

Abstract
PURPOSE
Colony-stimulating factor-3 receptor (CSF3R)-T618I is a recurrent activating mutation in chronic neutrophilic leukemia (CNL) and to a lesser extent in atypical chronic myeloid leukemia (aCML) resulting in constitutive JAK-STAT signaling. We sought to evaluate safety and efficacy of the JAK1/2 inhibitor ruxolitinib in patients with CNL and aCML, irrespective of CSF3R mutation status.
METHODS
We conducted a phase II study of ruxolitinib in 44 patients (21 CNL and 23 aCML). The primary end point was overall hematologic response rate (ORR) by the end of 6 continuous 28-day cycles for the first 25 patients enrolled. We considered a response as either partial (PR) or complete response (CR). We expanded accrual to 44 patients to increase our ability to evaluate secondary end points, including grade ≥ 3 adverse events, spleen volume, symptom assessment, genetic correlates of response, and 2-year survival.
RESULTS
ORR was 32% for the first 25 enrolled patients (8 PR [7 CNL and 1 aCML]). In the larger cohort of 44 patients, 35% had a response (11 PR [9 CNL and 2 aCML] and 4 CR [CNL]), and 50% had oncogenic CSF3R mutations. The mean absolute allele burden reduction of CSF3R-T618I after 6 cycles was greatest in the CR group, compared with the PR and no response groups. The most common cause of death is due to disease progression. Grade ≥ 3 anemia and thrombocytopenia were observed in 34% and 14% of patients, respectively. No serious adverse events attributed to ruxolitinib were observed.
CONCLUSION
Ruxolitinib was well tolerated and demonstrated an estimated response rate of 32%. Patients with a diagnosis of CNL and/or harboring CSF3R-T618I were most likely to respond.
INTRODUCTION
Chronic neutrophilic leukemia (CNL) and atypical chronic myeloid leukemia (aCML) are rare BCR-ABL1–negative myeloid neoplasms. The 2016 WHO diagnostic criteria for CNL and aCML incorporate recurrent genetic markers,1,2 which provide diagnostic clarity for these diseases.1,2 In particular, CSF3R (colony stimulating factor-3 receptor) mutations are present in the vast majority of patients with CNL,3,4 whereas RAS pathway–centric mutations are common in aCML and other myelodysplastic syndrome/myeloproliferative neoplasm (MDS/MPN) overlap neoplasms.5 CSF3R-T618I and -T615A are in the extracellular domain (known as membrane-proximal mutations), whereas the -T640N is in the transmembrane domain. These mutations result in ligand-independent dimerization and activation of CSF3R, leading to constitutive JAK/STAT signaling.6
There is no standard of care or approved therapy for CNL and aCML. The median survival for both diseases is approximately 2 years, with hemorrhage, marrow failure, and blastic transformation as common causes of death.7,8 Allogeneic hematopoietic stem-cell transplantation is recommended for eligible patients with donor options.9-11 Experience with nontransplant therapies is limited. In mouse models driven by oncogenic CSF3R, leukocytosis, splenomegaly, and deaths are significantly alleviated by JAK1/2 inhibition.12,13 On the basis of these preclinical data,3,12,13 we sought to determine safety and efficacy of single-agent ruxolitinib in patients with CNL or aCML.
METHODS
Patient Population
Patients with a diagnosis of CNL or aCML were eligible for this study. In cases where a single WHO 2008 criterion was not met, related to an arbitrary laboratory cutoff, qualitative evaluation of dysplasia, or requirement of hepatosplenomegaly, we favored the diagnosis of CNL or aCML if the leukemia was positive for CSF3R-T618I, -T615A, or -T640N and the other diagnostic criteria were met.14 The WHO 2016 criteria were not available at the time of study design. The coordinating center reviewed and confirmed eligibility on the basis of pathologic, laboratory, and genetic data.
Study Design and Treatment
This was an open-label, single-arm, phase II multicenter investigator-initiated clinical trial of ruxolitinib in patients with CNL and aCML (ClinicalTrials.gov identifier: NCT02092324). Simon’s 2-stage minimax design was used for the study. The accrual goal was at least 25 evaluable patients who had completed 6 cycles (1 cycle is 28 days). In the early conduct of the study, we observed that approximately one-third of patients did not complete 6 cycles, usually because of lack of response or disease progression. Therefore, we modified the protocol to evaluate all patients enrolled and kept the original accrual goal in place for other secondary end point analyses. Patients who failed to complete 6 cycles were recorded as having no response. Inclusion and exclusion criteria are in the Data Supplement. Ruxolitinib starting dose was guided in part by prescribing guidelines based on platelet counts, but investigator discretion was allowed.
Study Oversight
Oregon Health & Science University (OHSU) Knight Cancer Institute Data and Safety Monitoring Committee was responsible for ensuring that all member and affiliate investigators conducted this clinical study in compliance with local institutional review board standards, US Food and Drug Administration regulations, and National Institutes of Health policies, and in accordance with the Data and Safety Monitoring Plan. The OHSU coordinating center audited records and provided training at 6 other participating sites.
Study Objectives
The primary end point was to determine the proportion of patients with CNL and aCML with hematologic response to ruxolitinib by the end of cycle 6 (overall response rate [ORR]), defined as either partial response (PR) or complete response (CR). Secondary end points included evaluation of: 1) frequency of grade ≥ 3 hematologic and nonhematologic adverse events (AEs), 2) median percentage reduction of spleen volume and total symptom score using a myeloproliferative neoplasm symptom assessment form [MPN-SAF], defined in Table 1 footnote,15 3) International Working Group (IWG)–defined criteria for clinical benefit16 4) genetic correlates of hematologic responses, and 5) 2-year overall survival (OS).
TABLE 1.
Baseline Clinical Characteristics and Overall Study Outcomes

Response Criteria and Study Assessments
Because of the lack of validated response criteria for CNL and aCML, we designed response criteria using end point analyses considered clinically relevant in MPNs generally and taking into account that treatment with ruxolitinib and other JAK inhibitors is associated with hematologic adverse events of anemia and thrombocytopenia. We defined CR as normalization of WBC count and absolute neutrophil count (ANC), no evidence of granulocytic hyperplasia (CNL) or granulocytic dysplasia (aCML), and normal spleen by ultrasonography measurement (< 250 cm3)17 or by palpation. There is excellent correlation of spleen volume estimates using prolate ellipsoid method by ultrasonography and calculations by computed tomography.18 In patients who are not in CR, PR requires > 50% reduction of WBC, ANC, and granulocytic hyperplasia or dysplasia and > 25% reduction in spleen volume or palpable spleen length from the left midcostochondral line. Quantifying marrow dysplasia proved challenging among pathologists, so when a less-subjective criterion was not met for CR (eg, normalization of marrow cellularity or WBC/ANC), the patient was determined not to have a CR. Marrow cellularity and myeloid:erythroid ratio are measures of granulocytic hyperplasia. Details of response criteria are listed in the Data Supplement. All responses were assessed by an adjudication committee (J.G., M.M.N.D., S.T.O.), including the principal investigator (K-H.D.). We also evaluated clinical benefit as defined by MDS/MPN IWG.16 The data cutoff date was October 17, 2018.
Molecular Analyses
DNA from blood cells was sequenced using a custom panel of 76 genes recurrently mutated in leukemia (QIAseq; Qiagen, Hilden, Germany; Data Supplement). The lower limit of detection is 2% variant allele frequency (VAF), and the average coverage is 2,000×. After sequencing on the Illumina (San Diego, CA) NextSeq500, data were aligned against the hg19 reference genome. A custom bioinformatics analysis pipeline integrating multiple established variant calling tools (FreeBayes [arXiv, Cornell Univeristy, Ithaca, NY], MuTect2 [Broad Institute, Cambridge, MA], and Scalpel [http://scalpel.sourceforge.net/index.html]) and variant annotation tools (Oncotator, Broad Institute, Cambridge, MA) was used. The inter-run coefficient of variation of replicate sampling is 8%.
Statistical Analysis
For sample size calculation, we considered ruxolitinib promising if it demonstrated ORR of ≥ 30%.19 Other studies have considered this an acceptable ORR for investigational therapies in hematologic malignancies.20,21 The null hypothesis was 10% ORR, given that CNL and aCML are rare diseases with no best available therapy for comparison. On the basis of Simon’s 2-stage minimax design, 25 evaluable patients would provide 80% power at 5% significance level with an interim analysis on the availability of primary end point data for the first 15 patients. If 1 or 0 patients responded, the trial was stopped for futility. If ≥ 2 patients responded, accrual to the second stage was expanded to 25 patients. The study was subsequently amended to allow enrollment to 44 patients, because approximately one-third of the patients could not reach the end of cycle 6, a time point for evaluation of secondary end points. We report the primary end point (ORR) on the first 25 patients, and the ORR and other end points on the basis of the 44 enrolled patients. Summary statistics were used to describe demographic and clinical characteristics. To compare the baseline characteristics for patients, we used Kruskal-Wallis test for continuous variables and χ2 or Fisher’s exact test for categorical variables. Kaplan-Meier plot and log-rank test were used to visualize and compare 2-year OS between groups. Median follow-up was estimated using the reverse Kaplan-Meier method. Statistical analysis was performed using R 3.5.3, IBM SPSS Statistics 21 (SPSS, Armonk, NY), and GraphPad Prism 8 (GraphPad Software, San Diego, CA).
RESULTS
Patient Characteristics
At the time of data cutoff, 21 patients with CNL and 23 patients with aCML were enrolled. Patient characteristics are presented in Table 1. The median age was 73 years (range, 43-92 years). Patients with CNL were older (median, 74 years) than patients with aCML (median, 67 years; P = .026). As expected, CSF3R membrane-proximal and transmembrane mutations were more common in CNL (76%) than in aCML (26%; P = .002). The median WBC/ANC, hemoglobin, and platelet count at baseline were not statistically different (Table 1). Other clinical characteristics are summarized in Table 1, and ruxolitinib dosing is summarized in the Data Supplement.
Efficacy Analyses
For the first 15 patients in stage 1, 5/15 (33%) responded, allowing us to move on to stage 2 of the Simon’s design. Eight of the first 25 patients responded (32%; 8 PR and 0 CR). By diagnosis, ORR was 58% (7/12) in the CNL group and 8% (1/13) in the aCML group (P = .011). By CSF3R mutation status, ORR was 54% (7/13) in the CSF3R-mutated group and 8% (1/12) in the CSF3R–wild type group (P = .030). Although 25 patients will achieve adequate power for the primary end point, we enrolled 44 patients to allow a reasonable number of patients to reach the end of cycle 6 for evaluation of secondary end points. The data for the first 25 patients are provided in the Data Supplement.
In Table 1, we summarize overall study outcomes for all 44 patients except for ORR, which we reported for 43 patients because one patient (CNL-19) had a suboptimal bone marrow biopsy specimen at the end of cycle 6 and had a normal spleen volume at baseline. The adjudication committee deemed this case not evaluable for response. Among 43 patients evaluable for response, ORR was 35% (11 PR [9 CNL, 2 aCML] and 4 CR [CNL]). The Data Supplement summarizes the adjudicated CR cases.
Figure 1A provides a schematic of patients according to diagnosis, CSF3R mutation status, and disposition on study. Figure 1B depicts absolute changes in WBC, hemoglobin, platelet count, and spleen volume, and Table 2 summarizes median or mean change of these and other variables in patients who reached the end of cycle 6. Ruxolitinib produced greater reduction of median WBC/ANC in patients with CNL (v patients with aCML) and in CSF3R-mutated patients (v CSF3R–wild type patients). Spleen volume reduction ≥ 35% occurred more frequently in CSF3R-mutant patients (79% v 29% CSF3R–wild type patients; P = .056), but this did reach statistical significance. Overall, ruxolitinib therapy reduced mean hemoglobin and improved median platelet count (−0.5 [± 2.0 standard deviation] and +38 [range, −214 to 260], respectively).
FIG 1.

(A) Schematic of 44 patients enrolled by diagnosis and CSF3R mutation status. Mut, CSF3R-T618I (n = 20), -T615A (n = 1), or -T640N (n = 1); WT, CSF3R–wild type; NR, nonresponder, 15 of 44 (34.1%) withdrew from study before the end of cycle 6; > C6, patients reached the end of cycle 6, 29 of 44 (65.9%). (B) Waterfall plot of absolute change of various hematologic parameters according to individual patients. Numbering of patients is arbitrarily based on the order presented in the heat map by mutation profile in Figure 2. The flat end of the line represents baseline value and the diamond-head end represents the end of cycle 6 value. Presented in this manner, both minimal and significant changes can be assessed on an individual basis. (†) Incidences of complete normalization of WBC, hemoglobin (Hgb) increases by > 2 g/dL, platelet (Plt) increases by > 30 × 109/L, and spleen volume reduction by ≥ 35%. aCML, atypical chronic myeloid leukemia; CNL, chronic neutrophilic leukemia.
TABLE 2.
Change in Disease Characteristics, Baseline v End of Cycle 6

In Fig 2A, we summarize baseline mutation profile and individual responses. Comprehensive mutation data are provided in the Data Supplement. The mean baseline VAF of CSF3R-T618I was 0.44 (range, 0.09 to 0.89). Characteristics of compound mutations are summarized in the Data Supplement. We observed a higher frequency of DNMT3A mutations in CNL (24%) compared with aCML (4.3%). In contrast, TET2 and RAS mutations were more frequent in aCML (26% and 30%, respectively) compared with CNL (9.5% and 9.5%, respectively). ASXL1 and SETBP1 mutations were present in 89% and in 46% of patients, respectively, with no difference between CNL versus aCML.
FIG 2.

(A) Summary figure of patients according to mutation profile, response assessment, and clinical benefit (CB). The heat map displays the 11 most commonly mutated genes in this study population according to variant allele frequency (VAF; darkest blue 100% and white 0%). The protocol-defined and International Working Group (IWG)–defined response assessments are color-coded by response. CSF3R-T618I mutation status is indicated as well as other pertinent CSF3R variants. CB was assessed by IWG criteria with one modification.16 We added the CB criterion of decrease in spleen volume by ≥ 35% (used in myelofibrosis pivotal studies).34 CB was not evaluated in patients who had a complete response (CR) by IWG criteria or in patients who withdrew before the end of cycle 6 (denoted by gray shading). (B) Forest plot for the unadjusted odds ratio (OR) and 95% CI obtained from univariate logistic regression models, with some continuous variables scaled in units of 10 or 100 to produce reasonable range of ORs. Data separated by categorical and continuous variables. Analyses on 43 evaluable patients were performed based on various demographic and disease characteristics and also starting doses of ruxolitinib. Boldface indicates variables that were found to be statistically significant. We also explored the adjusted effects, potential confounders, and effect modifiers using multivariable logistic regression models. Multivariable models were not presented because diagnosis is the single dominating risk factor for response, even after controlling for other important risk factors (ie, CSF3R mutation status and spleen volume). Although CSF3R mutation and spleen volume showed individual confounding effects, the estimated effect of diagnosis from a multivariable logistic regression model with both covariates included was very close to that from the univariate model. aCML, atypical chronic myeloid leukemia; ANC, absolute neutrophil count; C6, cycle 6; CNL, chronic neutrophilic leukemia; Hgb, hemoglobin; MPN-SAF, myeloproliferative neoplasm symptom assessment form; NR, no response; Plt, platelet; PR, partial response; PS, palpable spleen; TSS, total symptom score.
Two patients attained CR by protocol-defined and IWG-defined criteria.16 In patients who did not obtain CR by IWG-defined criteria at the end of cycle 6, we performed clinical benefit analyses according to IWG criteria as defined in Figure 2A.16 Overall, 85% (23/27) of patients fulfilled criteria for ≥ 1 category of clinical benefit(s). In Figure 2B, univariate analyses were performed according to response by protocol-defined criteria. CNL diagnosis and CSF3R mutations were strongly correlated with response. No other variables were significantly correlated with response, including karyotype, number of mutations, and ASXL1 or SETBP1 mutations.
Patterns of Mutant CSF3R-T618I Allele Frequency Changes
Figure 3 shows examples of changes in CSF3R-T618I VAF during treatment. Two patients had CR with early VAF reduction (CNL-16, CNL-17). One patient had a PR at the end of cycle 6 but with continued treatment achieved a CR that correlated with VAF reduction (CNL-05). CNL-08 had a PR that tracked with progressive CSF3R-T618I reduction but developed disease progression with expansion of CSF3R-T618I–negative cells harboring STAT3-Y640F and STAG2-R305*. Although 2 patients with CR (CNL-16, CNL-17) maintained responses through cycle 19 and cycle 16 as of data cutoff, the other 2 patients with CR (CNL-07, CNL-18) maintained response for < 1 year. CNL-07 had early progression with expansion of STAT3-D427G– and CBL-W408C–mutant cells. The mean absolute VAF change for patients with CSF3R-T618I, -T615A, or -T640N was −0.26 for CR, −0.05 for PR, and +0.01 for no response, with 8% inter-run coefficient of variation (Data Supplement).
FIG 3.

Different patterns of CSF3R-mutation variant allele frequency (VAF) changes over time with ruxolitinib. The status of the patient and cycle they reached as of data cutoff are indicated. (A) CNL-16, on study C19. (B) CNL-17, on study C16. (C) CNL-05, on study C43. (D) CNL-08, off study C19. C, cycle; CNL, chronic neutrophilic leukemia; CR, complete response; PR, partial response.
Survival
Survival data were collected from the start of study drug and were censored on the day of hematopoietic stem-cell transplantation for 6 patients or last follow-up. All patients who did not die or proceed to transplant were followed for at least 2 years (median follow-up, 38.4 months). Median OS for all patients was 18.8 months (95% CI, 15.3 to 25.2 months). By response, median survival was 15.6 months for nonresponders and 23.1 months for responders. We expect the responders to have longer median survival because they had to survive at least 6 cycles of treatment to be evaluated as a potential responder. Figure 4 summarizes 2-year OS. Patients with lower International Prognostic Scoring System score as defined in Table 1 survived longer.22 Disease duration before study participation was not considered in survival analysis.
FIG 4.

Kaplan-Meier curves estimating probability of survival for all patients and subgroup analyses as indicated. The P values from log-rank (Mantel-Cox) tests are indicated on the graph. We currently have 2-year data for all 44 patients enrolled. For those with longer-term data, we censored the data at 2 years. Six patients were also censored on the date they underwent hematopoietic stem-cell transplantation. aCML, atypical chronic myeloid leukemia; CNL, chronic neutrophilic leukemia; IPSS, International Prognostic Scoring System; Mut, mutation; WT, wild type.
Treatment Status at the Data Cutoff Point and Safety Assessment
At data cutoff, 39 patients were off study. The main reasons for study discontinuation were disease progression with or without blastic transformation (n = 15) or provider preference, usually due to lack of clinical benefit (n = 11; Data Supplement). Thirty-one patients have died. The most common cause of death was disease progression (n = 13). Other causes of death are listed in the Data Supplement.
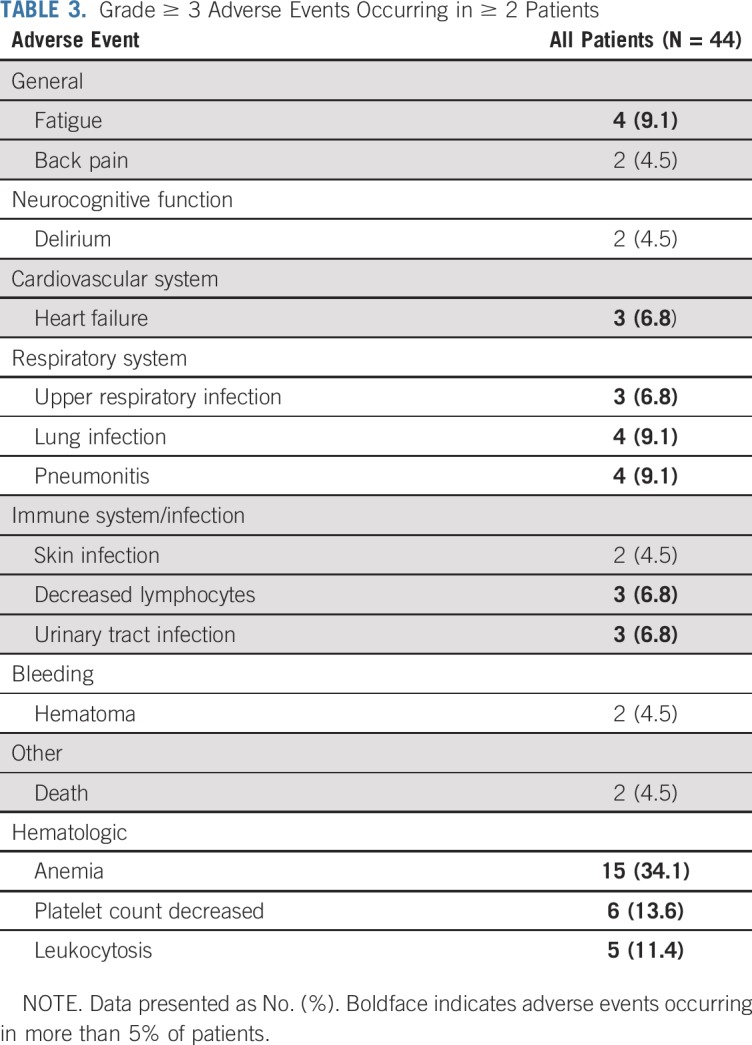
Because AEs of all grades with ruxolitinib have been reported previously,23,24 we focused on grade ≥ 3 AEs (Table 3). Fifty-five percent of patients experienced at least 1 grade ≥ 3 nonhematologic AE. Except 1 case of nutritional weight gain, no other nonhematologic grade ≥ 3 AEs were considered related or probably related to ruxolitinib. The expected hematologic AEs with ruxolitinib include anemia and thrombocytopenia. Grade ≥ 3 anemia and thrombocytopenia were observed in 34% and 14% of patients, respectively, occurring as a treatment effect or related to disease. In patients who experienced progressive leukocytosis, this was related to discontinuation of hydroxyurea and primary resistance or disease progression due to secondary resistance. All other grade ≥ 3 AEs were considered expected and/or were not related to ruxolitinib; therefore, no new serious AEs were identified.
TABLE 3.
Grade ≥ 3 Adverse Events Occurring in ≥ 2 Patients
DISCUSSION
Studies in cell lines, primary colony assays, and in vivo models suggest ruxolitinib targets signaling downstream of oncogenic CSF3R.3,12,13 Our clinical study provides evidence that ruxolitinib is a treatment option for a subset of patients with CNL and aCML. ORR by protocol-defined criteria for the first 25 patients was 32% (8/25 with PR). Previous experience with ruxolitinib in CNL and aCML has been limited to case reports with variable responses.25-29 Some responses are associated with allele burden reduction.27,28 Our data are consistent with these anecdotal experiences, including the observation that hematologic responses occur more often in CNL than in aCML. When evaluated by CSF3R mutation status, the CSF3R mutation effect was confounded by the strong association with CNL diagnosis (Fig 2B). We did not observe a high frequency of grade 3-4 anemia, as noted in patients with myelofibrosis treated with ruxolitinib, and actually observed increases in platelet counts in selected patients, a treatment benefit not typically experienced with hydroxyurea, the drug most commonly used to control proliferative myeloid neoplasms.
The majority of patients did not experience CR or PR and eventually succumbed to disease-related complications, but the potential for durable CR (including a molecular CR [CNL-16]) and IWG-defined clinical benefits is noteworthy. Two patients (CNL-16 and -17) with CR by protocol-defined criteria for > 1 year (and ongoing) presented with lower-risk features, suggesting that ruxolitinib may be more effective early in disease pathogenesis. These patients had mild/no cytopenias, had mild leukocytosis, lacked splenomegaly and overt symptoms, and had no other co-occurring mutations recurrently mutated in CNL and aCML. In the primary care setting, where neutrophilia may be ignored or attributed to reactive causes, increased awareness of the possibility of CNL may facilitate earlier diagnosis and therapy, when the disease could be more responsive to ruxolitinib. However, additional studies are required to determine whether early treatment could alter the natural history of CNL, whether combination therapies can improve ORR in CNL/aCML, and whether patients with durable responses could achieve long-term eradication of mutant cells. MEK inhibition in CSF3R-mutated CNL,30 CSF3R–wild type aCML,31 or MDS-MPN overlap neoplasms32 is another potential therapeutic consideration. In recent years, examples have emerged in chronic myeloid neoplasms indicating that complete responses, which are generally infrequent with noncytotoxic therapies, are not required for survival benefit (MDS/azacitidine33 and myelofibrosis/ruxolitinib34). Thus, novel therapies should be investigated for survival benefit even if they infrequently induce complete responses.
We highlight 2 novel genetic characteristics in this study population. First, we observed 3 patients with CSF3R-S799T. This variant occurs in the cytoplasmic domain and has uncertain oncogenic potential. In variant 3 CSF3R transcript, this site corresponds to S772, which is part of a motif that regulates receptor endocytosis. An S772A substitution results in increased cell surface expression,35 but it is not clear how a conserved substitution S772T would alter function. In one case, CSF3R-S799T occurred at < 5% VAF, and in the other 2 cases, after 6 cycles of ruxolitinib, CSF3R-S799T VAF was reduced from 12% to 0% (compound_T618I, CNL-13) and 21% to 0% (in isolation, CNL-15). CNL-15 was not evaluated in the CSF3R-mutated group, but these data suggest some degree of direct sensitivity to ruxolitinib, or alternatively, the S799T mutation exists in a clone sensitive to ruxolitinib by virtue of other genetic drivers. Second, ASXL1 and SETBP1 mutation status did not correlate with responses, but our sample size is too small to make definitive conclusions. Mutations in ASXL1 and SETBP1 are considered disease-modifying mutations and are associated with shortened survival in CNL and aCML.1,36 Anecdotally, we note that the two CR responses lasting > 1year harbored no ASXL1 or SETBP1 mutations (CNL-16 and -17). Meanwhile, PR responses can be durable for > 40 cycles even with these mutations, for example, CNL-05 (ASXL1 and SETBP1 mutations) and CNL-11 (ASXL1 mutation). We expect that the spectrum of responses we observed within the CSF3R-mutant subset can be at least partially explained by co-occurring mutations, which awaits additional preclinical and clinical modeling in larger studies.
In summary, our study offers evidence supporting the clinical efficacy of ruxolitinib in one-third of patients with CNL and aCML and provides new insights on genetic features of CNL and aCML in the context of ruxolitinib therapy.
ACKNOWLEDGMENT
We thank our patients and their families for participating in this study. We also thank the universities and medical centers that hosted this study and the Oregon Health & Science University Knight Cancer Institute for supporting the coordinating center’s function. We also thank our study teams, nurses, and colleagues for their help with this clinical trial. We thank Incyte Corporation for their funding of this work. J.R.G. acknowledges The Charles and Ann Johnson Foundation.
PRIOR PRESENTATION
Presented at the American Society of Hematology Annual Meeting, San Diego, CA, December 1-4, 2018.
SUPPORT
Supported by Incyte Corporation. National Institutes of Health National Cancer Institute grant No. 3P30CA069533-18S5 supported a subset of the genetic correlative studies.
Incyte Corporation had no role in data collection or analyses or writing of the manuscript.
CLINICAL TRIAL INFORMATION
AUTHOR CONTRIBUTIONS
Conception and design: Kim-Hien T. Dao, Jason Gotlib, Jorge E. Cortes, Robert H. Collins Jr, Samantha Savage Stevens, Yiyi Chen, Julia E. Maxson, Brian J. Druker, Jeffrey W. Tyner
Administrative support: Kim-Hien T. Dao, Stephen T. Oh, Anna Reister Schultz, Chase Brockett, Nan Subbiah, Cristina E. Tognon, Tara Macey
Provision of study material or patients: Kim-Hien T. Dao, Jason Gotlib, Michael M.N. Deininger, Stephen T. Oh, Jorge E. Cortes, Robert H. Collins Jr, Elliott F. Winton, Chase Brockett, Richard D. Press, Tara Macey, Jeffrey W. Tyner
Collection and assembly of data: Kim-Hien T. Dao, Michael M.N. Deininger, Stephen T. Oh, Jorge E. Cortes, Elliott F. Winton, Dana R. Parker, Hyunjung Lee, Anna Reister Schultz, Samantha Savage Stevens, Chase Brockett, Nan Subbiah, Michael Cascio, Catherine Degnin, Yiyi Chen, Tara Macey
Data analysis and interpretation: Kim-Hien T. Dao, Jason Gotlib, Stephen T. Oh, Jorge E. Cortes, Elliott F. Winton, Hyunjung Lee, Richard D. Press, Philipp W. Raess, Jennifer Dunlap, Yiyi Chen, Catherine Degnin, Cristina E. Tognon, Tara Macey, Brian J. Druker, Jeffrey W. Tyner
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Efficacy of Ruxolitinib in Patients With Chronic Neutrophilic Leukemia and Atypical Chronic Myeloid Leukemia
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/journal/jco/site/ifc.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Kim-Hien T. Dao
Honoraria: Incyte
Consulting or Advisory Role: Incyte
Research Funding: Incyte
Jason Gotlib
Consulting or Advisory Role: Blueprint Medicines, Deciphera Pharmaceuticals, Incyte, Kartos Therapeutics
Research Funding: Blueprint Medicines (Inst), Deciphera Pharmaceuticals (Inst), Incyte (Inst), Kartos Therapeutics (Inst), Promedior (Inst), CTI BioPharma (Inst), Gilead (Inst), Seattle Genetics (Inst), Novartis (Inst), Celgene (Inst)
Travel, Accommodations, Expenses: Blueprint Medicines, Deciphera Pharmaceuticals, Incyte, Celgene
Michael M.N. Deininger
Honoraria: Novartis, Bristol-Myers Squibb, Incyte, ARIAD Pharmaceuticals, Pfizer, Galena Biopharma, Blueprint Medicines
Consulting or Advisory Role: Novartis, Incyte, ARIAD Pharmaceuticals, Pfizer, Blueprint Medicines, Galena Biopharma, Ascentage Pharma, Humana, Adelphi, TRM Oncology, Fusion Pharmaceuticals, Takeda, Medscape, Incyte, Sangamo Therapeutics
Research Funding: Novartis (Inst), Bristol-Myers Squibb (Inst), Celgene (Inst), Gilead Sciences (Inst), Pfizer (Inst)
Expert Testimony: Bristol-Myers Squibb
Travel, Accommodations, Expenses: ARIAD Pharmaceuticals, Novartis, Pfizer, Galena Biopharma, Blueprint Medicines, Incyte
Stephen T. Oh
Consulting or Advisory Role: Incyte, Novartis, Blueprint Medicines
Research Funding: Incyte (Inst), Gilead Sciences (Inst), CTI BioPharma (Inst), Janssen (Inst), Kartos Therapeutics (Inst), Celgene (Inst)
Travel, Accommodations, Expenses: Incyte, Blueprint Medicines
Jorge E. Cortes
Consulting or Advisory Role: Bristol-Myers Squibb, BioLineRx, Novartis, Pfizer, Amphivena Therapeutics, Daiichi Sankyo, Bio-Path Holdings, Astellas Pharma, Takeda, Jazz Pharmaceuticals
Research Funding: Bristol-Myers Squibb (Inst), Novartis (Inst), Pfizer (Inst), Astellas Pharma (Inst), Immunogen (Inst), Sun Pharma (Inst), Takeda (Inst), Merus (Inst), Daiichi Sankyo (Inst), Tolero Pharmaceuticals (Inst), Trovagene (Inst), Jazz Pharmaceuticals (Inst)
Robert H. Collins Jr
Employment: University of Texas Southwestern Medical Center—Simmons Cancer Center
Research Funding: Agios, Oregon Health & Science University, MedImmune, Aeglea BioTherapeutics, Astellas
Elliott F. Winton
Research Funding: Incyte, Sierra Oncology, Samus Therapeutics, Blueprint Medicines
Richard D. Press
Consulting or Advisory Role: Cepheid, Guidepoint Global
Research Funding: Cepheid, AccuGenomics
Travel, Accommodations, Expenses: Cepheid
Yiyi Chen
Employment: Biotronik International (I)
Julia E. Maxson
Patents, Royalties, Other Intellectual Property: Licensing agreement with Vivid Biosciences
Brian J. Druker
Leadership: Amgen
Stock and Other Ownership Interests: MolecularMD, Blueprint Medicines, CTI BioPharma, GRAIL, Third Coast Therapeutics, Amgen
Honoraria: Aptose Biosciences
Consulting or Advisory Role: Blueprint Medicines, Gilead Sciences, Aptose Biosciences, MolecularMD, Beta Cat Pharmaceuticals, CTI BioPharma, GRAIL, Third Coast Therapeutics, Baxalta, Aileron Therapeutics, ALLCRON Pharma, Cepheid, Celgene, Monojul, Vivid Biosciences, The RUNX1 Research Program, Patient True Talk, Enliven Therapeutics, VB Therapeutics
Research Funding: Novartis (Inst), Bristol-Myers Squibb (Inst), Pfizer (Inst)
Patents, Royalties, Other Intellectual Property: Oregon Health & Science University patents, Merck
Travel, Accommodations, Expenses: Amgen
Uncompensated Relationships: Burroughs Wellcome Fund, Beat AML, CureOne
Jeffrey W. Tyner
Research Funding: Array BioPharma, Constellation Pharmaceuticals, Aptose Biosciences, AstraZeneca/MedImmune, Genentech, Incyte, Takeda, Janssen, Seattle Genetics, Syros, Takeda, Agios, Gilead
No other potential conflicts of interest were reported.
REFERENCES
- 1.Elliott MA, Tefferi A. Chronic neutrophilic leukemia 2016: Update on diagnosis, molecular genetics, prognosis, and management. Am J Hematol. 2016;91:341–349. doi: 10.1002/ajh.24284. [DOI] [PubMed] [Google Scholar]
- 2.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia Blood 1272391–2405.2016[Erratum: Blood 128:462-463, 2016] [DOI] [PubMed] [Google Scholar]
- 3.Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368:1781–1790. doi: 10.1056/NEJMoa1214514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pardanani A, Lasho TL, Laborde RR, et al. CSF3R T618I is a highly prevalent and specific mutation in chronic neutrophilic leukemia. Leukemia. 2013;27:1870–1873. doi: 10.1038/leu.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang SA, Hasserjian RP, Fox PS, et al. Atypical chronic myeloid leukemia is clinically distinct from unclassifiable myelodysplastic/myeloproliferative neoplasms. Blood. 2014;123:2645–2651. doi: 10.1182/blood-2014-02-553800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maxson JE, Luty SB, MacManiman JD, et al. Ligand independence of the T618I mutation in the colony-stimulating factor 3 receptor (CSF3R) protein results from loss of O-linked glycosylation and increased receptor dimerization. J Biol Chem. 2014;289:5820–5827. doi: 10.1074/jbc.M113.508440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elliott MA, Hanson CA, Dewald GW, et al. WHO-defined chronic neutrophilic leukemia: A long-term analysis of 12 cases and a critical review of the literature. Leukemia. 2005;19:313–317. doi: 10.1038/sj.leu.2403562. [DOI] [PubMed] [Google Scholar]
- 8.Breccia M, Biondo F, Latagliata R, et al. Identification of risk factors in atypical chronic myeloid leukemia. Haematologica. 2006;91:1566–1568. [PubMed] [Google Scholar]
- 9.Fu Y, Schroeder T, Zabelina T, et al. Postallogeneic monitoring with molecular markers detected by pretransplant next-generation or Sanger sequencing predicts clinical relapse in patients with myelodysplastic/myeloproliferative neoplasms. Eur J Haematol. 2014;92:189–194. doi: 10.1111/ejh.12223. [DOI] [PubMed] [Google Scholar]
- 10.Lim SN, Lee JH, Lee JH, et al. Allogeneic hematopoietic cell transplantation in adult patients with myelodysplastic/myeloproliferative neoplasms. Blood Res. 2013;48:178–184. doi: 10.5045/br.2013.48.3.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koldehoff M, Beelen DW, Trenschel R, et al. Outcome of hematopoietic stem cell transplantation in patients with atypical chronic myeloid leukemia. Bone Marrow Transplant. 2004;34:1047–1050. doi: 10.1038/sj.bmt.1704686. [DOI] [PubMed] [Google Scholar]
- 12.Fleischman AG, Maxson JE, Luty SB, et al. The CSF3R T618I mutation causes a lethal neutrophilic neoplasia in mice that is responsive to therapeutic JAK inhibition. Blood. 2013;122:3628–3631. doi: 10.1182/blood-2013-06-509976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maxson JE, Luty SB, MacManiman JD, et al. The Colony-stimulating factor 3 receptor T640N mutation is oncogenic, sensitive to JAK inhibition, and mimics T618I. Clin Cancer Res. 2016;22:757–764. doi: 10.1158/1078-0432.CCR-14-3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dao KT, Tyner JW, Gotlib J. Recent progress in chronic neutrophilic leukemia and atypical chronic myeloid leukemia. Curr Hematol Malig Rep. 2017;12:432–441. doi: 10.1007/s11899-017-0413-y. [DOI] [PubMed] [Google Scholar]
- 15.Scherber R, Dueck AC, Johansson P, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): International prospective validation and reliability trial in 402 patients. Blood. 2011;118:401–408. doi: 10.1182/blood-2011-01-328955. [DOI] [PubMed] [Google Scholar]
- 16.Savona MR, Malcovati L, Komrokji R, et al. An international consortium proposal of uniform response criteria for myelodysplastic/myeloproliferative neoplasms (MDS/MPN) in adults. Blood. 2015;125:1857–1865. doi: 10.1182/blood-2014-10-607341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Badran DH, Kalbouneh HM, Al-Hadidi MT, et al. Ultrasonographic assessment of splenic volume and its correlation with body parameters in a Jordanian population. Saudi Med J. 2015;36:967–972. doi: 10.15537/smj.2015.8.11809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paul FN, Taher A, Jahan M, et al: Splenic volume: Correlation between ultrasonogram and standard CT measurements. Bangladesh Med Res Counc Bull 43:58-62, 2017. [Google Scholar]
- 19. Simon RM: Design and analysis of clinical trials, in Davita VT, Lawrence TS, Rosenberg SA (eds): DeVita, Hellman, and Rosenberg’s Cancer: Principles & Practice of Oncology. Philadelphia, Lippincott Williams & Wilkins, 2008, pp 571-592. [Google Scholar]
- 20.Kantarjian HM, Giles F, Gattermann N, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is effective in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase following imatinib resistance and intolerance. Blood. 2007;110:3540–3546. doi: 10.1182/blood-2007-03-080689. [DOI] [PubMed] [Google Scholar]
- 21.Raza A, Reeves JA, Feldman EJ, et al. Phase 2 study of lenalidomide in transfusion-dependent, low-risk, and intermediate-1 risk myelodysplastic syndromes with karyotypes other than deletion 5q. Blood. 2008;111:86–93. doi: 10.1182/blood-2007-01-068833. [DOI] [PubMed] [Google Scholar]
- 22.Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113:2895–2901. doi: 10.1182/blood-2008-07-170449. [DOI] [PubMed] [Google Scholar]
- 23.Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366:799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cervantes F, Vannucchi AM, Kiladjian JJ, et al. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis Blood 1224047–4053.2013[Erratum: Blood 128:3013, 2016] [DOI] [PubMed] [Google Scholar]
- 25.Dao KH, Solti MB, Maxson JE, et al. Significant clinical response to JAK1/2 inhibition in a patient with CSF3R-T618I-positive atypical chronic myeloid leukemia. Leuk Res Rep. 2014;3:67–69. doi: 10.1016/j.lrr.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Freedman JL, Desai AV, Bailey LC, et al. Atypical chronic myeloid leukemia in two pediatric patients. Pediatr Blood Cancer. 2016;63:156–159. doi: 10.1002/pbc.25694. [DOI] [PubMed] [Google Scholar]
- 27.Gunawan AS, McLornan DP, Wilkins B, et al. Ruxolitinib, a potent JAK1/JAK2 inhibitor, induces temporary reductions in the allelic burden of concurrent CSF3R mutations in chronic neutrophilic leukemia. Haematologica. 2017;102:e238–e240. doi: 10.3324/haematol.2017.163790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nooruddin Z, Miltgen N, Wei Q, et al. Changes in allele frequencies of CSF3R and SETBP1 mutations and evidence of clonal evolution in a chronic neutrophilic leukemia patient treated with ruxolitinib. Haematologica. 2017;102:e207–e209. doi: 10.3324/haematol.2016.163089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szuber N, Finke CM, Lasho TL, et al. CSF3R-mutated chronic neutrophilic leukemia: Long-term outcome in 19 consecutive patients and risk model for survival. Blood Cancer J. 2018;8:21. doi: 10.1038/s41408-018-0058-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rohrabaugh S, Kesarwani M, Kincaid Z, et al. Enhanced MAPK signaling is essential for CSF3R-induced leukemia. Leukemia. 2017;31:1770–1778. doi: 10.1038/leu.2016.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khanna V, Pierce ST, Dao KH, et al. Durable disease control with MEK inhibition in a patient with NRAS-mutated atypical chronic myeloid leukemia. Cureus. 2015;7:e414. doi: 10.7759/cureus.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rocca S, Carrà G, Poggio P, et al. Targeting few to help hundreds: JAK, MAPK and ROCK pathways as druggable targets in atypical chronic myeloid leukemia. Mol Cancer. 2018;17:40. doi: 10.1186/s12943-018-0774-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009;10:223–232. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vannucchi AM, Kantarjian HM, Kiladjian JJ, et al. A pooled analysis of overall survival in COMFORT-I and COMFORT-II, 2 randomized phase III trials of ruxolitinib for the treatment of myelofibrosis. Haematologica. 2015;100:1139–1145. doi: 10.3324/haematol.2014.119545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aarts LHJ, Roovers O, Ward AC, et al. Receptor activation and 2 distinct COOH-terminal motifs control G-CSF receptor distribution and internalization kinetics. Blood. 2004;103:571–579. doi: 10.1182/blood-2003-07-2250. [DOI] [PubMed] [Google Scholar]
- 36.Shou LH, Cao D, Dong XH, et al. Prognostic significance of SETBP1 mutations in myelodysplastic syndromes, chronic myelomonocytic leukemia, and chronic neutrophilic leukemia: A meta-analysis. PLoS One. 2017;12:e0171608. doi: 10.1371/journal.pone.0171608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Damm F, Itzykson R, Kosmider O, et al. SETBP1 mutations in 658 patients with myelodysplastic syndromes, chronic myelomonocytic leukemia and secondary acute myeloid leukemias. Leukemia. 2013;27:1401–1403. doi: 10.1038/leu.2013.35. [DOI] [PubMed] [Google Scholar]
- 38.Laborde RR, Patnaik MM, Lasho TL, et al. SETBP1 mutations in 415 patients with primary myelofibrosis or chronic myelomonocytic leukemia: Independent prognostic impact in CMML. Leukemia. 2013;27:2100–2102. doi: 10.1038/leu.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Makishima H, Yoshida K, Nguyen N, et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet. 2013;45:942–946. doi: 10.1038/ng.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thol F, Suchanek KJ, Koenecke C, et al. SETBP1 mutation analysis in 944 patients with MDS and AML. Leukemia. 2013;27:2072–2075. doi: 10.1038/leu.2013.145. [DOI] [PubMed] [Google Scholar]
- 41.Hermans MH, Ward AC, Antonissen C, et al. Perturbed granulopoiesis in mice with a targeted mutation in the granulocyte colony-stimulating factor receptor gene associated with severe chronic neutropenia. Blood. 1998;92:32–39. [PubMed] [Google Scholar]