

Graphical abstract
Keywords: T-cell epitope, Computational prediction, Docking, Vaccine design
Highlights
-
•
Combining epitope prediction methods with molecular docking techniques to identify MHC class I restricted T-cell epitopes.
-
•
Based on available experimental data, the prediction accuracy is up to 90%.
-
•
Providing a valuable step forward for the design of better vaccines.
-
•
Better understanding the activation of T-cell epitopes by MHC binding peptides.
Abstract
In silico identification of T-cell epitopes is emerging as a new methodology for the study of epitope-based vaccines against viruses and cancer. In order to improve accuracy of prediction, we designed a novel approach, using epitope prediction methods in combination with molecular docking techniques, to identify MHC class I restricted T-cell epitopes. Analysis of the HIV-1 p24 protein and influenza virus matrix protein revealed that the present approach is effective, yielding prediction accuracy of over 80% with respect to experimental data. Subsequently, we applied such a method for prediction of T-cell epitopes in SARS coronavirus (SARS-CoV) S, N and M proteins. Based on available experimental data, the prediction accuracy is up to 90% for S protein. We suggest the use of epitope prediction methods in combination with 3D structural modelling of peptide-MHC-TCR complex to identify MHC class I restricted T-cell epitopes for use in epitope based vaccines like HIV and human cancers, which should provide a valuable step forward for the design of better vaccines and may provide in depth understanding about activation of T-cell epitopes by MHC binding peptides.
1. Introduction
Two major T-cell responses are involved in cellular immune response to pathogens: the first is mediated by MHC (major histocompatibility complex) class I-restricted CD8+ CTL (cytotoxic T lymphocytes), and the second is mediated by MHC class II-restricted CD4+ T helper responses (De Groot et al., 2001). The short peptides (~9 residues) primarily derived from intracellularly proteolysed proteins and long peptides (typically 12–28 residues) mainly derived from exogenous antigens are presented on the surface of infected cells to specific T-cell receptors (TCR) by MHC class I and II molecules respectively (Bian et al., 2003, Martin et al., 2003). Multi-epitope vaccine based on combination of different T-cell epitopes could induce potent multi-epitope-specific immune responses both in preventing viral infection and in blocking the possibility of immune evasion (Liu et al., 2003, Fayolle et al., 2001, Whitton et al., 1993). Crystallographic analyses of class I MHC/peptide complexes have shown that the charged peptide amino- and carboxyl-termini are coordinated to polar residues at the two ends of a binding groove with MHC residues, most of the central residues of the peptides are exposed in the MHC complexes and are recognized by TCRs (Garboczi et al., 1996). The rapid and reliable identification of T-cell epitopes is of great importance, as it will reduce significantly experimental workload for epitope-based vaccine design. Fortunately, many predictive methods are currently available (Doytchinova and Flower, 2002). However, each method has limited accuracy, none consistently outperforms the rest (Lundegaard et al., 2010, Roomp et al., 2010, Yu et al., 2002). The prediction based on primary sequence data alone is therefore not sufficient (Schueler-Furman et al., 1998).
In this study, we present a novel computational approach for the rapid and accurate selection of MHC class I restricted T-cell epitopes, to improve epitope-based vaccine design using HLA-A2/A0201-restricted epitopes in HIV p24 and influenza virus matrix proteins and S, M and N proteins of SARS-CoV as a model. Specifically, epitopes were selected following a sequential three-step strategy: (1) T-cell epitopes were predicted using available 12 epitope prediction methods, (2) molecular docking techniques were used to model the interactions between the selected peptide with MHC class I, and (3) peptide-loaded MHC class I molecule interaction with TCR α and β was modelled. While the first step allows to narrow down the candidate T-cell epitope list. The second and third allow to identify peptides resulting in negative free energy of peptide-MHC-TCR binding.
2. Methods
Protein sequences were downloaded from the NCBI web site: CAD36433 for HIV p24 protein (strain OOTCD in Chad), AAT12058 for influenza virus matrix protein (duck/Shanghai/H5N1), AAP41037 for SARS-CoV S protein (strain Tor2 in Canada), AAP41041 for SARS-CoV M protein, and P59595 for SARS-CoV N protein. The epitope prediction methods used here include: (1) ANNPred based on Artificial Neural Networks (ANNs) (Lata et al., 2007) (http://www.imtech.res.in/raghava/nhlapred/neural.html), (2) ComPred based on combination of Artificial Neural Networks (ANNs) and Quantitative Matrices (QM) (Lata et al., 2007) (http://www.imtech.res.in/raghava/nhlapred/comp.html), (3) BIMAS (BioInformatics & Molecular Analysis Section) based on quantitative matrix (Parker et al., 1994) (http://bimas.dcrt.nih.gov/molbio/hla_bind/). (4) CTLPred based on Artificial Neural Networks (ANNs), quantitative matrix (QM) and support vector machine (SVM) (Bhasin and Raghava, 2004) (http://www.imtech.res.in/raghava/ctlpred/index.html), (5) MHCPred based on Quantitative Structure Activity Relationship (QSAR) models generated by partial least squares (PLS) method (Guan et al., 2003) (http://www.jenner.ac.uk/MHCPred/), (6) NetMHC-3.0 based on neural network prediction of binding affinities (Nielsen et al., 2003) (http://www.cbs.dtu.dk/services/NetMHC/), (7) PREDEP based on the structural data of peptide-MHC complexes (Schueler-Furman et al., 2000) (http://bioinfo.md.huji.ac.il/marg/Teppred/mhc-bind/), (8) ProPred-I (Singh and Raghava, 2003) (http://www.imtech.res.in/raghava/propred1/), (9) RANKPED based on Position Specific Scoring Matrices (PSSMs) (Reche et al., 2002) (http://mif.dfci.harvard.edu/Tools/rankpep.html), (10) SMM based on linear programming (Peters et al., 2003) (http://zlab.bu.edu/SMM/), (11) SVMHC based on the Support Vector Machine (SVM) (Donnes and Elofsson, 2002) (http://www.sbc.su.se/svmhc/new.cgi), (12) SYFPEITHI based on peptide binding motif (Rammensee et al., 1999) (http://www.uni-tuebingen.de/uni/kxi/). The HLA-A2/A0201-restricted 9-residue epitopes of each protein were predicted by the above 12 methods. For each method and each protein we selected the top 20 predicted peptides, and the peptides identified by at least 4 methods were used for further analysis.
The tertiary structure of selected peptides was predicted using the PEPstr server (http://www.imtech.res.in/raghava/pepstr/). The molecular docking algorithm based on shape complementarity principles PatchDock (Schneidman-Duhovny et al., 2003) was used to model the interactions of the peptide with MHC and TCR molecules. The 3D coordinates of MHC (HLA-A0201) and TCR were downloaded from the Protein Database Bank (PDB ID: 1AO7). For the peptides obtained above, firstly, we dock the peptide into MHC molecule and extract top 60 conformations of MHC-peptide complexes. We consider those conformations similar to the native complex 1AO7, i.e. the peptide is docked into the peptide-binding groove with the same orientation as the native peptide in 1AO7 complex (the first amino acid (P1) of peptide is oriented towards C terminus of MHC molecule (residues 181–274)). The binding free energy score of each conformation is predicted by the Dcomplex program (Liu et al., 2004). Then from these conformations we select a conformation with the minimum binding free energy score as the theoretical model of peptide-MHC complex. Subsequently peptide-MHC complex is docked onto the TCR molecule using PatchDock. Similarly, we extract top 60 conformations of complex and take those conformations similar to the native complex 1AO7, in which the peptide interacts both with the TCR α chain and TCR β chain, the binding free energy scores of the two chains with the peptide are predicted by Dcomplex program. From these conformations we select the conformation that basically has the minimum binding free energy scores for the peptide with both chains, such a conformation is considered as the theoretical model of peptide–MHC–TCR complex. The visualization of all 3D structures was done in PROTEINEXPLORER (http://www.proteinexplorer.org).
3. Results and discussion
We employed 12 T-cell epitope prediction algorithms to conduct computational identification of HLA-A2/A0201-restricted T-cell epitopes for HIV p24 and influenza virus matrix proteins, for which class I restricted epitopes have been determined experimentally. The candidate T-cell epitopes identified by at least 4 prediction algorithms and their binding free energy scores with MHC and TCR are shown in Table 1, Table 2 , including 24 and 25 peptides in HIV p24 and influenza virus matrix proteins, respectively. Naturally, we consider the peptide without binding to either the TCR α or β chains as a non-epitope. On the other hand, we take the peptide, whose binding free energy scores with MHC, TCR α and β chains are all negative, as the T-cell epitopes. Hence, 7 peptides (p19, p20, p104, p117, p134, p141 and p214) are predicted as HLA-A2/A0201-restricted 9-mer T-cell epitopes of HIV p24 protein (Table 1), and 10 peptides (inf3, inf51, inf58, inf59, inf60, inf107, inf123, inf138, inf178 and inf211) are predicted as HLA-A2/A0201-restricted 9-mer T-cell epitopes of influenza virus matrix protein (Table 2). By searching the HIV Molecular Immunology Database (http://www.hiv.lanl.gov/content/immunology/) and MHCBN Database (http://www.imtech.res.in/raghava/mhcbn/), which consists of 20,000+ MHC binding and non-binding peptides, only 3 peptides (p19, p20 and p214) (Table 1) are HLA-A2/A0201-restricted 9-mer T-cell epitopes for HIV p24 protein, and 5 peptides (inf3, inf51, inf58, inf59 and inf60) (Table 2) are HLA-A2/A0201-restricted 9-mer T-cell epitopes for influenza virus matrix protein. Thus, using at least 4 epitope prediction methods alone, for HIV p24 protein only 3 out of 24 peptides (Table 1) are correctly predicted resulting in a prediction accuracy of 12.5%. For influenza virus matrix protein only 5 out of 25 peptides (Table 2) are correctly predicted resulting in a prediction accuracy of 20%. In particular, p144 is predicted to be an epitope by 11 methods (Table 1), but has been characterized as non-epitope experimentally (Sylvester-Hvid et al., 2004) (biochemical binding assay). On the other hand, when using at least 4 epitope prediction methods in combination with molecular docking technique, the prediction accuracy is greatly improved, the predictive results of 20 peptides are consistent with experiment among 24 peptides of HIV p24 protein and 25 peptides of influenza virus matrix protein (Table 1, Table 2), that is, the prediction accuracy is 83.3% for HIV p24 protein and 80% for influenza virus matrix protein.
Table 1.
HLA-A2/A0201-restricted T-cell epitopes of HIV p24 protein identified by at least 4 epitope prediction methods and molecular docking technique.
| Peptide | Sequence | No. of methods | Methodsa | Peptide binding free energy score |
Epitope |
|||
|---|---|---|---|---|---|---|---|---|
| MHC | TCR α | TCR β | Prediction | Experiment | ||||
| p15 | SLSPRTLNA | 9 | 1–4, 6, 8, 10–12 | −2.09 | No binding | 1.86 | No | No |
| p19 | RTLNAWVKV | 9 | 1–6, 8, 10, 12 | −0.91 | −0.42 | −0.66 | Yes | Yes |
| p20 | TLNAWVKVI | 9 | 1–4, 6,8, 9, 11, 12 | −3.38 | −0.5 | −0.32 | Yes | Yes |
| p36 | EVIPMFSAL | 6 | 1, 2, 4, 7–9 | −3.13 | 0.5 | 0.02 | No | No |
| p52 | DLNMMLNVV | 9 | 1–4, 6, 8, 9, 11, 12 | −3.94 | 1.58 | −0.12 | No | No |
| p66 | AMQMLKDTI | 6 | 2, 3, 8, 9, 11, 12 | −3.39 | 0.39 | 0.79 | No | No |
| p79 | AEWDRVHPV | 8 | 1–4, 8, 10–12 | −0.52 | −0.15 | 2.23 | No | No |
| p104 | DIAGTTSTL | 5 | 1, 2, 9, 11, 12 | −0.96 | −0.01 | −0.7 | Yes | No |
| p111 | TLQEQIGWM | 8 | 1, 3, 5, 6–8, 11, 12 | 0.81 | 3.69 | −0.36 | No | No |
| p117 | GWMTGNPPV | 4 | 2, 5, 7, 10 | −2.89 | −0.84 | −0.22 | Yes | No |
| p119 | MTGNPPVPV | 9 | 1–3, 5, 6, 8, 10–12 | −3.13 | 0.32 | 0.18 | No | No |
| p129 | DIYRRWIIL | 4 | 1, 7, 9, 12 | 0.39 | −1.48 | −1.73 | No | No |
| p131 | YRRWIILGL | 4 | 2, 5, 10, 12 | −3.25 | 1.09 | −0.64 | No | No |
| p134 | WIILGLNKI | 6 | 1–3, 8, 9, 12 | −1.82 | −0.56 | −1.16 | Yes | No |
| p135 | IILGLNKIV | 8 | 1–3, 5, 8, 9, 11, 12 | 0.75 | 0.4 | No binding | No | No |
| p138 | GLNKIVRMY | 4 | 1, 2, 9, 11 | −2.39 | No binding | −0.49 | No | No |
| p141 | KIVRMYSPV | 8 | 1–3, 5, 6, 8, 9, 12 | −0.91 | −0.43 | −0.64 | Yes | No |
| p144 | RMYSPVSIL | 11 | 1–6, 8–12 | −2.34 | 0.42 | 2.6 | No | No |
| p165 | YVDRFFKCL | 7 | 1–5, 8, 9 | −3.21 | No binding | −2.32 | No | No |
| p184 | NWMTETLLV | 4 | 1, 7, 9, 11 | −1.39 | 0.03 | −0.73 | No | No |
| p204 | KALGTGATL | 7 | 1–3, 8, 9, 11, 12 | −0.84 | 1.83 | −0.74 | No | No |
| p210 | ATLEEMMTA | 6 | 1–3, 6, 8, 10 | −1.93 | −0.46 | 0.35 | No | No |
| p211 | TLEEMMTAC | 8 | 1–3, 6–9, 11 | −3.65 | 1.68 | 1.24 | No | No |
| p214 | EMMTACQGV | 8 | 1–3, 6–9, 11 | −1.94 | −0.95 | −0.06 | Yes | Yes |
1: ANNPRED; 2: COMPRED; 3: BIMAS; 4: CTLPRED; 5: MHCPRED; 6: NETMHC; 7: PREDEP; 8: PROPRED-I; 9: RANKPED; 10: SMM; 11: SVMHC; 12: SYFPEITHI.
Table 2.
HLA-A2/A0201-restricted T-cell epitopes of influenza virus matrix protein identified by at least 4 epitope prediction methods and molecular docking technique.
| Peptide | Sequence | No. of methods | Methodsa | Peptide binding free energy score |
Epitope |
|||
|---|---|---|---|---|---|---|---|---|
| MHC | TCR α | TCR β | Prediction | Experiment | ||||
| inf3 | LLTEVETYV | 10 | 1–7, 10–12 | −0.86 | −0.04 | −0.15 | Yes | Yes |
| inf38 | DLEALMEWL | 6 | 2, 7–9, 11, 12 | −0.69 | 0.22 | 1.55 | No | No |
| inf41 | ALMEWLKTR | 8 | 1, 2, 5, 7, 8, 10–12 | −2.48 | 0.74 | 1.96 | No | No |
| inf47 | KTRPILSPL | 4 | 1, 2, 6, 12 | −0.41 | 0.17 | 1.22 | No | No |
| inf51 | ILSPLTKGI | 7 | 1–3, 5, 10–12 | −1.89 | −0.52 | −0.61 | Yes | Yes |
| inf54 | PLTKGILGF | 4 | 5, 7, 9, 11 | −2.02 | 0.69 | 0.34 | No | No |
| inf55 | LTKGILGFV | 4 | 1, 5, 11, 12 | −1.32 | 0.17 | −0.81 | No | No |
| inf58 | GILGFVFTL | 12 | 1–12 | −4.18 | −1.05 | −0.18 | Yes | Yes |
| inf59 | ILGFVFTLT | 6 | 1–3, 6, 10, 11 | −1.99 | −0.32 | −1.96 | Yes | Yes |
| inf60 | LGFVFTLTV | 4 | 3, 5, 10, 12 | −1.03 | −0.9 | −0.36 | Yes | Yes |
| inf107 | ITFHGAKEV | 4 | 3, 8, 11, 12 | −0.14 | −0.07 | −0.6 | Yes | No |
| inf116 | ALSYSTGAL | 7 | 1–3, 9–12 | −1.34 | 0.58 | −0.02 | No | No |
| inf123 | ALASCMGLI | 8 | 1–3, 5, 7, 10–12 | −2.06 | −1.11 | −0.75 | Yes | No |
| inf127 | CMGLIYNRM | 5 | 1–4, 6 | 2.58 | −0.26 | −0.17 | No | No |
| inf130 | LIYNRMGTV | 7 | 1–3, 6, 7, 10, 12 | −3.86 | No binding | 3.35 | No | No |
| inf134 | RMGTVTTEV | 10 | 1–6, 9–12 | −2.7 | −0.02 | 2.1 | No | No |
| inf138 | VTTEVAFGL | 8 | 1–4, 6, 7, 9, 11 | −3.26 | −1.51 | −0.13 | Yes | No |
| inf164 | QMATITNPL | 9 | 1–3, 5, 6, 9, 10–12 | −2.58 | No binding | 0.45 | No | No |
| inf178 | RMVLASTTA | 5 | 1–3, 6, 12 | −3.26 | −0.08 | −1.2 | Yes | No |
| inf180 | VLASTTAKA | 8 | 1–3, 5, 6, 10–12 | −0.41 | No binding | 2.01 | No | No |
| inf191 | QMAGSSEQA | 4 | 1, 2, 5, 8 | −1.39 | 0.21 | 2.5 | No | No |
| inf211 | QMVQAMRTI | 6 | 2, 3, 5, 7, 9, 11 | −2.06 | −0.43 | −0.28 | Yes | No |
| inf232 | NLLENLQAY | 6 | 1, 2, 6, 7, 9, 11 | −3.78 | 1.11 | 0.17 | No | No |
| inf236 | NLQAYQNRM | 6 | 1–4, 6, 11 | −3.98 | −0.27 | 0.46 | No | No |
| inf238 | QAYQNRMGV | 5 | 2–5, 7 | −1.65 | No binding | −0.88 | No | No |
1: ANNPRED; 2: COMPRED; 3: BIMAS; 4: CTLPRED; 5: MHCPRED; 6: NETMHC; 7: PREDEP; 8: PROPRED-I; 9: RANKPED; 10: SMM; 11: SVMHC; 12: SYFPEITHI.
Since such a novel approach is effective both for HIV p24 protein and for influenza virus matrix protein, we next used this approach to predict candidate T-cell epitopes for SARS-CoV S, N and M proteins, which are the major structural proteins of SARS-CoV. S mediates receptor binding and cell fusion, M is an integral membrane protein for virus budding, and N is important in providing nuclear-import signal and viral RNA packaging (Holmes, 2003).
The HLA-A2/A0201-restricted 9-mer T-cell epitopes identified by at least 4 methods for S, N and M proteins and their binding free energy scores with MHC and TCR are shown in Table 3, Table 4, Table 5 , including 18, 20 and 23 peptides in S, N and M, respectively. Based on the combination of epitope prediction methods and molecular docking technique and the same selection criteria used for HIV and Influenza proteins, 4 peptides (s208, s803, s1167 and s1202) were predicted for S protein (Table 3), 5 peptides for N protein (n139, n220, n313, n339 and n400) (Table 4) and 5 peptides for M protein (m14, m23, m25, m52 and m147) (Table 5) as HLA-A2/A0201-restricted T-cell epitopes. Among the 18 peptides identified for S protein, 10 peptides have been determined experimentally, that is, 2 peptides (s1167 and s1202) are T-cell epitopes and 8 peptides (s2, s131, s803, s897, s940, s958, s982 and s1174) are non-epitopes (Wang et al., 2004a, Wang et al., 2004b) (CTL assays both in vivo (transgenic mice and patients) and in vitro (human peripheral blood lymphocytes)). Thus, there are 9 out of 10 peptides predicted in our approach using at least 4 epitope prediction methods in combination with molecular docking technique do effectively correlate with experimental data (Table 3). When predicted by at least 4 epitope prediction methods is used as sole criteria, only 2 out of 10 peptides are correctly predicted for S protein (Table 3). The predicted 3D models of s1167/s1202-MHC-TCR complex are shown in Fig. 1 . In contrast to S no experimental data are available for N protein 9-mer epitopes. Among 23 peptides identified as T-cell epitope from M protein, only peptide m88 was determined experimentally as non-epitope (Sylvester-Hvid et al., 2004), which is consistent with the predictive result of our approach (Table 5), similar to p144 in HIV-1 p24 protein, which is predicted as epitope by 11 epitope prediction methods, but is a non-epitope when analysed experimentally (Table 1).
Table 3.
HLA-A2/A0201-restricted T-cell epitopes of SARS-CoV S protein identified by at least 4 epitope prediction methods and molecular docking technique.
| Peptide | Sequence | No. of methods | Methodsa | Peptide binding free energy score |
Epitope |
|||
|---|---|---|---|---|---|---|---|---|
| MHC | TCR α | TCR β | Prediction | Experiment | ||||
| s2 | FIFLLFLTL | 5 | 1, 3, 7, 11, 12 | −4.57 | −0.04 | 1.78 | No | No |
| s131 | ELCDNPFFA | 5 | 1–3, 6, 7 | −1.5 | No binding | −0.53 | No | No |
| s208 | DLPSGFNTL | 5 | 1, 2, 11, 12 | −3.14 | −0.36 | −0.36 | Yes | Unknown |
| s256 | YLKPTTFML | 9 | 1, 2, 5–7, 9–12 | −1.31 | No binding | 1.24 | No | Unknown |
| s282 | PLAELKCSV | 5 | 1, 7, 9, 11, 12 | −2.14 | 1.05 | 0.4 | No | Unknown |
| s404 | VIADYNYKL | 5 | 2, 6, 7, 10, 12 | −2.11 | −0.06 | 0.71 | No | Unknown |
| s673 | SIVAYTMSL | 4 | 5, 6, 9, 12 | 1.42 | −0.24 | No binding | No | Unknown |
| s803 | LLFNKVTLA | 4 | 3, 5, 6, 11 | −3.11 | −0.32 | −0.35 | Yes | No |
| s851 | MIAAYTAAL | 4 | 5, 9, 10, 12 | −2.96 | No binding | −2.12 | No | Unknown |
| s897 | VLYENQKQI | 4 | 3, 5, 9, 11 | −2.6 | 0.03 | −0.26 | No | No |
| s919 | SLTTTSTAL | 4 | 6, 8, 9, 11 | −2.48 | 1.05 | −0.13 | No | Unknown |
| s940 | ALNTLVKQL | 7 | 3–5, 8, 9, 11, 12 | −2.17 | 0.06 | No binding | No | No |
| s958 | VLNDILSRL | 8 | 1–4, 9–12 | −2.79 | −0.02 | 0.76 | No | No |
| s965 | RLDKVEAEV | 5 | 4, 5, 9, 11, 12 | −0.84 | 0.2 | −0.07 | No | Unknown |
| s982 | RLQSLQTYV | 5 | 3, 4, 6, 10, 11 | −0.44 | −0.19 | 1.14 | No | No |
| s1167 | RLNEVAKNL | 7 | 1–4, 8, 9, 11 | −0.86 | −0.36 | −0.03 | Yes | Yes |
| s1174 | NLNESLIDL | 7 | 1–4, 9, 11, 12 | −3.48 | −0.15 | 1.02 | No | No |
| s1202 | FIAGLIAIV | 6 | 1, 2, 9–12 | −2.33 | −0.12 | −0.15 | Yes | Yes |
1: ANNPRED; 2: COMPRED; 3: BIMAS; 4: CTLPRED; 5: MHCPRED; 6: NETMHC; 7: PREDEP; 8: PROPRED-I; 9: RANKPED; 10: SMM; 11: SVMHC; 12: SYFPEITHI.
Table 4.
HLA-A2/A0201-restricted T-cell epitopes of SARS-CoV N protein identified by at least 4 epitope prediction methods and molecular docking technique.
| Peptide | Sequence | No. of methods | Methodsa | Peptide binding free energy score |
Epitope |
|||
|---|---|---|---|---|---|---|---|---|
| MHC | TCR α | TCR β | Prediction | Experiment | ||||
| n45 | GLPNNTASW | 5 | 2, 7, 9, 11, 12 | 0.01 | −0.01 | 1.89 | No | Unknown |
| n49 | NTASWFTAL | 4 | 1, 2, 6, 9 | −3.7 | 1.71 | −0.35 | No | Unknown |
| n139 | ALNTPKDHI | 6 | 1–3, 9, 11, 12 | −2.97 | −0.57 | −0.28 | Yes | Unknown |
| n159 | VLQLPQGTT | 4 | 3, 6, 11, 12 | −1.69 | −0.12 | 3.43 | No | Unknown |
| n160 | LQLPQGTTL | 5 | 3, 2, 6, 8, 12 | −0.59 | −0.01 | 0.5 | No | Unknown |
| n217 | ETALALLLL | 5 | 2, 7–9, 12 | −3.55 | 0.13 | −0.09 | No | Unknown |
| n219 | ALALLLLDR | 6 | 1, 2, 5, 7, 11, 12 | −3.07 | −0.7 | 1.76 | No | Unknown |
| n220 | LALLLLDRL | 7 | 1–3, 5, 8, 9, 12 | −2.4 | −0.43 | −0.79 | Yes | Unknown |
| n223 | LLLDRLNQL | 11 | 1–7, 9–12 | −3.57 | 0.35 | −0.34 | No | Unknown |
| n227 | RLNQLESKV | 9 | 1–5, 9–12 | −3.74 | No binding | −0.14 | No | Unknown |
| n263 | RTATKQYNV | 8 | 1–4, 6, 10–12 | −1.19 | −0.35 | 0.11 | No | Unknown |
| n304 | QIAQFAPSA | 5 | 1, 3, 5, 10, 12 | −2.74 | −0.21 | No binding | No | Unknown |
| n313 | SAFFGMSRI | 4 | 1–3, 9 | −1.07 | −0.58 | −0.27 | Yes | Unknown |
| n317 | GMSRIGMEV | 7 | 1–3, 5, 6, 10–12 | −3.01 | 0.11 | 1.44 | No | Unknown |
| n332 | LTYHGAIKL | 8 | 1–3, 6, 8–10, 12 | −2.53 | 0.21 | −2.18 | No | Unknown |
| n339 | KLDDKDPQF | 4 | 5, 7, 9, 11 | −2.74 | −0.28 | −0.3 | Yes | Unknown |
| n352 | ILLNKHIDA | 6 | 1, 3, 7, 10–12 | −2.6 | 1.11 | −0.09 | No | Unknown |
| n388 | KKQPTVTLL | 4 | 1, 2, 6, 9 | 0.09 | No binding | 0.26 | No | Unknown |
| n400 | DMDDFSRQL | 8 | 1–3, 5, 7, 9, 11,12 | −2.34 | −0.1 | −0.43 | Yes | Unknown |
| n407 | QLQNSMSGA | 4 | 1, 3, 10, 11 | −1.97 | −0.09 | 1.4 | No | Unknown |
1: ANNPRED; 2: COMPRED; 3: BIMAS; 4: CTLPRED; 5: MHCPRED; 6: NETMHC; 7: PREDEP; 8: PROPRED-I; 9: RANKPED; 10: SMM; 11: SVMHC; 12: SYFPEITHI.
Table 5.
HLA-A2/A0201-restricted T-cell epitopes of SARS-CoV M protein identified by at least 4 epitope prediction methods and molecular docking technique.
| Peptide | Sequence | No. of methods | Methodsa | Peptide binding free energy score |
Epitope |
|||
|---|---|---|---|---|---|---|---|---|
| MHC | TCR α | TCR β | Prediction | Experiment | ||||
| m1 | MADNGTITV | 7 | 1, 2, 5, 9–12 | −2.33 | −1.32 | 1.28 | No | Unknown |
| m7 | ITVEELKQL | 4 | 2, 8, 9, 12 | −2.69 | 3.43 | 1.58 | No | Unknown |
| m14 | QLLEQWNLV | 6 | 3, 6, 7, 10–12 | −1.89 | −0.13 | −1.17 | Yes | Unknown |
| m15 | LLEQWNLVI | 6 | 1, 2, 7, 9, 11, 12 | −2.79 | 3.78 | −0.26 | No | Unknown |
| m20 | NLVIGFLFL | 9 | 1–4, 7–9, 11, 12 | −2.05 | 0.29 | 0.26 | No | Unknown |
| m23 | IGFLFLAWI | 4 | 3, 5, 8, 10 | −4.9 | −0.72 | −0.48 | Yes | Unknown |
| m25 | FLFLAWIML | 12 | 1–12 | −2.2 | −0.3 | −0.66 | Yes | Unknown |
| m26 | LFLAWIMLL | 6 | 1, 2, 6–9 | −3.99 | 0.03 | 1.8 | No | Unknown |
| m47 | IIKLVFLWL | 4 | 8, 9, 11, 12 | −1.53 | 0.59 | 0.98 | No | Unknown |
| m52 | FLWLLWPVT | 7 | 1–3, 5, 8, 10, 11 | −2.5 | −0.08 | −0.91 | Yes | Unknown |
| m54 | WLLWPVTLA | 9 | 1–3, 5, 6, 8, 10–12 | −2.8 | No binding | −0.76 | No | Unknown |
| m55 | LLWPVTLAC | 8 | 1–3, 5–8, 11 | −3.9 | 0.54 | −0.2 | No | Unknown |
| m58 | PVTLACFVL | 5 | 1, 2, 7–9 | −4.18 | −0.21 | 0.79 | No | Unknown |
| m60 | TLACFVLAA | 5 | 3, 4, 10–12 | −0.54 | 0.95 | 0.89 | No | Unknown |
| m64 | FVLAAVYRI | 10 | 1–6, 8–10, 12 | −2.88 | 0.11 | −0.34 | No | Unknown |
| m71 | RINWVTGGI | 4 | 9–12 | −2.66 | 0.59 | −0.25 | No | Unknown |
| m79 | IAIAMACIV | 4 | 1, 2, 6, 9 | −2.23 | No binding | −1.18 | No | Unknown |
| m88 | GLMWLSYFV | 12 | 1–12 | −4.47 | −0.29 | 1.72 | No | No |
| m89 | LMWLSYFVA | 4 | 3, 6, 7, 10 | −2.98 | 3.54 | −0.4 | No | Unknown |
| m95 | FVASFRLFA | 4 | 3–5, 10 | −2.29 | No binding | 2.68 | No | Unknown |
| m107 | SMWSFNPET | 8 | 1–3, 5, 6, 8–10 | −1.79 | No binding | −1.51 | No | Unknown |
| m131 | PLMESELVI | 6 | 1, 2, 5, 7–9 | −3.55 | 0.15 | −1.21 | No | Unknown |
| m147 | HLRMAGHSL | 4 | 2, 9, 11, 12 | −1.98 | −0.01 | −1.1 | Yes | Unknown |
1: ANNPRED; 2: COMPRED; 3: BIMAS; 4: CTLPRED; 5: MHCPRED; 6: NETMHC; 7: PREDEP; 8: PROPRED-I; 9: RANKPED; 10: SMM; 11: SVMHC; 12: SYFPEITHI.
Fig. 1.
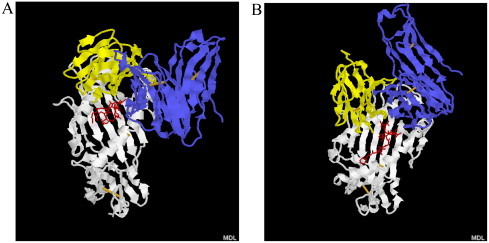
The predicted 3D models for the interactions of MHC molecule (HLA-A0201), T-cell receptor (TCR) and two epitopes: s1167 (RLNEVAKNL) (A) and s1202 (FIAGLIAIV) (B) in SARS coronavirus S protein. The white cartoon stands for MHC, yellow cartoon for TCR α chain, blue cartoon for TCR β chain, and red stick for the two epitopes. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
It must be pointed out that the evaluation of different epitope prediction methods is not the objective of this paper, although the difference of performance between the present method and the past prediction methods was reported in the text. In principle, we primarily use the previous methods to narrow down the selection of candidate epitopes for further docking studies. Here we restricted our selection process to those peptides identified by at least 4 prediction methods based on the following reasons. (a) More methods generate less candidate epitopes, which does not guarantee the accuracy of prediction (even all 12 methods, as mentioned above). (b) Too many candidate epitopes will require excessive computations. Such as S protein, all 12 methods identify about 140 different candidate epitopes in total. As stated above, the 3D structure of each peptide is first predicted, then the peptide is docked into MHC molecule and a complex with correct orientation and the minimum binding free energy score is chosen from 60 conformations, finally this complex is docked into TCR molecule and a complex, in which the peptide interacts with two chains of TCR with the minimum binding free energy score, is chosen also from 60 conformations. However we believe this should be cost-effective compared to experimental assay. On the other hand, due to the fact that the binding to the MHC groove is a typical feature of an epitope, hence, it is very critical to see whether or not peptides can be properly docked to the peptide-binding groove of MHC molecules. So the present strategy is first docking peptides to MHC and then to TCR. Moreover, the docking experiments demonstrated that it is not feasible for the docking in reverse order (i.e. first dock peptides to TCR and then to MHC), because it is very difficult to re-dock the peptide interacted with two chains TCR (TCR-alpha and TCR-beta) in the complex TCR-peptide to the peptide-binding groove of MHC molecules.
In conclusion, we developed a novel approach for the rapid and reliable identification of MHC class I T-cell epitopes by the combination of epitope prediction methods and molecular docking technique, which can dramatically increase the opportunity of successful identification and greatly reduce the number of peptides required for experimental assays. This approach is applicable to the whole viral genome, such as HIV, or other disease models (bacteria, cancer, etc.).
Conflict of interest statement
The authors declare that they have no competing financial interests.
References
- Bhasin M., Raghava G.P. Prediction of CTL epitopes using QM, SVM and ANN techniques. Vaccine. 2004;22(23–24):3195–3204. doi: 10.1016/j.vaccine.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Bian H., Reidhaar-Olson J.F., Hammer J. The use of bioinformatics for identifying class II-restricted T-cell epitopes. Methods. 2003;29(3):299–309. doi: 10.1016/s1046-2023(02)00352-3. [DOI] [PubMed] [Google Scholar]
- De Groot A.S., Bosma A., Chinai N., Frost J., Jesdale B.M., Gonzalez M.A., Martin W., Saint-Aubin C. From genome to vaccine: in silico predictions, ex vivo verification. Vaccine. 2001;19(31):4385–4395. doi: 10.1016/s0264-410x(01)00145-1. [DOI] [PubMed] [Google Scholar]
- Donnes P., Elofsson A. Prediction of MHC class I binding peptides, using SVMHC. BMC Bioinformatics. 2002;3:25. doi: 10.1186/1471-2105-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova I.A., Flower D.R. Quantitative approaches to computational vaccinology. Immunology and Cell Biology. 2002;80(3):270–279. doi: 10.1046/j.1440-1711.2002.01076.x. [DOI] [PubMed] [Google Scholar]
- Fayolle C., Osickova A., Osicka R., Henry T., Rojas M.J., Saron M.F., Sebo P., Leclerc C. Delivery of multiple epitopes by recombinant detoxified adenylate cyclase of Bordetella pertussis induces protective antiviral immunity. Journal of Virology. 2001;75(16):7330–7338. doi: 10.1128/JVI.75.16.7330-7338.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garboczi D.N., Ghosh P., Utz U., Fan Q.R., Biddison W.E., Wiley D.C. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature. 1996;384(6605):134–141. doi: 10.1038/384134a0. [DOI] [PubMed] [Google Scholar]
- Guan P., Doytchinova I.A., Zygouri C., Flower D.R. MHCPred: a server for quantitative prediction of peptide-MHC binding. Nucleic Acids Research. 2003;31(13):3621–3624. doi: 10.1093/nar/gkg510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes K.V. SARS coronavirus: a new challenge for prevention and therapy. Journal of Clinical Investigation. 2003;111(11):1605–1609. doi: 10.1172/JCI18819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lata S., Bhasin M., Raghava G.P.S. Application of machine learning techniques in predicting MHC binders. Methods in Molecular Biology. 2007;409:201–215. doi: 10.1007/978-1-60327-118-9_14. [DOI] [PubMed] [Google Scholar]
- Liu Z., Xiao Y., Chen Y. Epitope-vaccine strategy against HIV-1: today and tomorrow. Immunobiology. 2003;208:423–428. doi: 10.1078/0171-2985-00286. [DOI] [PubMed] [Google Scholar]
- Liu S., Zhang C., Zhou H., Zhou Y. A physical reference state unifies the structure-derived potential of mean force for protein folding and binding. Proteins. 2004;56(1):93–101. doi: 10.1002/prot.20019. [DOI] [PubMed] [Google Scholar]
- Lundegaard C., Lund O., Buus S., Nielsen M. Major histocompatibility complex class I binding predictions as a tool in epitope discovery. Immunology. 2010;130(3):309–318. doi: 10.1111/j.1365-2567.2010.03300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin W., Sbai H., De Groot A.S. Bioinformatics tools for identifying class I-restricted epitopes. Methods. 2003;29(3):289–298. doi: 10.1016/s1046-2023(02)00351-1. [DOI] [PubMed] [Google Scholar]
- Parker K.C., Bednarek M.A., Coligan J.E. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. Journal of Immunology. 1994;152:163–175. [PubMed] [Google Scholar]
- Peters B., Tong W., Sidney J., Sette A., Weng Z. Examining the independent binding assumption for binding of peptide epitopes to MHC-I molecules. Bioinformatics. 2003;19:1765–1772. doi: 10.1093/bioinformatics/btg247. [DOI] [PubMed] [Google Scholar]
- Rammensee H., Bachmann J., Emmerich N.P., Bachor O.A., Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50(3-4):213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- Reche P.A., Glutting J.P., Reinherz E.L. Prediction of MHC class I binding peptides using profile motifs. Human Immunology. 2002;63:701–709. doi: 10.1016/s0198-8859(02)00432-9. [DOI] [PubMed] [Google Scholar]
- Roomp K., Antes I., Lengauer T. Predicting MHC class I epitopes in large datasets. BMC Bioinformatics. 2010;11:90. doi: 10.1186/1471-2105-11-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D., Inbar Y., Polak V., Shatsky M., Halperin I., Benyamini H., Barzilai A., Dror O., Haspel N., Nussinov R., Wolfson H.J. Taking geometry to its edge: fast unbound rigid (and hinge-bent) docking. Proteins. 2003;52(1):107–112. doi: 10.1002/prot.10397. [DOI] [PubMed] [Google Scholar]
- Schueler-Furman O., Elber R., Margalit H. Knowledge-based structure prediction of MHC class I bound peptides: a study of 23 complexes. Folding and Design. 1998;3(6):549–564. doi: 10.1016/S1359-0278(98)00070-4. [DOI] [PubMed] [Google Scholar]
- Schueler-Furman O., Altuvia Y., Sette A., Margalit H. Structure-based prediction of binding peptides to MHC class I molecules: application to a broad range of MHC alleles. Protein Science. 2000;9(9):1838–1846. doi: 10.1110/ps.9.9.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H., Raghava G.P. ProPred1: prediction of promiscuous MHC class-I binding sites. Bioinformatics. 2003;19(8):1009–10014. doi: 10.1093/bioinformatics/btg108. [DOI] [PubMed] [Google Scholar]
- Sylvester-Hvid C., Nielsen M., Lamberth K., Roder G., Justesen S., Lundegaard C., Worning P., Thomadsen H., Lund O., Brunak S., Buus S. SARS CTL vaccine candidates; HLA supertype-, genome-wide scanning and biochemical validation. Tissue Antigens. 2004;63(5):395–400. doi: 10.1111/j.0001-2815.2004.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K., Petrovsky N., Schonbach C., Koh J.Y., Brusic V. Methods for prediction of peptide binding to MHC molecules: a comparative study. Molecular Medicine. 2002;8(3):137–148. [PMC free article] [PubMed] [Google Scholar]
- Wang B., Chen H., Jiang X., Zhang M., Wan T., Li N., Zhou X., Wu Y., Yang F., Yu Y., Wang X., Yang R., Cao X. Identification of an HLA-A*0201-restricted CD8+ T-cell epitope SSp-1 of SARS-CoV spike protein. Blood. 2004;104(1):200–206. doi: 10.1182/blood-2003-11-4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.D., Sin W.Y., Xu G.B., Yang H.H., Wong T.Y., Pang X.W., He X.Y., Zhang H.G., Ng J.N., Cheng C.S., Yu J., Meng L., Yang R.F., Lai S.T., Guo Z.H., Xie Y., Chen W.F., Yang H.H. T-cell epitopes in severe acute respiratory syndrome (SARS) coronavirus spike protein elicit a specific T-cell immune response in patients who recover from SARS. Journal of Virology. 2004;78(11):5612–5618. doi: 10.1128/JVI.78.11.5612-5618.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitton J.L., Sheng N., Oldstone M.B., McKee T.A. A “string-of-beads” vaccine, comprising linked minigenes, confers protection from lethal-dose virus challenge. Journal of Virology. 1993;67(1):348–352. doi: 10.1128/jvi.67.1.348-352.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]