

Abstract
Avian infectious bronchitis virus (IBV) is responsible of significant economic losses for poultry industry around the world, through evolution of its pathogenicity, inadequacy of vaccines, and virus evasion. Such evasion is related to the unstable nature of its RNA, in particular the S glycoprotein encoding gene, which raises great challenges with regard to the control of the disease, along with the lack of proof reading mechanisms of the RNA polymerase. The emergence of new variants might be a reason for the endemic outbreaks that are being reported in Tunisia, in addition to poor vaccination techniques and ineffective prophylactic programs.
In the present study, partial nucleotide sequences of the S1 glycoprotein gene and the 3′-untranslated region (UTR) of 2 Tunisian isolates, TN1011/16 and TN1012/16, identified in 2016, were determined. Specific mutations were found in S1 gene as well as in 3′UTR region. Phylogenetic analysis of the S1 nucleotide sequences showed that both isolates are closely related to the Algerian strains, and formed a common cluster within the genotype I. In addition, these isolates were non-recombinant ones, confirming that they are unique variants. Based on their S1 gene sequences, TN1011/16 and TN1012/16 strains were distant from the H120 vaccine strain, commercially used in Tunisia along with the variant vaccine 793B type (4/91). A comparison between nucleotide sequences of their 3′UTR region and S1 gene showed a difference in IBV classification. The obtained results have confirmed that the IBVsequence continues to drift and brings valuable information in relation with its evolution, vaccine development and better control of the disease.
Key words: infectious bronchitis virus, new variant, genotype I, S1 gene, Tunisia
INTRODUCTION
Infectious bronchitis (IB) is a disease caused by the IB virus (IBV), a member of the Gammacoronavirus genus, the Coronaviridae family, and the Nidovirales order (International Committee on Taxonomy of Viruses, 2009). IB has a significant economic impact with production losses related to poor weight gains, condemnation at processing and mortality in broilers, suboptimal egg production and egg downgrading in laying birds (Cook et al., 2012). Despite the applied intensive vaccination programs, outbreaks re-emerge as a consequence of infections with new variants that differ serologically from the strains used for vaccination (Chen et al., 2010; Leghari et al., 2016; Feng et al., 2018). Besides, in the absence of a “standard” vaccination program against IB, the program recommended by the National Committee for Avian Pathology (NCAP) remains an alternative to prevent avian disease infections. Unfortunately, it appears that there are programs as breeding as reported by Cherif et al. (2010). This largely explains the variability of protection outcomes and the sustainability of the disease, despite the availability of the latest generation of vaccines.
The IBV genome consists of a single-stranded positive-sense RNA molecule of approximately 27.6 kb, which encodes several nonstructural proteins involved in RNA transcription and replication and four structural proteins, known as small membrane (E), membrane (M), nucleoprotein (N), and spike glycoprotein (S) (Boursnell et al., 1987), which is formed by a globular (S1) subunit that is anchored in the membrane by the S2 subunit (Cavanagh, 2007). The S1 subunit is the major target for neutralizing antibodies and carries serotype-specific antigenic determinants (Liu et al., 2007). In addition to these roles, recently it was reported that S and 5a accessory gene are responsible for the attenuation of virulent IBV strains (Zhao et al., 2019). The genome of IBV includes the untranslated regions (UTR) at the 5′ and 3′ genome regions, which play a role in RNA synthesis (Sawicki et al., 2007) and classification of IBV (Hewson et al., 2009). The 3′-UTR is involved in the initiation of negative-strand RNA synthesis and has also been used to assess variations in emerging IBV strains and other members of coronavirus group 3 (Williams et al., 1993; Breslin et al., 1999).
In fact, IBV genotype may differ by up to 50% of the amino acids sequences of their S1 proteins (Sjaak de Wit et al., 2011); such variations have led to the emergence of new serotypes; for this, the S1 gene nucleotide sequence is being widely used for phylogenetic classification of IBV, especially the hypervariable regions HVR1, HVR2, and HVR3.
The emergence of variant strains may be related to extensive nucleotide and deduced amino acid sequence variabilities in the IBV genome, which are often responsible for IB outbreaks in vaccinated flocks (Liu et al., 2007; Roussan et al., 2008). The main aim of this study was to perform molecular characterization of Tunisian IBV isolates, based on the S1 gene compared to the 3′-UTR region.
MATERIALS AND METHODS
Study Population and Sample Collection
Samples were collected from commercial broilers of a flock housing 1,500 birds in the Eastern region of Tunisia (Nabeul), a major Tunisian industrial poultry region. They were aged 4 to 5 wk old, and showing respiratory symptoms. Tracheal and cloacal swabs were randomly collected from 10 live birds and separately pooled. Internal organs including liver, lungs, spleen, and tonsils were collected from eight necropsied birds. All chickens were previously vaccinated in the hatchery at 1 D old with a commercial IB Mass vaccine strain (H120) by spray and no samples from healthy animals were collected. All samples were brought to the Laboratory of Epidemiology and Veterinary Microbiology for diagnosis in the frame of the 2016 national surveillance program of myxoviruses. Organs were washed with PBS then grinded in presence of sterile Minimum Essential Media containing 5% antibiotics before centrifugation at 1500 rpm for 15 min at +4°C. Supernatants of grinded organs and swab samples were collected under sterile conditions. The virus suspension was then filtered through a 0.45 μm Millipore filter to avoid any bacterial contamination, divided into aliquots, and stored at −80°C.
Virus Isolation
A 0.2 ml volume of inoculum of pooled internal organs of each 1 of the 8 birds as well as the supernatants of tracheal and cloacal swabs of the pooled 10 live birds, were inoculated into the allantoic cavity of 11-day-old embryonated specific pathogen-free (SPF) hen eggs for amplification; 3 eggs were used for each sample. A maximum of 1 embryonated egg passage was used. After 48 to 72 h incubation period, the eggs were chilled and the allantoic fluids were harvested and specific embryonic lesions and mortalities were recorded. Collected allantoic fluids, organ, and swabs supernatants were then tested using a real time polymerase chain reaction (PCR), after virus RNA extraction.
RNA Extraction
Supernatants of grinded organs and swab samples as well as allantoic fluids of inoculated embryonated eggs were centrifuged at 12,000 rpm for 5 min at 4°C. The various collected supernatants (0.3 ml) were mixed with 1.0 ml TRIzol reagent (Invitrogen, Carlsbad, CA, USA), The lysates were then incubated at room temperature for 10 min to allow complete dissociation of the nucleoprotein complexes; and then, 0.2 ml chloroform was added and the mixtures were shaken vigorously for 15 s, followed by centrifugation at 12,000 rpm for 15 min at 4°C. The upper aqueous phase was carefully taken out into another microfuge tube and 0.8 ml isopropanol was added to precipitate total RNA. The extracted RNA was kept at −20°C overnight then taken out and centrifuged at 12,000 rpm for 30 min to pellet the RNA. After washing with 70% ethanol, the dried RNA pellet was dissolved in 20 μl RNase-free water and stored at −80°C until use.
Detection of IBV by REAL-TIME RT-PCR Assay
TaqMan real-time RT-PCR was performed to detect conserved (5′-UTR) region of IBV using AgPath-ID™ One-Step RT-PCR Kit Mix (Applied Biosystems) and specific primers and probes previously described by Callison et al. (2006). The cycling conditions for the quantitative RT-PCR were as follows: 1 cycle of 45°C for 10 min (reverse transcription), 95°C for 15 min (RT inactivation and activation of the AmpliTaq Gold DNA polymerase), and 40 cycles of 95°C for 15 s and 60°C for 1 min (annealing, extension step, and fluorescence data collection).
Detection of IBV by Conventional PCR Assay
The 3′-UTR region and the S1 gene were amplified for each isolate using primers reported previously (McFarlane and Verma, 2008; Hewson et al., 2009). One-step RT-PCR was performed using EasyScript One-Step RT-PCR (TransGen Biotech) that combines the first-strand cDNA synthesis with PCR reagents in the same tube to simplify reaction set-up and reduce any contamination. About 50 ng to 5 μg of total RNA of each sample were reverse transcribed at 45°C for 30 min and stopped at 94°C for 5 min followed by 36 cycles at 94°C for 30 s, 55°C/49°C for 30 s for the 3′-UTR and the S1 gene, respectively and then 72°C for 3 min, and a final extension at 72°C for 10 min. The PCR products were separated on 2 and 1% agarose gel and identified by ethidium bromide staining.
Sequencing of Partial S1 Gene and 3 UTR Region
The RT-PCR amplification products of S1-targeted gene were purified from the agarose gel using Easy Pure Genomic DNA Kit (TransGen Biotech). The PCR products were purified with shrimp alkaline phosphatase and exonuclease I to remove the excess dNTPs and primers as follows by adding 0.5 μl exonuclease I (20U/μl) and 0.5 μl shrimp alkaline phosphatase (1 U/μl) to each PCR sample (5 μl) in a final volume reaction of 10 μl and incubating in the thermal cycler at 37°C for 15 min then in 80°C for 15 min to inactivate the enzymes. Purified DNA was then sequenced using the BigDye Terminator version 3.1 Cycle Sequencing Kit that contains thermally stable AmpliTaq DNA polymerase, modified dNTPs, and a set of dye terminators labeled with high-senility dyes. The sequencing reaction was prepared as follows: 10 μl PCR product of each gene, 2 μl BigDye sequencing mix, 4 μl Buffer 5X, 1 μl of specific primer (10 μM), and 4 μl RNase-free H2 O. The sequencing program consisted of a pre-denaturation step at 96°C for 3 min followed by 25 cycles of 10 s denaturation at 96°C, 5 s annealing at 50°C, and 150 s elongation at 60°C. Overlapping DNA fragments were sequenced in both directions from 2 independent PCRs to generate a consensus sequence for each isolate. The finished sequencing reactions were washed with 50 μl of 80% ethanol, centrifuged at 13,000 rpm for 15 min, and dried at room temperature. The DNA was then resuspended in 20 μl RNase-free water. Sequencing reactions were run and analyzed in an ABI PRISM 3500 Genetic Analyzer (Applied Biosystems).
Phylogenetic Analysis
The quality of the obtained sequences was analyzed by BioEdit (version 7.2.5.0), and the sequence similarity was checked against sequences deposited in GenBank using a BLAST search at the National Center for Biotechnology Information (NCBI) site (http://www.ncbi.nlm.nih.gov). The ClustalW was used for sequence alignment and the phylogenetic trees were constructed with MEGA7 using the Neighbor-Joining method with 1,000 bootstrap replicates to assign confidence levels to branches. The obtained sequences of isolated virus were submitted to GenBank. For the analysis of recombination events, the RDP package v.4 software was used.
RESULTS
Clinical and Necropsy Findings
The birds of the suspected broiler flock have shown clinical signs characteristic of IB including gasping, coughing, tracheal rales, and nasal discharge. Mortality was about 60% following secondary Escherichia coli infections confirmed with the Api 20E system. Necropsy revealed increased tracheal mucus, tracheitis with congested lung, slight congestion and airsacculitis, catarrhal exudates in the nasal turbinate.
Virus Isolation
After the first passage in the allantoic cavity of embryonated SPF hen eggs, all inoculated embryos were dead between 48 and 72 h postinfection and showed hemorrhages and developed stunting, curling, and dwarfing, characteristic lesions of IBV infection.
Real Time and Conventional PCR for IBV Detection
Real time PCR was performed on the extracts from each collected organ of necropsied birds, pooled swabs of live birds and allantoic fluids. They were all shown positive with cycle threshold ranging from 15 to 31. No other viral respiratory infections such as avian influenza, metapneumovirus, and Newcastle diseases were detected, indicating the absence of any co-infection.
Out of all the tested extracts, only allantoic fluids of 2 (TN1011/16 and TN 1012/16) out 8 inoculated grinded supernatants were amplified by conventional PCR for both S1 gene and 3'UTR region. Positive amplification bands of 1,700 pb of S1 gene and 433 pb of 3’UTR region were visualized.
Analysis of Partial Sequences of S1 Gene and 3′ UTR Region
The obtained sequences of the 3′-UTR region and the S1 gene of TN1011/16 and TN1012/16 were submitted to GenBank under accession numbers KX290460, KX290461 and KX061458, KX061459, respectively.
To establish the phylogenetic relationships between these Tunisian isolates and 137 IBV reference strains, representative of 6 described genotypes, the standardized method elaborated by Valastro et al. (2016) was followed. Their partial sequences of S1 gene (800pb) were retrieved from GenBank and were compared to both isolates TN1011/16 and TN1012/16.
The results of sequenced S1 gene of the Tunisian isolates TN1011/16 and TN1012/16 showed 98.7% and 97% nucleotide and amino acid identities, respectively. Comparison of the partial S1 gene sequences showed that nucleotide identities between both TN1011/16 and TN1012/16 isolates with referenced IBV strains, ranged from 56.8 to 93%. Indeed, the TN1011/16 and TN1012/16 isolates showed the highest nucleotide sequence identity (92.8, 93.1, and 93.3%) with the Algerian strains: Algeria/26/b1 (KP892759), Algeria/26/b2 (KP892760), and Algeria/26/b3 (KP892761), respectively. Unexpectedly, the lowest nucleotide sequence identity was shown with the old Tunisian strain TN20/00 (56.8%), isolated in the year 2000.
Similarly, the S1 amino acid sequences analysis of TN1011/16 and TN1012/16 revealed its close relatedness to the Algerian strains. In fact, these 2 isolates showed the highest identity to Algeria/26/b1, Algeria/26/b2, and Algeria/26/b3 strains with 88.1, 88.1, and 89% amino acid sequence identities, respectively.
On the other hand, nucleotide identities between TN1011/16 and TN1012/16 strains and either the H120 (KF188436) or the 4/91 vaccine strains (KF377577) were either 78.6 and 78.5% or 74.9 and 74.5%, respectively. Furthermore, comparative analysis of their deduced amino acid sequences showed that they do not exceed 71.7 or 70.2% of identity, respectively.
Comparisons of S1 gene sequences of TN1011/16 and TN1012/16 showed differences in nucleotide and amino acid sequences with other reported strains. Specific mutations were shown for TN1011/16 and TN1012/16 and located in or nearly the hypervariable regions (HVR1 and HVR2), having close relationship with neutralizing epitopes. It was shown that DR-H and DR-P motifs between positions 14 and 17 were detected only in TN1011/16 and TN1012/16, and were different from all reference strains. Other mutations, specific to TN1011/16 and TN1012/16 were also found at positions 97 Q,202 R, and 272 G of S1 amino acid sequences.
The TN1011/16 isolate presented three specific differences in its amino acid sequence at positions250 F, 262 W, and281 G compared to all other strains. Besides, multiple amino acid sequence alignment of S1 gene revealed specific mutations within the genome of TN1012/16 strain at positions262 C,265 S,266 I, and 284 F, which are not found in any other IBV strains. With regard to the comparison of TN1011/16 and TN1012/16 with the Algerian strains, 5 common mutations in the amino acid sequences of the S1 glycoprotein were observed at positions 4 Q, 51 I,153 G, 207 S, and 244 K (Figure 1 ).
Figure 1.

Amino acid sequence alignment of the partial S1 glycoprotein gene. Sequences were aligned using the BioEdit program. The parameter for pairwise alignment was BLOSUM62 similarity matrix. The specific residues to TN1011/16 are boxed in green. The specific residues to TN1012/16 are boxed in blue. The identical residues of TN1011/16 and TN1012/16 are boxed in red. The identical residues to Algeria/26/b1, Algeria/26/b2, and Algeria/26/b3 are boxed in black.
Phylogenetic Analysis
The constructed phylogenetic tree deduced from the nucleotide sequences of the partial S1 glycoprotein gene of the Tunisian isolates (Figure 2 ) showed that they form a commun branch with the 3 Algerian IBV strains but clearly clustered into two distinct subgroups within the GI genotype. The sequence analysis of the partial S1 gene demonstrated that the Tunisian isolates represent a unique genotype as compared to other reference strains from various countries and clearly related to the Algerian strains with a high bootstrap value.
Figure 2.
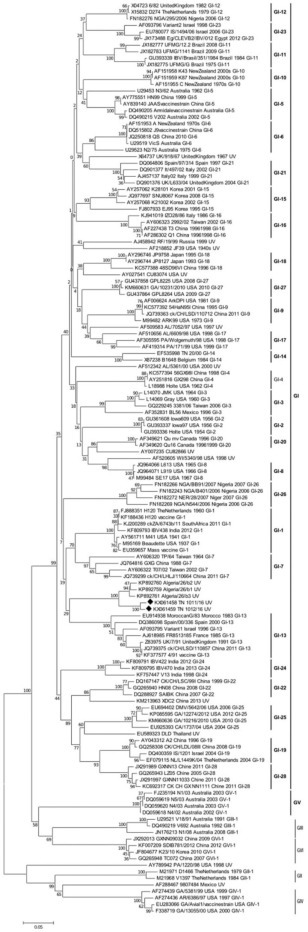
Phylogenetic tree of IBV based on the alignment of the nucleotide sequences of the S1 gene. Sequences were aligned using ClustalW with default settings in BioEdit (version 7.2.5.0). The phylogenetic tree was constructed by the Neighbor-Joining method with 1,000 bootstrap replicates using MEGA7. Tunisian isolates are marked in red square.
The phylogenetic tree allowed confirmation of the presence of unique variant of IBV circulating in Tunisia in 2016. According to the new proposed classification (Valastro et al., 2016), the generated phylogenetic tree showed the presence of 6 genotypes (GI-GVI) with 28 lineages into genotype I (GI), which contains the majority of IBV strains. Besides, 5 genotypes (GII-GVI) contain 1 lineage each and a number of unique variants that do not belong to any lineage (Valastro et al., 2016). The TN1011/16 and TN1012/16 isolates have not been grouped within any lineage and each one represents a unique variant. However, they may be grouped with the three Algerian strains since they share the highest nucleotide and amino acid identities. They are distant from the Tunisian strain isolated in 2000 (TN 20/00), which belongs to lineage 14 in GI. The Tunisian and the Algerian strains do not form a lineage yet since they were isolated from only two outbreaks (Valastro et al., 2016).
Using the RDP package v.4 software, developed by Martin et al. (2010), which identifies and characterizes individual recombination events, it was shown that no recombination events has occurred between the TN1011/16 and TN1012/16 isolates as compared to different sequences in the GeneBank database. This result supports the novelty of the strains by showing the absence of probable positions of break and recombination points. However, data analysis of the old Tunisian isolate TN20/00, showed that it is a recombinant strain with probable positions of breakpoint and recombination events at 583 and 612. Besides, its sequences came from the recombination of the major parent Alabama/10,902/97 (AF510334) with 76.4% similarity and the minor parent Spain/99/319 (DQ064810).
With regard to the 3′UTR, the nucleotide sequences of TN1011/16 and TN1012/16 isolates were aligned and compared to 16 representative 3′UTR strains, available in the GenBank, from the same reference strains used for the S1 gene classification. It appeared that the 3′UTR nucleotide sequences of both isolates were closely related to the 4/91 vaccine (KF377577) and the Ck/CH/LSD/110,857 (KP118885) strain, with 97.67 and 97.4% nucleotide identities respectively; such results were contrary to those obtained with the alignment of S1 gene. Unfortunately, the 3′UTR region was not sequenced for the Algerian strains showing highest sequence similarity based on the S1 gene. Moreover, the multiple nucleotide sequence alignment of the 3′UTR of TN1011/16 and TN1012/16 strains with all sequences available in GenBank revealed 2 new mutations 19 A and 117 C that have not been previously reported. Thus, the phylogenetic tree based on the 3'UTR region showed a completely different result with that obtained using the S1 gene. Indeed, GV and GI genotypes were grouped as one genotype. On the other hand, TN1011/16 and TN1012/16 strains were grouped in the lineage 13 within genotype I (GI-13) whereas they were classified as single variantwithin genotype I (GI) using the S1 gene (Figure 2).
DISCUSSION
The IBV has long emerged worldwide with several genotypes, serotypes and variants with no consistent basis for its classification. Recently, the study of Valastro et al. (2016) proposed a phylogenetic method that harmonizes the classification of IBV based on the S1 gene either in its entirety or through the hypervariable regions HVR1 and HVR2. Several new variants and genotypes have been identified in recent years. Very recently Chen et al. (2017) have identified a new lineage (GI-28) within the genotype I (GI) confirming the emergence of new genotypes and variants throughout the world. In this study, circulating IBV strains, in Tunisia, were examined to determine their genotype in comparison with 137 sequences of reference strains, representative of 6 described genotypes. The Tunisian isolates TN1011/16 and TN1012/16 have shown a distinct molecular background as compared to other genotypes and have been included in a new branch along with the Algerian strains with which they share the highest percentage of nucleotide identity (79.9%).
Based on the study of Valastro et al. (2016), which states that a genotype is defined only if a new branch appears and is composed of isolated strains from at least 3 different outbreaks. Since both Algerian and Tunisian isolates stemming from only 2 different outbreaks, our reported isolates were designated new unique variant. We notice that Tunisia is surrounded by countries where different genotypes that are quite distant from our isolates, most of which are of genotype G1-23, which includes Israelian and Egyptian variants (Even-Chen et al., 2014; Abdel-Sabour et al., 2017) as well as Turkish and Libyan variants that have been identified in 2011 and 2012, respectively (Kahya et al., 2013; Awad et al., 2014; Yilmaz et al., 2016). In Morocco, continuous monitoring of the IBV spread has shown the emergence of Italy 02 genotype, co-circulating with both Massachusetts and 4/91 serotypes clustered in the G1-21, G1-1, and G1-13, respectively (Fellahi et al., 2015; 2017), while Spanish IBV strains belong to the QX genotype (G1-19) (Moreno et al., 2017).
The present study confirms that new unique variant belonging to group I (GI) are represented by the 2 new IBV strains isolated in Tunisia, in 2016. The TN1011/16 and TN1012/16 strains were identified in broiler chickens suffering from severe respiratory distresses, using real-time RT-PCR. They were genotyped using the S1 gene and the 3′UTR region sequence analyses. The alignment of the S1 sequences of both Tunisian isolates along with 137 referenced IBV strains revealed a high homology and a close relationship with the Algerian strains, isolated in 2012 and 2013, but not with the old Tunisian variant isolated during 2000 to 2007 (Bourogâa et al., 2009a; Sid et al., 2015). Indeed, the first characterization of IBV in Tunisia has described a novel variant, serologically, very distant from the Mass H120 vaccine strain but closely related to CR88121 and D274 (Bourogâa et al., 2009a, 2009b). Between 2007 and 2010, other IBV variants have emerged showing between 50 and 77% similarities with variants isolated in 2000 and 2001 (Bourogâa et al., 2012).
In 2012, following variations in serotypes of IBV circulating in Tunisian flocks, combining CR88 (793B) with H120 (Mass type) vaccines was included in the natural vaccination program against IBV, for better protection. Indeed, Bourogaa et al. (2009b) conducted experimental trials to evaluate tracheal and renal cross protection in chickens vaccinated by oculo-nasal instillation. The results showed a weak protection when H120 strain was used alone. There were no reduction in tracheal and kidney damage after a challenge with the strain TN20/00. On the other hand, the combination of heterologous serotypes, based on the vaccine strains (H120) and CR88, allowed a higher protection. Thus, a vaccination protocol that combines CR88 with H120 has since been applied. The following years, the variant strain 4/91 has been also introduced.
The results obtained when using the 3′UTR sequences for the classification of the TN1011/16 and TN1012/16, isolates did not correlate with those obtained when using the S1 gene sequences. Phylogenetic analysis used to compare the nucleotide sequences of TN1011/16 and TN1012/16 with other reported strains revealed different clustering depending on whether the S1 gene or the 3′UTR is used for such analysis. Our results did not correlate with those of Hewson et al. (2009) who have found strong inter-strain correlation between the S1 gene and the hypervariable region in the 3′UTR and proposed the use of the 3′UTR for IBV strain differentiation. Williams et al. (1993) found that the 3′UTR contains a hypervariable region that may vary from 53 to 92.8% between IBV strains and may also be absent in other strains such as Mass 41 strain.
The comparison of the 3′UTR nucleotide sequences of TN1011/16 and TN1012/16 to that of 16 representative strains showed different results from those found with the S1 gene analysis. It is concluded that phylogenetic classification, using the 3'UTR region, does not correlate with that of the S1 gene, in particular with the new classification of IBV proposed by Valastro et al. (2016). For this reason, it is not recommended to use 3'UTR region in this regard.
Besides, the phylogenetic relationships of TN1011/16 and TN1012/16 strains with vaccine strains commonly used in Tunisia were evaluated. Indeed, the IBV H120 and 4/91 live attenuated vaccine strains; shared low similarities with the newly characterized strains and classified in fairly distant groups based on phylogenetic analysis of the S1 gene sequences. On the other hand, when phylogenetic analysis was based on the 3′UTR sequences, the attenuated vaccine strain 4/91 seemed to be closer to the new TN1011/16 and TN1012/16 strains.
The vaccine strains H120 and 4/91 currently used in Tunisia are classified in lineages GI-1 and GI-13 within genotype I, respectively. It is then clear that the new variants TN1011/16 and TN1012/16 are quite distant from these vaccine strains. Besides, Sid et al. (2015) have also reported that the Algerian strains are different from the new 4/91 strain and the commonly H120 strain used for a long time in the field.
The results trigger serious concerns about the efficacy of the vaccines used at this time in Tunisia (H120 at day old and 4/91 at 10 to 14 D as boosters). This newly proposed classification will help to facilitate monitoring and classification of newly emerged strains of IBV through epidemiological studies aimed at understanding and controlling the newly characterized. Tunisian isolates, shown to be new variants of IBV, highlighted the mutations detected in S1 gene and 3′UTR region.
Such classification would bring information about the genetic background necessary for the control and the surveillance of the disease. This result can be strongly supported by serological tests confirming the emergence of new serotypes. In vivo tests will also be useful to determine the pathogenicity of these strains and to test the efficacy of the H120 and 4/91 vaccines currently used against these strains.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
ACKNOWLEDGMENTS
This study was supported by the grant from the «Ministry of Higher Education and Scientific Research of Tunisia».
REFERENCES
- Abdel-Sabour M.A., Al-Ebshahy E.M., Khaliel S.A., Abdel-Wanis N.A., Yanai T. Isolation and molecular characterization of novel infectious bronchitis virus variants from vaccinated broiler flocks in Egypt. Avian Dis. 2017;61:307–310. doi: 10.1637/11566-121516-RegR. [DOI] [PubMed] [Google Scholar]
- Awad F., Baylis M., Ganapathy K. Detection of variant infectious bronchitis viruses in broiler flocks in Libya. Int. J. Vet. Sci. Med. 2014;2:78–82. [Google Scholar]
- Bourogâa H., Miled K., Gribâa L., El Behi I., Ghram A. Characterization of new variants of avian infectious bronchitis virus in Tunisia. Avian Dis. 2009;53:426–433. doi: 10.1637/8666-022609-Reg.1. [DOI] [PubMed] [Google Scholar]
- Bourogaa H., Miled K., Larbi I., Nsiri J., Gribaa L., El Behi I., Ben Rhouma W., Allagui E., Sassi H., Ghram A. Avian infectious bronchitis disease in Tunisia: seroprevalence, pathogenicity and compatibility studies of vaccine-field isolates. Archs. Inst. Pasteur Tunis. 2009;86:75–83. [PubMed] [Google Scholar]
- Bourogâa H., Hellal I., Hassen J., Fathallah I., Ghram A. S1 gene sequence analysis of new variant isolates of avian infectious bronchitis virus in Tunisia. Vet. Med. Res. Rep. 2012;3:41–48. doi: 10.2147/VMRR.S32498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boursnell M.E., Brown T.D., Foulds I.J., Green P.F., Tomley F.M., Binns M.M. Completion of the sequence of the genome of the coronavirus avian infectious bronchitis virus. J. Gen. Virol. 1987;68:57–77. doi: 10.1099/0022-1317-68-1-57. [DOI] [PubMed] [Google Scholar]
- Breslin J.J., Smith L.G., Fuller F.J., Guy J.S. Sequence analysis of the turkey coronavirus nucleocapsid protein gene and 3 untranslated region identifies the virus as a close relative of infectious bronchitis virus. Virus Res. 1999;65:187–193. doi: 10.1016/S0168-1702(99)00117-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callison S.A., Hilt D.A., Boynton T.O., Sample B.F., Robison R., Swayne D.E., Jackwood M.W. Development and evaluation of a real-time Taqman RT-PCR assay for the detection of infectious bronchitis virus from infected chickens. J. Virol. Methods. 2006;138:60–65. doi: 10.1016/j.jviromet.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D. Coronavirus avian infectious bronchitis virus. Vet. Res. 2007;38:281–297. doi: 10.1051/vetres:2006055. [DOI] [PubMed] [Google Scholar]
- Chen H.W., Huang Y.P., Wang C.H. Identification of intertypic recombinant infectious bronchitis viruses from slaughtered chickens. Poult. Sci. 2010;89:439–446. doi: 10.3382/ps.2009-00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Jiang L., Zhao W., Liu L., Zhao Y., Shao Y., Li H., Han Z., Liu S. Identification and molecular characterization of a novel serotype infectious bronchitis virus (GI-28) in China. Vet. Microbiol. 2017;198:108–115. doi: 10.1016/j.vetmic.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherif A., Bouslama A., Chakroun C., Turki I., Kaboudi K., Bouzouaia M. Suivi sérologique de la vaccination contre les principales viroses aviaires dans les élevages de reproducteurs en Tunisie. Revue Élev. Méd. vét. Pays trop. 2010;63:5–11. [Google Scholar]
- Cook J.K., Jackwood M., Jones R.C. The long view: 40 years of infectious bronchitis research. Avian Pathol. 2012;41:239–250. doi: 10.1080/03079457.2012.680432. [DOI] [PubMed] [Google Scholar]
- Even-Chen T., Barbakov M., Hiefetez S., Perelman B., Kin E., Ashash U., Finger A., Banet-Noach C. 8th International Symposium on Avian Corona- And Pneumoviruses And Complicating Pathogens. Rauischholzhausen; Germany: 2014. Development of new infectious bronchitis variant vaccines from local israeli isolates based on their prevalence in the field; pp. 215–218. [Google Scholar]
- Fellahi S., Ducatez M., El Harrak M., Guérin J.L., Touil N., Sebbar G., Bouaiti el A., Khataby K., Ennaji M.M., El-Houadfi M. Prevalence and molecular characterization of avian infectious bronchitis virus in poultry flocks in Morocco from 2010 to 2014 and first detection of Italy 02 in Africa. Avian Pathol. 2015;44:287–295. doi: 10.1080/03079457.2015.1044422. [DOI] [PubMed] [Google Scholar]
- Fellahi S., El Harrak M., Khayi S., Guerin J.L., Kuhn J.H., El Houadfi M., Ennaji M.M., Ducatez M. Phylogenetic analysis of avian infectious bronchitis virus isolates from Morocco: a retrospective study (1983 to 2014) Virol. Sin. 2017;32:155–158. doi: 10.1007/s12250-016-3885-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng K.Y., Chen T., Zhang X., Shao G.M., Cao Y., Chen D.K., Lin W.C., Chen F., Xie Q.M. Molecular characteristic and pathogenicity analysis of a virulent recombinant avain infectious bronchitis virus isolated in China. Poult. Sci. 2018;97:3519–3531. doi: 10.3382/ps/pey237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewson K., Noormohammadi A.H., Devlin J.M., Mardani K., Ignjatovic J. Rapid detection and non-subjective characterisation of infectious bronchitis virus isolates using high-resolution melt curve analysis and a mathematical model. Arch. Virol. 2009;154:649–660. doi: 10.1007/s00705-009-0357-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Committee on Taxonomy of Viruses (ICTV). Release. 2009. ICTV 9th Report; EC41: Leiden, June 2009; Email ratification 2009 (MSL #25). https://talk.ictvonline.org//taxonomy/p/taxonomy-history?taxnode_id=19980731.
- Kahya S., Coven F., Temelli S., Eyigor A., Carli K.T. Presence of IS/1494/06 genotype-related infectious bronchitis virus in breeder and broiler flocks in Turkey. Vet. J. Ankara Univ. 2013;60:27–31. [Google Scholar]
- Leghari R.A., Fan B., Wang H., Bai J., Zhang L., Abro S.H., Jiang P. Full-length genome sequencing analysis of avian infectious bronchitis virus isolate associated with nephropathogenic infection. Poult. Sci. 2016;95:2921–2929. doi: 10.3382/ps/pew259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Han Z., Chen J., Liu X., Shao Y., Kong X., Tong G., Rong J. S1 gene sequence heterogeneity of a pathogenic infectious bronchitis virus strain and its embryo-passaged, attenuated derivatives. Avian Pathol. 2007;36:231–234. doi: 10.1080/03079450701338730. [DOI] [PubMed] [Google Scholar]
- Martin D.P., Lemey P., Lott M., Moulton V., Posada D., Lefeuvre P. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26:2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane R., Verma R. Sequence analysis of the gene coding for the S1 glycoprotein of infectious bronchitis virus (IBV) strains from New Zealand. Virus Genes. 2008;37:351–357. doi: 10.1007/s11262-008-0273-6. [DOI] [PubMed] [Google Scholar]
- Moreno A., Franzo G., Massi P., Tosi G., Blanco A., Antilles N., Biarnes M., Majó N., Nofrarías M., Dolz R., Lelli D., Sozzi E., Lavazza A., Cecchinato M. A novel variant of the infectious bronchitis virus resulting from recombination events in Italy and Spain. Avian Pathol. 2017;46:28–35. doi: 10.1080/03079457.2016.1200011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussan D.A., Totanji W.S., Khawaldeh G.Y. Molecular subtype of infectious bronchitis virus in broiler flocks in Jordan. Poult. Sci. 2008;87:661–664. doi: 10.3382/ps.2007-00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki S.G., Sawicki D.L., Siddell S.G. A contemporary view of coronavirus transcription. J. Virol. 2007;81:20–29. doi: 10.1128/JVI.01358-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sid H., Benachour K., Rautenschlein S. Co-infection with multiple respiratory pathogens contributes to increased mortality rates in Algerian poultry flocks. Avian Dis. 2015;59:440–446. doi: 10.1637/11063-031615-Case.1. [DOI] [PubMed] [Google Scholar]
- Sjaak de Wit J.J., Cook J.K., van der Heijden H.M. Infectious bronchitis virus variants: a review of the history, current situation and control measures. Avian Pathol. 2011;40:223–235. doi: 10.1080/03079457.2011.566260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valastro V., Holmes E.C., Britton P., Fusaro A., Jackwood M.W., Cattoli G., Monne I. S1 gene-based phylogeny of infectious bronchitis virus: An attempt to harmonize virus classification. Infect. Genet. Evol. 2016;39:349–364. doi: 10.1016/j.meegid.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A.K., Wang L., Sneed L.W., Collisson E.W. Analysis of a hypervariable region in the 3' non-coding end of the infectious bronchitis virus genome. Virus Res. 1993;28:19–27. doi: 10.1016/0168-1702(93)90086-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz H., Altan E., Cizmecigil U.Y., Gurel A., Ozturk G.Y., Bamac O.E., Aydin O., Britton P., Monne I., Cetinkaya B., Morgan K.L., Faburay B., Richt J.A., Turan N. Phylogeny and S1 gene variation of infectious bronchitis virus detected in broilers and layers in Turkey. Avian Dis. 2016;60:596–602. doi: 10.1637/11346-120915-Reg.1. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Cheng J., Yan S., Jia W., Zhang K., Zhang G. S gene and 5a accessory gene are responsible for the attenuation of virulent infectious bronchitiscoronavirus. Virology. 2019;533:12–20. doi: 10.1016/j.virol.2019.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]