

Abstract
Ferroptosis is a recently defined, non‐apoptotic, regulated cell death (RCD) process that comprises abnormal metabolism of cellular lipid oxides catalysed by iron ions or iron‐containing enzymes. In this process, a variety of inducers destroy the cell redox balance and produce a large number of lipid peroxidation products, eventually triggering cell death. However, in terms of morphology, biochemistry and genetics, ferroptosis is quite different from apoptosis, necrosis, autophagy‐dependent cell death and other RCD processes. A growing number of studies suggest that the relationship between ferroptosis and cancer is extremely complicated and that ferroptosis promises to be a novel approach for the cancer treatment. This article primarily focuses on the mechanism of ferroptosis and discusses the potential application of ferroptosis in cancer therapy.
Keywords: cancer therapy, ferroptosis, GPx4, lipid peroxidation
1. INTRODUCTION
Death is inevitable for any living cell, but death is also a complicated process. The Nomenclature Committee on Cell Death (NCCD) has formulated guidelines for defining cell death in view of morphology, biochemistry and function. In recent recommendations, they listed more than 12 types of cell death, including apoptosis, necroptosis, pyroptosis, ferroptosis and autophagy‐dependent cell death.1 The high number of cell death forms can confuse but also inspire researchers to explore these mysteries. Publications on this subject have rapidly increased since the 1990s. However, most of the mechanisms underlying cell death are still veiled. Understanding the meaning and consequence of cell death, especially the “active” forms, are difficult, similar to the riddle raised by Douglas R. Green on this topic: How dispensable is something that is essential?2 Perhaps, as Douglas R. Green reminds us, we should look for answers in the consequences of cell death for the remaining living cells in the organism.3
Ferroptosis is an iron‐dependent, non‐apoptotic RCD process named by Scott J. Dixon in 2012. Small molecules, such as erastin and RSL3, can trigger ferroptosis, which is distinct from apoptosis, necrosis and autophagy‐dependent cell death in morphology, biochemistry and gene expression.4 Recently, ferroptosis has become a hot topic in a variety of diseases, especially cancer therapy.5, 6 For instance, new findings reveal that cell density can affect the sensitivity to ferroptosis, and another study showed that ferroptosis can spread through cell populations in a wave‐like manner.7, 8 These factors should be considered when ferroptosis is applied to cancer therapy. In addition, some groups have tried to use nanoparticles and exosomes as carriers of erastin and drugs to precisely induce ferroptosis in tumour tissues.9, 10 These new findings and treatment attempts enrich the study of ferroptosis. Therefore, it is meaningful to review the main mechanisms underlying ferroptosis and their potential treatment value.
2. A PREQUEL TO FERROPTOSIS
The cognition of ferroptosis is a cumulative process. Before Dixon defined ferroptosis, the key molecules associated with it had been reported. For example, the cystine and glutamate transport system (System Xc‐) was discovered in 1986, and scholars found that exposure to high levels of glutamate or low levels of cysteine could cause a decrease in glutathione and accumulation of intracellular peroxides.11, 12 Further, Dolma team used synthetic, lethal, high‐throughput screening to filtrate a mass of compounds for their potency to kill RAS‐mutated tumour cells and found one chemical compound, erastin, that could cause the death of cancer cells in a non‐apoptosis manner.13 Five years later, another two small molecules, named RSL3 and RSL5, were identified and found to lead to the death of RAS‐mutated cancer cells in an iron‐dependent, non‐apoptotic cell death manner.14 At the same time, a new finding emerged that GPx4 depletion caused tremendous lipid peroxidation and cell death with an unrecognized cell death pattern, which was 12/15‐lipoxygenase‐dependent and AIF‐mediated.15 Based on these studies, the Scott J. Dixon and team expanded, extended and systemically summarized this special type of cell death, naming it ferroptosis, which is a type of RCD caused by iron‐dependent lipid peroxides and shares none of the characteristic morphologic features associated with necrosis, apoptosis or autophagy‐dependent cell death.
3. MAIN MECHANISMS OF FERROPTOSIS
3.1. The role of lipid peroxides in ferroptosis
The most prominent feature of ferroptosis is iron‐dependent lipid peroxides. Lipid peroxides are generally viewed as eventual executioners of ferroptosis through their ability to cause plasma membrane damage.16 Physiologically, most intracellular oxygen is reduced to H2O via oxidative phosphorylation in the mitochondrial inner membrane.17 However, a small proportion of oxygen will participate in other physiological or biochemical activities, including phagocytosis, immune activation and xenobiotic metabolism, and result in harmful intermediates, such as reactive oxygen species (ROS).18, 19 Objectively speaking, a low and controlled ROS level is crucial for normal cellular and organismal function, and a moderately increased ROS level is beneficial for cancer development, but high ROS levels will cause cell injury and death.20
Current researches studied that accumulation of polyunsaturated fatty acid (PUFA) oxides is a hallmark of ferroptosis, but the accumulation process is complicated. The process involves inhibition of deoxidation, which mainly refers to glutathione peroxidase 4 (GPx4) and ferroptosis suppressor protein 1 (FSP1), and enhancement of hyperoxidation, which is catalysed by iron and a series of enzymes. Eventually, PUFA oxidation accumulates and leads to membrane integrity damage and ferroptosis.19, 21, 22, 23, 24 Lipoxygenases (LOXs) are the most important lipid oxidation enzymes for ferroptosis. LOX family members are iron‐incorporating enzymes that can catalyse the dioxygenation of PUFAs.25 Moreover, erastin‐induced cell death can be prevented if arachidonate lipoxygenase (ALOX) is silenced, and several LOX inhibitors can also have this effect.26, 27, 28
In addition, given that mitochondria are main site of redox reactions, it is rational to speculate that toxic ROS is generated by this organelle. However, Dixon's experiments found that mitochondria were not the source of the fatal ROS levels in erastin‐treated cells.4 To find the potential source of lethal ROS, they tested the role of the NADPH oxidase (NOX) family (NOX1–5, DUOX1, 2) in erastin‐induced ferroptosis. The results demonstrated that diphenylene iodonium (DPI, an inhibitor of canonical NOX), GKT137831 (a specific inhibitor of NOX1/4), and 6‐aminonicotinamde (6‐AN, an inhibitor of the NADPH‐generating pentose phosphate pathway) could strongly suppress erastin‐induced ferroptosis in Calu‐1 cells, a cell line that express a high level of NOX1 with RAS mutation.4, 29, 30 These results revealed the other source of lethal ROS, but this phenomenon was limited to several cell lines (Figure 1).3
Figure 1.
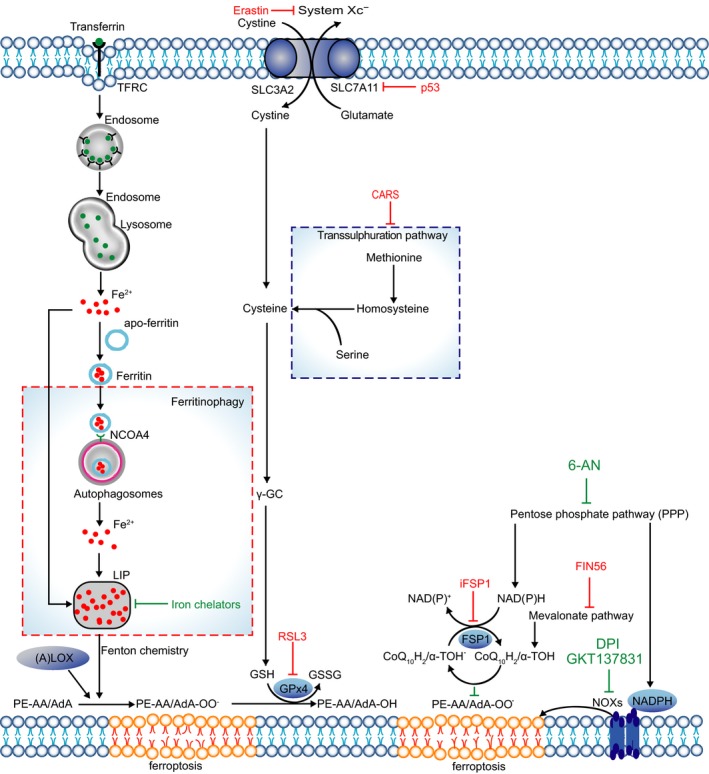
Main mechanisms of ferroptosis. TFRC‐mediated endocytosis promotes the uptake of transferrin. Ferritin can be degraded by autophagy through ferritinophagy. The LOXs, NOXs and increased labile iron result in lipid peroxide. Cysteine can be generated from uptake of cystine via system Xc‐ or transsulphuration pathway. GPx4 catalyses reduction reaction at the cost of GSH. CoQ10H2/α‐TOH traps lipid peroxyl radicals and FSP1 catalyses the regeneration of CoQ10
Molecular dynamics showed that increased lipid peroxidation directly enhances membrane permeability, changes membrane shape and curvature, and promotes increased accessibility to oxidants, eventually causing cell death.31, 32, 33 A recent study demonstrated that erastin and lipid peroxidation could activate the ASK1‐p38 pathway and trigger ASK1‐dependent cell death in a cell type‐specific manner. Further results indicated ASK1 was a downstream of lipid peroxidation in ferroptosis. However, these findings only apply in special cases.34
3.2. The role of glutathione in ferroptosis
System Xc− is important for the exchange of glutamate and cystine at a ratio of 1:1 across the plasma membrane.11 As early as 1989, it was reported that inhibition of system Xc− could increase oxidative stress and mediate the toxicity of glutamate.35 Subsequently, researchers discovered that small molecule compounds, erastin or other system Xc− inhibitors, could induce ferroptosis.4, 13, 36 Indeed, cystine, imported into the cytoplasm, will be catalysed to cysteine and then to GSH (glutathione) via a multistep enzyme‐catalysed reaction. GSH is an important substrate for GPx4 to reduce phospholipid hydroperoxide (PLOOH).37 Of note, another cysteine biosynthesis pathway, called the transsulphuration pathway, exists and generates cysteine via transfer of the sulphur atom of methionine to serine, and this pathway endows some cells with the ability to resist ferroptosis induced by system Xc− inhibitors. Further study revealed that knock‐down of CARS (cysteinyl‐tRNA synthetase) upregulated the transsulphuration pathway by causing cystathionine accumulation and upregulating genes associated with serine biosynthesis and transsulphuration, thereby antagonizing erastin‐induced ferroptosis (Figure 1).38
GPx4 is the articulation point of ferroptosis‐related glutathione metabolism and lipid peroxidation. GPx4 determines the fate of cells because it can directly reduce the lethal phospholipid hydroperoxides.39, 40 Ras‐selective lethal (RSL) small molecules, including RSL3 and erastin, can directly decrease the activity of GPx4 by binding to it or indirectly inhibiting the import of cystine, respectively, thus leading to ferroptosis. Interestingly, GPx4 can be classified into three types according to organelle location, mGPx4 (transported into mitochondria), nGPx4 (localized in nucleoli) and cGPx4 (localized in the cytosol and nucleus), and the three GPx4 types independently regulate local lipid hydroperoxides.40, 41, 42, 43 Mitochondrial GPx4 is associated with apoptosis by inhibiting the release of cytochrome c from mitochondria.40, 44, 45, 46 While cGPx4 is associated with ferroptosis. Importantly, the Dixon team found that cells with depleted mitochondrial DNA could still trigger potent ferroptosis.4 The organelle‐specific GPx4 in cells might provide an explanation for why mitochondria are dispensable for ferroptosis. However, a recent study revealed that mitochondria are involved in cysteine deprivation‐induced ferroptosis via the electron transfer chain (ETC) or TCA cycle. In addition, inhibition of glutaminolysis could lead to a parallel prohibitive effect. These findings indicate that mitochondria play a decisive role in cysteine deprivation‐induced ferroptosis but not in GPx4 inhibition‐induced ferroptosis.47
3.3. Iron metabolism in ferroptosis
Intracellular iron is under delicate regulation to sustain iron homoeostasis. Iron regulatory proteins (IRP1 and IRP2) modulate the cellular Fe2+ concentrations, and various proteins regulate iron import, storage, release and export.48 Most intracellular Fe2+ is stored in ferritin and iron‐containing proteins, and the amount of free Fe2+, also called the cellular labile iron pool (LIP), is very limited.31 In mammalian cells, a portion of the cellular iron can be distributed in mitochondria, the cytosol, the nucleus and lysosomes; although the amount is little, cells are sensitive to iron concentration, and a little fluctuation in concentration can cause a great response.49, 50
How to increase the level of labile iron in cells? First, uptake more iron from the extracellular environment. Dixon identified six high‐confidence genes in erastin‐induced ferroptosis, and IREB2 (iron response element binding protein 2) was one of them. If IREB2 was silenced, the expression of iron uptake, metabolism and storage genes, such as TFRC, ISCU, FTH1, and FTL, would decrease, and the cells exhibited resistance to erastin‐induced ferroptosis.4, 51, 52 Transferrin is an iron carrier protein in serum that can be transported into cells through TFRC‐mediated endocytosis. Silencing of TFRC significantly inhibited conditional serum‐induced ferroptosis.36 In other words, transferrin import is required for ferroptosis. The second way to release iron is autophagic degradation of ferritin, which is called ferritinophagy.53 This process is primarily associated with autophagy. A recent study found that silencing of Atg5 and Atg7 (autophagy‐related genes 5 and 7) decreased intracellular Fe2+ levels and lipid peroxidation and further limited erastin‐induced ferroptosis. Further study confirmed that during autophagy ferritin is degraded to release labile iron and the degradation process is regulated by NCOA4 (nuclear receptor coactivator 4), a selective cargo receptor for specific autophagy of ferritin. Blockage of autophagy or genetic inhibition of NCOA4 can inhibit ferritin degradation and suppress ferroptosis.54, 55, 56
How does increased labile iron result in lipid ROS? Indeed, it primarily depends on Fenton chemistry and LOXs. Fenton chemistry also refers to the Fenton reaction. For this reaction, there must be enough labile iron in this cycle. The increased labile iron portion (LIP) in ferroptosis exactly meets this condition. In this reaction, peroxides and Fe2+ are used to produce oxygen‐centred radicals.57 This was further confirmed by results showing that antioxidants could terminate the reaction.58 If iron chelators, such as deferoxamine (DFO), were applied to downregulate intracellular iron, then iron‐dependent lipid peroxidation formation is also restrained.4 The indirect way for iron to generate lipid ROS is through the catalytic activity of iron‐containing enzymes, and LOXs are the most important enzymes among these (Figure 1).59
3.4. FSP1‐NAD(P)H pathway in ferroptosis
Although GPx4 plays a crucial role in ferroptosis, certain cancer cell lines are resistant to ferroptosis caused by GPx4 inhibitors,60, 61 indicating that there might be additional factors that regulate ferroptosis. To unveil the underlying mechanism, Kirill Bersuker used a synthetic lethal CRISPR–Cas9 screen to distinguish genes in U‐2 OS osteosarcoma cells treated with a GPX4 inhibitor,62 and Sebastian Doll generated a cDNA expression library derived from an MCF7 ferroptosis‐resistant cell line and screened for genes complementing loss of GPX4.63 Coincidentally, they simultaneously found that ferroptosis suppressor protein 1(FSP1) is an important ferroptosis suppressor that parallels GPx4. In FSP1‐NAD(P)H pathway, coenzyme Q10 (CoQ10) can directly trap lipid peroxyl radicals to reduce lipid peroxides, FSP1 catalyses the regeneration of CoQ10 at the cost of NAD(P)H. Sebastian Doll also screened approximately 10,000 compounds to identify FSP1 inhibitors. iFSP1 was identified as a potent FSP1 inhibitor and can trigger ferroptosis in GPX4 knockout cells that overexpress FSP1 (Figure 1).63
4. ROLE OF KRAS IN FERROPTOSIS
RAS proteins play a causal role in human cancer, and the widespread prevalence of RAS mutations (KRAS, NRAS, HRAS) in human cancer has been recognized for many years. That mutations in KRAS alone account for approximately one million deaths per year worldwide have inspired multiple attempts to find KRAS inhibitors.13, 64 These attempts have led to the discovery of RSL. Ferroptosis was coined in this background. RAS‐mutant cancer cells are sensitive to erastin or RSL3‐induced ferroptosis. However, the underlying mechanism is unclear.
Indeed, oncogenic KRAS can regulate metabolic changes and alter cellular signalling, both of which can increase the production of intracellular reactive oxygen species (ROS), but at the same time, it also upregulates antioxidant systems to balance ROS to levels at which they are beneficial for tumour development and progression, while remaining below the threshold that causes cell death.65 KRAS can upregulate ROS through multiple mechanisms. For example, KRAS can regulate HIF‐1α and HIF‐2α target genes to modulate mitochondrial metabolism or regulate the transferrin receptor (TFRC) to modulate mitochondrial respiration and ROS generation.66, 67 Notably, TFRC is a regulator in ferroptosis, as mentioned above. KRAS can also activate Rac1‐NOX4 signalling to alter NADPH oxidase activities.68, 69 The NOX family also plays an important role in lipid peroxidation in ferroptosis. In addition, KRAS controls the regeneration of peroxiredoxins by inducing autophagy‐specific genes 5 and 7 (ATG5, ATG7) and repressing SESN3.70, 71 Autophagy is also important in ferroptosis. With regard to antioxidant systems to sustain redox homoeostasis in the presence of KRAS‐induced ROS production, a recent study showed that superoxide dismutase (Sod), glutathione peroxidase 4 (GPx4) and peroxiredoxin 3 (Prdx3) are indispensable to mitigate RAS‐induced ROS. Pim protein kinases are involved in multiple cellular processes, such as signal transduction, cell cycle progression, cell metabolism and tumour growth. 72, 73, 74 However, a recent study demonstrated that all three isoforms of Pim protein kinases knockout (TKO) reduced the expression of Sod, GPx4 and Prdx3, thus leading to mouse embryo fibroblasts (MEFs) becoming susceptible to killing by K‐RasG12V‐mediated ROS production.72 In contrast, transduction of the TKO cells with c‐Myc permitted KRAS‐induced cell growth by decreasing RAS‐induced ROS accumulation.72 In addition, KRAS also regulates cellular metabolism, especially the metabolism of glutamine and glutaminase,75, 76 which are associated with ferroptosis. In conclusion, small molecules that target system Xc−, GPx4 and autophagy disrupt the redox homoeostasis in RAS‐mutant cancer cells and eventually cause cell death in a manner referred to as ferroptosis. However, as a counterexample, overexpression of RAS (KRAS, HRAS, NRAS) in RMS13 rhabdomyosarcoma cells can protect cells from oxidative stress‐induced ferroptosis.77 Perhaps the effects of the RAF‐MEK‐ERK pathway on ferroptosis vary among cell lineages or with RAS‐mutant protein expression levels.78 This reminds us to consider genetic background in application of ferroptosis to treat cancer (Figure 2).
Figure 2.
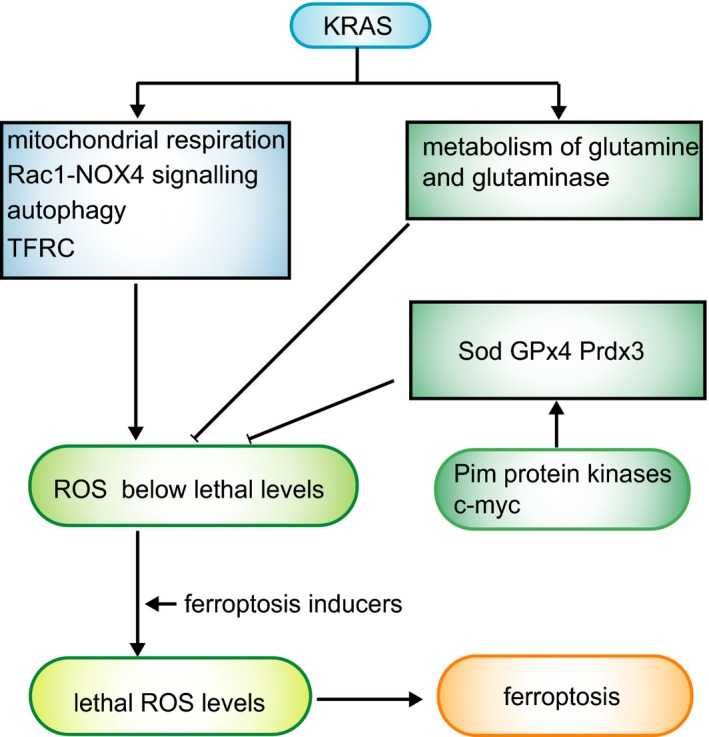
Roles of KRAS and p53 in ferroptosis. KRAS upregulates ROS by mitochondrial respiration, Rac1‐NOX4 signalling, autophagy and TFRC. Meanwhile, KRAS downregulates ROS by mediating metabolism of glutamine and glutaminase. In addition, Sod GPx4 Prdx3 help to eliminate ROS caused by KRAS. Ultimately, ROS is maintained at a moderate high level that below lethal levels. In this case, if cells were treated with ferroptosis inducers, this balance would be disrupted, thus leading to lethal ROS levels and cell death
5. ROLE OF P53 IN FERROPTOSIS
p53 is a crucial tumour suppressor that orchestrates specific cellular responses, such as transient cell cycle arrest, cellular senescence and apoptosis.79 A recent study revealed that p53 is also involved in regulation of ferroptosis. The Le Jiang and colleagues generated a tetracycline‐controlled (tet‐on) p53‐inducible cell line for microarray analysis to identify novel p53 target genes, and SLC7A11 (a component of system Xc−) was identified as a novel p53 target gene. p53 could transcriptionally suppress SLC7A11 and thereby inhibit cystine uptake and sensitize H1299, U2OS and MCF7 cells to ferroptosis. In addition, p533KR, an acetylation‐defective mutant that failed to induce cell cycle arrest, senescence and apoptosis, retained the ability to inhibit SLC7A11 expression and thereby downregulated cystine metabolism and ferroptosis upon ROS‐induced stress.80 Another study reported that p53 repressed the expression of SLC7A11 during erastin treatment by decreasing H2Bub1 occupancy on the SLC7A11 gene regulatory region.81 However, p53 regulation of SLC7A11 was cell line dependent. DPP4 (dipeptidyl‐peptidase‐4) can bind to NOX1, leading to lipid ROS and subsequent ferroptosis in p53‐deficient human colorectal cancer (CRC) cells. However, in p53 wild‐type cells, p53 induces DPP4 localization to the nucleus where DPP4 binds p53 to promote the expression of SLC7A11, which inhibits erastin‐induced ferroptosis, indicating that p53 has a pro‐survival function.82 Wild‐type p53 has been reported to delay the onset of ferroptosis in response to cystine deprivation via the p53‐p21 axis to mitigate the depletion of intracellular glutathione and reduce accumulation of lethal lipid ROS.83 In addition, certain mutant forms of p53 do not suppress the expression of SLC7A11. For example, a p53 that harboured mutations in the N‐terminal domain of the gene did not downregulate SLC7A11.84, 85
Conversely, p53 promotes ROS accumulation via regulation of cytochrome c oxidase 2 (SCO2), glucose transporter (GLUT)1, GLUT4 and glutaminase 2 (GLS2).86, 87, 88, 89 p53 can upregulate arachidonate 15‐lipoxygenase (ALOX15) via spermidine/spermine N1‐acetyltransferase 1 (SAT1), causing lipid peroxidation.86 Bo Chu and team performed an RNAi‐mediated loss‐of‐function screen to identify which lipoxygenases (ALOXE3, ALOX5, ALOX12, ALOX12B, ALOX15 and ALOX15B) affect p53‐dependent ferroptosis. The results demonstrated that inactivation of ALOX12 diminished p53‐mediated ferroptosis induced by ROS stress. SlC7A11 could bind to ALOX12 and thus inhibit the lipoxygenase activity of ALOX12. p53 can upregulate the enzymatic activity of ALOX12 by repressing SLC7A11 expression.90 In these conditions, p53 acts as a positive regulator of ferroptosis. Beyond doubt, this complicated genetic background of p53 will bring more challenge for cancer treatment.
6. PROGRESS IN FERROPTOSIS APPLICATION IN CANCER THERAPY
In the process of exploring how to kill RAS‐mutant cancer cells, ferroptosis was accidentally discovered. The ultimate purpose of elucidating the mechanism underlying ferroptosis is to obtain better cancer treatment options. Because, based on the molecular regulation mechanisms, we can specifically target the key regulators (such as system Xc−, GPx4, autophagy and iron) to trigger ferroptosis. Here, we list some new findings in the utilization of ferroptosis activators as weapons to treat cancer.
6.1. Death propagation
Although the forms of cell death vary in mechanism, from the perspective point of cellular population, the death of an individual cell can exert three types of effects on the population: first, a neutral effect on surrounding cells or a benefit that counteracts the harm; second, a benefit for neighbouring cells; and third, killing of neighbouring cells via a bystander effect.8 Apoptosis was initially identified as an active form of cell demise intended to degrade the cargos of doomed cells but that does not affect neighbouring cells (cell‐autonomous suicide); however, a recent study showed that these eliminable cells can trigger various signals that have a positive or negative influence on surrounding cells and tissues.91 Entosis is a process in which one cell (winner) engulfs another cell (loser); the loser is engulfed, or cannibalized while alive, and subsequently, undergoes cell death. The dead cells benefit the living cells.92 In contrast, ferroptosis can propagate among cells in a wave‐like manner, exhibiting a potent killing effect on neighbouring cells.8, 93, 94 Ultrasmall nanoparticle‐induced ferroptosis can cause rehabilitation of xenograft tumours, demonstrating that ferroptosis has potent tumour suppressive ability.93 The underlying mechanism of death propagation is unclear; however, the following factors might be involved. Large regions of dead cells that form in the interior of cancer lesions might increase the inner pressure that restricts key nutrients diffused from blood vessels and lead to energy deprivation. In this condition, necrosis of vast cell populations may occur secondary to a small portion of apoptotic cell death in the interior.8, 95, 96, 97 However, whether ferroptosis induces the demise of surrounding cells in this manner is unclear. Intriguingly, there some collective features shared by radiation and ferroptosis; for instance, they both destroy cells through lipid peroxidation that is regulated by glutathione.98, 99 Moreover, they both promote the expression of cyclooxygenase‐2 (COX‐2), and COX‐2 plays a vital role in the radiation‐induced bystander effect.8, 100, 101 Therefore, is there a mechanism in ferroptosis similar to the radiation‐induced bystander effect?
However, although we are glad that the propagation of cell death may contribute to cancer therapy, another study reported that a high cell density (>80% confluency) makes cells less sensitive to erastin‐induced ferroptosis via the Hippo pathway that regulates TAZ‐EMP1‐NOX4 axis (Figure 3).7 In addition, recent research has demonstrated that cell–cell contacts inhibit erastin‐induced ferroptosis and t‐BuOOH‐induced ferroptosis by decreasing basal and lipid peroxidation. Contact between cells can also antagonize ROS‐induced cell death by inhibiting the dissipation of MMPs and the increase in DNA DSBs.102 These findings seem to be incompatible with death propagation, but the underlying explanation for these phenomena may lie in the differences between in vivo and in vitro conditions.
Figure 3.
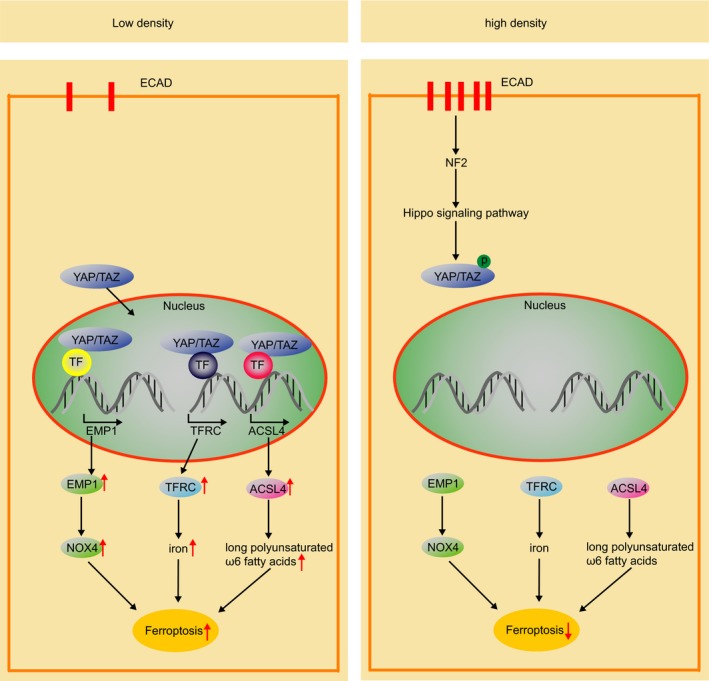
Hippo signalling pathway. Left, cells grow in low density; the Hippo signalling pathway is closed. YAP/TAZ enter nucleus to promote expression of EMP1, TFRC and ACSL4, thus cells are sensitive to ferroptosis. Right, cells grow in high density; the Hippo signalling pathway is activated. Phosphorylated YAP/TAZ is impeded in cytoplasm, thus cannot promote the expression of EMP1, TFRC and ACSL4 and cells exhibited resistance to ferroptosis
6.2. Molecular carrier
Although RSL has been studied for many years, they are not suited for direct use in clinical applications due to their poor water solubility, renal toxicity and other toxic side effects. Thus, enhancing the specificity and delivery efficiency of these drugs is a promising research direction. Recently, exosomes and nanosystems have been found to function well in this aspect.
Exosomes are nano‐sized extracellular vesicles (40‐150 nm) with a membrane lipid bilayer that are released by all living cells and efficiently enter other cells to transfer substances and signalling information.103, 104 Recently, a great number of studies have found that exosomes have better advantages in drug delivery due to their low immunogenicity, high biocompatibility and high efficiency.9 One team constructed an erastin‐loaded exosome formulation labelled with folate (FA) (erastin@FA‐exo) to target triple‐negative breast cancer (TNBC) cells with folate receptor overexpression. Compared with erastin@exo and free erastin, erastin@FA‐exo increased the uptake efficiency of erastin in MDA‐MB‐231 cells and promoted ferroptosis by inhibiting system Xc‐.9
In oestrogen receptor‐positive (ER+) breast carcinoma, mutation of the PI(3)K/Akt signalling pathway is frequent and causes excess intracellular glutathione (GSH) biosynthesis, leading to oxidative stress resistance and activation of tumour cell proliferation and infiltration.105 Researchers have developed a nanosystem called drug‐organics‐inorganics self‐assembled nanosystem (DFTA). In this nanosystem, doxorubicin (DOX) was used as a chemotherapeutic agent, ferric chloride (FeCl3) as a ferroptosis inducer and tannic acid (TA) as an activator of a superoxide dismutase (SOD)‐like reaction in an intracellular cascade. Photothermal excitation efficiently triggered drug release from the DFTA nanosystem. The results showed that combined use of the DFTA nanosystem and laser excitation significantly reduced the level of intracellular GSH through a series of reactions. This effect was further confirmed by evaluation of the antitumour efficiency in vivo, which revealed that the tumour inhibition ratio of in the DFTA nanosystem + laser group was as high as 93.38%, even though the dose of iron and DOX was reduced by approximately 9 and 1.5 times, respectively. In a word, the nanosystem based on triple combination therapy with chemotherapy, ferroptosis and PT greatly improved the treatment specificity, reduced the toxicity and enhanced the delivery efficiency.106
One study reported that ultrasmall (<10 nm in diameter) silica nanoparticles can trigger ferroptosis in starved cancer cells and cancer‐bearing mice. This nanosystem was called αMSH‐PEG‐C' dots and was found to be imported into lysosomes. The surface of this nanosystem was functionalized with αMSH (alpha‐melanocyte stimulating hormone), which recognizes a special surface receptor expressed on malignant melanoma cells (melanocortin‐1 receptor, MC1‐R), thereby orienting this system specifically to malignant melanoma cells. Treatment of amino acid‐starved cells with high concentrations of these nanoparticles induced ferroptosis, and the cell death propagated in a wave‐like manner.93
Another nanosystem called nanoparticle ferritin‐bound erastin and rapamycin (NFER) is an Abraxane‐inspired nanodrug. When NFER is engulfed by cancer cells, the nanosystem can release erastin and rapamycin, inhibiting system Xc‐ and autophagy, respectively, and eventually causing cell death via ferroptosis.10 In addition, a metal−organic network (MON) encapsulating p53 plasmid (MON‐p53) and combined with ferric ions can also induce ferroptosis by releasing iron and p53 plasmid to inhibit system Xc‐. In this study, MON‐p53 triggered a “bystander effect” to sensitize cancer cells to MON‐p53‐induced ferroptosis. In tumour‐bearing mice, MON‐p53 treatment both suppressed tumour growth and prolonged the mouse life span.107
To specifically release iron to tumour tissues, one team developed a nanoprobe that consisting of upconversion luminescence (UCL) nanoparticles as a core and a coordinated Fe3+/gallic acid complex as a shell. This nanoprobe can release Fe3+ only in tumour lesions in response to the lightly acidic tumour pH and thus triggered ferroptosis.108 Magnetosomes can also be iron carriers. Another team engineered a magnetosome with an Fe3O4 magnetic nanocluster (NC) as the core and pre‐engineered leucocyte membranes as the cloak. In addition, TGF‐β inhibitor (Ti) was loaded inside the membrane and PD‐1 antibody (Pa) was anchored on the membrane surface. In this way, the magnetosome induced ferroptosis and regulated immunomodulation to treat cancer (Table 1).109
Table 1.
List of carriers
| Carrier | Molecule | Target | Other components | Cell lines | Ref |
|---|---|---|---|---|---|
| Exosome | Erastin | System Xc‐ | Folate (FA) | Triple‐negative breast cancer cells | 9 |
| DFTA | FeCl3 | LIP |
DOX TA |
ER+.breast carcinoma | 106 |
| NFER | Erastin rapamycin | System Xc‐ autophagy | ‐ | Mouse breast cancer cell line | 10 |
| MON | Ferric ions p53‐plasmid |
LIP SLC7A11 |
‐ | HT1080 | 107 |
| UCNP | Fe3+ | LIP | Gallic acid | LS180 cells | 108 |
| Magnetosome | Fe3O4 | LIP |
TGF‐β inhibitor PD‐1 antibody |
B16F10 | 109 |
| Sal–AuNPs | Salinomycin | Mitochondrial | ‐ | Breast cancer stem cells (BCSCs) | 124 |
| CaP‐RSL3 |
Fe3+ RSL3 |
LIP GPx4 |
Ascorbate (Asc) | Murine.breast cancer cells (4 T1) | 134 |
| Ce6‐erastin | Erastin | System Xc‐ | Chlorin e6 (Ce6) | OTSCC | 135 |
6.3. Cancer stem cells
Cancer stem cells (CSCs) have self‐renewal and infinite proliferation capability and are vital to sustaining cancer proliferation, drug resistance, metastasis and recurrence. Cancer stem cells are also important therapeutic targets.110 The stemness of CSCs can be verified by detecting the expression of typical surface markers (CD133, CD44, CD24, CD38) and pluripotency factors (Sox2, Oct4, Nanog) and by their capacity to form spheres. William's team found a new compound that can inhibit system Xc‐ to induce ferroptosis in cancer stem cells.111 Different from general cancer cells, increased iron content in CSCs is a key feature in several types of tumours. Correspondingly, iron chelation can inhibit the stemness of CSCs, whereas iron supplementation can have the reverse effect. In other words, iron may function as a protective factor in CSCs.112, 113, 114, 115, 116, 117 However, iron deprivation has also been found to halt cell proliferation in mouse‐induced pluripotent cells and inhibit the expression of stemness markers.117 These findings indicate that iron may have a dual function in CSCs. Studies have shown that levels of both the iron storage protein ferritin and the iron exporter ferroportin (FPN) are decreased in cholangiocarcinoma CSCs and ovarian CSCs.112, 118 In addition, low H ferritin levels and high TFRC expression were detected in cholangiocarcinoma cells when grown in monolayers; however, when the same cell lines formed tumour spheres, there were high H ferritin levels and low TFR1 expression levels. This indicates that the labile iron that is generated from the degradation of H ferritin or mediated by TFRC is involved in the capacity to form spheres.118 H ferritin knock‐down inhibited glioblastoma CSC growth and impaired a stem‐like phenotype in breast cancer cells.115, 119 These studies showed that CSCs are iron‐rich and iron‐dependent. Indeed, sphere‐forming cholangiocarcinoma cells, despite higher levels of ROS and iron, show less susceptible to erastin‐induced ferroptosis than their counterparts grown in a monolayer. However, ovarian CSCs exhibited higher sensitivity to ferroptosis than non‐tumorigenic ovarian stem cells.118 These phenomena might be explained by the dual role of ferritin: in slow‐growing CSCs, iron is not required for proliferation and can be stored in ferritin, thereby preventing the generation of lipid peroxidation and explaining the resistance of cholangiocarcinoma cells with high ferritin content to erastin. Correspondingly, if ferritin degradation is initiated, ferritin may also represent a source of iron, thus causing ferroptosis.117
Salinomycin (Sal) is a polyether ionophore antibiotic that has been shown to kill CSCs in different types of human cancers.120 On the other hand, gold nanoparticles (AuNPs) are excellent drug carriers with good biocompatibility, easy synthesis and good drug functionalization capability.121, 122, 123 Studies have found that salinomycin‐loaded gold nanoparticles can induce ferroptosis in CSCs.124
6.4. Tumour microenvironment
The microenvironment is one of the most important features of tumour and plays a critical role in many aspects of tumorigenesis. Currently, the exploration of ferroptosis is primarily focused on vitro cell lines or xenogeneic tumour cell transplantation. Thus, the role of the tumour microenvironment in ferroptosis is being neglected. Iron is the key factor in ferroptosis. An aberrantly increased labile iron portion can originate from two main sources as mentioned above: import of transferrin and degradation of ferritin via ferritinophagy. TFR mediates the import of transferrin from the extracellular environment, while FPN1 is the only ferrous iron exporter. FPN1 is primarily expressed in iron‐recycling macrophage populations.125, 126 In the extracellular milieu, ferric iron is bound by apo‐transferrin (TF), which is the key iron transport protein in plasma. Hepcidin is an iron regulatory hormone that acts in a negative feedback manner through binding to FPN1.127, 128 Macrophages play an important role in sustaining iron homoeostasis. Although the mechanism of iron homoeostasis in the extracellular environment is clear, the changes in response to ferroptosis are unclear and whether these changes affect the efficiency of therapy remains to be unveiled.
In solid tumours, hypoxia and acidity characterize the microenvironment of cancer cells. Numerous iron metabolism‐associated genes, including iron transport (TFRC and SLC11A2) and iron storage (FTH, encoding ferritin heavy chain) genes, are under the control by hypoxia‐responsive elements (HRE).129, 130, 131 In addition, a recent study showed that hypoxia can protect cells from ferroptosis by upregulating the expression of carbonic anhydrase 9 (CA9). Carbonic anhydrases (CAs) represent a superfamily of metalloenzymes that equilibrate the reactions among CO2, bicarbonate and H+.132 Under hypoxia conditions, increased CA9 can enhance the formation of FTL and FTH, leading to a corresponding decrease in the protein levels of TFRC and FPN‐1 and in the labile iron portion, thus inhibiting iron‐dependent lipid peroxidation.133
However, despite the protective function against ferroptosis, two new therapy strategies target the acidic and hypoxic environment in tumour tissues. One study reported that ascorbate (Asc) can selectively kill cancer cells via hydrogen peroxide (H2O2) accumulation only in tumour extracellular fluids. The team synergized the action of Asc with a lipid‐coated calcium phosphate (CaP) hybrid nanocarrier that can concurrently load polar Fe3+ and non‐polar RSL3. Interestingly, the hybrid nanocarrier showed accelerated drug release under acidic conditions (pH 5.0). In addition, this method produced significantly elevated levels of hydroxyl radicals, lipid peroxides and depleted glutathione under hypoxia, which was accompanied by strong cytotoxicity. Moreover, this system also functioned well in a tumour‐bearing xenograft mouse model.134 On the other hand, the non‐invasive nature of photodynamic therapy (PDT) is an increasingly important therapeutic method with spatiotemporal selectivity for the treatment of cancer and thus enables preservation of organ function in cancer patients. However, hypoxia limits the supply of O2 that is required for PDT. Considering that Fenton reaction in ferroptosis will produce reactive oxygen species (ROS) and sustainably supply O2, another team constructed a Ce6‐erastin nanoparticle system to combine ferroptosis with PDT. Their results showed that the Ce6‐erastin nanoparticles exhibited low cytotoxicity to normal tissues but led to the death of cancer cells and achieved satisfactory antitumour effects in a xenograft tumour mouse model upon irradiation.135
7. CONCLUSION AND PERSPECTIVE
Although the main mechanisms underlying ferroptosis have been revealed, there are still many uncertain factors affecting the destiny of cancer cells, such as genetic background, cell type and cell density. Moreover, the situation is more complicated if extended to cancer therapy. For instance, if the expression of cysteinyl‐tRNA synthetase (CARS) is low in cancer cells, the transsulphuration pathway would be activated due to the accumulation of cystathionine and expression of genes associated with serine biosynthesis and transsulphuration, and in this case, using erastin alone will not achieve the expected effect.38 In addition, cell density is a negative regulator of ferroptosis. When cells grow at high density, E‐cadherin (ECAD) expression is increased, and ferroptosis is suppressed in epithelial cells. However, cancer cells with mesenchymal or metastatic properties, including a low level of ECAD expression, are highly sensitive to ferroptosis.136, 137 Jiao Wu's team found that ECAD expression increased as cell density increased and then activated intracellular NF2 and the Hippo signalling pathway to mediate the density‐dependent resistance to ferroptosis. Regarding the mechanism, NF2 can inhibit the expression of TFRC and acyl‐CoA synthetase long‐chain family member 4 (ACSL4) by suppressing the activity of YAP, and both TFRC and ACSL4 are crucial mediators of ferroptosis. This finding has clear implications because of the cancer therapies. Because cadherin–NF2–Hippo signalling axis is frequently mutated in cancer, these mutations endow cancer cells with sensitivity to ferroptosis (Figure 3).137 Acyl‐CoA synthetase long‐chain family member 4 (Acsl4) could potentiate the kill effect of GPx4 inhibitor (RSL3) via enriching cellular membranes with long polyunsaturated ω6 fatty acids. Moreover, Acsl4 could predict cancer cells’ sensitivity to ferroptosis for therapeutic approach due to its preferential expression in a panel of basal‐like breast cancer cell lines.138 Concerning how to precisely induce ferroptosis in vivo, the key regulators of ferroptosis, which have been uncovered, provide vital therapeutic targets, such as targeting system Xc−, GPx4, autophagy and iron. Researchers have found a number of small molecules that can induce ferroptosis by targeting these therapeutic targets. However, these compounds are not currently suited for direct use in vivo, but further application of cutting‐edge technology may bring new options for cancer treatment based on ferroptosis.
CONFLICT OF INTEREST
The authors have declared no conflicting interests.
AUTHOR CONTRIBUTION
YQ conceived of the presented idea; ZY, WL and QZ wrote the manuscript; QH, ML, QS, ZZ, GF, WX and SJ searched the literature; XX and XY supervised the project.
Ye Z, Liu W, Zhuo Q, et al. Ferroptosis: Final destination for cancer?. Cell Prolif. 2020;53:e12761 10.1111/cpr.12761
Ye, Liu and Zhuo contributed equally to this work.
Funding information
This work was supported by the National Science Foundation of China (grant number 81871950, 81702871 and 81602085).
Contributor Information
Xianjun Yu, Email: yuxianjun@fudanpci.org.
Yi Qin, Email: qinyi@fudanpci.org.
Xiaowu Xu, Email: xuxiaowu@fudanpci.org.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Green DR. The coming decade of cell death research: five riddles. Cell. 2019;177:1094‐1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Green DR, Victor B. The pantheon of the fallen: why are there so many forms of cell death? Trends Cell Biol. 2012;22:555‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell. 2012;149:1060‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xu T, Ding W, Ji X, et al. Molecular mechanisms of ferroptosis and its role in cancer therapy. J Cell Mol Med. 2019;23:4900‐4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yu H, Guo P, Xie X, Wang Y, Chen G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med. 2017;21:648‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang W‐H, Ding C‐KC, Sun T, et al. The hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep. 2019;28:2501‐2508.e2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Riegman M, Bradbury MS, Overholtzer M. Population dynamics in cell death: mechanisms of propagation. Trends Cancer. 2019;5:558‐568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yu M, Gai C, Li Z, et al. Targeted exosomes‐encapsulated erastin induced ferroptosis in the triple negative breast cancer cells. Cancer Sci. 2019;110:3173‐3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li Y, Wang X, Yan J, et al. Nanoparticle ferritin‐bound erastin and rapamycin: a nanodrug combining autophagy and ferroptosis for anticancer therapy. Biomater Sci. 2019;7:3779‐3787. [DOI] [PubMed] [Google Scholar]
- 11. Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem. 1986;261:2256‐2263. [PubMed] [Google Scholar]
- 12. Sato H, Tamba M, Okuno S, et al. Distribution of cystine/glutamate exchange transporter, system x(c)‐, in the mouse brain. J Neurosci. 2002;22:8028‐8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype‐selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285‐296. [DOI] [PubMed] [Google Scholar]
- 14. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron‐dependent, nonapoptotic cell death in oncogenic‐RAS‐harboring cancer cells. Chem Biol. 2008;15:234‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seiler A, Schneider M, Förster H, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15‐lipoxygenase dependent‐ and AIF‐mediated cell death. Cell Metab. 2008;8:237‐248. [DOI] [PubMed] [Google Scholar]
- 16. Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165‐176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wilson DF. Oxidative phosphorylation: regulation and role in cellular and tissue metabolism. J Physiol. 2017;595:7023‐7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Galaris D, Pantopoulos K. Oxidative stress and iron homeostasis: mechanistic and health aspects. Crit Rev Clin Lab Sci. 2008;45:1‐23. [DOI] [PubMed] [Google Scholar]
- 19. Galaris D, Barbouti A, Pantopoulos K. Iron homeostasis and oxidative stress: an intimate relationship. Biochim Biophys Acta Mol Cell Res. 2019;1866:118535. [DOI] [PubMed] [Google Scholar]
- 20. Galadari S, Rahman A, Pallichankandy S, Thayyullathil F. Reactive oxygen species and cancer paradox: to promote or to suppress? Free Radic Biol Med. 2017;104:144‐164. [DOI] [PubMed] [Google Scholar]
- 21. Doll S, Conrad M. Iron and ferroptosis: a still ill‐defined liaison. IUBMB Life. 2017;69:423‐434. [DOI] [PubMed] [Google Scholar]
- 22. Pryor WA, Porter NA. Suggested mechanisms for the production of 4‐hydroxy‐2‐nonenal from the autoxidation of polyunsaturated fatty acids. Free Radic Biol Med. 1990;8:541‐543. [DOI] [PubMed] [Google Scholar]
- 23. Davies SS, Guo L. Lipid peroxidation generates biologically active phospholipids including oxidatively N‐modified phospholipids. Chem Phys Lipids. 2014;181:1‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maiorino M, Conrad M, Ursini F. GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid Redox Signal. 2018;29:61‐74. [DOI] [PubMed] [Google Scholar]
- 25. Haeggstrom JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev. 2011;111:5866‐5898. [DOI] [PubMed] [Google Scholar]
- 26. Friedmann Angeli JP, Schneider M, Proneth B, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang WS, Kim KJ, Gaschler MM, et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. 2016;113:E4966‐4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xie Y, Song X, Sun X, et al. Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem Biophys Res Commun. 2016;473:775‐780. [DOI] [PubMed] [Google Scholar]
- 29. Kamata T. Roles of Nox1 and other Nox isoforms in cancer development. Cancer Sci. 2009;100:1382‐1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Laleu B, Gaggini F, Orchard M, et al. First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J Med Chem. 2010;53:7715‐7730. [DOI] [PubMed] [Google Scholar]
- 31. Nakamura T, Naguro I, Ichijo H. Iron homeostasis and iron‐regulated ROS in cell death, senescence and human diseases. Biochim Biophys Acta Gen Subj. 2019;1863:1398‐1409. [DOI] [PubMed] [Google Scholar]
- 32. Feng H, Stockwell BR. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018;16:e2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Agmon E, Solon J, Bassereau P, Stockwell BR. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep. 2018;8:5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hattori K, Ishikawa H, Sakauchi C, Takayanagi S, Naguro I, Ichijo H. Cold stress‐induced ferroptosis involves the ASK1‐p38 pathway. EMBO Rep. 2017;18:2067‐2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547‐1558. [DOI] [PubMed] [Google Scholar]
- 36. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ingold I, Berndt C, Schmitt S, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide‐induced ferroptosis. Cell. 2018;172:409‐422.e21. [DOI] [PubMed] [Google Scholar]
- 38. Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl‐tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23:270‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med. 2003;34:145‐169. [DOI] [PubMed] [Google Scholar]
- 40. Imai H, Matsuoka M, Kumagai T, Sakamoto T, Koumura T. Lipid peroxidation‐dependent cell death regulated by GPx4 and ferroptosis. Curr Top Microbiol Immunol. 2017;403:143‐170. [DOI] [PubMed] [Google Scholar]
- 41. Arai M, Imai H, Sumi D, et al. Import into mitochondria of phospholipid hydroperoxide glutathione peroxidase requires a leader sequence. Biochem Biophys Res Commun. 1996;227:433‐439. [DOI] [PubMed] [Google Scholar]
- 42. Nakamura T, Imai H, Tsunashima N, Nakagawa Y. Molecular cloning and functional expression of nucleolar phospholipid hydroperoxide glutathione peroxidase in mammalian cells. Biochem Biophys Res Commun. 2003;311:139‐148. [DOI] [PubMed] [Google Scholar]
- 43. Imai H, Sumi D, Hanamoto A, Arai M, Sugiyama A. Molecular cloning and functional expression of a cDNA for rat phospholipid hydroperoxide glutathione peroxidase: 3'‐untranslated region of the gene is necessary for functional expression. J Biochem. 1995;118:1061‐1067. [DOI] [PubMed] [Google Scholar]
- 44. Arai M, Imai H, Koumura T, et al. Mitochondrial phospholipid hydroperoxide glutathione peroxidase plays a major role in preventing oxidative injury to cells. J Biol Chem. 1999;274:4924‐4933. [DOI] [PubMed] [Google Scholar]
- 45. Nomura K, Imai H, Koumura T, Arai M, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase suppresses apoptosis mediated by a mitochondrial death pathway. J Biol Chem. 1999;274:29294‐29302. [DOI] [PubMed] [Google Scholar]
- 46. Korytowski W, Basova LV, Pilat A, Kernstock RM, Girotti AW. Permeabilization of the mitochondrial outer membrane by Bax/truncated Bid (tBid) proteins as sensitized by cardiolipin hydroperoxide translocation: mechanistic implications for the intrinsic pathway of oxidative apoptosis. J Biol Chem. 2011;286:26334‐26343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gao M, Yi J, Zhu J, et al. Role of mitochondria in ferroptosis. Mol Cell. 2019;73:354‐363.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Andrews NC, Schmidt PJ. Iron homeostasis. Annu Rev Physiol. 2007;69:69‐85. [DOI] [PubMed] [Google Scholar]
- 49. Zabrodina LV, Gaponova EB, Gorokhova LG, et al. Indices of lipid metabolism and peroxidation in normal newborn infants. Lab Delo. 1991;9:16‐18. [PubMed] [Google Scholar]
- 50. Petrat F, de Groot H, Rauen U. Subcellular distribution of chelatable iron: a laser scanning microscopic study in isolated hepatocytes and liver endothelial cells. Biochem J. 2001;356:61‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Salahudeen AA, Thompson JW, Ruiz JC, et al. An E3 ligase possessing an iron‐responsive hemerythrin domain is a regulator of iron homeostasis. Science. 2009;326:722‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vashisht AA, Zumbrennen KB, Huang X, et al. Control of iron homeostasis by an iron‐regulated ubiquitin ligase. Science. 2009;326:718‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tang M, Chen Z, Wu D, Chen L. Ferritinophagy/ferroptosis: Iron‐related newcomers in human diseases. J Cell Physiol. 2018;233:9179‐9190. [DOI] [PubMed] [Google Scholar]
- 54. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021‐1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425‐1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Torii S, Shintoku R, Kubota C, et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J. 2016;473:769‐777. [DOI] [PubMed] [Google Scholar]
- 57. Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett. 1995;82–83:969‐974. [DOI] [PubMed] [Google Scholar]
- 58. Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482:419‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shah R, Shchepinov MS, Pratt DA. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent Sci. 2018;4:387‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kagan VE, Mao G, Qu F, et al. Arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bersuker K, Hendricks JM, Li Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature, 2019;575(7784), 688‐692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione‐independent ferroptosis suppressor. Nature, 2019;575(7784):693‐698. [DOI] [PubMed] [Google Scholar]
- 64. Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170:17‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Storz P. KRas, ROS and the initiation of pancreatic cancer. Small GTPases. 2017;8:38‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chun SY, Johnson C, Washburn JG, Cruz‐Correa MR, Dang DT, Dang LH. Oncogenic KRAS modulates mitochondrial metabolism in human colon cancer cells by inducing HIF‐1alpha and HIF‐2alpha target genes. Mol Cancer. 2010;9:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jeong SM, Hwang S, Seong RH. Transferrin receptor regulates pancreatic cancer growth by modulating mitochondrial respiration and ROS generation. Biochem Biophys Res Commun. 2016;471:373‐379. [DOI] [PubMed] [Google Scholar]
- 68. Kong B, Qia C, Erkan M, Kleeff J, Michalski CW. Overview on how oncogenic Kras promotes pancreatic carcinogenesis by inducing low intracellular ROS levels. Front Physiol. 2013;4:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ogrunc M, Di Micco R, Liontos M, et al. Oncogene‐induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014;21:998‐1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kim MJ, Woo S‐J, Yoon C‐H, et al. Involvement of autophagy in oncogenic K‐Ras‐induced malignant cell transformation. J Biol Chem. 2011;286:12924‐12932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zamkova M, Khromova N, Kopnin BP, Kopnin P. Ras‐induced ROS upregulation affecting cell proliferation is connected with cell type‐specific alterations of HSF1/SESN3/p21Cip1/WAF1 pathways. Cell Cycle. 2013;12:826‐836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Song JH, An N, Chatterjee S, et al. Deletion of Pim kinases elevates the cellular levels of reactive oxygen species and sensitizes to K‐Ras‐induced cell killing. Oncogene. 2015;34:3728‐3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Verbeek S, van Lohuizen M, van der Valk M, Domen J, Kraal G, Berns A. Mice bearing the E mu‐myc and E mu‐pim‐1 transgenes develop pre‐B‐cell leukemia prenatally. Mol Cell Biol. 1991;11:1176‐1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. van Lohuizen M, Verbeek S, Krimpenfort P, et al. Predisposition to lymphomagenesis in pim‐1 transgenic mice: cooperation with c‐myc and N‐myc in murine leukemia virus‐induced tumors. Cell. 1989;56:673‐682. [DOI] [PubMed] [Google Scholar]
- 75. Gaglio D, Metallo CM, Gameiro PA, et al. Oncogenic K‐Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 2011;7:523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Son J, Lyssiotis CA, Ying H,et al. Glutamine supports pancreatic cancer growth through a KRAS‐regulated metabolic pathway. Nature. 2013;496:101‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schott C, Graab U, Cuvelier N, Hahn H, Fulda S. Oncogenic RAS mutants confer resistance of RMS13 rhabdomyosarcoma cells to oxidative stress‐induced ferroptotic cell death. Front Oncol. 2015;5:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. 2016;73:2195‐2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53‐mediated tumour suppression. Nat Rev Cancer. 2014;14:359‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jiang L, Kon N, Li T, et al. Ferroptosis as a p53‐mediated activity during tumour suppression. Nature. 2015;520:57‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang Y, Yang L, Zhang X, et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 2019;20:e47563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Xie Y, Zhu S, Song X,et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20:1692‐1704. [DOI] [PubMed] [Google Scholar]
- 83. Tarangelo A, Magtanong L, Bieging‐Rolett KT, et al. p53 Suppresses metabolic stress‐induced ferroptosis in cancer cells. Cell Rep. 2018;22:569‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Jiang L, Hickman JH, Wang SJ, Gu W. Dynamic roles of p53‐mediated metabolic activities in ROS‐induced stress responses. Cell Cycle. 2015;14:2881‐2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Latunde‐Dada GO. Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta Gen Subj. 2017;1861:1893‐1900. [DOI] [PubMed] [Google Scholar]
- 86. Zhang W, Gai C, Ding D, Wang F, Li W. Targeted p53 on small‐molecules‐induced ferroptosis in cancers. Front Oncol. 2018;8:507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Matoba S, Kang JG, Patino WD, et al. p53 regulates mitochondrial respiration. Science. 2006;312:1650‐1653. [DOI] [PubMed] [Google Scholar]
- 88. Bensaad K, Tsuruta A, Selak MA, et al. TIGAR, a p53‐inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107‐120. [DOI] [PubMed] [Google Scholar]
- 89. Schwartzenberg‐Bar‐Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down‐regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004;64:2627‐2633. [DOI] [PubMed] [Google Scholar]
- 90. Chu BO, Kon N, Chen D, et al. ALOX12 Is required for p53‐mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21(5):579‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Perez‐Garijo A, Steller H. Spreading the word: non‐autonomous effects of apoptosis during development, regeneration and disease. Development. 2015;142:3253‐3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sun Q, Luo T, Ren Y, et al. Competition between human cells by entosis. Cell Res. 2014;24:1299‐1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kim SE, Zhang L, Ma K, et al. Ultrasmall nanoparticles induce ferroptosis in nutrient‐deprived cancer cells and suppress tumour growth. Nat Nanotechnol. 2016;11:977‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Linkermann A, Skouta R, Himmerkus N, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA. 2014;111:16836‐16841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lipponen P. Apoptosis in breast cancer: relationship with other pathological parameters. Endocr Relat Cancer. 1999;6:13‐16. [DOI] [PubMed] [Google Scholar]
- 96. Moinfar F, Mannion C, Man YG, Tavassoli FA. Mammary, "comedo"‐DCIS: apoptosis, oncosis, and necrosis: an electron microscopic examination of 8 cases. Ultrastruct Pathol. 2000;24:135‐144. [DOI] [PubMed] [Google Scholar]
- 97. Steinbach JP, Wolburg H, Klumpp A, Probst H, Weller M. Hypoxia‐induced cell death in human malignant glioma cells: energy deprivation promotes decoupling of mitochondrial cytochrome c release from caspase processing and necrotic cell death. Cell Death Differ. 2003;10:823‐832. [DOI] [PubMed] [Google Scholar]
- 98. Azzam EI, de Toledo SM, Little JB. Oxidative metabolism, gap junctions and the ionizing radiation‐induced bystander effect. Oncogene. 2003;22:7050‐7057. [DOI] [PubMed] [Google Scholar]
- 99. Narayanan PK, Goodwin EH, Lehnert BE. Alpha particles initiate biological production of superoxide anions and hydrogen peroxide in human cells. Cancer Res. 1997;57:3963‐3971. [PubMed] [Google Scholar]
- 100. Zhou H, Ivanov VN, Gillespie J, et al. Mechanism of radiation‐induced bystander effect: role of the cyclooxygenase‐2 signaling pathway. Proc Natl Acad Sci USA. 2005;102:14641‐14646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chai Y, Lam RK, Calaf GM, Zhou H, Amundson S, Hei TK. Radiation‐induced non‐targeted response in vivo: role of the TGFbeta‐TGFBR1‐COX‐2 signalling pathway. Br J Cancer. 2013;108:1106‐1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Wenz C, Faust D, Linz B, Turmann C, Nikolova T, Dietrich C. Cell‐cell contacts protect against t‐BuOOH‐induced cellular damage and ferroptosis in vitro. Arch Toxicol. 2019;93:1265‐1279. [DOI] [PubMed] [Google Scholar]
- 103. Kowal J, Tkach M, Thery C. Biogenesis and secretion of exosomes. Curr Opin Cell Biol. 2014;29:116‐125. [DOI] [PubMed] [Google Scholar]
- 104. Kamerkar S, LeBleu VS, Sugimoto H, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546:498‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lien EC, Lyssiotis CA, Juvekar A, et al. Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt‐driven breast cancer. Nat Cell Biol. 2016;18:572‐578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Xiong H, Wang C, Wang Z, Jiang Z, Zhou J, Yao J. Intracellular cascade activated nanosystem for improving ER+ breast cancer therapy through attacking GSH‐mediated metabolic vulnerability. J Control Release. 2019;309:145‐157. [DOI] [PubMed] [Google Scholar]
- 107. Zheng DW, Lei Q, Zhu J‐Y, et al. Switching apoptosis to ferroptosis: metal‐organic network for high‐efficiency anticancer therapy. Nano Lett. 2017;17:284‐291. [DOI] [PubMed] [Google Scholar]
- 108. Zhang P, Hou Y, Zeng J, et al. Coordinatively unsaturated Fe(3+) based activatable probes for enhanced MRI and therapy of tumors. Angew Chem Int Ed Engl. 2019;58:11088‐11096. [DOI] [PubMed] [Google Scholar]
- 109. Zhang F, Li F, Lu G‐H, et al. Engineering magnetosomes for ferroptosis/immunomodulation synergism in cancer. ACS Nano. 2019;13:5662‐5673. [DOI] [PubMed] [Google Scholar]
- 110. Toh TB, Lim JJ, Chow EK. Epigenetics in cancer stem cells. Mol Cancer. 2017;16:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Taylor WR, Fedorka SR, Gad I, et al. Small‐molecule ferroptotic agents with potential to selectively target cancer stem cells. Sci Rep. 2019;9:5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Raggi C, Gammella E, Correnti M, et al. Dysregulation of iron metabolism in cholangiocarcinoma stem‐like cells. Sci Rep. 2017;7:17667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Chanvorachote P, Luanpitpong S. Iron induces cancer stem cells and aggressive phenotypes in human lung cancer cells. Am J Physiol Cell Physiol. 2016;310:C728‐739. [DOI] [PubMed] [Google Scholar]
- 114. Bisaro B, Mandili G, Poli A, et al. Proteomic analysis of extracellular vesicles from medullospheres reveals a role for iron in the cancer progression of medulloblastoma. Mol Cell Ther. 2015;3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Schonberg DL, Miller TE, Wu Q, et al. Preferential iron trafficking characterizes glioblastoma stem‐like cells. Cancer Cell. 2015;28:441‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kanojia D, Zhou W, Zhang J, et al. Proteomic profiling of cancer stem cells derived from primary tumors of HER2/Neu transgenic mice. Proteomics. 2012;12:3407‐3415. [DOI] [PubMed] [Google Scholar]
- 117. Recalcati S, Gammella E, Cairo G. Dysregulation of iron metabolism in cancer stem cells. Free Radic Biol Med. 2019;133:216‐220. [DOI] [PubMed] [Google Scholar]
- 118. Basuli D, Tesfay L, Deng Z, et al. Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene. 2017;36:4089‐4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Chitambar CR, Al‐Gizawiy MM, Alhajala HS, et al. Gallium maltolate disrupts tumor iron metabolism and retards the growth of glioblastoma by inhibiting mitochondrial function and ribonucleotide reductase. Mol Cancer Ther. 2018;17:1240‐1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Naujokat C, Steinhart R. Salinomycin as a drug for targeting human cancer stem cells. J Biomed Biotechnol. 2012;2012:950658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Murphy CJ, Gole AM, Stone JW, et al. Gold nanoparticles in biology: beyond toxicity to cellular imaging. Acc Chem Res. 2008;41:1721‐1730. [DOI] [PubMed] [Google Scholar]
- 122. Daniel MC, Astruc D. Gold nanoparticles: assembly, supramolecular chemistry, quantum‐size‐related properties, and applications toward biology, catalysis, and nanotechnology. Chem Rev. 2004;104:293‐346. [DOI] [PubMed] [Google Scholar]
- 123. Guo J, Rahme K, He Y, Li LL, Holmes JD, O'Driscoll CM. Gold nanoparticles enlighten the future of cancer theranostics. Int J Nanomedicine. 2017;12:6131‐6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Zhao Y, Zhao W, Lim YC, Liu T. Salinomycin‐loaded gold nanoparticles for treating cancer stem cells by ferroptosis‐induced cell death. Mol Pharm. 2019;16:2532‐2539. [DOI] [PubMed] [Google Scholar]
- 125. Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090‐2093. [DOI] [PubMed] [Google Scholar]
- 126. Vulpe CD, Kuo Y‐M, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. 1999;21:195‐199. [DOI] [PubMed] [Google Scholar]
- 127. Pfeifhofer‐Obermair C, Tymoszuk P, Petzer V, Weiss G, Nairz M. Iron in the tumor microenvironment‐connecting the dots. Front Oncol. 2018;8:549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Aschemeyer S, Qiao B, Stefanova D, et al. Structure‐function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood. 2018;131:899‐910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Tacchini L, Bianchi L, Bernelli‐Zazzera A, Cairo G. Transferrin receptor induction by hypoxia. HIF‐1‐mediated transcriptional activation and cell‐specific post‐transcriptional regulation. J Biol Chem. 1999;274:24142‐24146. [DOI] [PubMed] [Google Scholar]
- 130. Qian ZM, Mei Wu X, Fan M, et al. Divalent metal transporter 1 is a hypoxia‐inducible gene. J Cell Physiol. 2011;226:1596‐1603. [DOI] [PubMed] [Google Scholar]
- 131. Huang BW, Miyazawa M, Tsuji Y. Distinct regulatory mechanisms of the human ferritin gene by hypoxia and hypoxia mimetic cobalt chloride at the transcriptional and post‐transcriptional levels. Cell Signal. 2014;26:2702‐2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Supuran CT. Carbonic anhydrases and metabolism. Metabolites. 2018;8:E25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Li Z, Jiang LI, Chew SH, Hirayama T, Sekido Y, Toyokuni S. Carbonic anhydrase 9 confers resistance to ferroptosis/apoptosis in malignant mesothelioma under hypoxia. Redox Biol. 2019;26:101297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. An Y, Zhu J, Liu F, et al. Boosting the ferroptotic antitumor efficacy via site‐specific amplification of tailored lipid peroxidation. ACS Appl Mater Interfaces. 2019;11:29655‐29666. [DOI] [PubMed] [Google Scholar]
- 135. Zhu T, Shi L, Yu C, et al. Ferroptosis promotes photodynamic therapy: supramolecular photosensitizer‐inducer nanodrug for enhanced cancer treatment. Theranostics. 2019;9:3293‐3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Viswanathan VS, Ryan MJ, Dhruv HD, et al. Dependency of a therapy‐resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547(7664):453‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Jiao WU, Minikes AM, Gao M, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2‐YAP signalling. Nature. 2019;572(7769):402‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 13(1), 91‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.