

Abstract
A virulent infectious bronchitis virus (IBV), designated as CK/CH/GD/QY16 (referred as QY16), was isolated from a diseased chicken farm in Guangdong province, China, in 2016. The complete genome of the strain was sequenced and analyzed. The results show that the genome of QY16 consists of 27,670 nucleotides, excluding poly (A) tail, and that its genome organization is 5’ UTR-1a-1b-S-3a-3b-E-M-4b-4c-5a-5b-N-6b-3’ UTR-poly (A) tail. Sequence comparison among QY16 and other IBV strains was conducted and its results demonstrate that the S1 gene of QY16 has the highest nucleotide sequence identity with that of 4/91, and the other part of its genome is highly similar to that of YX10. The results of the phylogenic analysis show that the entire genome of QY16 and most of the QY16 genes are located in the same cluster as those of YX10, except for the S1 gene which is located in the same cluster with that of 4/91. It has been further confirmed by the RDP and SimPlot analysis that QY16 is a recombinant strain deriving from YX10 (as the major parental sequence) and 4/91 (as the minor parental sequence), and that the recombination occurs in a region which includes the 3’-terminal 1b sequence (85 nt) and the 5’-terminal S1 protein gene sequence (1,466 nt). The results of the vaccination-challenge test suggest that QY16 is a nephropathogenic strain of IBV and that the vaccine strains–H120 and 4/91—cannot provide effective protection against it. These results indicate that the continuing evolution of IBV strains by genetic drift and genetic recombination may lead to IBV outbreaks even among the vaccinated chickens in China.
Key words: infectious bronchitis virus, recombinant, complete genome, vaccination-challenge test
INTRODUCTION
Avian infectious bronchitis (IB) is an acute and highly contagious disease caused by the avian infectious bronchitis virus (IBV). It has a major impact on the poultry industry worldwide and causes severe economic losses. IBV affects chickens of all ages and varieties, and causes upper respiratory diseases, nephritic syndromes, egg production decrease, and secondary bacterial infections (Yu et al., 2001).
IBV belongs to the genus Coronavirus of the family Coronaviridae in the order Nidovirales. Its viral genome consists of a linear, non-segmented, positive-sense, and single-stranded RNA of approximately 27.6 kilobases (kb), The genome of IBV contains 6 genes (gene 1 to 6) and can be transcribed into a nested set of mRNAs (mRNA 1 to 6) by a unique discontinuous transcription mechanism of coronavirus (Sawicki and Sawicki, 1998). The gene 1 encodes 2 polyproteins—polyprotein 1a (pp 1a) and polyprotein 1ab (pp 1ab), which is an extension of pp 1a by a minus 1 frameshift translation mechanism (Brierley et al., 1987). The 2 polyproteins can be cleaved by viral proteases into 15 non-structural proteins (nsp) that participate in the genome replication, transcription, and viral infection (Hodgson et al., 2004; Armesto et al., 2009). The gene 2 encodes a structural protein spike (S) glycoprotein that can be translated into a precursor spike glycoprotein (S0) and later be cleaved into 2 subunits S1 and S2 by cellular proteases (Cavanagh et al., 1992). The S1 subunit forms the tip of a spike, whereas the S2 subunit anchors the S1 to the viral membrane. The S1 subunit, with virus neutralizing epitopes and serotype-specific sequences (Cavanagh, 1983; Cavanagh et al., 1986), plays an important role in viral attachment to host cells and the induction of neutralizing antibodies (Koch et al., 1990). The gene 3 encodes nsp 3a and 3b, and a structural protein envelope protein (E), whereas the gene 4 encodes a structural protein membrane glycoprotein (M) and nsp 4b and 4c. The gene 5 encodes nsp 5a and 5b. The M and E are 2 membrane-binding proteins, whereas the 4 nsps (3a, 3b, 5a, and 5b) have been proved to be dispensable for virus replication but affect the efficiency of the replication and immunogenicity of the virus (Hodgson et al., 2006). The gene 6 encodes a structural protein nucleocapsid protein (N) and a nsp 6b. The N protein has the ability to bind the ribonucleoprotein generated by the viral RNA of IBV and plays a key role in viral replication and assembly as well as in cellular immunity (Ignjatovic and Galli, 1994; Ignjatovic and Galli, 1995). The nsp 4b, 4c, and 6b have rarely reported in literature although they were present in most IBVs and Turkey coronavirus (TCoV) (Cao et al., 2008; Gomaa et al., 2008; Thor et al., 2011; Xue et al., 2012; Reddy et al., 2015).
Although IBV vaccines have been used worldwide in most of the commercial chickens, IB still breaks out frequently and causes severe production problems. There are several reasons for the difficulties in preventing and controlling IBV. Firstly, the viral RNA-dependent RNA polymerase (RdRp) of IBV is not able to proofread its RNA, leading to RNA insertion, deletion, and substitution in the process of RNA replication. Secondly, due to the unique template switch during discontinuous RNA synthesis in IBV transcription, the nature recombination of the viral RNA frequently occurs among different strains (Zuniga et al., 2010). In addition, there were at least 30 serotypes of IBV identified worldwide, yet most of the available IBV vaccines can hardly provide effective cross-protection against strains of different serotype (Cook et al., 2012). As such, variants of the virus that varies in pathogenicity or serotype incessantly emerge, leading to different outbreaks of the disease.
In China, IBVs have been identified since 1982 and have struck all provinces of the country (Han et al., 2011). In previous studies, the phylogenetic analysis of the S1 gene of IBV indicates that the strains of IBV isolated in China could be divided into at least 7 genotypes including QX-type, 4/91-type, TW-type, Mass-type, HN08-type, LDT3-type, and TC07–2-type (Feng et al., 2014). Furthermore, there are IBV variants isolated as a result of gene recombination whose serotypes or pathogenicity may be different from any one of the parental strains (Feng et al., 2017). Despite the extensive use of attenuated or inactivated IBV vaccines (Cook et al., 2012; Jackwood, 2012), IB still breaks out frequently even among the vaccinated flocks. It can be attributed to the low efficacy of the current vaccines against the emergence of new serotypes or antigenic variants of IBV in China.
In this study, we sequenced the complete genome of a novel virulent IBV strain CK/CH/GD/QY16 (hereafter referred to as QY16). Sequence comparison, phylogenetic analysis, and recombination analysis were carried out to compare QY16 with other reference strains including. An animal experiment was conducted to explore the efficacy of 2 vaccines in protecting the chickens against the new strain. The results of this study present a valuable insight into the evolution of IBV in China, give a better understanding of the virus at the molecular level, and demonstrate the needs to improve the efficacy of the vaccines against the IBV infection in the poultry industry.
MATERIAL AND METHODS
Virus Isolation and Amplification
The IBV CK/CH/GD/QY16 strain (referred as QY16) was isolated from the kidneys of IBV-infected yellow feather broilers in a farm in Qingyuan city Guangdong province, China, in November 2016. The infected chickens were mainly less than 30-day-old, and typical clinical sign was nephritis that caused 30% mortality rate were identified. The specimens were frozen and thawed 3 times before treated with phosphate-buffered saline containing 200 U/mL penicillin and 200 μg/mL streptomycin and then centrifuged at 10,000 × g for 5 min. After keeping in 4°C for 3 h, the viruses were propagated through blind passage 5 times. During each blind passage process, 9-day-old embryos of specific pathogen-free (SPF) chickens were inoculated with 0.2 mL of the supernatant (first blind passage) or allantoic fluid (second to fifth blind passage) via the allantoic cavity. After incubation at 37°C for 48 h post-inoculation, the allantoic fluids were harvested and stored at –70°C. The virus in the eggs after the fifth passage was selected for genome sequencing, pathogenicity, and vaccination-challenge test. The virus isolation and follow-up experiments were carried out in biosafety level 2 biocontainment laboratory.
Viral RNA Extraction, RACE, and RT-PCR
The genomic RNA of the strain was extracted through using the AxyPrep Body Fluid Viral DNA/RNA Miniprep Kit (Axygen, China) according to the manufacturer's instructions. Reverse transcription (RT) was performed using PrimeScript II 1st Strand cDNA Synthesis Kit (Takara, Japan) following the manufacturer's protocol. Polymerase chain reaction (PCR) was conducted using a set of primers. The S1 gene of the stain was amplified using a sense primer 5′-AAG ACT GAA CAA AAG ACC GAC T-3′ and an antisense primer 5′-CAA AAC CTG CCA TAA CTA ACAT A-3′ (Supplementary Table S1). The size of the amplified segment was about 1,760 bp, including the entire S1 gene and the protease cleavage motif. The complete genome sequence of the strain, excluding the 5′ and the 3′ terminal, was amplified as 22 overlapping genome fragments using IBV consensus primer sets (Supplementary Table S1) designed based on the alignment of the genome nucleotide sequences of IBV strains. PCR was conducted in a 50 μL reaction volume containing 2 μL of cDNA, 2 μL of the specific primer (10 pmol of each), 21 μL of dH2 O, and 25 μL of PrimeSTAR Max Premix (2×) (Takara, Japan). The thermocycler was set at 35 cycles. During the process, the cDNA was desaturated at 98°C for 10 s, annealed at 55°C for 5 s, and extended at 72°C for 20 s. After the 35 cycles, a final 10 min DNA extension step was conducted at 72°C. The 5′ and 3′ rapid amplification of cDNA ends (RACE) was performed using designed RACE primer (Supplementary Table S1) and the SMARTer RACE 5′/3′ Kit (Clontech) following the manufacturer's instructions.
Cloning and Nucleotide Sequencing
The PCR products were tested by electrophoresis in 1% agarose gels and purified by using the AxyPrep DNA Gel Extraction Kit (Axygen, China). The gel-purified PCR products were ligated to pCloneEZ Topoisomerase cloning vector (Clonesmarter) and transformed into DH5a Escherichia coli competent cells (Takara, Japan). Cells carrying the recombinant plasmid were selected on Luria-Bertani agar plates containing ampicillin (100 μg/mL). Positive clones were screened by PCR following the same steps as the above-mentioned PCR amplification. Three positive clones were sequenced by Sang-gong Technology & Services Co., Ltd (Shanghai, China).
Genome Sequence Comparison and Phylogenetic Analysis
The obtained 24 overlapped nucleotide sequences of the strain (including 5′ and 3′ untranslated region [UTR]) were assembled into a complete genome sequence using Seqman program in the Lasergene package (DNAStar, Madison, WI). Comparison and analysis of the genome sequences, open reading frames (ORFs), and the deduced proteins of the strain and the reference IBV strains were conducted using EditSeq and MegAlign programs also in this package. After aligning the genome sequence of the strain with the reference IBV sequences using the ClustalW multiple alignment algorithm, phylogenetic analysis of the nucleotide sequences of the strain's complete genome and specific genes was run with the neighbor-joining method using MEGA version 7.0 (www.megasoftware.net). The bootstrap values were determined from 1,000 replicates of the original data.
Recombination Analysis
The putative recombinant sequence of the strain and its parental strains were detected by using recombination detection program (RDP) software version 4.80 with the recombination detection methods (RDP, GENECONV, BootScan, MaxChi, Chimaera, and SiScan). The detection of recombination breakpoints through at least 5 of these methods was taken as confirmatory for any putative recombination event. The potential recombination events and breakpoints were further verified by SimPlot version 3.5.1. The nucleotide identity comparison was carried out using the Kimura (2-parameter) method with a transition-transversion ratio of 2, and the window width and step size were 200 and 20 bp, respectively.
Pathogenicity and Vaccination-Challenge Test
Forty 3-day-old SPF chickens were divided into 4 groups of 10 and kept in 4 isolators with negative pressure. Food and water were provided ad libitum. Chickens in group 2 and 3 were vaccinated oculonasally with commercially available H120 and 4/91 vaccines, respectively, with a dose of 103.5 EID50 per chick. Chickens in group 1 and the control group were mock inoculated with sterile allantoic fluid. At 14 d post-inoculation (dpi), chickens in groups 1, 2, and 3 were challenged with strain of QY16 by the oculonasal administration with a dose of 105.0 EID50 per chicken, whereas those in the control group were mock inoculated again with sterile allantoic fluid. All chickens were observed daily to track the morbidity and mortality among them for 20 d after challenge. Dead chickens were dissected for the observation of their organs. Blood samples from all the chickens in each group were collected at 7 and 14 d post-vaccination and at 7, 14, and 20 d post-challenge, and the serum was stored at –80°C before the test. The serum samples were assayed at a single dilution using a commercial enzyme-linked immunosorbent assay designed for the detection of total antibodies (IDEXX Corporation) according to the manufacturer's instructions and the seropositivity rates were calculated. Cloacal swabs collected from all the chickens in each group at 7, 14, and 20 d post-challenge were used for virus recovery. Each swab sample was prepared as 10% w/v tissue suspension in phosphate-buffered saline, clarified by centrifugation at 12,000 g at 4°C for 10 min, and filtered through 0.22 mm membrane filters (Millipore). Three 9-day-old SPF chicken embryos were inoculated with 0.2 mL filtered supernatant of each sample. The inoculated eggs were incubated at 37°C and were candled daily. The allantoic fluid was collected from 2 of the inoculated embryos 72 h post-inoculation. Detection of the virus was performed by extracting RNA from the allantoic fluid and amplifying S1 gene by RT-PCR. The RT-PCR procedure uses the PrimeScript One Step RT-PCR Kit (Takara, Japan) according to the manufacturer's instructions. The RT-PCR products were analyzed on 1.0% agarose gels.
IBV Strains Published in Genbank
Forty-four representative IBV strains published in Genbank were selected for the sequence and phylogenetic analysis of the S1 gene of IBVs, and 16 IBV strains were used for the complete genome and specific genes (besides S1 gene) alignment, sequence comparison, phylogenetic, and recombination analysis. The accession numbers of these reference strains are listed in Supplementary Table S2.
RESULTS
Sequence and Phylogenetic Analysis of S1 Gene
The S1 gene of QY16 is composed of 1,617 nt with cleavage motifs His-Arg-Arg-Arg-Arg (HRRRR). Basic local alignment search tool was used to compare the nucleotide and protein sequences of QY16 with the sequence databases. The S1 gene of QY16 has the highest nucleotide identity and amino acid (aa) identity with that of 4/91 (97.5% and 96%, respectively).
The S1 gene of QY16 was compared to that of 44 available representative IBV strains and the nucleotide identity range from 60 to 97.5%. The QY16 showed high-sequence identity of S1 gene with 4/91-type strains (4/91, TA03, 7/93, CK/CH/LHLJ/140,906, and CK/CH/LHB/130,630), and the nucleotide identity ranges from 95 to 97.5%, whereas the amino acid (aa) identity ranges from 94 to 96%. The nucleotide identities between the S1 gene of QY16 and those of the vaccine strains used in China including H120, H52, and LDT3-A are respectively 78.7%, 77.6%, and 85%, whereas the aa identities between them are respectively 76%, 76%, and 84.3%. The QY16 showed lowest sequence identity of S1 gene with strain DE072 and the nucleotide identity is only 60%.
Phylogenetic analysis was conducted using the nucleotide sequences of the full S1 gene of QY16 and those of 44 reference strains (Figure 1 ). It is shown that the Chinese strains can be divided into 6 genotypes according to the previous studies (Feng et al., 2017). The strain QY16 clusters to the 4/91-type that includes reference strains such as 4/91, TA03, and 7/93, and distinct from other genotypes.
Figure 1.
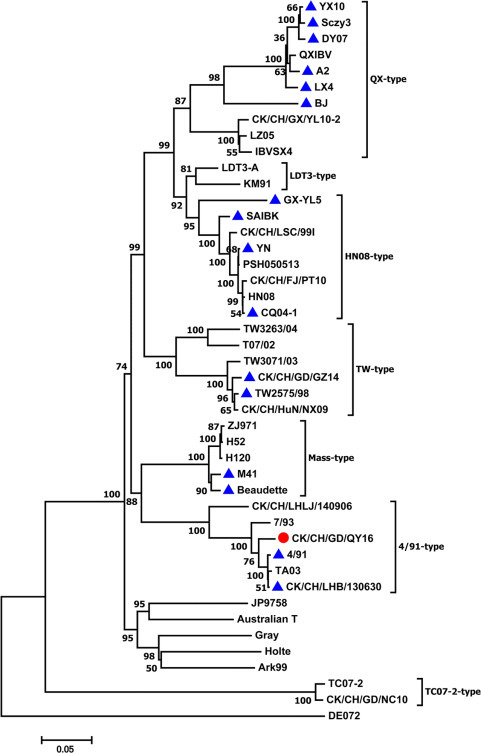
Consensus phylogenetic tree resulting from the analysis of the nucleotide sequences of the S1 gene of QY16 (red dots) and 44 reference strains. The trees were computed using the neighbor-joining method with 1,000 bootstrap replicates by the MEGA7 program. The 16 strains labeled with blue triangles were used for the complete genome sequence comparison, the phylogenetic analysis, and the recombination analysis.
Complete Genome and Organization of Strain QY16
The complete genome sequence (including the 5′-and 3′-terminal segments) of QY16 was obtained through assembling 24 overlapping sequences via the DNAstar software. The complete genome sequence has been submitted to GenBank under the accession no.MG197727.
The complete genomic sequences of QY16 are comprised of 27,670 nt, excluding poly (A) tail, and there are also 6 different genes and 13 ORFs flanked by 5′ and 3′ UTRs. The genome organization and location is as follows. 5’ UTR (1 to 525 nt), gene 1[1a (526 to 12,387 nt), 1b (12,462 to 20,420 nt)], gene 2[S (20,371 to 23,865 nt)], gene 3 [3a, (23,865 to 24,038 nt), 3b (24,038 to 24,229 nt), E (24,213 to 24,539 nt)], gene 4 [M (24,511 to 25,188 nt), 4b(25,189 to 25,473 nt),4c (25,394 to 25,555)], gene 5 [5a (25,552 to 25,749 nt), 5b (25,746 to 25,994 nt)], and gene 6 [N (25,937 to 27,166 nt), 6b (27,175 to 27,396)], 3′ UTR (27,397 to 27,670 nt) poly (A) tail.
Gene 1 has 19,892 nt, consisting of 2 overlapping ORFs, ORF1a (11,859 nt) and ORF1b (7,959 nt), and it encodes 2 polymerase proteins including1a and 1ab, which can be incised into 15 nsps and are related to RNA replication and transcription. The predicted papain-like proteinase (PLpro) and 3C-like proteinase (3CLpro) cleavage sites that define the nsp2 to 16 boarders are shown in Table 1 . There are 50 nt overlaps between gene 1 and gene 2. With a length of 3,498 nt, gene 2 has a single ORF, and it encodes a spike (S) glycoprotein that is cleaved into 2 subunits S1 and S2, which are with 1,617 and 1,878 nt, respectively (encoding 539 and 625aa). Between gene 2 and gene 3, there is only 1 nt overlap. There are 675 nt in gene 3 that encodes 2 nsps including 3a and 3b as well as a structural protein E. 3a, 3b, and protein E are with 174, 192, and 327 nt, respectively (encoding 57, 63, and 108aa). The number of nt overlaps between 3a and 3b, and 3b and E are respectively 1 and 20. There are 29 nt overlaps between gene 3 and gene 4. With a length of 1,045 nt, gene 4 contains 3 ORFs, and it encodes a structural protein M protein, and 2 nsps 4b and 4c which are with 678, 285, and 162, nt respectively (encoding 225aa, 94aa, and 53aa). Overlaps (80 nt) are identified between 4b and 4c. As for gene 4 and gene 5, it has been found that there are 4 nt overlaps. Containing 443 nt, gene 5 consists of 2 overlapping ORFs, ORF5a and ORF5b, which are 198 and 249 nt in length, respectively (encoding 65 and 82aa). There are 58 nt overlaps between gene 5 and gene 6. Gene 6 encodes N protein and nsp6b which are respectively with 1,230 and 222 nt (encoding 409 and 73aa). The 5′ and 3′ UTR of the genome are 525 and 274 nt in length, respectively.
Table 1.
The nsp2–15 produced by protease processing polyprotein 1a/1ab.
| Cleavage products | Position on genome | Position on polyprotein | C-end cleavage | Size (amino acid) | Cleavage by | Possible motif2 |
|---|---|---|---|---|---|---|
| nsp2 | 526–2,547 nt | M1-G674 | AG/GK1 | 674 | PLpro | |
| nsp3 | 2,548–7,323 nt | G675–G2266 | AG/GV | 592 | PLpro | PLpro |
| nsp4 | 7,324–8,865 nt | G2267–Q2780 | LQ/AG | 514 | 3CLpro | TM |
| nsp5 | 8,866–9,786 nt | A2781–Q3087 | LQ/SS | 307 | 3CLpro | 3CLpro |
| nsp6 | 9,787–10,668 nt | S3088–Q3381 | VQ/SK | 296 | 3CLpro | TM |
| nsp7 | 10,669–10,917 nt | S3382–Q3464 | LQ/LV | 83 | 3CLpro | |
| nsp8 | 10,918–11,547 nt | L3465–Q3674 | LQ/NN | 210 | 3CLpro | |
| nsp9 | 11,548–11,880 nt | N3675–Q3785 | LQ/SK | 111 | 3CLpro | |
| nsp10 | 11,881–12,315 nt | S3786–Q3930 | VQ/SD | 145 | 3CLpro | GFL |
| nsp11 | 12,316–12,384 nt | S3931–G3953 | – | 23 | 3CLpro | |
| nsp12 | 12,316–15,134 nt | S3931–Q4870 | LQ/SC | 940 | 3CLpro | RdRp |
| nsp13 | 15,135–16,934 nt | S4871–Q5470 | LQ/GT | 600 | 3CLpro | HEL |
| nsp14 | 16,935–18,497 nt | G5471–Q5991 | LQ/SI | 521 | 3CLpro | ExoN |
| nsp15 | 18,498–19,511 nt | S5992–Q6329 | LQ/SA | 338 | 3CLpro | NendoU |
| nsp16 | 19,512–20,420 nt | S6330–M6631 | – | 302 | 3CLpro | 2-O-MT |
A alanine, C cysteine, D aspartic acid, G glycine, K Lysine, L leucine, N asparagine, Q glutamine, S serine, T threonine, V valine.
PLpro, papain-like protease; TM, transmembrane domain; 3CLpro, 3C-like proteinase; GFL: growth factor-like domain; RdRp: RNA-dependant RNA polymerase; HEL, helicase domain; ExonN, exoribonuclease; NendoU, nidoviral uridylate-specific endoribonuclease; 2’ O-MT, 2’ -O-ribose methyltransferase.
Genome Sequence Comparison
The complete genome sequence of QY16 was compared to the complete genomic sequences of 16 IBV reference strains available in GenBank. As shown in Table 2 , the sequence comparison analysis reveals that the nucleotide identity of complete genome between QY16 and these strains ranges from 85.9 to 96.4%. QY16 has the highest resemblance to YX10 with 96.4% identity and is least similar to M41 with 85.9% identity. The sequence identity between QY16 and reference strains in terms of their different genes or regions was also calculated in Table 2; the nucleotide identity of the 5′-UTR, 1a, 1b, S1, S2, 3ab, E, M, 4bc, 5ab, N, 6b, and the 3′-UTR between QY16 and other IBV strains were 90 to 100%, 84.5 to 97.1%, 86.3 to 97.2%, 75.5 to 96.7%, 85 to 97.6%, 79.2 to 99.7%, 77.5 to 98.9%, 90.2 to 99.6%, 70.8 to 100%, 80.6 to 99.8%, 86.4 to 99.4%, 97.2 to 99.4%, and 97.1 to 99.6%, respectively. QY16 has the highest nucleotide identity with strain YX10 in most of their genes or regions (5′-UTR, 1a, 1b, S2, 3ab, E, M, 4bc, 5ab, and the 3′-UTR). In terms of the 6b gene, QY16 is most similar to the strain DY07. In terms of the N protein, it has the highest identity with SAIBK. In terms of full S gene, QY16 has the highest identity (90.5%) with that of the 4/91. With regard to the S1 gene, QY16 has the highest identity (97.5%) with the 4/91 and 80.7% identity with the YX10, whereas regarding the S2 gene, it has the highest identity (97.6%) with YX10 and 85% identity with the 4/91. The putative cleavage site of the QY16 S protein is His-Arg-Arg-Arg-Arg (HRRRR) which is similar to that of the QX-type strains of IBV (DY07, GX-LY5, LX4, Sczy3, and YX10).
Table 2.
Percentage of sequence identities of the complete genome and different regions of QY16 compared with those of other IBV strains.
| Gene 1 |
Gene 2 |
Gene 3 |
Gene 4 |
Gene 5 |
Gene 6 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Strain | Genome | 5′-UTR | 1a | 1b | S | S1 | S2 | 3ab | E | M | 4bc | 5ab | N | 6b | 3′-UTR |
| LX4 | 89.7 | 95.7 | 88.4 | 90.5 | 86.1 | 79.9 | 91.5 | 95 | 90.3 | 92.7 | 90.4 | 91.6 | 86.4 | – | 98.9 |
| YX10 | 96.41 | 100 | 97.1 | 97.2 | 89.6 | 80.7 | 97.6 | 99.7 | 98.9 | 99.6 | 100 | 99.8 | 96.8 | 97.9 | 99.6 |
| Sczy3 | 94.5 | 99.4 | 95.1 | 95.4 | 89.5 | 80.7 | 97.2 | 99.5 | 98.6 | 99.2 | 92.9 | 96.1 | 88.1 | 98.7 | 99.3 |
| A2 | 89.4 | 96.4 | 87.5 | 89.9 | 85 | 76.4 | 92.5 | 88.1 | 91.4 | 91.8 | 87.9 | 89.9 | 87.6 | 91.9 | 98.9 |
| DY07 | 94.4 | 98.5 | 95.5 | 95.3 | 89.6 | 80.6 | 97.4 | 99.5 | 98.8 | 99.4 | 88.8 | 94.1 | 88.7 | 98.7 | 98.5 |
| CQ04–1 | 92.5 | 97 | 92.1 | 93.9 | 86 | 76.6 | 94.4 | 87.7 | 77.5 | 86.8 | 81.1 | 84.0 | 95.7 | 95.7 | 97.5 |
| GX-YL5 | 93.7 | 99.4 | 95.5 | 95.7 | 85.3 | 75.5 | 93.9 | 80.3 | 85.6 | 90.2 | 80.1 | 85.2 | 96 | 95.3 | 99.3 |
| SAIBK | 88.9 | 90 | 89.4 | 89.4 | 85.7 | 76.9 | 93.5 | 86.4 | 87.2 | 91.4 | 80.4 | 85.9 | 99.4 | – | 97.8 |
| BJ | 89 | 96.2 | 87.4 | 89.8 | 81.9 | 77.1 | 86 | 89.1 | 88.3 | 92.4 | 91.9 | 92.2 | 87.6 | 95.3 | 99.3 |
| YN | 89.8 | 92.7 | 89.2 | 90.1 | 85.6 | 76.8 | 93.4 | 86.6 | 87.8 | 93.1 | 80.7 | 86.9 | 96 | 95.3 | 98.2 |
| 4/91 | 86.9 | 92.1 | 84.5 | 86.3 | 90.5 | 97.5 | 85 | 80.5 | 81.4 | 90.9 | 85.4 | 88.2 | 86.9 | 74 | 99.3 |
| CK/CH/LHB/130,630 | 86.8 | 92.1 | 84.5 | 86.3 | 90.4 | 96.5 | 85 | 80.5 | 81.4 | 90.9 | 85.4 | 88.2 | 86.8 | 74 | 98.9 |
| CK/CH/GD/GZ14 | 94.6 | 99.4 | 96.4 | 96.9 | 86.5 | 76.2 | 95.5 | 79.2 | 92.5 | 94.9 | 80.1 | 87.5 | 88.4 | 96.2 | 98.9 |
| TW2575/98 | 86.2 | 93.8 | 83.9 | 86.3 | 83.1 | 76.7 | 88.7 | 81.7 | 89.2 | 91.8 | 81.4 | 86.6 | 88.9 | 83.8 | 97.1 |
| M41 | 85.9 | 93.6 | 85.4 | 87.1 | 82.2 | 78.1 | 85.8 | 82.1 | 88.3 | 90.2 | 85.4 | 87.8 | 87.1 | – | 98.9 |
| Beaudette | 86.1 | 93.2 | 85.2 | 86.9 | 82 | 78 | 85.6 | 81.7 | 88.3 | 90.4 | 70.8 | 80.6 | 86.7 | 69.4 | 99.5 |
“–” indicates the absence of ORF 6b.
The highest nucleotide identities of different regions are indicated in bold values.
In addition, the analysis of the nucleotide identity between nsps encoded by the gene 1 of QY16 and other IBV strains was conducted (Table 3 ). The nsp3, nsp5, and nsp12 were predicted as PLpro, 3CLpro, and RdRp, respectively. As shown in the Table 3, the nsp2, nsp3, nsp4, nsp5, and nsp14 of QY16 have the highest identity with the those proteins encoded by YX10, whereas its nsp6, nsp7, nsp9, and nsp11 have the highest identity with the those proteins encoded by GX-YL5. The nsp12, nsp15, and nsp16 encoded by QY16 have the highest identity with those of strain CK/CH/GD/GZ14, whereas its nsp10 and nsp13 have the highest identity with those of Sczy3. The nsp8 and nsp11 of QY16 have the highest identity with those of BJ and YN, respectively.
Table 3.
Percentage of sequence identity of the QY16 Gene 1 fragments compared with other IBV strains.
| Strain | nsp2 | nsp3 | nsp4 | nsp5 | nsp6 | nsp7 | nsp8 | nsp9 | nsp10 | nsp11 | nsp12 | nsp13 | nsp14 | nsp15 | nsp16 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LX4 | 90.3 | 84.8 | 91.8 | 87 | 88.5 | 95.6 | 92.5 | 93.1 | 94.7 | 98.6 | 93.9 | 95.7 | 92.4 | 92.5 | 90.3 |
| YX10 | 98.51 | 97.3 | 99.6 | 98.9 | 96.4 | 97.2 | 94 | 93.4 | 95.6 | 98.6 | 96.9 | 97.6 | 98.7 | 98.1 | 90.9 |
| Sczy3 | 95.8 | 95.2 | 92.9 | 94.5 | 98 | 96.8 | 93.5 | 93.1 | 95.9 | 97.1 | 96.5 | 98.4 | 95.7 | 93.2 | 90 |
| A2 | 83.8 | 84.5 | 91.2 | 91.1 | 92.3 | 94 | 94.4 | 91.3 | 89.9 | 97.1 | 94.3 | 94.7 | 93.3 | 91.8 | 88.2 |
| DY07 | 96.4 | 95.7 | 94.2 | 93.7 | 97.8 | 97.6 | 93.7 | 93.1 | 96.3 | 98.6 | 96.5 | 96.6 | 93.6 | 93.7 | 90.4 |
| CQ04–1 | 95.9 | 91.1 | 85.4 | 89.3 | 98.1 | 97.6 | 93.3 | 93.7 | 97.2 | 100 | 97.1 | 97.1 | 94.3 | 97.8 | 95 |
| GX-YL5 | 95.9 | 96.5 | 92 | 93.8 | 98.1 | 97.6 | 93 | 94 | 95.9 | 100 | 96.6 | 97.3 | 95.1 | 93.7 | 92.8 |
| SAIBK | 93.6 | 90.1 | 83.9 | 86.4 | 87.9 | 92.4 | 86.3 | 90.1 | 91.7 | 92.8 | 89.9 | 89.5 | 88.7 | 89.5 | 89.7 |
| BJ | 83.7 | 84.7 | 90.8 | 90.8 | 92 | 95.6 | 95.1 | 91 | 87.8 | 91.3 | 93.3 | 94.1 | 92.7 | 92 | 90.8 |
| YN | 93.6 | 89.3 | 84.5 | 85.5 | 88.1 | 94.8 | 87.3 | 91.6 | 92.2 | 100 | 94.3 | 89.5 | 89.1 | 89.2 | 91.7 |
| 4/91 | 85.8 | 81.9 | 84.1 | 85.3 | 85.5 | 92.8 | 86.3 | 91 | 90.6 | 92.8 | 89.3 | 89 | 88.4 | 87.1 | 89.3 |
| CK/CH/LHB/130,630 | 85.8 | 81.9 | 84.1 | 85.2 | 85.5 | 92.8 | 86.3 | 91 | 90.6 | 92.8 | 89.1 | 89 | 88.4 | 87.2 | 89.4 |
| CK/CH/GD/GZ14 | 97 | 96.9 | 91.8 | 95.3 | 96.3 | 96.8 | 95.9 | 98.8 | 99.1 | 98.6 | 98 | 97.5 | 98.3 | 98.7 | 93.8 |
| TW2575/98 | 86.4 | 80 | 85.4 | 84.9 | 87.1 | 91.2 | 87 | 90.4 | 87.4 | 94.2 | 89.4 | 90.2 | 89.4 | 89 | 89.5 |
| M41 | 84.6 | 83.9 | 84.6 | 88.2 | 86.3 | 92.4 | 86.5 | 88 | 90.3 | 95.7 | 89.7 | 88.9 | 90.6 | 87.3 | 88.6 |
| Beaudette | 84.3 | 84 | 83.7 | 86.4 | 86.5 | 92.8 | 85.9 | 89.5 | 91.7 | 95.7 | 88.8 | 89.4 | 91.2 | 87.7 | 88.2 |
The highest nucleotide identities of different regions are indicated in bold values.
The transcription-regulating sequence (TRS) comparison shows that the core sequence (CS) CTT/GAACAA is conserved for most IBVs. The CS of the gene 1, gene 4, and gene 6 of QY16 is CTTAACAA, whereas the CS of its gene 2 and gene 3 is CTGAACAA. However, it has been detected that the CSs of the gene 5 of QY16, LX4, A2, BJ, and Sczy3 have an additional 6 nucleotide (G/AAGAAA) insertion and 2 nucleotide (CG) deletion (Figure 2 ), whereas the CSs of the gene 5 of CK/CH/GD/GZ14, SAIBK, DY07, CQ04–1, GX-YL5, and YN have an additional 3 nucleotide (AAA) insertion compared to the homologous sequences of other IBV strains (Figure 2). In addition, the overseas strains 4/91, Beaudette, and M41 were found to have no insertion. The CS is located in the initiation codons of the genes 1 to 6 of QY16 upstream 461, 52, 23, 77, 7, and 93 nt, respectively.
Figure 2.
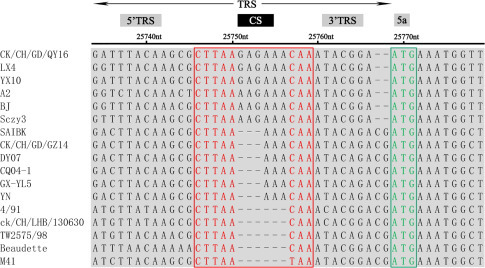
Results of the sequence comparison of QY16 with 16 reference strains in terms of the TRS of their gene 5. In the TRS of gene 5, the CSs of QY16, LX4, YX10, A2, BJ, and Sczy3 have a 6 nt insertion (G/AAGAAA) and 2 nt deletion (CG). Strains SAIBK, CK/CH/GD/GZ14, DY07, CQ04–1, GX-YL5, and YN have a 3 nt (AAA) insertion (AAA) compared to 4/91, CK/CH/LHB/130,630, Beaudette, and M41 without insertion.
In order to study whether the insertions of CS in TRS of gene 5 exist extensively in Chinese strains, the comparison of TRS of gene 5 among QY16, 42 Chinese isolates, and 36 oversea isolates was conducted (Supplementary Figure S1). The result shows that 44% (19/43) of Chinese strain have 5 to 6 nt insertion (G/AA/CGAAA or CTTAA) and 2 nt (CG) deletion, 37% (16/43) of Chinese strain have 3 to 4 nt insertion (AAAA or AAA) without deletion, and only 8 strains without insertion. By contrast, only a British and a Korean strains have 6 nt insertion (ACTAAA or GAGAAA) and 2 nt (CG) deletion, 3 strains isolated from Thailand have 3 nt insertion (AAA), most overseas strains are without insertion. In addition, the overseas strains with insertion in TRS of gene 5 mainly isolated from the countries surrounding China (Korea and Thailand), and the strains may have close relationship with Chinese IBV strains. It suggests that an insertion in the TRS of the gene 5 is common among the Chinese IBV but uncommon among oversea IBV strains.
Phylogenetic Analysis
In order to assess the genetic relatedness between QY16 and the reference IBV strains, phylogenetic trees were constructed for the nucleotide sequences of the complete genome and specific genes of QY16 and the 16 reference strains. As shown in Figure 3 and Supplementary Figure S2, the complete genome, 1a, 1b, S2, E, M, N, 3ab, 4bc, 5ab, and 6c, of QY16 is in the same cluster as those of YX10, whereas the full S gene, similar to the S1 gene, is located in the same cluster as those of 4/91 and evolutionarily distant from those of YX10. With the combination of the results of the sequence comparison and phylogenetic analysis, it is reasonable to assume that QY16 has a close evolutionary relationship with IBV YX10 and 4/91.
Figure 3.
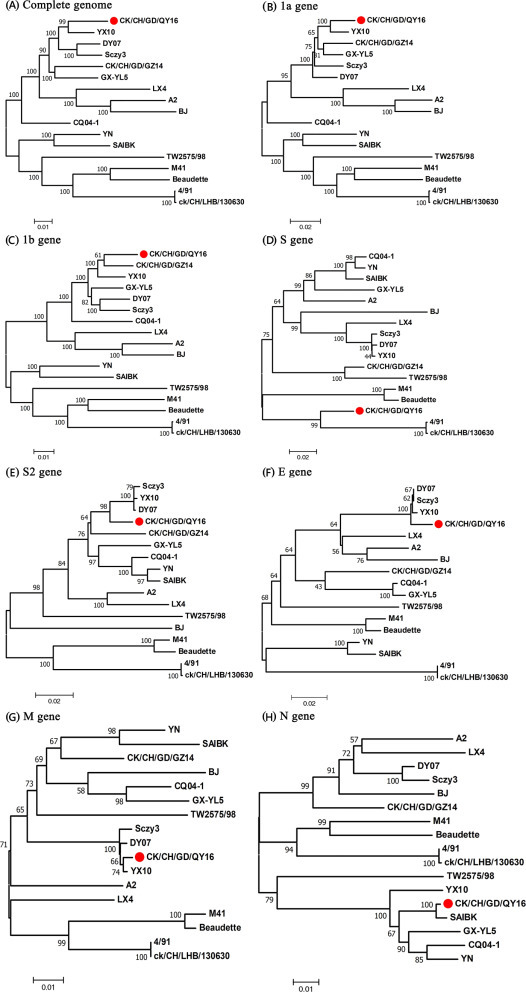
Phylogenetic trees constructed by using the neighbor-joining method based on the complete genome and different regions of the genome of the strains (bootstrapping for 1,000 replicates with its value >60%). (A) complete genomes, (B) 1a gene, (C) 1b gene, (D) S gene, (E) S2 gene, (F) E gene, (G) M gene, (H) N gene. The QY16 sequence is labeled with red dots.
Recombination Analysis
In order to detect possible recombination events within the QY16 genomic sequence, RDP analysis was conducted and the complete genomes of 16 reference strains were used as putative parent strains. The results indicate that QY16 is possibly a recombinant strain formed by a major parent strain YX10 and a minor parent strain 4/91. It has been proved that the recombination breakpoints of QY16 are located at the 20, 311 to 20, 343, and 21,810 to 21, 844 nt. The region 1 to 20,310 and 21,845 to 27,678 nt of QY16 is 97.5% similar to that of YX10, and the region 20,334 and 21,811 nt of QY16 showed 98.9% similarity with that of 4/91. The P-value of RDP method is 3.97 × 10−217.
To confirm the results of the RDP analysis, genomic sequences analysis of QY16, YX10, and 4/91 was carried out using the Simplot software. The result indicates that the recombination signal and the breakpoint of QY16 are located at 20,336 and 21,836 nt. Furthermore, the recombination region consists of a 3′-terminal ORF 1b with 85 nt and a 5′-terminal S1 protein gene with 1466 nt (there are 50 nt overlaps between the2 genes), and the region between these 2 breakpoints is a putative recombinant region, which is also consistent with the RDP results (Figure 4A ).
Figure 4.
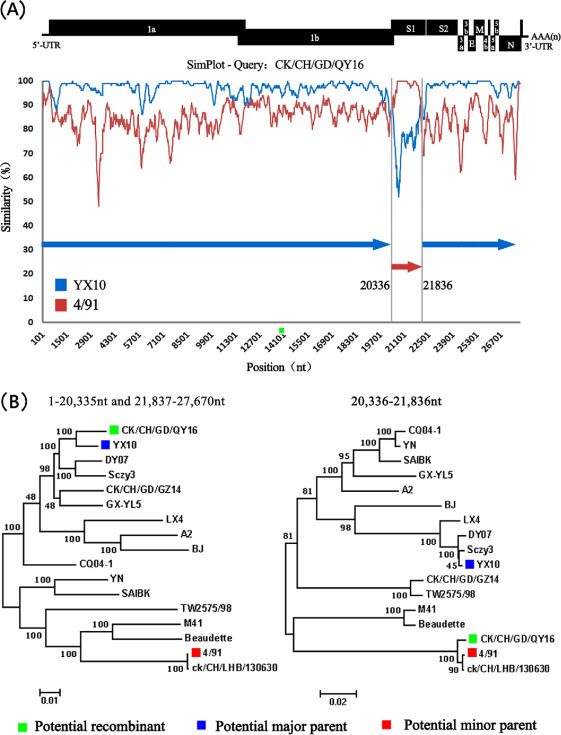
Results of the recombination analysis of QY16. SimPlot analysis was performed with QY16 as the query sequence and YX10 (blue) and 4/91(red) as putative parental strains. (A). The dummy lines show the deduced breakpoints at the genome position 20,336 and 21,886 nt. Bars at the top demonstrate the relative positions of the genome of IBV. The recombination region corresponding to the genome includes the 3′-terminal ORF 1b and most of the 5′-terminal S1 protein gene. The y-axis shows the percentage of similarity within a sliding window of 200 bp centered on the position plotted, with a step size between plots of 20 bp. (B). Phylogenetic trees of the genome regions 1 to 20,335, 21,837 to 27,670, and 20, 336 to 21,836 nt among QY16, YX10, 4/91, and other 14 reference strains. Phylogenetic trees were constructed using the neighbor-joining method (bootstrapping for 1,000 replicates). The recombinant strain QY16 was labeled with green squares, the major parental strain YX10 with blue squares, and the minor parental strain 4/91 with red squares.
The phylogenetic tree of the complete genomic sequences of the strains and that of their detected recombinant regions were also constructed and analyzed, demonstrating that there is a topological alternation in the recombinant region of QY16, which further confirms the recombination event (Figure 4B).
Pathogenicity Study and Vaccination-Challenge Tests
All the chickens were observed clinical signs after the vaccination or the challenge, and none of the chickens showed any clinical signs before the challenge. As shown in Table 4 , at 3 to 14 d post-challenge, chickens in group 1 showed severe clinical signs such as depression, ruffled feathers, sagging head and wings, diarrhea, lassitude, and huddling. However, no obvious respiratory sign or muscle lesions appeared in them. Four chickens died during 5 to 12 d post-challenge with gross lesions present as typical nephritis symptoms such as swollen specked kidney, distended ureter filled with uric acid, severe dehydration, and weight loss. It suggests that QY16 is a nephropathogenic IBV strain. At 15 d post-challenge, there was neither clinical symptom nor death in the remaining chickens. However, half (3/6) of cloacal swabs collected from the survival chickens in group 1 were positive at 20 d post-challenge, and the virus recovery result indicates that the survival chickens had been releasing the virus incessantly through the cloaca.
Table 4.
Results of pathogenicity and vaccination-challenge test using QY16.
| Group1 | Morbidity | Mortality | Antibody2 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Days post-vaccination |
Days post-challenge |
Virus recovery3 |
||||||||
| 7 | 14 | 7 | 14 | 20 | 74 | 14 | 20 | |||
| 1 | 10/10 | 4/10 | 0/10 | 0/10 | 4/7 | 6/6 | 6/6 | 7/7 | 5/6 | 3/6 |
| 2 | 9/10 | 4/10 | 3/10 | 9/10 | 8/8 | 6/6 | 6/6 | 8/8 | 4/6 | 2/6 |
| 3 | 4/10 | 2/10 | 4/10 | 10/10 | 9/9 | 8/8 | 8/8 | 5/9 | 2/8 | 1/8 |
| Control | 0/10 | 0/10 | 0/10 | 0/10 | 0/10 | 0/10 | 0/10 | 0/10 | 0/10 | 0/10 |
Chickens in groups 2 and 3 were vaccinated with H120 and 4/91 vaccines, respectively, and challenged with QY16. Chickens in group 1 were only challenged with QY16. Chickens in the control group were not exposed to any viruses.
Number seroconverted/number survive.
Number of virus-positive chickens in virus recovery tests after challenge/the number of chickens used for attempted virus recovery after challenge.
Days after challenge.
As shown in Table 4, groups 2 and 3 are with different levels of morbidity rate, mortality rate, and virus recovery rate. The morbidity rate, mortality rate, and virus-positive rate (at 20 d post-challenge) are respectively 90, 40, and 33% in group 2 where chickens were inoculated with the H120 vaccine and are respectively 40, 20, and 13% in group 3 where chickens were inoculated with the 4/91 vaccine. The results suggest that both the H120 vaccine and the 4/91 vaccine do not provide effective and full protection against QY16. As expected, none of the chickens was either ailed or present as virus positive in the control group.
The results of the serological responses induced by QY16 and the other 2 IBV vaccines are presented in Table 4. Only 30% of chickens in group 2 and 40% in group 3 had seroconverted at 7 d post-vaccination, but at 14 d post-vaccination more than 90% of the vaccinated chickens in group 2 and group 3 had seroconverted. After 14 d post-challenge, all the survival chickens in group 1 had seroconverted.
DISCUSSION
The frequent outbreaks of IBV cause severe problems to the poultry industry in China, despite the intensive use of anti-IBV vaccines. Due to its vast territory, China is caught in complicated situations where multiple IBV genotypes and serotypes coexist (Liu et al., 2006). The analysis of sequence of IBV is main focus on S1 gene in previous studies. Although it plays an important role in the viral attachment to host cells, and the induction of neutralizing antibodies (Koch et al., 1990), the S1 gene is only a small part of the IBV genome and thus cannot represent all the biological characteristics of the virus. It is, therefore, necessary to sequence and analyze the complete genome of some isolated strains, especially the recombinant ones.
In this study, the IBV strain QY16 was isolated from yellow feather broilers vaccinated with H120 and 4/91 vaccines. It has been found that the S1 gene of QY16 is highest identical to 4/91 and is located in the same cluster with it in the phylogenic tree. Whether QY16 is re-isolated from the 4/91 vaccine strain or is the recombinant result originating from 4/91, it needs to be further confirmed by sequencing and analyzing its complete genome.
The genome organization of QY16 is 5′UTR-1a-1b-S-3a-3b-E-M-4b-4c-5a-5b-N-6b-3′UTR, which is different from that of most IBV genomes (5′UTR -1a-1b-S-3a-3b-E-M-5a-5b-N-3′UTR) as previously reported. Although 4b, 4c, and 6b are present in the most of the IBV genomes, they have rarely been studied, and their functions were thus unknown (Reddy et al., 2015). The gene 1 accounts for two-thirds of the genome, and encodes 2 overlapping proteins pp1a and pp1ab. Pp1ab is an extension of pp1a by a minus 1 frameshifting translation mechanism (Brierley et al., 1987). The pp1a/1ab is cleaved into 15 nsps (nsp2 to nsp16) by PLpro and 3CLpro encoded by the virus. These nsps, forming a transcription/replication complex that involves in the viral genome's transcription, replication, and the viral infection, are important functional proteins for the virus. The cleavage products and cleavage sites of the pp1a/1ab of QY16 are highly identical to those of reported reference IBV strain A2 (Liu et al., 2009), Sczy3 (Zhao et al., 2013), and TCoV (Gomaa et al., 2008). It suggests that pp1a/1ab is highly conserved in IBV. The CS (CTT/GAACAA) of the TRS is another conserved sequence of IBV. The CS can be the binding center of the leader sequence and the subgenomic mRNA, and can promote the transcription of mRNA2 to 6(Sola et al., 2005). In this study, the CS sequence of QY16 is found to be conserved as CTT/GAACAA except for the gene 5 which is with a 6 nt insertion (GAGAAA) and 2 nt deletion (CG). It has been found in QY16, LX4, and YX10, whereas another 6 nt (AAGAAA) insertion was reported in the IBV strain (Liu et al., 2009)–A2 and BJ, and Sczy3. Furthermore, a 3 nt (AAA) insertion was found in the strains including SAIBK, CK/CH/GD/GZ14, DY07, CQ04–1, GX-YL5, and YN which are all Chinese isolates. In addition, the overseas strains 4/91, Beaudette, and M41 were found to have no insertion. The further comparison among QY16, 42 Chinese isolates, and 36 overseas isolates suggests that an insertion in the CS of TRS of the gene 5 is common among the Chinese IBV but uncommon among oversea IBV strains. Whether these insertions could be a genetic marker between Chinese and overseas strains needs to be further verified.
The results of the sequence comparison and phylogenetic analysis show that QY16 has the highest identity with 4/91 in terms of the S1 gene and is located in the same cluster with it as well. Nevertheless, its complete genome and most specific genes (except for S1) are of high similarity to YX10 rather than 4/91. These results suggest that QY16 may be a recombinant strain of 4/91 and YX10. The recombinant analysis including RDP, SimPlot, and phylogenetic analysis further confirms that QY16 originates from the recombination of a major parent strain YX10 and a minor parent strain 4/91. The QX-type strain YX10 was isolated in Zhejiang province, China, in 2010, and it is reported to be involved in recombinant variant in recent years (Wu et al., 2016; Feng et al., 2017). The 4/91 pathogenic strain was isolated in the UK and causes tremendous economic losses to the poultry industry in European (Gough et al., 1992; Cook et al., 1996). The 4/91 vaccine obtained through attenuating the pathogenic strain has been widely used in the global poultry industry. Despite the fact that the 4/91 strain has become an important gene donor in genetic evolution of IBV in China (Han et al., 2011; Zhang et al., 2015), it is difficult to identify the 4/91 pathogenic or vaccine strain involve in the formation the recombinant IBV strains. In this study, the 4/91 vaccine is proved to be a minor parent strain of QY16 and the recombination region is predicted to include the 3′ 1b terminal (85 nt) and most part of the S1 gene (1,466 nt). It is believed that the recombination “hot spots,” including C(T/G)TAACAA (Jia et al., 1995; Lee and Jackwood, 2000), CTTTTG (Wang et al., 1993), CTTTT(C/T) (Mondal and Cardona, 2007), and other A-T-rich regions (Zhao et al., 2013), are located adjacent to the putative crossover sites. In our study, these putative crossover sites are found to be nowhere near the breakpoint, and it is difficult to determine whether these motifs are associated with the recombination.
As the recombination region of QY16 includes most of the S1 gene, the pathogenicity study and vaccination-challenge tests were conducted to examine the efficacy of the H120 and 4/19 vaccines against QY16. The result shows QY16 causes 40% death among 17-day-old SPF chickens compared with the reported mortality rate of 25% among 15-day-old SPF chickens caused by its major parent strain YX10 (Xue et al., 2012; Feng et al., 2015). The pathogenicity of QY16 has apparently enhanced in this regard as this result shows. The clinical symptoms of the chickens infected with QY16 are typical nephritis symptoms which are similar to the situations caused by YX10 but different from the muscle lesions caused by the 4/91 pathogenic strain. Meanwhile, it has been proved by our study that the H120 vaccine cannot provide effective protection against the QY16 challenge, and it is probably because of the difference between their serotypes. The diversity of serotypes among the Mass-type vaccines (H120, H52, W93, etc.) and Chinese isolates has been confirmed by previous studies (Li et al., 2012; Feng et al., 2017), which can provide a possible explanation for H120's failure in serving as an effective vaccine against QY16. Our results also prove that the 4/91 vaccine cannot provide complete protection against QY16 challenge either. On one hand, recombinant QY16 and 4/91 may have difference in serotypes. Although the S1 gene of QY16 is highly similar to that of 4/91, the difference in the S1 gene and other genes may still lead to the great change in the serotype and pathogenicity. On the other hand, a broader degree of cross-protection provided by a vaccine against different viral strains is related to the cell-mediated immune responses to the shared T-cell epitopes among some heterologous strains (Collisson et al., 2000; Seo et al., 2000). As most of the T-cell epitopes of IBV remain in the M and N protein (Ignjatovic and Galli, 1994), the diversity in M and N genes may contribute to low cross-protection efficacy, in this case 4/91 vaccine's low protection efficacy against QY16.
In summary, the complete genome and specific gene sequences of QY16 have different levels of sequence identity with other IBV strains. The recombinant strain QY16 is proved by our study to derive from YX10 (as the major parental sequence) and 4/91 (as the minor parental sequence). It can be inferred from the results of the pathogenicity study and vaccination-challenge test that CK/CH/GD/QY16 is a type of nephropathogenic strain and that the vaccine strains H120 and 4/91 cannot provide complete protection against it. These results indicate that the continuing evolution of IBV strains by genetic drift and genetic recombination may lead to IBV outbreaks even among the vaccinated chickens in China, which calls for the further development of new vaccines to help prevent and control the disease.
Acknowledgments
This study was supported by the National Modern Agricultural Industry Technology System Project of China (CARS-41), Guangdong Province Agricultural Industry Technology System Project (2016LM1112), and Science and Technology Program of Guangzhou (201607010363).
CONFLICT OF INTEREST
The authors declare that there are no conflicts of interest.
Footnotes
Supplementary Table S1. Primers used for QY16 genome amplification.
Supplementary Table S2. The accession numbers of IBVs used for S1 gene, complete genome and specific genes alignment, sequence comparison, phylogenetic and recombination analysis,
Supplementary Figure S1. Results of the sequence comparison of QY16, 42 Chinese isolates, and 36 overseas isolates in terms of the TRS of their gene 5.
Supplementary Figure S2. Phylogenetic trees constructed by using the neighbor-joining method based on the complete genome and different regions of the genome of the strains (bootstrapping for 1,000 replicates with its value >60%). (a) 3ab, (b) 4bc, (c) 5ab, and (d) 6b. The QY16 sequence is labeled with red dots.
Supplementary Material
References
- Armesto M., Cavanagh D., Britton P. The replicase gene of avian coronavirus infectious bronchitis virus is a determinant of pathogenicity. PLoS One. 2009;4:e7384. doi: 10.1371/journal.pone.0007384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley I., Boursnell M.E., Binns M.M., Bilimoria B., Blok V.C., Brown T.D., Inglis S.C. An efficient ribosomal frame-shifting signal in the polymerase-encoding region of the coronavirus IBV. EMBO J. 1987;6:3779–3785. doi: 10.1002/j.1460-2075.1987.tb02713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J., Wu C.C., Lin T.L. Complete nucleotide sequence of polyprotein gene 1 and genome organization of turkey coronavirus. Virus Res. 2008;136:43–49. doi: 10.1016/j.virusres.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D. Coronavirus IBV: structural characterization of the spike protein. J. Gen. Virol. 1983;64:2577–2583. doi: 10.1099/0022-1317-64-12-2577. [DOI] [PubMed] [Google Scholar]
- Cavanagh D., Davis P.J., Cook J.K., Li D., Kant A., Koch G. Location of the amino acid differences in the S1 spike glycoprotein subunit of closely related serotypes of infectious bronchitis virus. Avian Pathol. 1992;21:33–43. doi: 10.1080/03079459208418816. [DOI] [PubMed] [Google Scholar]
- Cavanagh D., Davis P.J., Darbyshire J.H., Peters R.W. Coronavirus IBV: virus retaining spike glycopolypeptide S2 but not S1 is unable to induce virus-neutralizing or haemagglutination-inhibiting antibody, or induce chicken tracheal protection. J. Gen. Virol. 1986;67:1435–1442. doi: 10.1099/0022-1317-67-7-1435. [DOI] [PubMed] [Google Scholar]
- Collisson E.W., Pei J., Dzielawa J., Seo S.H. Cytotoxic T lymphocytes are critical in the control of infectious bronchitis virus in poultry. Dev. Comp. Immunol. 2000;24:187–200. doi: 10.1016/s0145-305x(99)00072-5. [DOI] [PubMed] [Google Scholar]
- Cook J.K.A., Jackwood M., Jones R.C. The long view: 40 years of infectious bronchitis research. Avian Pathol. 2012;41:239–250. doi: 10.1080/03079457.2012.680432. [DOI] [PubMed] [Google Scholar]
- Cook J.K., Orbell S.J., Woods M.A., Huggins M.B. A survey of the presence of a new infectious bronchitis virus designated 4/91 (793B) Vet. Rec. 1996;138:178–180. doi: 10.1136/vr.138.8.178. [DOI] [PubMed] [Google Scholar]
- Feng K., Wang F., Xue Y., Zhou Q., Chen F., Bi Y., Xie Q. Epidemiology and characterization of avian infectious bronchitis virus strains circulating in southern China during the period from 2013–2015. Sci. Rep. 2017;7:6576. doi: 10.1038/s41598-017-06987-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng K., Xue Y., Wang J., Chen W., Chen F., Bi Y., Xie Q. Development and efficacy of a novel live-attenuated QX-like nephropathogenic infectious bronchitis virus vaccine in China. Vaccine. 2015;33:1113–1120. doi: 10.1016/j.vaccine.2015.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng K., Xue Y., Wang F., Chen F., Shu D., Xie Q. Analysis of S1 gene of avian infectious bronchitis virus isolated in southern China during 2011–2012. Virus Genes. 2014;49:292–303. doi: 10.1007/s11262-014-1097-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomaa M.H., Barta J.R., Ojkic D., Yoo D. Complete genomic sequence of turkey coronavirus. Virus Res. 2008;135:237–246. doi: 10.1016/j.virusres.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough R.E., Randall C.J., Dagless M., Alexander D.J., Cox W.J., Pearson D. A ’new’ strain of infectious bronchitis virus infecting domestic fowl in Great Britain. Vet. Rec. 1992;130:493–494. doi: 10.1136/vr.130.22.493. [DOI] [PubMed] [Google Scholar]
- Han Z., Sun C., Yan B., Zhang X., Wang Y., Li C., Zhang Q., Ma Y., Shao Y., Liu Q. A 15-year analysis of molecular epidemiology of avian infectious bronchitis coronavirus in China. Infect. Genet. Evol. 2011;11:190–200. doi: 10.1016/j.meegid.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson T., Britton P., Cavanagh D. Neither the RNA nor the proteins of open reading frames 3a and 3b of the coronavirus infectious bronchitis virus are essential for replication. J. Virol. 2006;80:296–305. doi: 10.1128/JVI.80.1.296-305.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson T., Casais R., Dove B., Britton P., Cavanagh D. Recombinant infectious bronchitis coronavirus Beaudette with the spike protein gene of the pathogenic M41 strain remains attenuated but induces protective immunity. J. Virol. 2004;78:13804–13811. doi: 10.1128/JVI.78.24.13804-13811.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignjatovic J., Galli L. The S1 glycoprotein but not the N or M proteins of avian infectious bronchitis virus induces protection in vaccinated chickens. Arch. Virol. 1994;138:117–134. doi: 10.1007/BF01310043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignjatovic J., Galli U. Immune responses to structural proteins of avian infectious bronchitis virus. Avian Pathol. 1995;24:313–332. doi: 10.1080/03079459508419072. [DOI] [PubMed] [Google Scholar]
- Jackwood M.W. Review of infectious bronchitis virus around the world. Avian Dis. 2012;56:634–641. doi: 10.1637/10227-043012-Review.1. [DOI] [PubMed] [Google Scholar]
- Jia W., Karaca K., Parrish C.R., Naqi S.A. A novel variant of avian infectious bronchitis virus resulting from recombination among three different strains. Arch. Virol. 1995;140:259–271. doi: 10.1007/BF01309861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch G., Hartog L., Kant A., van Roozelaar D.J. Antigenic domains on the peplomer protein of avian infectious bronchitis virus: correlation with biological functions. J. Gen. Virol. 1990;71:1929–1935. doi: 10.1099/0022-1317-71-9-1929. [DOI] [PubMed] [Google Scholar]
- Lee C.W., Jackwood M.W. Evidence of genetic diversity generated by recombination among avian coronavirus IBV. Arch. Virol. 2000;145:2135–2148. doi: 10.1007/s007050070044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Wang X.Y., Wei P., Chen Q.Y., Wei Z.J., Mo M.L. Serotype and genotype diversity of infectious bronchitis viruses isolated during 1985–2008 in Guangxi, China. Arch. Virol. 2012;157:467–474. doi: 10.1007/s00705-011-1206-6. [DOI] [PubMed] [Google Scholar]
- Liu X., Su J., Zhao J., Zhang G. Complete genome sequence analysis of a predominant infectious bronchitis virus (IBV) strain in China. Virus Genes. 2009;38:56–65. doi: 10.1007/s11262-008-0282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.W., Zhang Q.X., Chen J.D., Han Z.X., Liu X., Feng L., Shao Y.H., Rong J.G., Kong X.G., Tong G.Z. Genetic diversity of avian infectious bronchitis coronavirus strains isolated in China between 1995 and 2004. Arch. Virol. 2006;151:1133–1148. doi: 10.1007/s00705-005-0695-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S.P., Cardona C.J. Genotypic and phenotypic characterization of the California 99 (Cal99) variant of infectious bronchitis virus. Virus Genes. 2007;34:327–341. doi: 10.1007/s11262-006-0014-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy V., Theuns S., Roukaerts I., Zeller M., Matthijnssens J., Nauwynck H. Genetic characterization of the Belgian nephropathogenic infectious bronchitis virus (NIBV) reference strain B1648. Viruses. 2015;7:4488–4506. doi: 10.3390/v7082827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki S.G., Sawicki D.L. A New Model for Coronavirus Transcription. Springer; USA: 1998. [DOI] [PubMed] [Google Scholar]
- Seo S., Pei J.W., Dzielawa J., Collisson E. Adoptive transfer of infectious bronchitis virus primed alphabeta T cells bearing CD8 antigen protects chicks from acute infection. Virology. 2000;269:183–189. doi: 10.1006/viro.2000.0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola I., Moreno J.L., Zuniga S., Alonso S., Enjuanes L. Role of nucleotides immediately flanking the transcription-regulating sequence core in coronavirus subgenomic mRNA synthesis. J. Virol. 2005;79:2506–2516. doi: 10.1128/JVI.79.4.2506-2516.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thor S.W., Hilt D.A., Kissinger J.C., Paterson A.H., Jackwood M.W. Recombination in avian gamma-coronavirus infectious bronchitis virus. Viruses. 2011;3:1777–1799. doi: 10.3390/v3091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Junker D., Collisson E.W. Evidence of natural recombination within the S1 gene of infectious bronchitis virus. Virology. 1993;192:710–716. doi: 10.1006/viro.1993.1093. [DOI] [PubMed] [Google Scholar]
- Wu X., Yang X., Xu P., Zhou L., Zhang Z., Wang H. Genome sequence and origin analyses of the recombinant novel IBV virulent isolate SAIBK2. Virus Genes. 2016;52:509–520. doi: 10.1007/s11262-016-1337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y., Xie Q., Yan Z., Ji J., Chen F., Qin J., Sun B., Ma J., Bi Y. Complete genome sequence of a recombinant nephropathogenic infectious bronchitis virus strain in china. J. Virol. 2012;86:13812–13813. doi: 10.1128/JVI.02575-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L., Jiang Y., Low S., Wang Z., Nam S.J., Liu W., Kwangac J. Characterization of three infectious bronchitis virus isolates from China associated with proventriculus in vaccinated chickens. Avian Dis. 2001;45:416–424. [PubMed] [Google Scholar]
- Zhang T., Han Z., Xu Q., Wang Q., Gao M., Wu W., Shao Y., Li H., Kong X., Liu S. Serotype shift of a 793/B genotype infectious bronchitis coronavirus by natural recombination. Infect. Genet. Evol. 2015;32:377–387. doi: 10.1016/j.meegid.2015.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F., Zou N., Wang F., Guo M., Liu P., Wen X., Cao S., Huang Y. Analysis of a QX-like avian infectious bronchitis virus genome identified recombination in the region containing the ORF 5a, ORF 5b, and nucleocapsid protein gene sequences. Virus Genes. 2013;46:454–464. doi: 10.1007/s11262-013-0884-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga S., Cruz J.L., Sola I., Mateos-Gomez P.A., Palacio L., Enjuanes L. Coronavirus nucleocapsid protein facilitates template switching and is required for efficient transcription. J. Virol. 2010;84:2169–2175. doi: 10.1128/JVI.02011-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.