

Abstract
The outbreak of severe acute respiratory syndrome (SARS) in 2002 affected thousands of people and an efficient diagnostic system is needed for accurate detection of SARS coronavirus (SARS CoV) to prevent or limit future outbreaks. Of the several SARS CoV structural proteins, the nucleocapsid protein has been shown to be a good diagnostic marker. In this study, an ssDNA aptamer that specifically binds to SARS CoV nucleocapsid protein was isolated from a DNA library containing 45-nuceotide random sequences in the middle of an 88mer single-stranded DNA. After twelve cycles of systematic evolution of ligands by exponential enrichment (SELEX) procedure, 15 ssDNA aptamers were identified. Enzyme-linked immunosorbent assay (ELISA) analysis was then used to identify the aptamer with the highest binding affinity to the SARS CoV nucleocapsid protein. Using this approach, an ssDNA aptamer that binds to the nucleocapsid protein with a Kd of 4.93 ± 0.30 nM was identified. Western blot analysis further demonstrated that this ssDNA aptamer could be used to efficiently detect the SARS CoV nucleocapsid protein when compared with a nucleocapsid antibody. Therefore, we believe that the selected ssDNA aptamer may be a good alternative detection probe for the rapid and sensitive detection of SARS.
Key words: SARS coronavirus, nucleocapsid, aptamer, ELISA, Western blot analysis
Severe acute respiratory syndrome (SARS) emerged in November 2002 and claimed about 800 lives worldwide (1). Although no reoccurrence has yet been reported, SARS continues to be of great concern because no vaccine or clinical drug is currently available. SARS was caused by a novel coronavirus, SARS coronavirus (SARS CoV). The virus is a (+) sense single-stranded RNA virus with a genome containing 29,727 nucleotides 2., 3. which consist of four structural proteins: spike (S), envelope (E), membrane (M), and nucleocapsid (N) (4). Similar to other coronaviruses, the S and N proteins are highly expressed during viral infection and are a useful target for immune detection 5., 6.. The SARS CoV N protein is of particular interest because it can be a potential target for vaccine development (7) as well as a diagnostic marker for SARS CoV infection 8., 9.. The N protein has been shown to stimulate immune responses in the host, indicating that it has strong antigenicity. Moreover, it is expressed predominantly during the early stage of viral infection (10) and thus is an attractive target for diagnostics. It has also been reported that the convalescent sera from SARS patients strongly reacted with the N protein.
Many attempts have been made to develop diagnostic assay systems for the detection of viral infections such as RT-PCR, immunoblotting, and ELISA. Especially for viral detection of the N protein by ELISA, it is very important that a high affinity monoclonal antibody specific to the N protein be developed. In fact, many researchers have been trying to develop efficient monoclonal antibodies that can be used to detect the N protein by ELISA during the early stage of SARS CoV infection 1., 11., 12.. Recently, a novel method for the electrical detection of the N protein using nanowire biosensors with an antibody mimic protein was reported (13).
Recent progress in the selection of nucleic acid aptamers is noteworthy because of their potential use in diagnostics as well as therapeutics. Nucleic acid aptamers that are produced by systematic evolution of ligands by exponential enrichment (SELEX) can bind to target proteins with high affinities. Therefore, aptamers are regarded as a complement to monoclonal antibodies (14).
In the present study, we identified single-stranded DNA (ssDNA) aptamers that could specifically bind to SARS CoV N protein and the ssDNA aptamer that bound with the highest affinity was selected for further studies. Western blot analysis using the selected ssDNA aptamer demonstrated that the aptamer was a good alternative probe to monoclonal antibodies. When considering the advantages of aptamers relative to antibodies, including their stability and facile synthesis, the selected ssDNA aptamer may be useful for the detection of SARS CoV N protein.
Materials and methods
Expression and purification of SARS-CoV N protein
SARS-CoV N protein expression vector was constructed as described previously (15) and transformed into Escherichia coli BL21 cells. Cells were grown at 37°C until A 600 = 0.7 in LB broth. N protein expression was induced by the addition of 0.5 mM isopropyl-β-thiogalactopyranoside (IPTG), and the cells were incubated overnight at 28°C. The cells were then harvested by centrifugation and lysed by sonication. Cell lysate was subjected to Q-sepharose ion-exchange chromatography (GE Healthcare) and the flow through fractions containing the N protein were collected. The N proteins were further purified by Ni chelating affinity chromatography (GE Healthcare) and Sephadex G-75 (Sigma) gel filtration chromatography, sequentially. The protein concentration was determined by absorbance measurements at 280 nm in 8 M urea (the extinction coefficient is 43,890 M−1 cm−1) and by using a Bio-Rad protein assay system (Bio-Rad) with bovine serum albumin as a standard.
Preparation of random DNA library and PCR condition
The DNA library consisted of two primer regions and a 45-base random region (5′-GCAATGGTACGGTACTTCC-N45-CAAAAGTGCACGCTACTTTGCTAA-3′), where N45 represents 45 nucleotides with equimolar incorporation of A, G, C, and T at each position. The 5′-FAM™ labeled forward primer (5′-FAM-GCAATGGTACGGTACTTCC-3) and the 5′-phosphorylated reverse primer (5′-P-TTAGCAAAGTAGCGTGCACTTTTG-3′) were used for PCR. All PCR reactions were carried out in a volume of 100 μL with forward primer and reverse primer as follows. The PCR mixture (100 μL) contained 0.2 mM dNTPs, 0.5 μM each primer, 10 nM template and 2.5 U Taq DNA polymerase. The mixture was thermally cycled 30 times through 95°C for 1 min, 37°C for 30 s, and 58°C for 40 s, which was followed by a 5 min extension step at 58°C. All synthetic DNAs were purchased from Integrated DNA Technologies.
ssDNA generation
After PCR using the 5′-phosphorylated reverse primer, purified dsDNA was incubated with 25 U λ-exonuclease (NEB, Germany) in a total reaction volume of 50 μL at 37°C for 3 h (16). The reaction was then terminated by incubation at 75°C for 10 min. The products of the digested strand were analyzed by electrophoresis in a 10% polyacrylamide/8 M urea TBE gel and the band was purified from the gel for the next round of selection.
SELEX procedure
Selection of DNA aptamers specific to the recombinant N protein was performed as described previously (17) with slight modifications. The initial ssDNA pool was heated at 90°C for 10 min, and immediately cooled on ice for 10 min, and then 5 μg of the DNA library was pre-incubated with 100 μL of Ni–NTA sepharose beads in 100 μL of binding buffer (50 mM Tris/Cl, pH 8.0, 150 mM NaCl, 1.5 mM MgCl2, 2 mM dithiothreitol (DTT) and 1% (w/v) BSA) for 30 min at room temperature with occasional shaking. The nonspecific DNA-bead complexes were precipitated and discarded. The pre-cleared supernatant was then incubated with 2 μg of His-tagged N protein in 100 μL binding buffer for 30 min at room temperature and 100 μL of Ni chelating sepharose (GE Healthcare) was added. The mixture was further incubated for 30 min at room temperature and washed five times with 500 μL of binding buffer. The N protein complexed with DNA was separated from the Ni chelating sepharose by adding 200 μL of elution buffer (binding buffer plus 0.4 M imidazole). The eluted supernatants of each round were precipitated with phenol/chloroform/isoamyl alcohol (PCI) treatment and then ethanol precipitation. Twelve sequential selection rounds were repeated using the same procedure. A more stringent condition was employed by reducing the protein concentration from round 8; 1 μg (round 8), 0.5 μg (round 9), 0.25 μg (round 10), and 0.125 μg (rounds 11–12) in 100 μL binding buffer. After the 12th round, ssDNA was amplified by PCR and cloned into a linearized pGEM T vector (Promega). After subcloning and transformation into E. coli DH5α, plasmid DNA was isolated from individual clones and the DNA sequences were determined. Bioinformatic analysis by ClustalW2 was performed to align the multiple sequences of the aptamers (18). The secondary structures of selected ssDNA aptamers were predicted using the MFold program, which was based on the Zuker algorithm (19).
Measurement of the N protein–biotinylated ssDNA aptamer interaction by ELISA
5′-Biotinylated aptamers were generated by PCR using a 5′-biotinylated forward primer instead of the 5′-FAMTM labeled forward primer. This was followed by λ-exonuclease digestion as described above. Ni-coated 96-well plates (Pierce) were coated with purified N protein (100 nM/100 μL per well) for 1 h at room temperature while shaking at 180 rpm. The wells were washed with PBST (0.1% Tween 20 in PBS; pH 7.4) three times and blocked with 5% BSA in PBST at room temperature for 1 h. After washing, various concentrations of the 5′-biotinylated aptamers were denatured at 90°C for 10 min, immediately put on ice, added to wells, and incubated at room temperature for 1 h. The bound aptamers were detected using a streptavidin-conjugated horseradish peroxidase (HRP) (1:1000 in PBST, Pierce). The color developing reaction was initiated by adding OPD (o-phenylenediamine) solution (Pierce), and terminated by adding 2.5 N H2SO4. The absorbance of each well was measured at 492 nm using a TRIAD microplate reader (Dynex Technologies).
Western blot analysis
The purified N protein was separated by SDS-PAGE and transferred to a PVDF membrane. The membranes were blocked with 5% BSA in PBST at room temperature for 1 h, and then incubated with 5′-biotinylated ssDNA aptamer in PBST for 1 h. After 4 washes, the membranes were incubated with streptavidin-HRP for 1 h. After washing, the membranes were visualized by enhanced chemiluminescence (ECL) reaction and exposed to LAS 4000 (GE Healthcare). The same experiments were repeated using His probe-conjugated HRP (Pierce) or anti-N protein (Santa Cruz Biotechnology) instead of the selected ssDNA aptamer to compare the efficacy of the selected aptamer with commercial antibodies. After the blotting, blocking, and washing steps, the membranes were incubated with His probe-HRP or Anti-N protein. In the case of incubation with His probe-HRP, membranes were visualized by the ECL reaction without further treatment. When the membrane was incubated with Anti-N protein, the membrane was also reacted with the goat anti-mouse IgG-HRP prior to the ECL reaction step.
Results and discussion
Selection of ssDNA aptamers to recognize N protein of SARS CoV
The N protein for selection of ssDNA aptamer was purified to homogeneity through sequential chromatography steps including Q-sepharose, Ni-IDA, and Sephadex G-75. The purified N protein was then subjected to SDS-PAGE and Western blotting using His probe-HRP (data not shown). The 88mer ssDNA library containing 45-nucleotide long random sequences was constructed and screened by SELEX to discover ssDNA aptamers that specifically bound to the SARS CoV N protein (Fig. 1 ). To generate the ssDNA after PCR, λ-exonuclease was added to digest the phosphorylated DNA strand and the ssDNA band was confirmed by native PAGE at each cycle (data not shown).
Fig. 1.
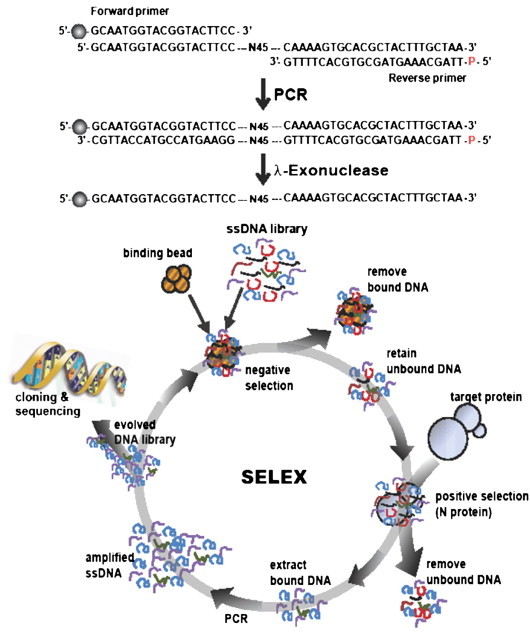
A schematic showing the sequence of the ssDNA library used for in vitro selection and the design of the SELEX procedure. A random ssDNA library was obtained by PCR, which contains 45 random sequences, using 5′-FAMTM labeled forward primer and 5′-phosphorylated reverse primer, which was followed by λ-exonuclease digestion. Ball symbol at the 5′ end of the forward primer represents FAMTM. After eliminating non-specific binding to Ni–NTA beads, the ssDNA pool was incubated with the His-tagged N proteins, which were immobilized on Ni–NTA sepharose beads. Unbound DNA was discarded, and bound DNA was eluted by the addition of imidazole. The DNA was enriched after twelve cycles of selection, and the selected DNA was amplified by PCR, cloned and sequenced.
To isolate the ssDNA aptamers that displayed a high binding affinity for the N protein, a more stringent selection condition was employed at round no. 8 by decreasing the protein concentration as described in Materials and methods. Enrichment of ssDNA aptamers specific to the N protein was monitored based on the N protein–biotinylated ssDNA aptamer interaction by ELISA (Fig. 2A). ssDNA pools isolated after rounds 6, 8, 11, and 12 were tested for binding to the N protein. In these experiments, the absorbance at 492 nm increased significantly at round no. 11. After twelve cycles of selection, we obtained 15 different ssDNA aptamers, and the nucleotide sequences of these aptamers were determined (Fig. 2B). Subsequent bioinformatic analysis was performed by ClustalW2, and the ssDNA aptamers were classified into four groups and five unrelated individual ssDNA aptamers as shown in Fig. 2B. Groups 1–4 contained characteristic clusters of nucleotides in the 45 random sequence region (indicated by asterisk). In group 1, four different ssDNA aptamers were shown to contain three nucleotides (TTG) followed by GC rich conserved sequences in the 45-base random region. Groups 2 and 3 were shown to have (CTTG) followed by (CAGTTCG) and (CAATA), respectively. In addition, several nucleotides were matched in group 4, but it was difficult to identify any characteristic clusters of nucleotides in the five unrelated individual ssDNA aptamers.
Fig. 2.
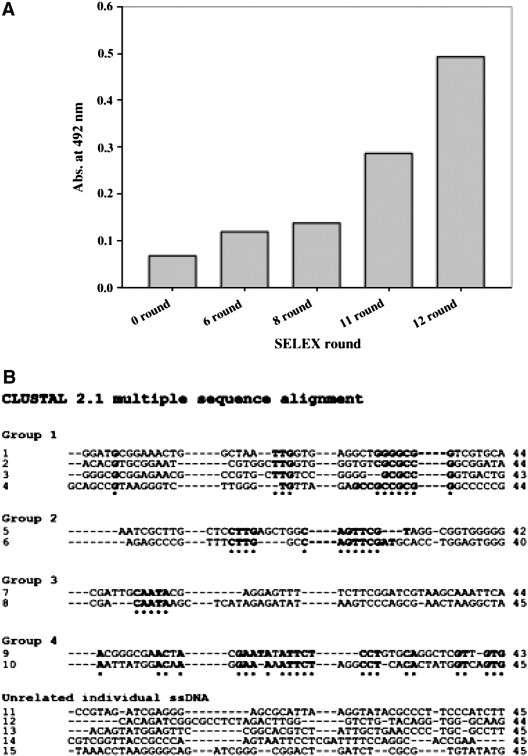
Specific binding activity of ssDNA pools and sequences of ssDNA aptamers after twelve rounds of selection. (A) Enrichment of ssDNA aptamers specific to the N protein. Specific binding of the DNA pool was measured by ELISA after 6, 8, 11, and 12 rounds of selection. (B) Fifteen different ssDNA aptamer sequences in the DNA pool after twelve rounds of selection are shown and classified into four groups and five unrelated individual ssDNA aptamers by ClustalW2 analysis. Asterisks indicate the conserved nucleotide sequences in each group.
Binding affinity measurements of selected ssDNA aptamers with N protein
ELISA experiments were then performed using the 5′-biotinylated aptamer candidates identified above to assess the binding affinities of these aptamers to the N protein. The N protein was immobilized on Ni-coated plates and various concentrations of the 5′-biotinylated aptamer candidates were added. After addition of streptavidin-HRP, the absorbance at 492 nm was measured. The binding curve was fit to a hyperbolic equation based on Michaelis–Menten kinetics, and we were able to obtain the amplitude and K d value (Table S1). Of the 15 aptamer candidates, the K d values of two aptamers, named aptamer 1 and aptamer 11, that displayed high affinity were determined from the binding curve (Fig. 3 A). Although both aptamers showed high binding affinities to the N protein, aptamer 1 (K d = 4.93 ± 0.30 nM) exhibited a slightly higher affinity than aptamer 11 (K d = 9.02 ± 1.89 nM). When we compared the K d values of the ssDNA aptamers with RNA aptamer (equilibrium dissociation constant of 1.65 nM), which was recently published (15), they were quite comparable. However, the nucleotide sequence and secondary structure of the selected RNA aptamer are different from the ones of our selected DNA aptamers. This suggests that the nucleotide component is not important for high affinity binding to the N protein. Therefore, proper binding to the protein might be more important for nucleic acid aptamers even though the exact binding site is unknown at present. In addition, the fact that natural RNA is very liable to digest by nuclease attack makes DNA a more suitable candidate for target protein detection. Based on the ELISA results, aptamer 1 was chosen for further bioinformatic analysis and the MFOLD program was used to predict secondary structures (Fig. 3B). In this analysis, the 45 random sequence region of aptamer 1 (shaded nucleotides) was shown to consist of a major loop with a small hairpin and base-paired nucleotides with the defined sequences flanking the random region. The GC rich conserved sequence, as described in group 1 aptamers, was also found to be located at the stem-loop region (indicated by black line).
Fig. 3.
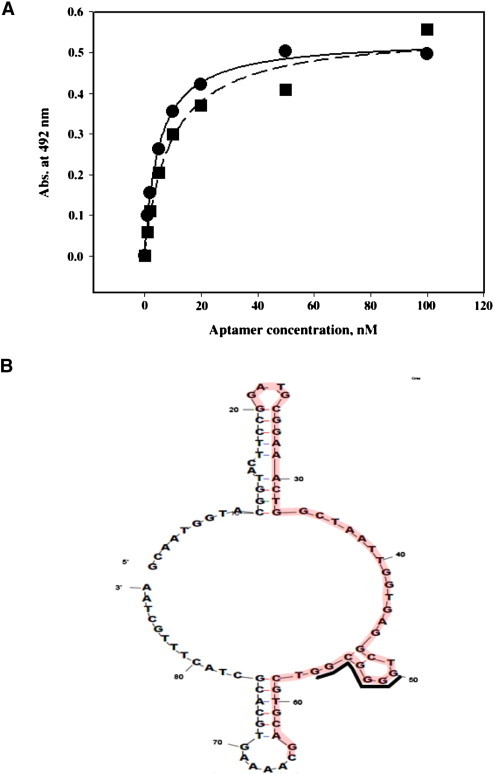
Affinity measurements and predicted secondary structure of aptamer 1. (A) Affinity measurement of N protein-selected ssDNA aptamers interaction by ELISA. The N protein was incubated with increasing concentrations of 5′-biotinylated aptamer 1 and 11. After addition of streptavidin-HRP, the amount of N protein-aptamer complex was calculated and graphed as a function of aptamer concentration. The graph was fit to the Michaelis–Menten equation and the Kd was calculated (4.93 ± 0.30 nM (aptamer 1) and 9.02 ± 1.89 nM (aptamer 11)). (B) Predicted secondary structure of aptamer 1 using Zuker's MFOLD program.
Western blot analysis using selected ssDNA aptamer
Western blot analysis was performed to determine if the selected ssDNA aptamer 1 can be used as a substitute for the N protein antibody. When various amounts of N protein (0–18.4 μg) were analyzed by Western blotting with 500 ng/mL of 5′-biotinylated aptamer 1, we found that the band intensities increased as the amount of N protein increased (Fig. 4A). This indicates that the selected aptamer 1 bound to the N protein in dose dependent manner, and could be used as an alternative to antibodies for the detection of the N protein. We also performed a comparative Western blotting experiments with commercial His probe-HRP and Anti-N protein followed by secondary antibody conjugated HRP (Fig. 4B). The result showed that the band intensities generated using the anti-N protein and aptamer 1 were similar to each other (lanes 2 and 3 in Fig. 4B). However, the detection sensitivity was not significant when the His probe-HRP was used (lane 1 in Fig. 4B) under the reaction condition described above. Our results suggest that the ssDNA aptamer isolated in this study could be used as an alternative to the antibody for the detection of the N protein. Because aptamers can bind to specific target proteins with high affinity, they have been referred to as ‘chemical antibodies’. It is also worth noting that DNA can be synthesized easily while antibody generation relies on the immune system of animals. In addition, the fact that aptamers have higher stability than protein antibodies makes aptamers more useful in the development of diagnostic systems, such as viral infection and cancers (20).
Fig. 4.
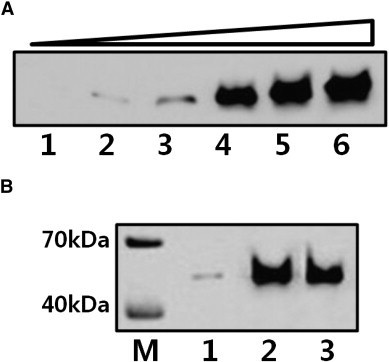
Western blot analysis with ssDNA aptamer, His-probe HRP, and anti-N protein. (A) Western blot analysis using aptamer 1. Various amounts of N protein were separated by SDS-PAGE and incubated with 5′-biotinylated aptamer 1 followed by streptavidin-HRP. The bands were visualized using the ECL reaction. Lane 1, 0 μg; lane 2, 0.92 μg; lane 3, 1.84 μg; lane 4, 4.6 μg; lane 5, 9.2 μg; and lane 6, 18.4 μg N protein. (B) Western blot analysis was performed to compare the efficacy of the His-probe HRP, anti-N protein, and aptamer 1. 9.2 μg of N protein was separated by SDS-PAGE and incubated with His-probe HRP (lane 1), anti-N protein (lane 2), and 5′-biotinylated aptamer 1 (lane 3), respectively. Lane M is the molecular weight marker. The method used for band visualization was described in Materials and methods.
Considering the importance of the N protein to SARS CoV infection, a method for the fast and reliable detection of the N protein should be established. In the present study, we developed a new ssDNA aptamer that specifically bound to the SARS CoV N protein. It is expected that the selected ssDNA aptamer 1 can be used for the sensitive diagnosis of SARS.
The following are the supplementary materials related to this article.
Western blot analysis with SARS coronavirus spike protein using selected ssDNA aptamer 1. Western blot analysis was performed to determine if the selected ssDNA aptamer 1 specific to N protein shows non-specific binding to other protein. SARS coronavirus spike (S) protein (molecular weight is 25 kDa) was chosen because SARS coronavirus contains S protein as well as N protein (molecular weight is 46 kDa). When various amounts of S protein (0–10 μg) were analyzed by Western blotting with 500 ng/mL of 5′-biotinylated aptamer 1 as described in Materials and Methods, we found that the aptamer 1 did not bind to S protein (Fig. S1, lanes 1–5). Lane 6 of Fig. S1 is a control experiment using 10 μg of N protein. Lane 1, 0 μg; lane 2, 1 μg; lane 3, 2 μg; lane 4, 5 μg; lane 5, 10 μg S protein; and lane 6, 10 μg N protein as a control. Lane M is the molecular weight marker.
Kd values of selected 15 clones after twelve rounds of SELEX.
Acknowledgments
This study was supported by a grant of the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare & Family Affairs, Republic of Korea (A090410).
References
- 1.He Q., Du Q., Lau S., Manopo I., Lu L., Chan S.W., Fenner B.J., Kwang J. Characterization of monoclonal antibody against SARS coronavirus nucleocapsid antigen and development of an antigen capture ELISA. J. Virol. Methods. 2005;127:46–53. doi: 10.1016/j.jviromet.2005.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., and other 48 authors The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 3.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., and other 25 authors Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 4.Stadler K., Masignani V., Eickmann M., Becker S., Abrignani S., Klenk H.D., Rappuoli R. SARS—beginning to understand a new virus. Nat. Rev. Microbiol. 2003;1:209–218. doi: 10.1038/nrmicro775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haynes L.M., Miao C., Harcourt J.L., Montgomery J.M., Le M.Q., Dryga S.A., Kamrud K.I., Rivers B., Babcock G.J., and other 11 authors Recombinant protein-based assays for detection of antibodies to severe acute respiratory syndrome coronavirus spike and nucleocapsid proteins. Clin. Vaccine Immunol. 2007;14:331–333. doi: 10.1128/CVI.00351-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tripp R.A., Haynes L.M., Moore D., Anderson B., Tamin A., Harcourt B.H., Jones L.P., Yilla M., Babcock G.J., Greenough T., and other 17 authors Monoclonal antibodies to SARS-associated coronavirus (SARS-CoV): identification of neutralizing and antibodies reactive to S, N, M and E viral proteins. J. Virol. Methods. 2005;128:21–28. doi: 10.1016/j.jviromet.2005.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu M.S., Pan Y., Chen H.Q., Shen Y., Wang X.C., Sun Y.J., Tao K.H. Induction of SARS-nucleoprotein-specific immune response by use of DNA vaccine. Immunol. Lett. 2004;92:237–243. doi: 10.1016/j.imlet.2004.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li F.Q., Xiao H., Tam J.P., Liu D.X. Sumoylation of the nucleocapsid protein of severe acute respiratory syndrome coronavirus. FEBS Lett. 2005;579:2387–2396. doi: 10.1016/j.febslet.2005.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y.H., Li J., Liu X.E., Wang L., Li T., Zhou Y.H., Zhuang H. Detection of the nucleocapsid protein of severe acute respiratory syndrome coronavirus in serum: comparison with results of other viral markers. J. Virol. Methods. 2005;130:45–50. doi: 10.1016/j.jviromet.2005.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Che X.Y. Nucleocapsid protein as early diagnostic marker for SARS. Emerg. Infect. Dis. 2004;10:1947–1949. doi: 10.3201/eid1011.040516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shang B., Wang X.Y., Yuan J.W., Vabret A., Wu X.D., Yang R.F., Tian L., Ji Y.Y., Deubel V., Sun B. Characterization and application of monoclonal antibodies against N protein of SARS-coronavirus. Biochem. Biophys. Res. Commun. 2005;336:110–117. doi: 10.1016/j.bbrc.2005.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu G., Hu S., Hu Y., Chen P., Yin J., Wen J., Wang J., Lin L., Liu J., You B., and other 13 authors The C-terminal portion of the nucleocapsid protein demonstrates SARS-CoV antigenicity. Genomics Proteomics Bioinformatics. 2003;1:193–197. doi: 10.1016/S1672-0229(03)01024-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishikawa F.N., Chang H.K., Curreli M., Liao H.I., Olson C.A., Chen P.C., Zhang R., Roberts R.W., Sun R., Cote R.J., Thompson M.E., Zhou C. Label-free, electrical detection of the SARS virus N-protein with nanowire biosensors utilizing antibody mimics as capture probes. ACS Nano. 2009;3:1219–1224. doi: 10.1021/nn900086c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.James W. Aptamers in the virologists' toolkit. J. Gen. Virol. 2007;88:351–364. doi: 10.1099/vir.0.82442-0. [DOI] [PubMed] [Google Scholar]
- 15.Ahn D.G., Jeon I.J., Kim J.D., Song M.S., Han S.R., Lee S.W., Jung H., Oh J.W. RNA aptamer-based sensitive detection of SARS coronavirus nucleocapsid protein. Analyst. 2009;134:1896–1901. doi: 10.1039/b906788d. [DOI] [PubMed] [Google Scholar]
- 16.Avci-Adali M., Paul A., Wilhelm N., Ziemer G., Wendel H.P. Upgrading SELEX technology by using lambda exonuclease digestion for single-stranded DNA generation. Molecules. 2010;15:1–11. doi: 10.3390/molecules15010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng C., Dong J., Yao L., Chen A., Jia R., Huan L., Guo J., Shu Y., Zhang Z. Potent inhibition of human influenza H5N1 virus by oligonucleotides derived by SELEX. Biochem. Biophys. Res. Commun. 2008;366:670–674. doi: 10.1016/j.bbrc.2007.11.183. [DOI] [PubMed] [Google Scholar]
- 18.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R., and other 3 authors Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 19.Zuker M. Computer prediction of RNA structure. Methods Enzymol. 1989;180:262–288. doi: 10.1016/0076-6879(89)80106-5. [DOI] [PubMed] [Google Scholar]
- 20.Tombelli S., Minunni M., Mascini M. Aptamers-based assays for diagnostics, environmental and food analysis. Biomol. Eng. 2007;24:191–200. doi: 10.1016/j.bioeng.2007.03.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Western blot analysis with SARS coronavirus spike protein using selected ssDNA aptamer 1. Western blot analysis was performed to determine if the selected ssDNA aptamer 1 specific to N protein shows non-specific binding to other protein. SARS coronavirus spike (S) protein (molecular weight is 25 kDa) was chosen because SARS coronavirus contains S protein as well as N protein (molecular weight is 46 kDa). When various amounts of S protein (0–10 μg) were analyzed by Western blotting with 500 ng/mL of 5′-biotinylated aptamer 1 as described in Materials and Methods, we found that the aptamer 1 did not bind to S protein (Fig. S1, lanes 1–5). Lane 6 of Fig. S1 is a control experiment using 10 μg of N protein. Lane 1, 0 μg; lane 2, 1 μg; lane 3, 2 μg; lane 4, 5 μg; lane 5, 10 μg S protein; and lane 6, 10 μg N protein as a control. Lane M is the molecular weight marker.
Kd values of selected 15 clones after twelve rounds of SELEX.