass tolerance <100 ppm. In MS/MS mode, the fragment ion mass accuracy was set to <0.7 Da.
2.7. The phosphorylation of SARS-CoV N protein by PKCα
Approximately 2 μg purified N proteins were incubated with the phosphorylation reaction buffer containing 50 mM Tris/HCl, pH 7.5, 10 mM MgCl2, 2 mM CaCl2, 1 mM DTT, 10 μM ATP, 50 μg/ml phosphatidyl serine, and 1 μg/ml phorbol-12-myristate-13-acetate (PMA) for 30 min followed by adding 0.1 μg PKCα (Promega). The reaction was stopped after 1 h incubation.
3. Results
3.1. The electrophoretic behavior of the SARS-CoV N protein on 2DE
The technique of 2DE is still a powerful tool to separate the modified proteins. To monitor the possible modification forms of the N protein, the proteins obtained from different expression systems were loaded to 2DE followed by dye staining as well as immuno-assay. As depicted in Fig. 1 , the 2DE pattern of the N protein expressed from yeast was different from that of the bacterial recombinants. The N protein from E. coli exhibited a single spot located at alkalic side, whereas one from yeast appeared in string spots slightly close to acidic range. Since the expression of the N protein in mammal cells was too low to detect by dye staining, the N proteins expressed from prokaryote and eukaryote was further examined with 2DE Western blot using the anti-N antibody as the primary antibody (Fig. 2 ). Both N proteins expressed in yeast and HEK293 cells remained the similar patterns with string spots on 2DE even though the N proteins from HEK293 cells distributed along a wider pH range with more spots close to acidic pH side. The N protein from E. coli was only recognized as a single spot in Western blot, being in agreement with the 2DE result stained by Coomassie Blue. These results demonstrated that the status of the expressed N proteins was dependent on the host environment and the post-translational modifications of the N proteins could lead to the changes of the apparent pI values, but without significant change in apparent molecular mass. According to theoretical analysis as well as other research reports, phosphorylated modification was an ideal candidate to illustrate the phenomenon.
Fig. 1.
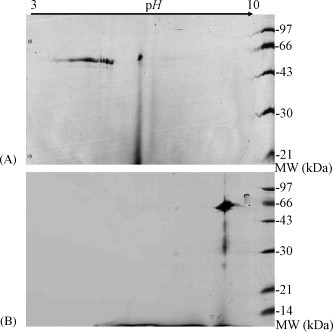
The 2DE images for the N proteins expressed in yeast and E. coli system. A, the GST fusioned N protein expressed in Y258 yeast cells; B, the thioredoxin fusioned N protein expressed in E. coli.
Fig. 2.
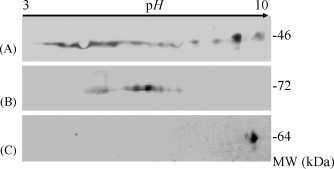
Western blot analysis to identify the N proteins expressed in different cells. A, the HEK293 cells transfected by pCDNA-N; B, the Y258 yeast cells transfected by pEGH-GST-N; C, the E. coli cells transformed by pET32-N.
3.2. Identification of 2DE spots by MALDI-TOF/TOF MS
All the string 2DE spots ranged pI 4–6 from yeast cells were excised and processed for protein identification using MALDI-TOF/TOF MS. The typical spectra of MALDI-TOF/TOF MS were represented in Fig. 3 . These mass signals were analyzed with MasCot for peptide search, resulting in a fragment of the SARS-CoV N protein with amino acid sequence, RGPEQTQGNFGDQNGGR. The results of mass identification were totally in agreement with the conclusion achieved from 2DE-Western blot. All the string 2DE spots contained the fragments of the SARS-CoV N protein and these identified peptides occupied over 70% of the entire amino acid sequence of the N protein. Next question was if these forms of the N protein could be derived from phosphorylation modification. As shown in Table 1 , the careful calculations upon the data of MALDI-TOF/TOF MS indicated that all the satisfied mass signals well matched with the theoretical peptide predication but without 80 delton shift, which was a symbol of phosphorylation. Furthermore, the digested products were treated with IMAC for enrichment of the phosphorylated peptides, and the enriched peptides still released the poor signals for the phosphorylated peptides of the N protein.
Fig. 3.
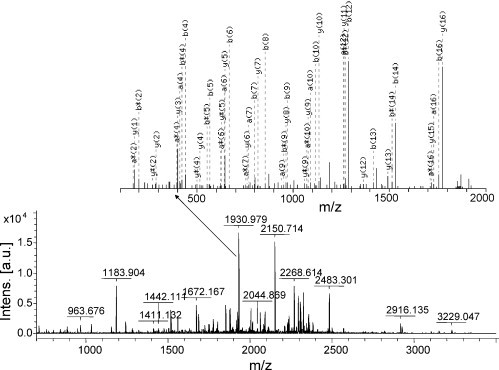
The 2DE spot from the Y258 yeast cells was identified by MALDI-TOF/TOF MS.
Table 1.
The peptides of the N protein completely digested by trypsin
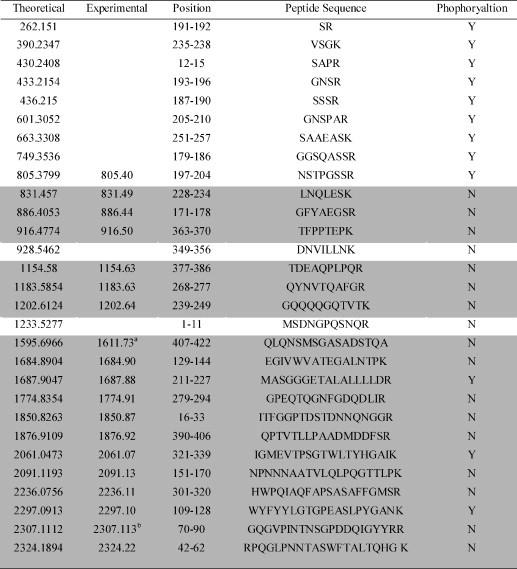
aOxidation (M). bThe peptide contains the putative phosphorylation site.
The peptides in the shadow part were identified by MALDI TOF/TOF MS in the experiments.
What is a proper explanation to the conflict results from 2DE and mass spectrometry for the N protein? The NetPhos 2.0 software was adopted to analyze the putative sites of phosphorylation in the N protein. The prediction results revealed that 22 serine, 8 threonine and 3 tyrosine residues could be phosphorylated in this protein (Fig. 4 ). It was obvious in Fig. 4 that a region around 180–210 aa occupied 60% putative phosphorylated serine residues, so called “dense serine” island. On the other hand, this island also contained six arginine residues. After a completely tryptic digestion, thus, the N fragment located 180–210 aa could generate a series of short peptides, which were not easily detected by MALDI-TOF/TOF MS. In most N proteins of coronaviruses, phosphorylation of tyrosine and threonine was rarely reported. So serine phosphorylation became a target in this study. Table I listed all the putative sites of phosphorylated serine and their corresponding mass signals. Not surprisingly, these tryptic peptides located at the “dense serine” island were so short that they were not identified by MALDI-TOF/TOF MS. Identification of the phosphorylation sites through mass spectrometry, therefore, seemed infeasible in a complete digestion with trypsin to the N protein. More experimental designs were required to access these phosphorylated sits on the N protein.
Fig. 4.

Prediction of the putative phosphorylation sites on the N protein with NetPhos 2.0. The upper panel shows the primary amino acid sequence of the protein, and in the lower panel, the tyrosines (Y), threonines (T), or serines (S) are the sites that could be potentially phosphorylated. The shadow region is “dense serine” island.
3.3. Identification of the phosphorylation regions of the N protein with the strategy of truncated recombinants
To test the hypothesis described above, three expression vectors were constructed, which were able to express the N-full, ΔN124 and ΔN256 proteins (Table 2 ). The N-full protein contained the full length of amino acid sequence of the N protein, the ΔN124 protein lacked 124 amino acids at the N-terminus of the N protein but contains 298 amino acids at C-terminus, and the ΔN256 protein only had 166 amino acids at the C-terminus of the N protein. Specifically, N-full and ΔN124 remained 180–210 aa regions, whereas ΔN256 lost this region. All the three recombinants were incubated with PKCα and the reacted products were detected by 2DE Western blot. As shown in Fig. 5 , several immunostained spots of N-full and ΔN124 shifted to acidic pH side, conversely, ΔN256 remained at a single spot located around its theoretical pI of 6.8 after PKCα phosphorylation was induced. This convincingly demonstrated that the “dense serine” island, at least at the region of 1–256 aa at the N-terminus of the N protein, was likely to be the phosphorylation region of the N protein. Moreover, the next question was how to precisely locate the phosphorylated residues in this region.
Table 2.
The physiochemical information of the truncated N proteins
| Protein | Position | MW (kDa) | pI |
|---|---|---|---|
| N-full | 1–420 aa | 64 | 9.48 |
| ΔN124 | 125–420 aa | 50 | 9.06 |
| ΔN256 | 257–420 aa | 36 | 6.77 |
Fig. 5.

Western blot analysis to identify the truncated N proteins treated with/without PKC. A and B, the N-full protein fusioned with thioredoxin expressed in E. coli was treated with/without PKCα; C, the ΔN124 protein fusioned with thioredoxin in E. coli was treated with PKCα; D, the ΔN256 protein fusioned with thioredoxin expressed in E. coli was treated with PKCα.
3.4. Identification of phosphorylation sites of the N protein with the strategy of partially tryptic digestion
As described above, these short peptides at the “dense serine” island generated by a completely tryptic digestion should be avoided for the experiments of mass spectrometry. A strategy was introduced, in which the N protein expressed from yeast were excised and partially digested with trypsin, thus, these relatively long fragments of the N protein were achieved for peptide identification by mass spectrometry. The two kinds of the N recombinants, either from E. coli or from yeast, were used in the partially digestive experiments. Compared of the mass signals in the N peptides from E. coli, two unique mass peaks appeared in the yeast N recombinant with the significant neutral loss. As shown in Fig. 6 , a new PMF peak at 2113.90 was detected and further analyzed by MS/MS to match with the peptide, GFYAEGSRGGSQASSRSSpSR, located at 171–190 aa. The residue of Ser189 was possibly phosphorylated. This fragment contained the two miss-cleaved tryptic sites, which were never found in the completely digestive products of the N protein expressed in E. coli. More importantly, when the N protein from E. coli was phosphorylated by PKCα and the phosphorylated N protein was treated with partial digestion of trypsin, the same PMF as well as MS/MS spectra were monitored consistently. Another similar phenomenon was observed at PMF peak at 2371.10, which was further identified by tandem MS to confirm the N fragment, GNSGNSTPGSSRGNSpPARMASGGGK, located at 193–217 aa (spectra not shown). The residue of Ser207 was likely phosphorylated. The mass spectrometry data, thus, provided undoubted evidence that some serine residues located at the “dense serine” island were the substrates for the protein kinases in host cells. These data also coincided with the prediction of NetPhos 2.0 described in Fig. 4. Furthermore, the phenomenon of the multiple 2DE spots of the N protein expressed from yeast could be partially explained by these dada. Although the MasCot search gave the results indicating Ser189 and Ser207 to be the phosphorylation sites, the phosphorylation possibility for other serine residues around Ser189 and Ser207 could not be excluded, because the neutral loss of phosphoric acid (H3PO4) could be generated from other neighbor serines but fail in detection by mass spectrometry. Therefore, enrichment of phosphorylated peptides and site-directed mutagenesis are required in further investigation to precisely define the locations for all serine candidates of phosphorylation in the N protein.
Fig. 6.
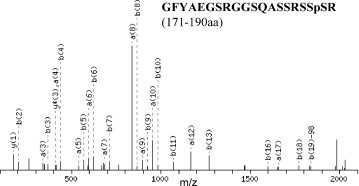
MS/MS spectra of the detected PMF peak at 2311.90.
4. Discussion
Protein phosphorylation is a ubiquitous modification in eukaryotic cells responding to extracellular stimuli or intracellular metabolic changes, and the modified proteins can perform multiple functions, particularly in signaling transduction [15]. Taking a closer look at the sequenced coronaviral genomes, the N proteins contain high percentage of serine residues with multiple theoretical phosphorylation sites. Experimentally, the phosphorylation of the N protein for other coronaviruses was demonstrated long time ago. Siddell et al. reported that the N protein of coronavirus JHM was phosphorylated during infection, specifically for serine phosphorylation [16]. Interestingly, a protein kinase that phosphorylates the N protein had a strong association with this virion, and was co-purified with the coronavirus, suggesting this viral N protein mainly existed as the phosphorylated status in the infected cells. The PRRSV N protein was confirmed to be a phosphorylated protein [17]. Notably, the phosphorylation of the N protein was not only observed in the infected cells, but also detected in the N gene transfected eukaryote cells, indicating that the phosphorylation was independent from viral components, and was associated with the amino acid sequences of the N protein or the environment of the host cells. On the other hand, even though the phenomenon of the phosphorylated N protein was discovered long time ago, the studies on these modified proteins have not been extensively carried out so far. There have been few reports regarding the phosphorylated sites in the N proteins, in particular the accurate identification based upon biochemical or molecular biological approaches [18]. Moreover, the phosphorylated N protein in coronavirus is likely to play different roles in the infected host cells. For instance, the phosphorylated N protein in MHV appeared in strong affinity to genomic RNA, the dephosphorylated MHV N protein would resulted in the viral RNA releasing [19]. However, the localization of N protein from PRRSV was not regulated by its phosphorylation [17]. Using double radiolabeling (35S and 32Pi), an equivalent distribution of the phosphorylated N protein was observed in both cytoplasmic and nuclear fractions in the PRRSV infected host cells. To understand the phophorylaion status of the SARS-CoV N protein could extend our knowledge how SARS virus effectively infects human and causes a spread infection.
Based on the data from mass spectrometry, Ying et al. declared that there was no evidence indicative for the phosphorylated residues in the SARS-CoV N protein [12]. The observation was challenged by theoretical prediction and experimental evidence. First of all, the mass data, which was obtained from a proteomic survey to the SARS-CoV proteins, was not favorable to aim at specific searching for the phosphorylated peptides. If the N protein contains multiple phosphorylation sites and not all the sites phophorylated simultaneously, the average signals for these phosphorylated peptides are expected in lowabundance. On the other hand, the phosphorylated peptides are usually suppressed in the intensity of mass signals [20]. Hence, as compared with so many high abundance non-phosphorylated peptides, these phosphorylated peptides are possible in miss-detection in such proteomic survey. Secondly, as briefly described in Section 3, the complete tryptic digestion, which was a digestion approach adopted by the two groups for proteomic search of SARS-CoV protein [11], [12], can produce several short peptides around the “dense serine” island which are not easily detected by mass spectrometry. Thirdly, using 2DE and Western blot, a series of string spots corresponding to the N protein were detected from the sera of the SARS patients and the Vero E6 cells infected by SARS-CoV, suggesting that this N protein had several modification forms due to infection (unpublished observations from our group). Furthermore, Lal et al. provided the direct evidence for the phosphorylation of the N protein [13], [14]. Just based upon the logical deduction, the systematical investigation was initiated to specifically address this issue regarding the phosphorylation sites of the N protein, and the results presented here firmly supported the hypothesis that indeed the SARS-CoV N protein was phosphorylated in the host eukaryote cells.
Using amino acid sequence of the “dense serine” island to blast against the protein database in NCBI, the blasted results demonstrated that not only in the SARS-CoV N protein, but the similar domains also widely existed in other N proteins from several viruses, such as bat coronavirus, porcine epidemic diarrhea virus, heliothis zea virus, and human herpesvirus. So the phenomenon of multiple serine phosphorylation sites located at a specific region may represent the significant features for the functions of these viruses. Identifying these phosphorylation sites is likely to partially elucidate the infection mechanisms. In the “dense serine” island, total of 12 serine residues could be phosphorylated. If these residues share the equal opportunities for phosphorylation, the possible modification forms are 4095 upon combination calculation. The theoretical calculation seems to well match with the experimental observations because the string 2DE spots imply many modified forms of the N protein. However, there were only two sites of the SARS-CoV N proteins identified in this study. According to the combination estimation, the two sites could only generate three forms in maximum. Obviously, the current results are still limit to fully explain the 2DE behavior of the N protein. The extensive investigation in near future should focus on the enrichment of the phosphorylated peptides and generation of site-directed mutants or truncated N proteins, which will supply the precise identification to the phosphorylation candidates in the N protein.
Acknowledgements
The authors thank the efforts at the initial stage of this project contributed from Mr. Hao Wang and Miss Shuting Li. Grant support: 863 Program of China (grant no. 2006AA02Z492) and CAS International Partnership Program for Creative Research Teams.
Contributor Information
Rong Wang, Email: rong.wang@mssm.edu.
Siqi Liu, Email: siqiliu@genomics.org.cn.
References
- 1.Ksiazek T.G., Erdman D., Goldsmith C.S. N. Engl. J. Med. 2003;348:1953. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 2.WHO Wkly. Epidemiol. Rec. 2003;78:81. [Google Scholar]
- 3.Rota P.A., Oberste M.S., Monroe S.S. Science. 2003;300:1394. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 4.W. Jian, L. Wei, X. Zuyuan, et al. Chin. Sci. Bull., 48 (2003), 941.
- 5.Masters P.S. Adv. Virus Res. 2006;66:193. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drosten C., Preiser W., Gunther S. Trends Mol. Med. 2003;9:325. doi: 10.1016/S1471-4914(03)00133-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai M.M., Cavanagh D. Adv. Virus Res. 1997;48(1997):1. doi: 10.1016/S0065-3527(08)60286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faaberg K.S., Murtaugh M.P., Yuan S. Adv. Exp. Med. Biol. 2001;494:37. doi: 10.1007/978-1-4615-1325-4_5. [DOI] [PubMed] [Google Scholar]
- 9.Yang J., Koprowski H., Dietzschold B. J. Virol. 2002;73:1661. doi: 10.1128/jvi.73.2.1661-1664.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu X., Gong X., Foley H.D. J. Virol. 2002;76:4153. doi: 10.1128/JVI.76.9.4153-4161.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O. Krokhin, Y. Li, A. Andonov, et al. Mol. Cell. Proteomics 2 (2003): 346. [DOI] [PMC free article] [PubMed]
- 12.Ying W., Hao Y., Zhang Y. Proteomics. 2004;4:492. doi: 10.1002/pmic.200300676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Surjit M., Kumar R., Mishra R.N. J. Virol. 2005;79:11476. doi: 10.1128/JVI.79.17.11476-11486.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Surjit M., Kumar R., Mishra R.N. J. Biol. Chem. 2006;281:10669. doi: 10.1074/jbc.M509233200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munter S., Way M., Frischknecht F. Sci. STKE. 2006;16:re5. doi: 10.1126/stke.3352006re5. [DOI] [PubMed] [Google Scholar]
- 16.Siddell S., Wege H., ter Meulen V. Curr. Top. Microbiol. Immunol. 1982;99:131. doi: 10.1007/978-3-642-68528-6_4. [DOI] [PubMed] [Google Scholar]
- 17.Rowland R.R., Yoo D. Virus Res. 2003;95:23. doi: 10.1016/S0168-1702(03)00161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banerjee S., Narayanan K., Mizutani T., Makino S. J. Virol. 2002;76:5937. doi: 10.1128/JVI.76.12.5937-5948.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bednar V., Verma S., Blount A., Hogue B.G. Adv. Exp. Med. Biol. 2006:127. doi: 10.1007/978-0-387-33012-9_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salih E. Mass Spectrom. Rev. 2005;24:828. doi: 10.1002/mas.20042. [DOI] [PubMed] [Google Scholar]