

Abstract
Background
Formation and maintenance of appropriate neural networks require tight regulation of neural stem cell proliferation, differentiation, and neurogenesis. microRNAs (miRNAs) play an important role in brain development and plasticity, and dysregulated miRNA profiles have been linked to neurodevelopmental disorders including autism, schizophrenia, or intellectual disability. Yet, the functional role of miRNAs in neural development and postnatal brain functions remains unclear.
Methods
Using a combination of cell biology techniques as well as behavioral studies and brain imaging, we characterize mouse models with either constitutive inactivation or selectively hippocampal knockdown of the neurodevelopmental disease-associated gene Mir146a, the most commonly deregulated miRNA in developmental brain disorders (DBD).
Results
We first show that during development, loss of miR-146a impairs the differentiation of radial glial cells, neurogenesis process, and neurite extension. In the mouse adult brain, loss of miR-146a correlates with an increased hippocampal asymmetry coupled with defects in spatial learning and memory performances. Moreover, selective hippocampal downregulation of miR-146a in adult mice causes severe hippocampal-dependent memory impairments indicating for the first time a role for this miRNA in postnatal brain functions.
Conclusion
Our results show that miR-146a expression is critical for correct differentiation of neural stem cell during brain development and provide for the first time a strong argument for a postnatal role of miR-146a in regulating hippocampal-dependent memory. Furthermore, the demonstration that the Mir146a−/− mouse recapitulates several aspects reported in DBD patients, including impaired neurogenesis, abnormal brain anatomy, and working and spatial memories deficits, provides convincing evidence that the dysregulation of miR146a contributes to the pathogenesis of DBDs.
Keywords: Neurodevelopmental disorders, MicroRNA, miR-146a, Neurodevelopment, Neural stem cells differentiation, Hippocampus-dependent cognitive functions
Background
MicroRNAs (miRNAs) are endogenous short single-stranded non-coding RNAs of ~20–22 nucleotides that play a key role in gene expression regulation by targeting specific messenger RNA (mRNA) for degradation or translation repression [1]. miRNA genes are first transcribed as primary transcripts that consist of one or more hairpin structures. This transcript is cleaved into short pre-miRNA transcript that contains a stem-loop and subsequently exported to the cytoplasm. Following export, pre-miRNA is processed by the endonuclease Dicer to generate a mature miRNA duplex and both strands derived from this duplex (-5p and -3p) can be loaded into the Argonaute proteins so as to generate the effector RISC complex. miRNAs function as master regulators as each of them interacts with hundreds of mRNA transcripts. miRNAs are expressed throughout embryonic development and life of most eukaryotic organisms and take part in almost every cellular and developmental process investigated so far.
miRNAs are particularly important for proper brain development as demonstrated by the dramatic apoptotic phenotypes of mouse mutants abolishing all miRNAs synthesis [2]. Fifty percent of all identified miRNAs are expressed in the mammalian brain [3] where they are differentially distributed in distinct regions and cell types. miRNAs regulate multiple aspects of brain development, including neural stem cell proliferation and differentiation, cell subtype determination, neurogenesis, and synapse formation [4]. Moreover, synapse stability and plasticity are regulated by local interactions between miRNAs and target mRNAs. Despite rapid progress in miRNA discovery, understanding the in vivo implication of single or subnetworks of miRNAs in specific cellular contexts, and the biological relevance of their predicted miRNA–mRNAs interactions remain challenging. The use of conventional genetic approaches is difficult, in particular in mammals, because of redundancy (multiple miRNA loci encoding the same mature form) and the small size of the loci, making them poor targets for mutation. Thereby, most insights into the role of miRNAs in neural development were obtained, in cell culture and animal models, by investigating the effect of miRNAs silencing by miRNA antagonist or the impact of miRNA overexpression by miR Mimics.
Over the past decade, dysregulated processing and altered abundance of a wide range of miRNAs have been associated with neurodegenerative disorders [5] and neuroblastoma [6]. Widespread changes in miRNA levels have also been observed in developmental brain disorders (DBDs) including autism spectrum disorders (ASD) [7], schizophrenia [8], intellectual disability (ID) [9], and epilepsy [10]. However, the precise miRNA-based mechanisms during brain development and in higher-order brain processing, including learning and memory and cognition, are still poorly understood.
Among the differentially expressed miRNAs identified by transcriptomic analysis in DBD patients, miR-146a-5p (referred to as miR-146a from now on) is one of the most commonly observed [7]. In ASD cases, it has been reported in five different cohorts and tissue types including post-mortem brain [11, 12] olfactory mucosal stem cells, fibroblasts [9], and lymphoblastoid cell line [13]. Its abnormal expression has also been reported in ID [9] and epilepsy [14]. miR-146a is 100% conserved between humans and mice; it is expressed broadly in the brain during embryonic development and restricted to neurons at postnatal stages. Importantly, modulating the expression of this particular miRNA was shown to reduce the latency, the duration as well as the intensity of the induced epilepsy in rodent models of telencephalon epilepsy [15, 16].
miR-146a is known for regulating NFκB [17], NOTCH [18], and WNT/β-catenin [19, 20] pathways in a tissue and developmental stage-dependent manner. Our group has also demonstrated the pro-neuronal effects of miR-146a overexpression in a H9 model of human neural stem cells [12]. Moreover, a body of in vitro works also suggests a role of miR-146a in neuron survival and apoptosis [21, 22], axonal growth [23], and AMPA receptor endocytosis [24]. Taken together, these data support the importance of miR-146a in cultured cells but give no indication either on its mechanism of action or on its physiological functions in vivo during neurodevelopment and at postnatal stages.
To investigate these aspects, we decided to study the impact of abnormal miR-146a expression level on mouse brain development and at adult stages. We focused on a mutant mouse line bearing a targeted deletion of the miR-146a gene [25] and characterized this Mir146a−/− mouse model using a combination of imaging and cell biology techniques as well as behavioral studies. We first showed that loss of miR-146a leads to transient neurodevelopmental defects in the differentiation of apical radial glia (aRG) and neurogenesis. We next found that loss of miR-146a correlates with an increased hippocampal asymmetry in the adult brain coupled with defects in spatial learning and memory performances. Lastly, we showed that selective knockdown of miR-146a in the adult hippocampus leads to severe learning and memory impairments associated with a reduction of adult hippocampal neurogenesis, demonstrating for the first time that miR-146a play an important role in adult brain functions. Together, these results highlight the key roles of small non-coding RNA in the brain for the control of neuronal development and cognition and provide new insight into the mechanisms linking impaired miR-146a expression to DBDs.
Methods
Animal housing and ethics
For this study, we used the mouse line B6.Cg-Mir146tm1.1Bal/J (stock no. 016239) from Jackson Laboratory. Mice were housed and handled in accordance with current best practices. Their well-being was monitored daily by a contractor as a paid service (Animalliance). Homozygous mice at 6–8 months of age start to develop lymphoproliferative and myeloproliferative diseases, subsequently leading to anemia, thrombocytopenia, and lymphopenia ([25] and Jackson Laboratory). As such, all molecular and behavioral analyses described in this study were performed before mice reach 6 months old (4 months old at the latest for behavioral tests), before the appearance of any inflammatory disease signs. All experiments performed were approved by an independent ethics committee of Paris Descartes University (approval number 2015101215207380) and declared to the French Ministry of Research.
ELISA analysis
Two ELISA kits were used to assess the concentration of Il-1β (15571957, Fischer Scientific) and TNF-α (15562017, Fischer Scientific). We tested the hippocampus and cortex of the first batch of mice used for the behavioral studies. Cells were lysed using the FastPrep system (MPbio) in EBC lysis buffer, followed by a 15-min centrifugation at 4 °C to remove debris. The supernatant containing the proteins were used for the tests that were performed following the manufacturer’s protocol.
Brain magnetic resonance imaging
MRI were performed on a 4.7 Tesla MR scanner (Biospec 47/20 BrukerBiospin, Ettlingen, Germany) with a combination of a transmit volume coil and a surface receive branch coil. Anesthesia was induced with 4% isoflurane and maintained at 1% during imaging. The parameters of the spin-echo TurboRARE T2-weighted sequences are as follows: field of view (FOV) 2 × 1.5 cm; Matrix 200 × 150; resolution 100 × 100 μm; slice thickness 350 μm; 30 slices for the sagittal slices with TR/TE 5096/60 ms (11 min, 28 s); 46 slices for the axial slices with TR/TE 7850/60 ms (17 min, 40 s); RARE factor 9. All four mutant mice were males whereas the control group was constituted of one female and two males.
BrdU injection and detection
E14.5 heterozygous Mir146a+/− pregnant mice were injected with BrdU (Thermo Fisher) at 28 mg/kg and dissected 2 h after injection for pulse chase or at P7 for birth dating analysis. Brains were fixed in 4% PFA (Antigenfix, Diapath) during 24 h at 4 °C, washed 3 times in PBS and incubated 24h in PBS–30% sucrose. Brains were then frozen in O.C.T. compound (Tissue-Tek SAKURA) and stored at – 80 °C. Forteen micrometers sagittal sections were cut using a cryostat (Thermo Fisher) and mounted onto Fisher Superfrost Plus slides. Sections were incubated in 2N HCl for DNA denaturation at 37 °C for 30 min then washed 3 times with 0.1% Triton X-100 (Sigma) in PBS. Blocking was realized using 10% Normal Goat Serum (Clinisciences) and 0.1% Triton X-100 in PBS. Slides were incubated overnight at 4 °C with a rat anti-BrdU antibody (ab6326, Abcam). Slides were then washed 3 times with 0.1% Triton X-100 in PBS and incubated with Alexa fluor 488 donkey anti-rat antibody (A21208, Thermo Fisher) at room temperature for 2 h. Slides were finally washed three times in PBS, mounted using the Prolong gold antifade reagent with DAPI (P36935, Thermo Fisher) and scanned in a Nanozoomer 2.0 (Hamamatsu). 250μm × 250 μm images were taken in the neocortex and horizontally binned in 5 layers of equal size; for birth dating 1000 × 800 μm images were used. BrdU+ cells were counted in each bin or layer.
PH3, PAX6, TBR2, and NeuN immunofluorescence and quantification
Pups were sacrificed at E14.5 and the embryonic brains were fixed, frozen, and cut into sagittal sections as previously described. An antigen retrieval step was performed by incubating the slides with 10mM Sodium Citrate buffer 10 min at 100 °C. Slides were then allowed to cool down and rinsed once with PBS before blocking 1h at room temperature with 10%NGS and 0.1% Triton X-100 in PBS. Anti-PH3 antibody (06-570, Merck Millipore) and anti-NeuN (ab177487, Abcam) were used alone while anti-PAX6 (901301, Biolegend) and anti-TBR2 (14-4875-80, ThermoFisher) antibodies were used concomitantly for co-staining. In all conditions, primary antibody was incubated overnight at 4 °C. After three washes with 0.1% Triton X-100 in PBS, slides were incubated with a matching secondary antibody (Alexa fluors, Thermo Fisher). Slides were then washed three times in PBS and mounted and scanned as previously described. We counted the PH3+ cells at the apical surface and in the SVZ independently in images of 650 μm of width and calculated the percentage of PH3+ cells at the SVZ. 250μm × 250 μm images were taken in the neocortex for the PAX6-TBR2 co-staining. Each cell type was counted independently. To analyze the NeuN labeling at E14.5, the thickness of the NeuN+ layer was measured at least four times along the neocortex.
Doublecortin (DCX) immunohistochemistry
For DCX immunohistochemistry, brain sections (25 μm) were washed in 0.1 M PBS and then quenched in 0.3% H2O2 in 0.1 M PBS/CH3OH (1:1) for 15 min at room temperature. Sections were then washed and blocked in 10% normal donkey serum in 0.1 M PBS with 0.5% Triton X-100 for 2 h at room temperature. Incubation with primary antibody was performed at 4 °C overnight (Rabbit Anti-Doublecortin antibody (ab18723)) in 0.1 M PBS with 0.5% Triton X-100. Sections were then washed in 0.1 M PBS and incubated with a biotinylated secondary antibody (donkey anti-rabbit; 1:250, Jackson ImmunoResearch, West Grove, PA) for 2 h at room temperature. Sections were then washed in 0.1 M PBS and treated next with avidin-biotin-peroxidase complex (ABC Elite Kit, Vector Labs, Burlingame, CA) followed by a 3,3′diaminobenzidine as a substrate for staining (Vector, Burlingame, CA).
Primary neuronal culture and neurite outgrowth measurement
Primary cortical neurons were extracted from E17.5 embryos using as previously described. Approximately 30,000 cells were plated per well in the 96-well plate for live cell imaging (IncuCyte® system, Essen Bioscience). For each well, 9 images were taken at fixed spots every 3 h during 6 days. Media was change 1 day after plating to remove debris, then every 3 days thereafter. In post analysis, a mask was designed using the inbuilt Neurotrack software module to detect neurites and cell body. The same mask was applied to all time points and all repeats to record neurite extension over time.
Adeno-associated viruses expressing shRNA
Adeno-associated viruses (AAV) expressing shRNA were purchased from Applied Biological Materials Inc (Richmond, Canada). shRNAs specific to mir146a (mmu-miR-146a-5p AAV miRNA Virus, serotype 9), or scrambled non-targeting negative control (AAV-miR-Off-Blanck Control Virus, serotype 9) were injected in a volume of 1 μl (bilaterally), 3 weeks prior to behavioral tests. The AAV titers were > 1 × 109GC/ml.
Stereotaxic surgery
Mice were anesthetized by intraperitoneal injection of ketamine hydrochloride (20 mg/ml BW) (1000 Virbac) and xylazine (100mg/ml BW) (Rompun 2%; Bayer) and placed in a stereotaxic frame (900SL-KOPF). Ophthalmic eye ointment was applied to the cornea to prevent desiccation during surgery. The area around the incision was trimmed and Vétoquinol was applied. AAV shRNA were injected bilaterally into the dorsal hippocampi using the following coordinates from Bregma (Paxinos and Franklin, 2008): X = ± 1.4 mm, Y = 2.0 mm, and Z = − 1.33 mm. A 1-μl volume of AAV was injected stereotaxically over 4 min (injection rate 0.25 μl per min). To limit reflux along the injection track, the needle was maintained in situ for 4 min between each 1 μl injection.
3-foot shock contextual fear conditioning (CFC)
Mice were transported a short distance from the holding mouse facility to the testing room in their home cages and left undisturbed for at least 1 h before the beginning of the test. The conditioning chambers were obtained from Bioseb (France) with internal dimensions of 25 × 25 × 25 cm. Each chamber was located inside a larger, insulated plastic cabinet that provided protection from outside light and noise (67 × 55 × 50 cm, Bioseb, France), and mice were tested individually in the conditioning boxes. Floors of the chamber consisted of 27 stainless steel bars wired to a shock generator with scrambler for the delivery of foot shock. Signal generated by the mice movements was recorded and analyzed through a high sensitivity weight transducer system. The analog signal was transmitted to the Freezing software module through the load cell unit for recording purposes and analysis of time active/time immobile (Freezing) was performed. The CFC procedure took place over two consecutive days. On day 1, mice were placed in the conditioning chamber, and received 3 foot-shocks (1 s, 0.5 mA), which were administrated at 60, 120, and 180 s after the animals were placed in the chamber. They were returned to their home cages 60 s after the final shock. Contextual fear memory was assessed 24 h after conditioning by returning the mice to the conditioning chamber and measuring freezing behavior during a 4-min retention test. Freezing was scored and analyzed automatically using Packwin 2.0 software (Bioseb, France). Freezing behavior was considered to occur if the animals froze for a period of at least two seconds. Behavior was scored by the Freezing software and analyzed by two observers blind to mouse treatment (LSM and SM).
Novel object recognition paradigm (NOR)
We used a modified version of the NOR task described by Ennaceur and Delacour in 1988 [26]. Mice were transported a short distance from the holding mouse facility to the testing room in their home cages and left undisturbed for at least 1 h before the beginning of the test. The testing room was lit with two 60 W light bulbs and behavior sessions were recorded with a camera above the testing arena (grey plastic box (60 × 40 × 32 cm)). Mice could not contact or see each other during the exposures. The light intensity was equal in all parts of the arena (approximately 20 lx). Two different objects were used, available in triplicate. The objects were (1) a blue ceramic pot (diameter 6.5 cm, maximal height 7.5 cm) and (2) a clear, plastic funnel (diameter 8.5 cm, maximal height 8.5 cm). The objects that serve as a novel object, as well as the left/right localization of the objects, were counterbalanced within each group. The objects elicited equal levels of exploration as determined in pilot experiments and training phase. Between exposures, mice were held individually in standard cages, the objects and arenas were cleaned with phagosphore, and the bedding replaced.
The NOR paradigm consists of three phases (over 3 days): a habituation phase, a training phase, and a testing phase. Mice were always placed in the center of the arena at the start of each exposure. On day 1, the habituation phase, mice were given 5 min to explore the arena, without any objects and were then taken back to their home cage. On day 2, the training phase, mice were allowed to explore, for 10 min, two identical objects arranged in a symmetric opposite position from the center of the arena and were then transported to their home cage. On day 3, the testing phase, mice were given 15 min to explore two objects: a familiar object and a novel one, in the same arena, keeping the same object localization.
The following behaviors were considered as exploration of the objects: sniffing, licking, or touching the object with the nose or with the front legs or directing the nose to the object at a distance ≤ 1 cm. Investigation was not scored if the mouse was on top of the object or completely immobile. The preference index for the novel object was calculated as (time spent exploring the new object/the total time spent exploring both objects), and the discrimination index was calculated as (time spent exploring the new object − time spent exploring the familiar object)/(total time spent exploring both objects). As a control, preference index for the (right/left) object location or for the object A versus B during the training phase of the NOR was measured in all groups of mice exposed to the test. We confirm here that no initial preference for any exposed object (A or B) or any orientation (right/left) was observed in any groups. The locomotion was assessed for each mouse. Behavior was scored on videos by two observers blind to treatment and the total exploration time of the objects was quantified in the training and testing phases (AP and SM).
Object location memory test (OLM)
For the object location memory task, all procedures were identical to the novel object recognition task except that during the testing phase, rather than presenting a novel object, mice encountered both familiar objects, with one object located in a different place in the arena. The time and frequency of exploration of the novel/relocated object is measured as an index of memory. As a control, preference index for the (right/left) object location during the training phase of the OLM was measured in all groups of mice. We confirm here that no initial preference for any orientation (right/left) was observed in any groups. Behavior was scored on videos by two observers blind to treatment and the total exploration time of the objects was quantified in the training and testing phases (LSM and SM).
Light-to-dark transition test (D/LT)
This test is based on the innate aversion of rodents to brightly illuminated areas and on their spontaneous exploratory behavior in response to the stressor that light represents. The test apparatus consists of a dark, safe compartment and an illuminated, aversive one. The lit compartment was brightly illuminated with an 8 W fluorescent tube (1000 lx). Naive mice were placed individually in the testing chamber in the middle of the dark area facing away from the doorway to the light compartment. Mice were tested for 10 min, and two parameters were recorded: time spent in the lit compartment and the number of transitions between compartments, indices of anxiety-related behavior and exploratory activity. Behavior was scored using an infrared light beam activity monitor using actiMot2 Software (PhenoMaster Software, TSE), and it was statistically analyzed using Prism program.
Open-field est (OFT)
This test takes advantage of the aversion of rodents to brightly lit areas. Each mouse is placed in the center of the OFT chamber (43 × 43 cm chamber) and allowed to explore for 30 min. Mice were monitored throughout each test session by infrared light beam activity monitor using actiMot2 Software (PhenoMaster Software, TSE). The overall motor activity was quantified as the total distance traveled (ambulation). Anxiety was quantified by measuring the time and distance spent in the centerversus periphery of the open-field chamber.
Results
Altered balance between aRG and IP populations in the Mir146a−/− neocortex
miR-146a has been shown to regulate the balance of cell cycle progression and differentiation of cultured progenitor cells in vitro [12]. Yet, we observed that the number of cortical neurons is unaltered in the adult Mir146a−/− brain (Figure S1, Additional file 1), suggesting that, in vivo, there is no major defect in proliferation of neural progenitors. To resolve this discrepancy, we quantified the ratio between the two main neuronal progenitor cell types that coexist in the mouse necortex at E14.5: apical radial glia (aRG) and intermediate progenitors (IP) (Figure S2, Additional file 1). We performed immuno-stainings of the neocortical slices for PAX6 and TBR2 in order to label aRG and IP, respectively (Fig. 1a). Mir146a−/− embryos showed an imbalance of aRG/IP cell populations as they exhibit a 10% decrease of PAX6+ cells (Fig. 1b), and a 10% increase of TBR2+ cells (Fig. 1c) compared to control littermates. Consequently, the difference in ratio of TBR2+/PAX6+ cells was significantly (p = 0.00012) altered in mutant mice in comparison to controls (Fig. 1d).
Fig. 1.

Altered aRG and IP population size in the Mir146−/− brains. a Sagittal sections from Mir146a+/+and Mir146a−/− brains at E14.5 labeled with PAX6, TBR2, and merge. b Boxplot showing the number of PAX6+ cells (±S.D.) counted using 3 images from comparable plane per mouse. c Boxplot showing the quantification of TBR2+ cells (±S.D.) from the same images as b. d Boxplot showing the ratio of TBR2+/PAX6+ cells. e Phospho-histone 3 (PH3) stains dividing cells in the neocortex at E14.5 (white arrows). f Graph showing the average number of PH3+ cells (±S.D.) in the SVZ. *P < 0.05 by Student’s unpaired 2-tailed t test. g BrdU staining in the neocortex at E14.5 after 2h pulse chase; images were divided into five equal bins for counting. h Percentage of BrdU+ cells (±S.D.) per bin (normalized against the total number of labeled cells). NS not significant; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 by Mann-Whitney U test
aRG and IP undergo mitosis at distinct locations as RG cells divide in the ventricular zone (VZ) to produce IP cells, while IP cells divide in the subventricular zone (SVZ) to produce pairs of daughter neurons. We thus stained the neocortex for the mitosis marker Phospho-Histone 3 (PH3) (Fig. 1e). In the mutant neocortex, there was no difference in the number of PH3+ cells at the apical surface (where aRG divide) (Figure S3, Additional file 1). However, we observed a significant increase of PH3+ cells in the SVZ (where IP divide) (Figure 1f). To further corroborate these findings, we assessed the position of the cells that are progressing through S phase by BrdU pulse chase. We counted their distribution in the neocortex in five equal bins and observed a shift towards the basal lamina in the mutants compared to WTs (Fig. 1g). Quantification of BrdU+ cells in each bin showed a significant increase in bin III and a significant decrease in bin V (Fig. 1h), corresponding to the SVZ and the apical surface, respectively. Collectively, these results suggest an altered balance between the self-renewal of radial glial stem cells and the production of their descendent progenitor cells in Mir146a−/− neocortex.
Loss of miR-146a causes defects in neuronal differentiation and neurite outgrowth
As neurogenesis relies on fine coordination between proliferation and differentiation of aRG, we tested whether the loss of miR-146a could impact the number and/or properties of newborn neurons in the neocortex. We, thus, stained the neocortical slices at E14.5 for the neuronal post-mitotic factor NeuN and measured the thickness of the neuronal layer (Fig. 2a). Consistent with previous results, a significantly enlarged NeuN+ cells layer (30% increase) was observed in the mutant neocortex compared to controls (Fig. 2b). As previously mentioned, this difference does not persist throughout brain development, as in the cortex of P30 and P60 of mutants and control Mir146a+/+ littermates, we did not detect any difference between the number of NeuN+ cells (Figure S1, Additional file 1). To investigate whether the absence of miR-146a may alter the neurite outgrowth, we used live cell imaging to monitor neurite extension in cultured primary neuron for 1 week (Fig. 2c and Additional file 2: Tables S1). Mir146a−/− neurons had an average neurite length that was constantly longer than the length of the neurites of the control neurons (Fig. 2d, e).
Fig. 2.

Altered neurogenesis in the Mir146a−/− brains. a Sagittal section of E14.5 neocortex of Mir146a+/+ and Mir146a−/− brain stained with DAPI and the post-mitotic neuronal marker NeuN; white arrows show the differentiated neuron layer where the measurements were made. b Graph comparing the average thickness (±S.D.) of the NeuN+ cells layer between WT and mutant embryos; at least 3 images from comparable plane per mouse were used for the analysis; **p < 0.01 by Student’s unpaired 2-tailed t test. c Automated phase contrast image segmentation of neurites (pink) and cell body clusters (CBC in blue) of Mir146a−/− neurons at day 3 using the Neurotrack software module. d Graph shows the total length of neurites (±S.E.M) of neurons from Mir146a+/+ and (n = 7) Mir146a−/− embryos (n = 6). Images were taken by the Incucyte S3 at 9 different spots per well (replicate) every 3 h during 6 days of culture; post analysis was performed automatically using the pre-designed mask; there are at least 4 technical replicates per embryo. e Graph shows the average of slopes (±S.D.) of neurite extension of corresponding graph d. **p < 0.01 by Wilcoxon ranked sum test
Neuronal differentiation and migration are two closely related processes essential for the formation of the multilayered cortical structure. Therefore, we assessed whether this defect in neurogenesis interferes with the migrating trajectory of neurons by BrdU birth dating technique. We labeled with BrdU newly born neurons at E14.5, which are destined for layer IV of the cortex [27], and studied their distribution in each cortical layer at P7. This analysis did not detect any obvious difference in the distribution of BrdU+ cells in any cortical layer between mutants and controls (Figure S4, Additional file 1). Taken together, these results suggest a pro-neurogenic impact of the loss of miR146a in the Mir146a−/− neocortex, which do not lead to obvious malformation of the cortical structure.
Impaired hippocampal anatomy in the Mir146a−/− mice
A wide spectrum of structural brain anomalies is frequently observed in DBD patients [28–30]. This includes white matter and/or temporal lobe abnormalities in children with non-syndromic ASD, mega cisterna magna in low-functioning autistic patients, brain hemispheric asymmetry in children with high-functioning autism and developmental language disorder, corpus callosum defects, cortical and/or cerebellar atrophy, and hippocampal alteration in intellectually delayed children. Structural brain anomalies were also described in mouse models of DBDs. Indeed, reduced volume of cerebellum as well as reduced thickness of corpus callosum were observed in Mecp2-null mice [31], while the Fragile X syndrome mouse model exhibits an increased volume of the parieto-temporal lobe and a smaller striatum [32]. To investigate whether such anatomical defects are present in the Mir146a−/− mice, we performed magnetic resonance imaging (MRI) studies at P30, the earliest stage that allows us to measure the volume of the cortex and the hippocampus. While no significant difference was observed in the overall volume of these two regions (Fig. 3a, b) between mutant and control littermates, we observed a high hippocampal asymmetry between the two hemispheres in the mutant brains (Fig. 3c). In the Mir146a−/− brains, one side is 15% bigger than the other with a random distribution regarding the enlarged side (2 mice with right > left and 2 mice with left > right) (Fig. 3d). The same analysis was also performed at P60 and similar results were observed (Figure S5, Additional file 1).
Fig. 3.

Increased hippocampus asymmetry in the Mir146a−/− brain at P30. Volume (±S.D.) of the hippocampus (a) of the mutant and WT mice as measured by brain MRI at P30. The surface area of each region was measured from MRI images at 100 μM apart (5 images for the hippocampus and 15 for the cortex) and reconstructed using Image J to obtain final volume. b Brain MRI showing coronal sections. White arrows point towards the hippocampi; the larger white arrow point towards the enlarged hippocampus in the mutant. c Graph showing the volumes difference between two hippocampal hemispheres in Mir146a+/+ and Mir146a−/− brains. *p < 0.05 by Student’s unpaired 2-tailed t test. On average, one hippocampus side is 15% larger than the other in Mir146a−/− brains with random distribution (2 right > left and 2 left > right)
Loss of miR-146a results in learning and memory deficits
In human, abnormal expression of miR-146a is observed in patients with cognitive deficits. Our MRI analyses performed in the Mir146a−/− mice demonstrated a severe asymmetry of the hippocampus, a region of the brain playing a critical role in cognitive processes, such as learning and memory. Therefore, we subjected 3-month-old Mir146a−/− mice and their controls littermates to a series of behavioral tests, that is thought to reflect behavioral functions related to hippocampus, measuring associative (one-trial contextual fear conditioning, CFC), episodic (novel object recognition test, NOR), and spatial (Object localization memory OLM) learning and memory (Fig. 4). In CFC, mutant mice exhibited no differences in baseline freezing time. However, ablation of miR-146a resulted in decreased context-elicited freezing time during the testing phase compared to their WT littermates, indicating that contextual fear memory is impaired in the mutant mice (Fig. 4a). Next, we used a modified version of the NOR paradigm [26, 33] that measures the rodent’s ability to recognize a novel object in the environment. WT mice are capable of differentiating novel objects from familiar ones and tend to explore novel ones for longer time. As shown in (Fig. 4b), 3-month-old Mir146a−/− mice explored significantly less the novel object than WT controls. The same impairments were observed when memory was analyzed through the OLM task (assessing spatial learning and memory in rodents). Indeed, mutant mice explored significantly less the relocated object than WT littermates during the testing phase (Fig. 4c). Of note, mutant and WT mice had comparable performance in the open-field test (OFT) and light/dark paradigm (L/DT) (Figure S6, Additional file 1), indicating that their locomotion and anxious state were intact. Neuroinflammation is a well-recognized etiological component in ASD [34]. Given the role of miR-146a in innate and adaptive immune responses, we asked whether the behavioral anomalies observed in mutant mice could be related to altered neuro-immunoregulation. To that end, we tested by ELISA assays the level of inflammatory markers IL1-β and TNF-α in hippocampal and cortical brain lysates from the mice after the behavioral tasks. We did not detect any difference between the mutants and WT, excluding the confounding effects of inflammation in the observed behavioral defects (Figure S7, Additional file 1). Taken together, these data demonstrate that miR-146a constitutive inactivation significantly influences hippocampal-dependent spatial learning and memory performances, episodic memory and associative fear memory acquisition through a mechanism independent of the role of this miRNA in the regulation of inflammatory response.
Fig. 4.

Behavioral analyses of the Mir146a−/− mice.a 3-foot shock CFC performed in a 3-month-old Mir146a−/− mice (n = 16) and their control littermates (n = 15). Percent freezing was measured in CFC for each group during the training (as a control for the basal level of mouse freezing) and testing phases (to assess memory performances). b Novel object recognition test (NOR) performed in a 3-month-old Mir146a−/− mice (n = 16) and their control littermates (n = 15). Discrimination and preference indexes were measured for each group during the testing phase. c Object location memory (OLM) task performed in a 3-month-old Mir146a−/− mice (n = 16) and their control littermates (n = 15). Discrimination and preference indexes were measured for each group during the testing phase
Postnatal knockdown of miR-146a affect adult neurogenesis and causes behavioral deficits
Adult neurogenesis is known to play an important role in the maintenance of hippocampal memory capacity. We thus assessed the impact of postnatal mir-146a-knockdown on cognitive functions. To modulate miR-146a expression, we performed stereotactic injections of adeno-associated viruses (AAV) expressing shRNAs directed against mir-146a in the hippocampus of 2-months-old WT mice (Fig. 5a) and subjected mice to behavioral analyses 3 weeks after injections. As a result, we observed behavioral deficits in NOR, CFC, and OLM tests similar to those seen in Mir146a−/− mice (Fig. 5b–d). Last, to get insight into the mechanisms underlying the observed behavioral phenotypes, we investigated the impact of miR-146a knockdown on hippocampal adult neurogenesis. The microtubule-associated protein doublecortin (DCX) is transiently expressed in proliferating progenitor cells and newly generated neuroblasts and is a reliable marker of adult neurogenesis level. As shown in Fig. 5e, we detected a drastic decrease of the number of DCX+ cells in the dentate gyrus of the mice locally injected with AAV-shRNA miR-146, demonstrating a reduced number of immature neurons in this region. Taken together, these findings demonstrate for the first time that the role of mir-146a in the brain is not restricted to neurodevelopmental processes, but that it also plays an important postnatal role in hippocampal neurons.
Fig. 5.
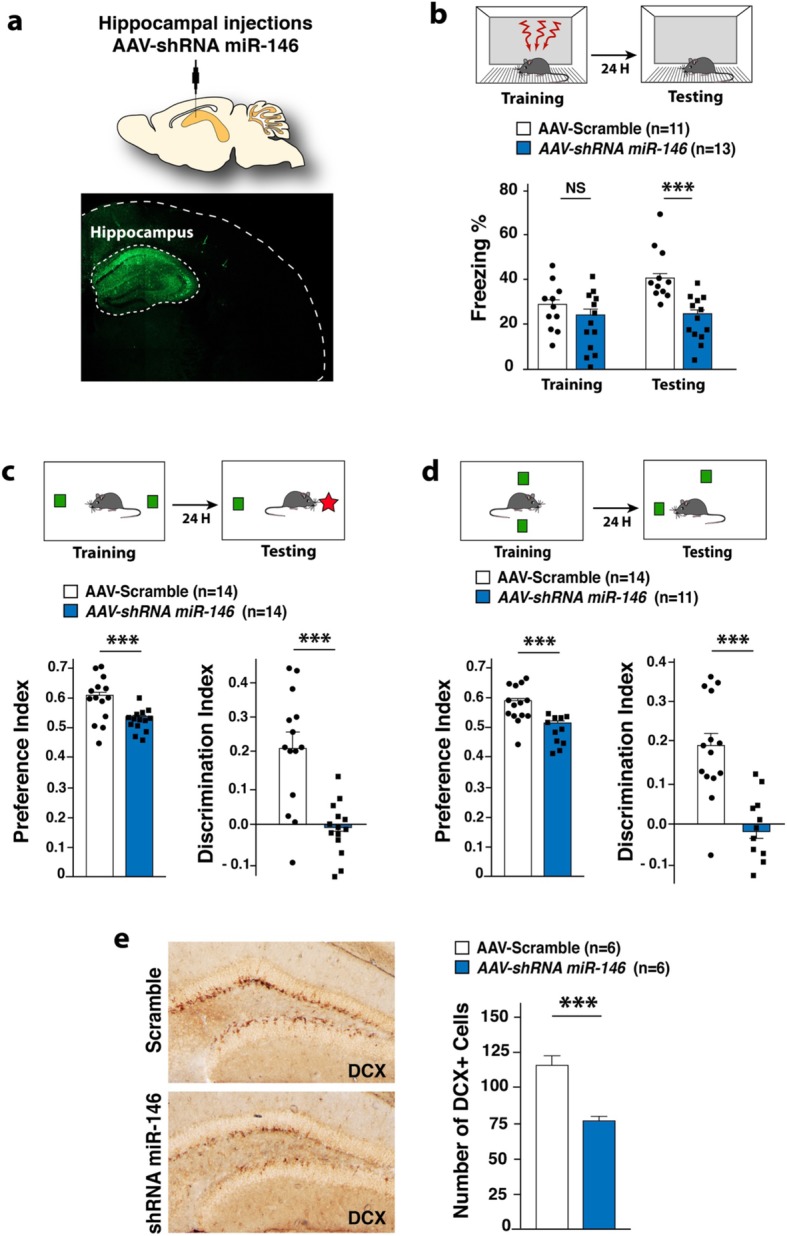
Behavioral analyses after selective hippocampal downregulation of Mir146.a Fluorescent microscopy image of a coronal brain section collected 3 weeks after local stereotactic injections with AAV expressing shRNAmir for Mir146a gene. The construct expresses eGFP, which allow us to verify the site and the efficacy of infection. A Panoramic photo of an entire brain region was obtained by automatically aligning and stitching 50 tiled images into a mosaic (using the Zen Light Zeiss LSM software package). b–d Behavioral analyses (CFC (E), NOR (F), OLM (G)) performed in 3-month-old mice, 3 weeks after hippocampal stereotactic injections with either: AAV-Mir146a-shRNA or AAV-Scramble-shRNA. e Microtubule-associated protein doublecortin (DCX) immunohistochemistry showing a significantly lower number of DCX+ cells (mean number of DCX+ cells in the dentate gyrus (DG)/brain slides) in the DG of the mice locally injected with AAV-shRNA miR 146a in comparison to the one injected with AAV-shRNA scramble. All behavioral tests were performed on at least two independent experiments for each experiment: n6 mice per group. Results are given as mean ± s.e.m. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, NS: not significant; by Student’s t test compared to 3-month-old mice littermates. All behavioral tests were performed on at least two independent experiments for each experiment: n6 mice per group. Results are given as mean ± s.e.m. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, NS: not significant; by Student’s t test compared to 3-month-old mice littermates
Discussion
While many in vitro studies have been reported, the physiological role of miRNAs in brain development, cognitive functions and diseases has remained largely unexplored. Elucidating the specific cells and gene network regulated by each brain-expressed microRNAs is crucial for our understanding of both the mechanisms of neural circuit development and the etiology of neurodevelopmental disorders. A recent report demonstrated that in mouse partial loss of miR-137, a locus linked to autism and intellectual disability, impairs postnatal development and regulates synaptic plasticity by targeting the cyclic nucleotide phosphodiesterase family member Pde10a [35]. Here, we used mouse models with either constitutive inactivation or selectively hippocampal knockdown of the neurodevelopmental disease-associated gene Mir146a to evaluate the consequences of this inactivation on the differentiation and migration of neural stem cells, as well as on brain anatomy and cognition.
We first show that proper miR-146a expression is critical for correct differentiation of neural stem cell during brain development (Fig. 6). These results are in line with the previously published in vitro studies demonstrating that increased miR-146a level enhanced neuronal differentiation in mouse NSC [36] and in H9 human neural stem cells [12]. Yet, while miR-146a overexpression in cultured NSC halts cell cycle progression and promotes neuronal differentiation through NOTCH pathway, how loss of miR-146a causes defective neuronal differentiation and which miR-146a target(s) may mediate these effects in the mutant brains is still an open question. In this regard, the adaptor protein NUMB protein is an excellent candidate. NUMB plays a key role in asymmetric division and cell fate determination in many cell types, including neural progenitors [37, 38]. NUMB is a validated direct target of miR-146a [39], and miR-146a was reported to facilitate activation of SHH signaling by targeting NUMB expression [40]. Various Numb transcript isoforms have been characterized that differ by the inclusion or not of exon3 and exon9. These isoforms display distinct developmental expression pattern and functions in the neuronal lineage [41]. The expression of the long NUMB isoform starts at E7, peaks at E10, and becomes undetectable by E13. By contrast, short isoform is present throughout development and in the adult brain. Moreover, while the long isoform promotes proliferation without affecting neuronal differentiation, the short one inhibits proliferation of the stem cells and promotes neuronal differentiation. Testing for NUMB transcript expression level in E14.5 neocortex and NUMB protein level by Western blot of aRG protein extracts did not reveal a significant difference between Mir146a+/+ and Mir146a−/− samples. However, given the complexity of NUMB's function in brain development, and the role of NUMB isoform switch for neuronal development, we propose that loss of miR-146a in earlier stages of brain development may affect either short isoform NUMB protein level or NUMB isoform switch.
Fig. 6.
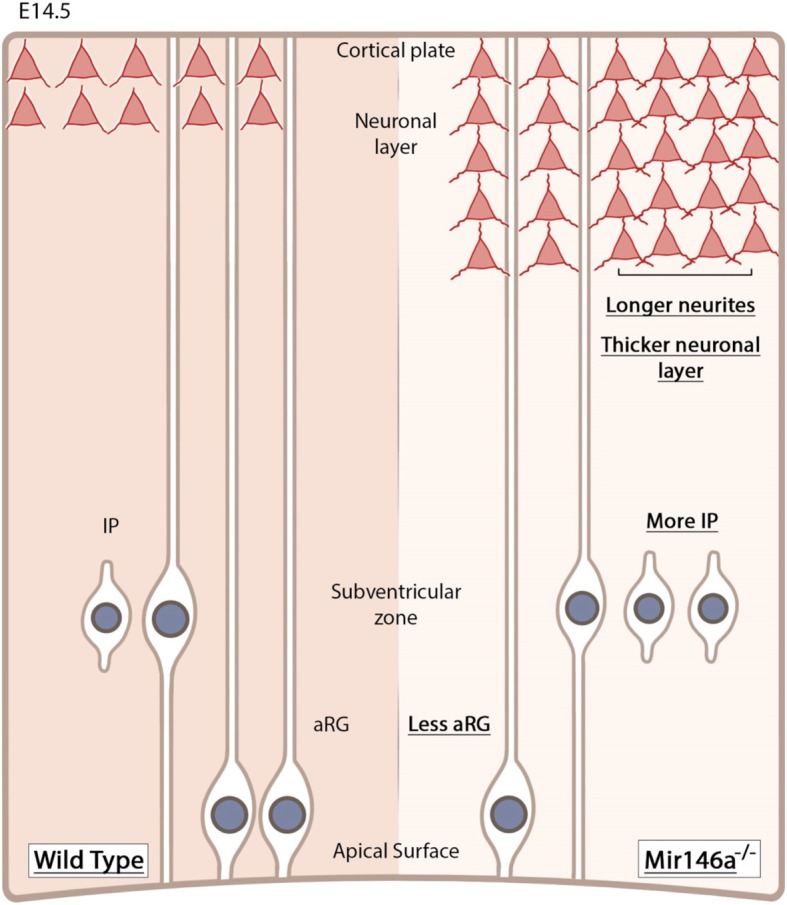
miR-146a promotes neuronal differentiation at E14.5. During neurogenesis, loss of miR-146a leads to imbalance of radial glial cells (aRG) and intermediate progenitor (IP) and enhances neuronal differentiation and neurite branching
Besides the neurogenesis defects observed in early stages of brain development, no noticeable difference was observed in the number of neurons, the size, and the structure of the cortex between control and mutant brains at postnatal stages. This result suggests the establishment of compensatory mechanisms that prevent the production of supernumerary neurons and major structural abnormalities. However, we observed that Mir146a−/− mice display an abnormal hippocampus structure with an increased hemispheric asymmetry that was not associated with any particular brain hemisphere. In human, the hippocampal volume is slightly larger in the right hemisphere both in children and in adults [42, 43]. While this asymmetry is sometimes lost in pathologies such as Williams syndrome [44] or Alzheimer’s disease [45], to our knowledge, an increased asymmetry with randomized distribution between right and left hemispheres was never reported. Although the mechanism underlying this defect remains unknown, especially its link with impaired radial glia differentiation and neurogenesis in the neocortex, this finding suggests that loss of miR-146a causes subtle alterations in brain anatomy. Perhaps the most intriguing finding is that Mir146a−/− mice exhibited severe impairments in learning and memory functions without any locomotion or anxiety-related behavioral phenotype. Mechanistically, the deficit in learning and memory performances can be explained, at least in part, by the brain anatomical and neuronal differentiation defects observed in the mutant mice. However, our observation that postnatal downregulation of mir-146a specifically in the hippocampus region is sufficient to impair learning and memory capacities and causes altered neurogenesis indicates for the first time that this miR also have a postnatal role and is involved in hippocampus-dependent cognitive functions.
Conclusion
Collectively, our data demonstrate that miR-146a inactivation alters processes involved in neural stem cell differentiation and provide for the first time a strong argument for a postnatal role of miR-146a in regulating hippocampal-dependent memory. The demonstration that the Mir146a−/− mouse recapitulates several aspects reported in DBD patients, including impaired neurogenesis, abnormal brain anatomy, and working and spatial memories deficits, provides convincing evidence that impaired expression of miR-146a contributes to the pathogenesis of DBDs. Last, the relevance of the miR146a-/- mouse model to study DBD opens the way for future investigation in the role of this miRNA in other key aspects of neurodevelopment such as glial cell biology, cognitive disorders and adult neurogenesis.
Supplementary information
Additional file 1: Figure S1. The number of cortical neurons is unaltered in the adult Mir146a-/- brain. (a) Cortical slides of WT (top panel) and Mir146a-/- mouse (bottom panel) stained for NeuN to label post-mitotic neurons at P60. Number of NeuN+ cells normalized to area counted (±S.D.) in layer 2 (b), layers 3-4 (c), layer 6 (d) and all layers (e) in the WT and Mir146a-/- cortical slices. At least 2 images at comparable plane per mouse were analyzed. The number of NeuN+ cells at P30 and P60 are indistinguishable, thus, we combined the results for each genotype. Figure S2. Neurogenesis in the mouse neocortex. Neuroepithelial cells (NE) proliferate up to E11 before differentiating into apical radial glia (aRG). aRG are located in the ventricular zone (VZ) where their nucleus undergo interkinetic nuclear migration (INM) and divide at the apical surface. aRG either self-replicate or give rise to a neuron or an intermediate progenitor (IP) after an asymmetric division. IP remain in the subventricular zone (SVZ) and can complete one or two cell cycles before differentiating into neurons. Neurons coming from aRG or IP migrate towards the intermediate zone (IZ) and the cortical plate (CP) where they acquire their final position and identity and start expressing post-mitotic markers such as NeuN. Figure S3. Number of PH3+ cells at apical surface of the E14.5 neocortex is unaltered in the Mir146a-/-. Graph showing the number of PH3+ cells (±S.D.) in images shown in Fig. 1e. NS, not significant by Student’s 2-tailed T-Test. Figure S4. Loss of miR-146a does not affect cortical layers organization. (a) BrdU signal at P7 labeled neurons born at E14.5. The white box shows the analyzed area. (b) Zoom of the white box from (a). The 6 layers of the mature cortex were determined and BrdU+ cells were counted using 3 images from comparable plane. (c) Graph shows the percentage of cells in each layer for the two genotypes, normalized against the total number of labeled cells. Figure S5. Increased hippocampus asymmetry in the Mir146a-/- mice at P60. Volume (±S.D.) of the cortex (a) and the hippocampus (b) of the mutant and WT mice as measured by brain MRI at P60. (c) Scatterplot comparing the ratio between volumes of two hippocampal hemispheres in mutant and WT mice. NS: not significant Student’s unpaired 2-tailed T-Test. On average, one hippocampus side is 15% larger than the other in mutant brains with random distribution (2 right>left and 2 left>right). Figure S6. Locomotion and anxious state of the Mir146a-/- mice are not affected. (a) Open field test (OFT) performed in 3-month old Mir146a-/- mice (n=16) and their control littermates (n=15). Total distance (meter), % of the distance traveled in the center versus periphery, and % of the time spent in the center versus periphery were measured. (b) Light/Dark transition Test (L/DT) performed in 3-month old Mir146a-/- mice (n=16) and their control littermates (n=15). The number of entries in the lit compartment, and % of the distance traveled and time spent in the lit compartment were measured. All behavioral tests were performed on at least two independent experiments, for each experiment: n≥6 mice per group. Results are given as mean ± s.e.m. *P ≤ 0.05, **P≤ 0.01, ***P ≤ 0.001, NS: not significant; by Student’s t-test compared to 3-month-old mice littermates. Figure S7. The Mir146a-/- brain shows no sign of inflammation. Expression level (±S.D.) of inflammatory markers IL1-β (a and b) and TNF- α (c and d) in brain lysates of 8 WT and 8 mutant mice that were used for behavior studies. Technical quadruplicates were performed.
Acknowledgements
We would like to thank the core platforms of the Imagine Institute, notably the Histology Platform. We also wish to thank Dr. Fiona Francis and her group for their precious advice during experimental setup. We deeply acknowledge Dr Fabiola Terzi for the gift of the Mir146a+/− mouse line.
Abbreviations
- DBD
Developmental brain disorders
- ASD
Autism spectrum disorders
- ID
Intellectual disability
- hNSC
Human neural stem cell
- miRNA
MicroRNA
- INM
Interkinetic nuclear migration
- PH3
Phospho-histone 3
- PAX6
Paired box 6 protein
- TBR2
T-box brain protein 2
- BrdU
Bromo-desoxy-uridine
- DEG
Differentially expressed gene
- CNS
Central nervous system
- aRG
Radial glial cell
- AAV
Adeno-associated viruses
- IP
Intermediate progenitor
- VZ
Ventricular zone
- SVZ
Subventricular zone
- WT
Wild type
Authors’ contributions
FO and LC conceived the project. JF, SM, and LSN performed the experiments. AP helped to perform the IHC experiments. JF, LSN, SM, FO, and LC wrote the manuscript. All other authors provided feedback on the manuscript. All author(s) read and approved the final manuscript.
Funding
This work received a state subsidy managed by the National Research Agency under the “Investments for the Future” program bearing the reference ANR-10-IHU-01 and the “ANR-SAMENTA 2012” program. This work was also supported by the Fondation pour la Recherche Médicale (DEQ20160334938) and the Fondation de France. LC is supported by the Centre National de la Recherche Scientifique. JF is supported by Crédit Agricole d'Ile-de-France Mécénat. FO is supported by the Fondation pour la Recherche Médicale (FRM), Human Frontier Scientific Program-CDA, ATIP-AVENIR Program, AGEMED INSERM program, ANR, Fondation Schlumberger pour l’Education et la Recherche (FSER), and Emergence de la Ville de Paris.
Ethics approval and consent to participate
All experiments performed with mice were approved by an independent ethic committee of Paris Descartes University (approval number 2015101215207380) and declared to the French Ministry of Research.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Julien Fregeac and Stéphanie Moriceau contributed equally to this work.
Contributor Information
Julien Fregeac, Email: julien.fregeac@inserm.fr.
Stéphanie Moriceau, Email: stephanie.moriceau@inserm.fr.
Antoine Poli, Email: antoine.poli@gmx.fr.
Lam Son Nguyen, Email: lam-son.nguyen@inserm.fr.
Franck Oury, Email: franck.oury@inserm.fr.
Laurence Colleaux, Email: laurence.colleaux@inserm.fr.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13229-020-00328-3.
References
- 1.Regulation of microRNA biogenesis, (2014).
- 2.Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, et al. Dicer is essential for mouse development. Nat Genet. 2003;35(3):215–217. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- 3.O'Carroll D, Schaefer A. General principals of miRNA biogenesis and regulation in the brain. Neuropsychopharmacology. 2013;38(1):39–54. doi: 10.1038/npp.2012.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rajman M, Schratt G. MicroRNAs in neural development: from master regulators to fine-tuners. Development. 2017;144(13):2310–2322. doi: 10.1242/dev.144337. [DOI] [PubMed] [Google Scholar]
- 5.MicroRNAs and neurodegeneration: Role and impact, (2013). [DOI] [PMC free article] [PubMed]
- 6.The roles of microRNAs in neuroblastoma, (2014). [DOI] [PubMed]
- 7.Fregeac J, Colleaux L, Nguyen LSLS. The emerging roles of MicroRNAs in autism spectrum disorders. Neuroscience and Biobehavioral Reviews. 2016;71:729–738. doi: 10.1016/j.neubiorev.2016.10.018. [DOI] [PubMed] [Google Scholar]
- 8.Gibbons A, Udawela M, Dean B. Non-coding RNA as novel players in the pathophysiology of schizophrenia. Noncoding RNA. 2018;4(2). [DOI] [PMC free article] [PubMed]
- 9.Nguyen LS, Lepleux M, Makhlouf M, Martin C, Fregeac J, Siquier-Pernet K, et al. Profiling olfactory stem cells from living patients identifies miRNAs relevant for autism pathophysiology. Mol Autism. 2016;7:1. doi: 10.1186/s13229-015-0064-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Functions of noncoding RNAs in neural development and neurological diseases., (2011). [DOI] [PMC free article] [PubMed]
- 11.Mor M, Nardone S, Sams DS, Elliott E. Hypomethylation of miR-142 promoter and upregulation of microRNAs that target the oxytocin receptor gene in the autism prefrontal cortex. Mol Autism. 2015;6:46. doi: 10.1186/s13229-015-0040-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen LS, Fregeac J, Bole-Feysot C, Cagnard N, Iyer A, Anink J, et al. Role of miR-146a in neural stem cell differentiation and neural lineage determination: relevance for neurodevelopmental disorders. Mol Autism. 2018;9:38. doi: 10.1186/s13229-018-0219-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Talebizadeh Z, Butler MG, Theodoro MF. Feasibility and relevance of examining lymphoblastoid cell lines to study role of microRNAs in autism. Autism Res. 2008;1(4):240–250. doi: 10.1002/aur.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.An N, Zhao W, Liu Y, Yang X, Chen P. Elevated serum miR-106b and miR-146a in patients with focal and generalized epilepsy. Epilepsy Research. 2016. [DOI] [PubMed]
- 15.He F, Liu B, Meng Q, Sun Y, Wang W, Wang C. Modulation of miR-146a/complement factor H mediated inflammatory responses in a rat model of temporal lobe epilepsy. Biosci Rep. 2016. [DOI] [PMC free article] [PubMed] [Retracted]
- 16.Tao H, Zhao J, Liu T, Cai Y, Zhou X, Xing H, et al. Intranasal delivery of miR-146a mimics delayed seizure onset in the lithium-pilocarpine mouse model. Mediators Inflamm. 2017;2017:6512620. doi: 10.1155/2017/6512620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103(33):12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang C, Liu XJ, QunZhou XJ, Ma TT, Meng XM, et al. MiR-146a modulates macrophage polarization by inhibiting Notch1 pathway in RAW264.7 macrophages. Int Immunopharmacol. 2016;32:46–54. doi: 10.1016/j.intimp.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 19.Ghahhari NM, Babashah S. Interplay between microRNAs and WNT/β-catenin signalling pathway regulates epithelial-mesenchymal transition in cancer. European Journal of Cancer. 2015. [DOI] [PubMed]
- 20.Hwang SJ, Seol HJ, Park YM, Kim KH, Gorospe M, Nam DH, et al. MicroRNA-146a suppresses metastatic activity in brain metastasis. Mol Cells. 2012. [DOI] [PMC free article] [PubMed]
- 21.Wang L, Chopp M, Szalad A, Zhang Y, Wang X, Zhang RL, et al. The role of miR-146a in dorsal root ganglia neurons of experimental diabetic peripheral neuropathy. Neuroscience. 2014;259:155–163. doi: 10.1016/j.neuroscience.2013.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou X, Su S, Li S, Pang X, Chen C, Li J, et al. MicroRNA-146a down-regulation correlates with neuroprotection and targets pro-apoptotic genes in cerebral ischemic injury in vitro. Brain Research. 1648;2016:136–143. doi: 10.1016/j.brainres.2016.07.034. [DOI] [PubMed] [Google Scholar]
- 23.Jia L, Wang L, Chopp M, Zhang Y, Szalad A, Zhang ZG. MicroRNA 146a locally mediates distal axonal growth of dorsal root ganglia neurons under high glucose and sildenafil conditions. Neuroscience. 2016. [DOI] [PMC free article] [PubMed]
- 24.Chen Y-L, Shen C-KJ. Modulation of mGluR-dependent MAP1B translation and AMPA receptor endocytosis by microRNA miR-146a-5p. J Neurosci. 2013;33:9013–9020. doi: 10.1523/JNEUROSCI.5210-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, et al. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell. 2010;142(6):914–929. doi: 10.1016/j.cell.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav Brain Res. 1988;31(1):47–59. doi: 10.1016/0166-4328(88)90157-X. [DOI] [PubMed] [Google Scholar]
- 27.Molyneaux BJ, Arlotta P, Menezes JRL, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nature Reviews Neuroscience. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- 28.Boddaert N, Zilbovicius M, Philipe A, Robel L, Bourgeois M, Barthélemy C, et al. MRI findings in 77 children with non-syndromic autistic disorder. PLoS ONE. 2009. [DOI] [PMC free article] [PubMed]
- 29.Erbetta A, Bulgheroni S, Contarino VE, Chiapparini L, Esposito S, Annunziata S, et al. Low-functioning autism and nonsyndromic intellectual disability: magnetic resonance imaging (MRI) findings. J Child Neurol. 2015. [DOI] [PubMed]
- 30.Herbert MR, Ziegler DA, Deutsch CK, O'Brien LM, Kennedy DN, Filipek PA, et al. Brain asymmetries in autism and developmental language disorder: a nested whole-brain analysis. Brain. 2005;128(Pt 1):213–226. doi: 10.1093/brain/awh330. [DOI] [PubMed] [Google Scholar]
- 31.Saywell V, Viola A, Confort-Gouny S, Le Fur Y, Villard L, Cozzone PJ. Brain magnetic resonance study of Mecp2 deletion effects on anatomy and metabolism. Biochem Biophys Res Commun. 2006. [DOI] [PubMed]
- 32.Ellegood J, Pacey LK, Hampson DR, Lerch JP, Henkelman RM. Anatomical phenotyping in a mouse model of fragile X syndrome with magnetic resonance imaging. NeuroImage. 2010. [DOI] [PubMed]
- 33.Denny CA, Burghardt NS, Schachter DM, Hen R, Drew MR. 4- to 6-week-old adult-born hippocampal neurons influence novelty-evoked exploration and contextual fear conditioning. Hippocampus. 2012;22(5):1188–1201. doi: 10.1002/hipo.20964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Onore C, Careaga M, Ashwood P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav Immun. 2012;26(3):383–392. doi: 10.1016/j.bbi.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng Y, Wang ZM, Tan W, Wang X, Li Y, Bai B, et al. Partial loss of psychiatric risk gene Mir137 in mice causes repetitive behavior and impairs sociability and learning via increased Pde10a. Nat Neurosci. 2018;21(12):1689–1703. doi: 10.1038/s41593-018-0261-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mei J, Bachoo R, Zhang CL. MicroRNA-146a inhibits glioma development by targeting Notch1. Mol Cell Biol. 2011;31(17):3584–3592. doi: 10.1128/MCB.05821-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rasin MR, Gazula VR, Breunig JJ, Kwan KY, Johnson MB, Liu-Chen S, et al. Numb and Numbl are required for maintenance of cadherin-based adhesion and polarity of neural progenitors. Nat Neurosci. 2007;10(7):819–827. doi: 10.1038/nn1924. [DOI] [PubMed] [Google Scholar]
- 38.Shen Q, Zhong W, Jan YN, Temple S. Asymmetric Numb distribution is critical for asymmetric cell division of mouse cerebral cortical stem cells and neuroblasts. Development (Cambridge, England) 2002;129:4843–4853. doi: 10.1242/dev.129.20.4843. [DOI] [PubMed] [Google Scholar]
- 39.Kuang W, Tan J, Duan Y, Duan J, Wang W, Jin F, et al. Cyclic stretch induced miR-146a upregulation delays C2C12 myogenic differentiation through inhibition of Numb. Biochem Biophys Res Commun. 2009;378(2):259–263. doi: 10.1016/j.bbrc.2008.11.041. [DOI] [PubMed] [Google Scholar]
- 40.Ghorpade DS, Sinha AY, Holla S, Singh V, Balaji KN. NOD2-nitric oxide-responsive microRNA-146a activates Sonic hedgehog signaling to orchestrate inflammatory responses in murine model of inflammatory bowel disease. J Biol Chem. 2013;288:33037–33048. doi: 10.1074/jbc.M113.492496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dho SE, French MB, Woods SA, McGlade CJ. Characterization of four mammalian numb protein isoforms. Identification of cytoplasmic and membrane-associated variants of the phosphotyrosine binding domain. J Biol Chem. 1999;274(46):33097–33104. doi: 10.1074/jbc.274.46.33097. [DOI] [PubMed] [Google Scholar]
- 42.Giedd JN, Vaituzis AC, Hamburger SD, Lange N, Rajapakse JC, Kaysen D, et al. Quantitative MRI of the temporal lobe, amygdala, and hippocampus in normal human development: Ages 4-18 years. J Comparative Neurol. 1996. [DOI] [PubMed]
- 43.Pruessner JC. Volumetry of hippocampus and amygdala with high-resolution MRI and three-dimensional analysis software: minimizing the discrepancies between laboratories. Cerebral Cortex. 2000. [DOI] [PubMed]
- 44.Sampaio A, Sousa N, Férnandez M, Vasconcelos C, Shenton ME, Gonçalves ÓF. Williams syndrome and memory: a neuroanatomic and cognitive approach. J Autism Dev Disord. 2010. [DOI] [PubMed]
- 45.Barnes J, Scahill RI, Schott JM, Frost C, Rossor MN, Fox NC. Does Alzheimer’s disease affect hippocampal asymmetry? Evidence from a cross-sectional and longitudinal volumetric MRI study. Dementia and Geriatric Cognitive Disorders. 2005. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. The number of cortical neurons is unaltered in the adult Mir146a-/- brain. (a) Cortical slides of WT (top panel) and Mir146a-/- mouse (bottom panel) stained for NeuN to label post-mitotic neurons at P60. Number of NeuN+ cells normalized to area counted (±S.D.) in layer 2 (b), layers 3-4 (c), layer 6 (d) and all layers (e) in the WT and Mir146a-/- cortical slices. At least 2 images at comparable plane per mouse were analyzed. The number of NeuN+ cells at P30 and P60 are indistinguishable, thus, we combined the results for each genotype. Figure S2. Neurogenesis in the mouse neocortex. Neuroepithelial cells (NE) proliferate up to E11 before differentiating into apical radial glia (aRG). aRG are located in the ventricular zone (VZ) where their nucleus undergo interkinetic nuclear migration (INM) and divide at the apical surface. aRG either self-replicate or give rise to a neuron or an intermediate progenitor (IP) after an asymmetric division. IP remain in the subventricular zone (SVZ) and can complete one or two cell cycles before differentiating into neurons. Neurons coming from aRG or IP migrate towards the intermediate zone (IZ) and the cortical plate (CP) where they acquire their final position and identity and start expressing post-mitotic markers such as NeuN. Figure S3. Number of PH3+ cells at apical surface of the E14.5 neocortex is unaltered in the Mir146a-/-. Graph showing the number of PH3+ cells (±S.D.) in images shown in Fig. 1e. NS, not significant by Student’s 2-tailed T-Test. Figure S4. Loss of miR-146a does not affect cortical layers organization. (a) BrdU signal at P7 labeled neurons born at E14.5. The white box shows the analyzed area. (b) Zoom of the white box from (a). The 6 layers of the mature cortex were determined and BrdU+ cells were counted using 3 images from comparable plane. (c) Graph shows the percentage of cells in each layer for the two genotypes, normalized against the total number of labeled cells. Figure S5. Increased hippocampus asymmetry in the Mir146a-/- mice at P60. Volume (±S.D.) of the cortex (a) and the hippocampus (b) of the mutant and WT mice as measured by brain MRI at P60. (c) Scatterplot comparing the ratio between volumes of two hippocampal hemispheres in mutant and WT mice. NS: not significant Student’s unpaired 2-tailed T-Test. On average, one hippocampus side is 15% larger than the other in mutant brains with random distribution (2 right>left and 2 left>right). Figure S6. Locomotion and anxious state of the Mir146a-/- mice are not affected. (a) Open field test (OFT) performed in 3-month old Mir146a-/- mice (n=16) and their control littermates (n=15). Total distance (meter), % of the distance traveled in the center versus periphery, and % of the time spent in the center versus periphery were measured. (b) Light/Dark transition Test (L/DT) performed in 3-month old Mir146a-/- mice (n=16) and their control littermates (n=15). The number of entries in the lit compartment, and % of the distance traveled and time spent in the lit compartment were measured. All behavioral tests were performed on at least two independent experiments, for each experiment: n≥6 mice per group. Results are given as mean ± s.e.m. *P ≤ 0.05, **P≤ 0.01, ***P ≤ 0.001, NS: not significant; by Student’s t-test compared to 3-month-old mice littermates. Figure S7. The Mir146a-/- brain shows no sign of inflammation. Expression level (±S.D.) of inflammatory markers IL1-β (a and b) and TNF- α (c and d) in brain lysates of 8 WT and 8 mutant mice that were used for behavior studies. Technical quadruplicates were performed.