

Abstract
Background. Highly pathogenic avian H5N1 influenza viruses preferentially infect alveolar type II pneumocytes in human lung. However, it is unknown whether this cellular tropism contributes to high viral virulence because the primary target cells of other influenza viruses have not been systematically studied.
Methods. We provide the first comparison of the replication, tropism, and cytokine induction of human, highly pathogenic avian influenza A virus subtype H5N1 and other animal influenza A viruses in primary human lung organ cultures.
Results. Subytpe H5N1 and human-adapted subtype H1N1 and H3N2 viruses replicated efficiently in the lung tissue, whereas classic swine and low-pathogenicity avian viruses propagated only poorly. Nevertheless, all viruses examined were detected almost exclusively in type II pneumocytes, with a minor involvement of alveolar macrophages. Infection with avian viruses that have a low and high pathogenicity provoked a pronounced induction of cytokines and chemokines, while human and pandemic H1N1-2009 viruses triggered only weak responses.
Conclusions. These findings show that differences in the pathogenic potential of influenza A viruses in the human lung cannot be attributed to a distinct cellular tropism. Rather, high or low viral pathogenicity is associated with a strain-specific capacity to productively replicate in type II pneumocytes and to cope with the induced cytokine response.
Influenza A viruses are prototypical members of the Orthomyxoviridae family that have a segmented RNA genome and circulate mainly in human, porcine, and avian hosts [1]. The socioeconomic impact of influenza A viruses in humans is significant because they cause acute severe respiratory disease resulting in 3000–49 000 annual deaths in the United States alone [2]. The avian and porcine host reservoirs directly influence the epidemiology of human influenza because novel strains with some or all of their genes originating from animal viruses occasionally emerge and spread in the population. The most devastating of these pandemics was the Spanish Flu in 1918, which claimed up to 40 million deaths worldwide [3]. In 2009, a novel influenza A subtype H1N1 virus with gene segments from the porcine host reservoir emerged in North America and rapidly spread worldwide, causing the first influenza pandemic of this century [4].
The replication of most avian influenza viruses in the human respiratory tract is attenuated [5, 6], which was initially thought to be linked to distinct receptor specificity. The hemagglutinin (HA) molecule of avian viruses preferentially binds sialic acids with an α-2,3 linkage to galactose (α-2,3SA), which corresponds to the high expression of α-2,3SA in the intestine of infected birds, the main replication site for avian viruses [7]. In contrast, the HA of typical human strains binds primarily to α-2,6SA, which is highly expressed in the human trachea [8]. However, it was recognized recently that both α-2,3SA and α-2,6SA receptor determinants are present in human lung and that both avian and human viruses can attach to alveolar epithelial cells [9–11]. These findings indicated that factors in addition to a specific receptor must limit the replication of avian viruses in the human host.
Highly pathogenic avian influenza viruses of the H5N1 subtype (HPAIV-H5N1) are exceptional in that they cause fatal respiratory disease in humans, with a mortality rate close to 60%, but the precise reasons for this are far from being understood [12, 13]. HPAIV-H5N1 viruses induce high levels of cytokines and chemokines, which possibly contribute to enhanced immunopathology [14–18]. However, the significance of this hypercytokinemia for disease severity has recently been debated on the basis of controversial findings from an autopsy [19] and from animal models [20–22].
The main pathological finding in deceased patients who had H5N1 infection is diffuse alveolar damage, and viruses from such patients were indeed shown to have infected alveolar epithelia [12, 13]. This led to the suggestion that a distinct cellular tropism could be at least partly responsible for the high pathogenicity of these viruses [11, 12, 23]. Various cell types that reside in the human lung, including goblet cells, type I and type II pneumocytes, and alveolar macrophages, are believed to react differently toward a viral challenge [24]. Type I pneumocytes are flattened nondividing cells that make up 8% of all cells in the alveolar tissue but cover >95% of the alveolar surface and are responsible for gas exchange and fluid homeostasis. In contrast, the cuboidal type II pneumocytes, which account for 15% of total cells, produce surfactant and may serve as stem cells that differentiate into type I pneumocytes [24]. While type II pneumocytes display large amounts of mitochondria and endoplasmic reticulum in their cytoplasm, these organelles are only sparely represented in type I pneumocytes [25, 26], indicating high and low metabolic activities, respectively. Significantly, HPAIV-H5N1 was shown to mainly bind to and infect type II pneumocytes [10, 23, 27]. In contrast, seasonal influenza viruses initially infect cells in the upper respiratory tract, but in a minority of cases they also penetrate into the lungs, causing severe respiratory complications [28, 29]. In fixed slices of human lung tissue, they attached primarily to type I pneumocytes [11]. However, it has not been established whether seasonal strains also productively infect these cells, because there is a scarcity of autopsy reports and uncertainties about the informative value of tissue culture systems and animal models.
To improve our understanding of the mechanisms that support or restrict the growth of influenza viruses in the lower respiratory tract of humans, we used a human ex vivo lung model. This allowed, for the first time, a systematic comparison of influenza A viruses from human, avian, and porcine hosts for their replication, cell tropism, and cytokine induction in an experimental model that retains the 3-dimensional structure and cell diversity of human lung tissue. Most strikingly, all examined viruses predominantly infected type II pneumocytes, although we observed large differences between the strains with regard to replication and cytokine induction. These findings suggest that the pathogenicity of influenza viruses in human lung is not determined by cell tropism, but is rather associated with the extent of replication in type II pneumocytes and with the capacity to cope with induced cytokine responses.
MATERIALS AND METHODS
Lung Explants, Tissue Culture, and Viruses
Fresh lung explants were obtained from patients undergoing lung resection at thoracic surgery centers in Berlin. Written informed consent was obtained from all patients, and the study was approved by the ethics committee at the Charité clinic (projects EA2/050/08 and EA2/023/07). Tumor-free normal lung tissue was immediately stamped into small cylinders (height ∼3; diameter 8 mm) and incubated in Roswell Park Memorial Institute 1649 medium (containing 0.3% bovine serum albumin, 2 mM L-glutamine, and antibiotics) at 37°C with 5% CO2. After overnight incubation, lung organ cultures were inoculated with 4 × 105 plaque-forming units (PFU) of virus for 1 hour, followed by 3 washing steps with phosphate-buffered saline to remove excess virus. For growth-curve analysis, supernatants were titrated on Madin-Darby canine kidney (MDCK) cells by standard plaque assay, as described elsewhere [30]. MDCK cells were grown at 37°C with 5% CO2 in minimum essential medium (containing 10% fetal calf serum, 2 mM L-glutamine, and antibiotics). The viruses used for this study included the HPAIV A/Thailand/1(Kan-1)/2004 (Thai/04[H5N1]) strain isolated from a fatal human case, the pandemic A/Bayern/63/2009 (Bay/09[H1N1pdm]) strain, the seasonal A/New Caledonia/20/1999 (NC/99[H1N1]) and A/Panama/2007/1999 (Pan/99[H3N2]) strains, the low-pathogenicity avian virus A/duck/Alberta/60/76 (Dk/Alb[H12N5]) strain, and the classic swine virus A/swine/Wisconsin/1/67 (Sw/Wis[H1N1]) strain (Table 1). We used a recombinant version of the influenza A/Panama/2007/1999 strain generated by reverse genetics, which will be described elsewhere. The human virus strains were grown on MDCK cells, whereas the duck and swine isolates were propagated in 11-day-old embryonated chicken eggs. Virus stocks were aliquoted, stored at –80°C and titrated on MDCK cells by a plaque assay. All experiments with H5N1 virus were performed in a biosafety level 3 containment laboratory approved by local authorities for work with these viruses.
Table 1.
Influenza A Viruses (IAV) Used in This Study
| Name | Subtype | Abbreviation | Property |
|---|---|---|---|
| A/Panama/2007/1999 | H3N2 | Pan/99(H3N2) | Seasonal human IAV |
| A/New Caledonia/20/1999 | H1N1 | NC/99(H1N1) | Seasonal human IAV |
| A/Bayern/63/2009 | H1N1 | Bay/09(H1N1pdm) | Pandemic H1N1-2009 IAV |
| A/Thailand/1(Kan-1)/2004 | H5N1 | Thai/04(H5N1) | Highly pathogenic avian IAV |
| A/duck/Alberta/60/1976 | H12N5 | Dk/Alb(H12N5) | Low-pathogenicity avian IAV |
| A/swine/Wisconsin/1/1967 | H1N1 | Sw/Wis(H1N1) | Classic swine IAV |
Immunohistochemistry and Confocal Microscopy
Lung tissue was fixed in 4% paraformaldehyde (Sigma). After deparaffination, tissue sections were heated in citrate buffer (10 mM sodium citrate; pH 6.0) at 95°C for 30 minutes for antigen retrieval and permeabilized with 1% Triton-X-100 for 15 minutes. The slides were blocked with 5% adequate serum for 30 minutes and incubated with diluted Alexa Fluor 488–labeled influenza A virion–specific antibody (Serotec OBT1551; 1:30) overnight at 4°C. Influenza A virion antibodies were labeled using the DyLight 488 Microscale Antibody Labeling Kit (Thermo Scientific). If desired, α-2, 3– and α-2, 6–linked sialic acids were detected by Vector Red staining of lectins. Tissue sections were then incubated overnight at 4°C with primary antibodies for prosurfactant protein C (Chemicon AB3786; 1:800), EMP2 (Sigma HPA014711; 1:50), and/or CD68 (Abcam ab955-500; 1:50), respectively. Afterward, the slides were incubated with corresponding Alexa 546– or Alexa 555–conjugated secondary antibodies (Invitrogen), mounted in Mowiol, and analyzed with LSM 780 or LSM5 Pascal confocal microscopes (objectives 40×, Plan-Neofluar/oil, NA 1.3; 63×, Plan-Apochromat/oil, NA 1.4; Zeiss, Jena, Germany).
Transmission Electron Microscopy
Human lung explants were infected with virus for 24 hours and were subsequently fixed by immersion in 2.5% glutaraldehyde (in 50 mM HEPES) and postfixed with 1% OsO4 (for 1 hour), 0.1% tannic acid (in 50 mM HEPES for 30 minutes), and 2% uranyl acetate (for 2 hours). Samples were dehydrated in graded ethanols and embedded in Epon resin. Thin sections were cut using an ultramicrotome (Ultracut S, Leica, Wetzlar, Germany) and were counterstained with 2% uranyl acetate (for 20 minutes) and lead citrate (for 3 minutes). Sections were examined with a TEM 902 microscope (Carl Zeiss SMT AG). Images were digitized using a slow-scan charge-coupled-device camera (Pro Scan, Scheuring, Germany).
Cytokine and Chemokine Measurement
The concentrations of IP-10 and MIP-1β in supernatants of infected lung explants were determined with the Procarta Cytokine Assay Kit (Panomics) after inactivation of virus during 1-hour incubation with 0.15% TNBP/Triton X-100 at 4°C. Interferon β (IFN-β) and interleukin 1β (IL-1β) levels in supernatants were analyzed by using commercial enzyme-linked immunosorbant assay kits (Fujirebio, Tokyo, and BD Biosciences, Heidelberg).
RESULTS
Human Lung Explants Support Replication of Influenza A Viruses
To systematically compare the replication of human and animal influenza A viruses (Table 1) in human lungs, we established an ex vivo explant model. The lung specimens mainly consisted of complex alveolar tissue derived from the lower pulmonary lobe (Figure 1A). Infection of the explants with the prototypic seasonal Pan/99(H3N2) virus resulted in productive infection with a strong increase in the number of infectious viral particles in the culture supernatant (Figure 1B). The viability of the specimens remained stable for up to 96 hours as judged by constantly low values for released lactate dehydrogenase (data not shown). Hence, infections of explants from the same donor at 0 hours or 1 day later resulted in only minor differences between the viral growth curves (Figure 1B). The addition of trypsin did not increase virus titers, suggesting that protease activity required for cleavage activation of the viral HA was preserved in the lung explants (data not shown).
Figure 1.
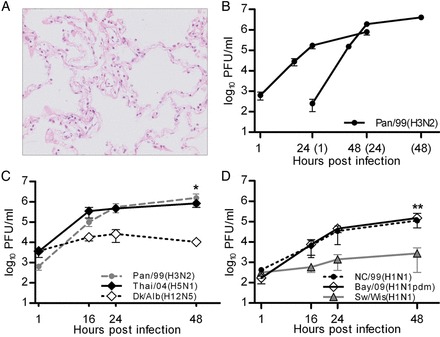
Influenza virus replication in human lung explants. A, Hematoxylin-eosin staining of human lung tissue 48 hours after mock infection illustrating the preserved tissue structure. B, Human lung explants were either infected at 0 hours or 24 hours later with 4 × 105 plaque-forming units (PFU) of Pan/99(H3N2) virus and collected supernatants were titrated on Madin-Darby canine kidney (MDCK) cells. C and D, Human lung explants were either infected with Pan/99(H3N2), Thai/04(H5N1), and Dk/Alb(H12N5) (C) or with Bay/09(H1N1pdm), NC/99(H1N1), and Sw/Wis(H1N1) (D). Supernatants were collected at indicated time points and titrated on MDCK cells. Mean values ± standard error of the mean of triplicates from 1 (B), 6 (C), and 3 (D) independent experiments are shown. Asterisks indicate significant differences between Dk/Alb(H12N5) and Thai/04(H5N1) (B) and between Sw/Wis(H1N1) and NC/99(H1N1) (C). *P < .05 and **P < .01, by the 2-tailed Student t test.
Systematic comparison of replication efficiency revealed remarkable and reproducible differences between the strains: the seasonal Pan/99(H3N2) and highly pathogenic Thai/04(H5N1) viruses propagated efficiently, whereas the low-pathogenicity avian Dk/Alb(H12N5) virus replicated 2 orders of magnitude lower (Figure 1C). Similarly, the seasonal and pandemic H1N1 viruses NC/99(H1N1) and Bay/09(H1N1pdm) replicated to comparable titers, while the growth of the classic swine influenza virus Sw/Wis(H1N1) was significantly attenuated (Figure 1D). The observed differences were unlikely the result of a specific receptor preference because lectin histochemical analysis identified both α-2,3SA and α-2,6SA receptor determinants in the alveolar compartment (Supplementary Figure 1), in confirmation of a previous report [9]. Overall, these data demonstrated substantial differences in the growth capacities of human and animal influenza A viruses in human lung tissue. Interestingly, there appeared to be a correlation between the ability of the different influenza A viruses to replicate in the explants and their capacity to cause lower respiratory tract infections in humans.
Highly Pathogenic H5N1 and Seasonal and Pandemic Viruses Predominantly Infect Type II Pneumocytes
The observed growth restriction of porcine and low-pathogenicity avian influenza viruses could have been caused by several factors, including a lack of initial infection, low viral gene expression, strong activation of innate immune responses, or differences in the cellular tropism. To identify possible differences between the human and animal virus strains on a cellular level, we infected human lung explants and performed immunohistochemical analyses (Figure 2). Despite poor propagation in human lung tissue, viral antigen was detected in cells infected with low-pathogenicity avian and classic swine virus at similar rates. Interestingly, costaining of virion antigens and the type II marker surfactant protein C precursor [25] revealed that the vast majority of cells infected with Sw/Wis(H1N1), Dk/Alb(H12N5), Pan/99(H3N2), NC/99(H1N1), Thai/04(H5N1), or Bay/09(H1N1pdm) virus represented type II pneumocytes (Figure 2A and 2B). Conversely, we failed to detect any of these viruses in type I pneumocytes, as judged by costaining for the marker antigens EMP2 (Figure 3) and caveolin 1 (data not shown), although lectin staining confirmed the presence of receptors for human and avian strains on both pneumocyte types (Figure 3C). Double staining of viral antigen and the CD68 marker also detected influenza virus antigen–positive alveolar macrophages for all viruses tested (Supplementary Figure 2). Most likely, this indicated an ongoing infection of macrophages because no influenza A virus–positive cells were detected after incubation with ultraviolet light (UV)–inactivated viruses (data not shown), although it cannot be excluded that parts of the signals were caused by phagocytosed viral antigen. Quantification of the infected cell types on tissue cross sections showed that alveolar macrophages accounted only for 4%–11% of the antigen-positive cells, whereas 89%–96% were type II pneumocytes, depending on the virus strain (Figure 4).
Figure 2.
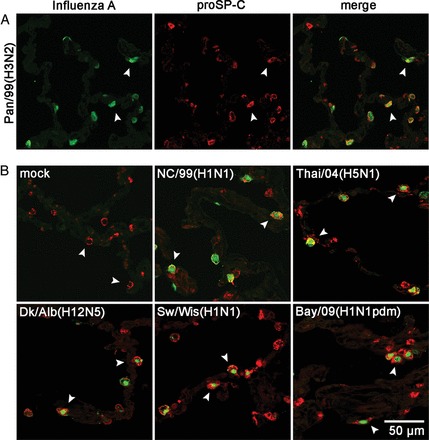
Seasonal, high- and low-pathogenicity avian, porcine, and pandemic 2009 viruses infect mainly type II pneumocytes. A, Lung explants were infected with Pan/99(H3N2) for 24 hours. Fixed and deparaffinated tissue sections were incubated with fluorescently labeled anti–influenza A virus antibody (green channel) and anti–pro-SP-C antibody (red channel) to detect virus-infected type II pneumocytes by confocal microscopy. The overlay of both channels (merge) demonstrated staining of both antigens in the same cells (white arrowheads). B, Lung explants were not infected (mock) or infected with the strains NC/99(H1N1), Thai/04(H5N1), Dk/Alb(H12N5), Sw/Wis(H1N1), and Bay/09(H1N1pdm) for 24 hours and processed as described in A. Representative pictures of at least 3 independent experiments are shown.
Figure 3.
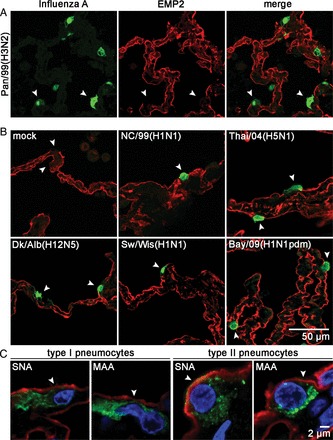
Seasonal, high- and low-pathogenicity avian, porcine, and pandemic 2009 viruses do not infect type I pneumocytes. A, Human lung tissue was infected with Pan/99(H3N2) for 24 hours. Slices were incubated with fluorescently labeled anti–influenza A virus antibody (green channel) to detect virus-infected cells (white arrowheads) and anti-EMP2 antibody (red channel) to detect type I pneumocytes. B, Lung explants were not infected (mock) or were infected with the strains NC/99(H1N1), Thai/04(H5N1), Dk/Alb(H12N5), Sw/Wis(H1N1), and Bay/09(H1N1pdm) for 24 hours and processed as described in A. Micrographs show fields with merged red and green channels. Representative pictures of at least 3 independent experiments are shown. C, Detection of human (α-2,6SA) and avian (α-2,3SA) influenza virus receptors on type I and type II pneumocytes. Sections of uninfected human lung tissue were incubated with alkaline phosphatase–conjugated lectin from Maackia amurensis (MAA; α-2,3SA specificity) or Sambucus nigra agglutinin (SNA; α-2,6SA specificity) and developed by Vector Red staining (red channel), followed by incubation with anti–caveolin 1 (green channel) to detect type I pneumocytes or anti–prosurfactant protein C (green channel) to detect type II pneumocytes. Nuclei were stained with DAPI (blue channel). The slides were analyzed by confocal microscopy by spectral imaging, utilizing the fluorescent properties of the Vector Red alkaline phosphatase substrate and unmixing the different spectra of Alexa 488, tissue autofluorescence, and Vector Red (objective 63 × , Plan-Apochromat, NA 1.4).
Figure 4.
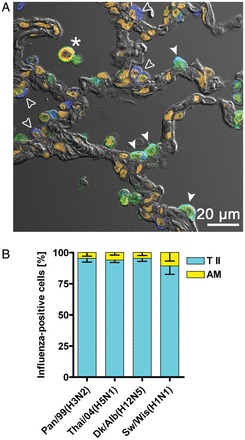
Quantification of influenza A virus–infected cells. A, Cross sections from infected lung explants were stained for influenza virus antigen (green channel), prosurfactant protein C (blue channel) to detect type II pneumocytes (T II; white arrowheads for infected cells [cyan]; open arrowheads for uninfected cells [blue]), and CD68 (red channel) to detect alveolar macrophages (AM; asterisk indicates yellow-stained infected cell). Nuclear staining with DAPI is shown in dark orange, and lung structure is visualized with differential interference contrast. The stains were visualized by confocal microscopy and tissue autofluorescence was separated from specific fluorescence by spectral unmixing. B, At least 50 influenza virus antigen–positive cells from each of 3 patient probes infected with Thai/04(H5N1), Pan/99(H3N2), Dk/Alb(H12N5), or Sw/Wis(H1N1) were counted and assigned as AM or T II cells as indicated. AM counted for 4%–11% and T II cells for 89%–96% of all influenza virus–positive cells, respectively.
To evaluate our immunohistochemical data regarding cellular tropism on the ultrastructural level, lung explants were infected and processed for transmission electron microscopy. Figure 5 depicts alveolar epithelial cells from lung explants 24 hours after infection with Pan/99(H3N2). Budding of virus particles was observed on the apical side of the cells that were identified as type II pneumocytes by the typical presence of surfactant-filled multilamellar bodies (Figure 5A and 5B). In none of our specimens did we observe virus budding from cells with hallmarks of type I pneumocytes. These data show that human and animal influenza A viruses share the same cell tropism for type II pneumocytes in human lung tissue, although they remarkably differ in their capacity to propagate in these cells.
Figure 5.
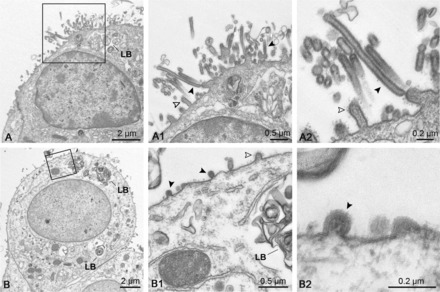
Transmission electron microscopy confirms virus budding from type II pneumocytes. Lung explants were infected with Pan/99(H3N2) for 24 hours and processed for transmission electron microscopy. A and B, Infected cells with indicated multilamellar bodies (LB) typically found in type II pneumocytes. The enlarged frames (A1, A2, B1, and B2) show virus budding and released viral particles (black arrowheads), as well as cellular microvilli (open arrowheads) on the cell surface. Scale bars are indicated.
Differential Induction of Cytokines and Chemokines in Human Lung Tissue by Human and Animal Influenza A Viruses
It is unclear whether the enhanced cytokine levels observed in H5N1-infected patients and mice [16, 20, 22] are derived mainly from the primarily infected epithelial cells and alveolar macrophages or from recruited immune cells. Thus, we determined the induction of cytokines in the lung tissue by HPAIV and other viruses of the panel. Infection of human lung tissue with the seasonal NC/99(H1N1) and Pan/99(H3N2) viruses and with the pandemic Bay/09(H1N1pdm) virus provoked only very weak secretions of IP-10, MIP-1β, IFN-β, and IL-1β (Figure 6). In contrast, the Thai/04(H5N1) virus elicited a strong induction of these cytokines and chemokines. Surprisingly, low-pathogenicity avian virus Dk/Alb(H12N5) also provoked a pronounced induction of these cytokines and chemokines, reaching 3–13-fold higher levels than were observed for the seasonal NC/99(H1N1). This upregulation hardly occurred when the tissue was probed with UV-inactivated virus or allantoic fluid, indicating that cytokine induction was triggered by viral infection (Supplementary Figure 3). The classic swine virus Sw/Wis(H1N1) caused a weak cytokine response, with only IP-10 reaching levels significantly higher than for the seasonal viruses. In conclusion, these findings indicate that avian influenza viruses with low pathogenicity and those with high pathogenicity induce a considerably stronger cytokine response in the alveolar tissue than human-adapted viruses, even in the absence of recruited immune cells.
Figure 6.
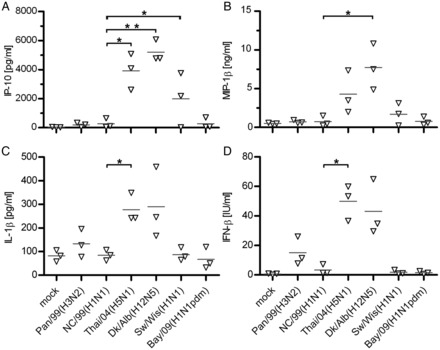
Chemokine and cytokine release by different influenza A viruses in human lung explants. Human lung explants were infected with the indicated viruses, and supernatants were collected at 24 hours (A–C) and 48 hours (D) after infection for determination of cytokine/chemokine levels by multiplex cytokine assay or enzyme-linked immunosorbant assay. Values from 3 independent experiments (each done in triplicate) are shown individually, and mean values are indicated (grey line). IFN-β, interferon β; IL-1β, interleukin-1β. *P < .05 and **P < .01 by the 2-tailed Student t test.
DISCUSSION
Influenza is a prime example of a major human infectious disease originating from an animal reservoir that continues to contribute viral genes to the generation of novel human virus strains with unique biological or antigenic properties [1, 31]. Influenza viruses acquire additional genetic diversity through their error-prone RNA polymerase, which can rapidly generate mutant variants with a selective advantage in a new host. Hence, mutations in the viral polymerase and the HA molecule of avian influenza viruses are known to promote adaptation to a mammalian host [6]. However, a complete understanding of the host factors facilitating or restricting the growth of influenza viruses in humans is just beginning to emerge [6, 32].
One of our goals is to gain a better understanding of the processes associated with adaptation of influenza viruses to the lower human respiratory tract that is strongly affected in severe influenza. Here, we systematically compared tropism, replication, and cytokine induction of influenza A viruses from human, avian, and porcine hosts in explanted human lung tissue. The suitability of the lung organ cultures for our study was supported by the findings that HPAIV-H5N1 and human-adapted seasonal and pandemic viruses propagated efficiently. In contrast, low-pathogenicity avian and classic swine viruses replicated only poorly, which may reflect the strong attenuation of most avian viruses in humans and the rare detection of human patients with swine influenza virus infection [5, 33].
Mathematical modeling suggests that a specific cell tropism could strongly influence influenza severity [34]. In fact, differences in the target cell type correlated with the magnitudes in replication of avian and human influenza A viruses in reconstituted human tracheobronchial epithelium [35, 36]. Similarly, it had been proposed that preferential infection of type II pneumocytes by H5N1 viruses in human lung may explain high viral loads and strong pathologic responses in patients and nonhuman primates [10, 11, 23]. This suggestion was based on the fast metabolism of this cell type and its essential roles in maintaining alveolar surface tension and the initiation of repair processes [12, 27, 37]. However, the immunohistochemical and electron microscopy analyses of explanted lung tissues in essence debilitated this hypothesis as HPAIV-H5N1 and all other examined viruses showed a strong preference to infect type II pneumocytes, although they varied up to several orders of magnitude in their progeny titers. These results confirm and extend previous reports on the detection of viral antigen from pandemic and seasonal strains in type II pneumocytes, which however, did not address the production of infectious virus particles in this cell type [38, 39]. We found it intriguing that at least one other important respiratory viral pathogen, SARS coronavirus, has evolved to preferentially replicate in type II pneumocytes although it clearly uses a different receptor for cell entry than influenza viruses [40–42]. This indicates that type II pneumocytes are highly susceptible to viral infections and this might be important to consider in the development of future antiviral therapies.
In line with the large overlaps of influenza virus and type II marker antigen staining, we did not detect viral gene expression in type I pneumocytes suggesting that infection did not advance beyond an early stage in these cells. Seasonal influenza viruses were previously shown to primarily attach in fixed lung sections to cells morphologically resembling type I pneumocytes, but it was not addressed whether this initiated a viral replication cycle [11]. Interestingly, type I cells were reported to become infectable by influenza virus after purification from lung tissue [43, 44]. Clearly, further work is required to resolve the question why this was not observed in lung organ culture in which type II pneumocytes are also present.
In the infected lung specimens, we also detected viral antigen in some alveolar macrophages, regardless which strain was tested. This finding is consistent with recent reports showing that isolated alveolar macrophages are susceptible to infection with human and avian influenza viruses [44, 45]. Nevertheless, macrophages constituted only a very minor fraction of all infected cells, and they most likely did not contribute a great extent to virus growth but may well have contributed to the production of cytokines. The seasonal H3N2 and H1N1 viruses induced only a moderate cytokine response, indicating that these viruses avoid or suppress a vigorous activation in epithelial lung tissue [46]. Also, the pandemic H1N1-2009 virus provoked a weak cytokine response, which confirms previous reports involving infected human macrophages, dendritic cells, and primary alveolar cells [47, 48]. The low level of cytokine induction may have contributed to the successful propagation and rapid spread of the H1N1-pdm virus in humans. H5N1 virus replicated at least as well as seasonal H3N2 virus but induced strong cytokine responses in resident alveolar cells in human lung explants. Because recruited immune cells are absent in the ex vivo infection model, it is likely that the pronounced cytokine induction by HPAIV-H5N1 in infected patients [16] is already initiated at the level of the infected alveolar cells.
Interestingly, low-pathogenicity avian virus also triggered a strong cytokine response in lung explants despite poor viral growth. This finding indicates that this virus either produces a high amount of RNA that leads to enhanced activation of intracellular receptors controlling cytokine induction or that the ligation of avian receptor determinants provides a signal for the expression of cytokine genes. The latter possibility would be in line with recent findings demonstrating equal or even higher concentrations of MIP-1α and IL-1β in the lungs of mice infected with low-pathogenicity avian H5N1 virus as compared to HPAIV-H5N1 virus [22]. Furthermore, Ramos et al recently showed that an avian receptor binding preference contributes to strong cytokine induction in various human cells, but the precise reasons for that are currently unclear [49]. The classic swine virus triggered in comparison only low levels of cytokine secretion. Thus, poor growth of an animal influenza virus in human lung is not necessarily caused by a strong cytokine induction but may involve other viral and/or cellular factors.
The identification of type II pneumocytes as a common target cell for influenza viruses, the selective support for replication of human and highly pathogenic avian H5N1 strains, and the ability to initiate cytokine responses highlights the value of human lung organ cultures for providing insights into pathophysiological processes. Whether the permissiveness of human lung tissue for a given influenza virus depends on its inherent replicative properties in type II pneumocytes, a specific adaptation to withstand the induced innate responses [50], or a combination thereof remains to be determined. One should keep in mind that no ex vivo or animal model can completely reproduce the complex structures and immunological responses in the human respiratory tract. Nevertheless, the experimental findings obtained in this study closely mirror clinical and pathological observations and will therefore allow more detailed investigations regarding factors of species adaptation, innate immune responses, and therapeutic interventions in future analyses.
Supplementary Material
Notes
Acknowledgments. We thank Andrea Zöhner, Gudrun Heins, and Doris Stoll, for excellent technical support, and Matthias Budt, for critical comments on the manuscript.
Financial support. This work was supported by the FluResearchNet, which is funded by the German Federal Ministry of Research and Education (BMBF grants 01 KI 07131 to S. H. and grants 01 KI 07132 and 01 KI 1006 C to T. W.) by PROGRESS (Pneumonia Research Network on Genetic Resistance and Susceptibility for the Evolution of Severe Sepsis), which is funded by the BMBF (grant C8 to A. C. H.); and by the Transregional Collaborative Research Center SFB-TR84 of the Deutsche Forschungsgemeinschaft (grants B2 to T. W., Z1a to A. C. H., and Z1b to A. D. G.). J. K. was supported by a stipend of the International Max Planck Research School for Infectious Diseases and Immunology.
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Wright PF, Neumann G, Yoshihiro K. Orthomyxoviruses. 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007. Fields Virology; pp. 1691–740. [Google Scholar]
- 2.Thompson MG, Shay DK, Zhou H, et al. Centers for Disease Control and Prevention. Morb Mortal Wkly Rep. 2010;59:1057–62. [Google Scholar]
- 3.Patterson KD, Pyle GF. The geography and mortality of the 1918 influenza pandemic. Bull Hist Med. 1991;65:4–21. [PubMed] [Google Scholar]
- 4.Neumann G, Noda T, Kawaoka Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature. 2009;459:931–9. doi: 10.1038/nature08157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beare AS, Webster RG. Replication of avian influenza viruses in humans. Arch Virol. 1991;119:37–42. doi: 10.1007/BF01314321. [DOI] [PubMed] [Google Scholar]
- 6.Taubenberger JK, Kash JC. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe. 2010;7:440–51. doi: 10.1016/j.chom.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito T, Couceiro JN, Kelm S, et al. Molecular basis for the generation in pigs of influenza A viruses with pandemic potential. J Virol. 1998;72:7367–73. doi: 10.1128/jvi.72.9.7367-7373.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Couceiro JN, Paulson JC, Baum LG. Influenza virus strains selectively recognize sialyloligosaccharides on human respiratory epithelium; the role of the host cell in selection of hemagglutinin receptor specificity. Virus Res. 1993;29:155–65. doi: 10.1016/0168-1702(93)90056-s. [DOI] [PubMed] [Google Scholar]
- 9.Shinya K, Ebina M, Yamada S, Ono M, Kasai N, Kawaoka Y. Avian flu: influenza virus receptors in the human airway. Nature. 2006;440:435–6. doi: 10.1038/440435a. [DOI] [PubMed] [Google Scholar]
- 10.van Riel D, Munster VJ, de Wit E, et al. H5N1 virus attachment to lower respiratory tract. Science. 2006;312:399. doi: 10.1126/science.1125548. [DOI] [PubMed] [Google Scholar]
- 11.van Riel D, Munster VJ, de Wit E, et al. Human and avian influenza viruses target different cells in the lower respiratory tract of humans and other mammals. Am J Pathol. 2007;171:1215–23. doi: 10.2353/ajpath.2007.070248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korteweg C, Gu J. Pathology, molecular biology, and pathogenesis of avian influenza A (H5N1) infection in humans. Am J Pathol. 2008;172:1155–70. doi: 10.2353/ajpath.2008.070791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu H, Gao Z, Feng Z, et al. Clinical characteristics of 26 human cases of highly pathogenic avian influenza A (H5N1) virus infection in China. PLoS One. 2008;3:e2985. doi: 10.1371/journal.pone.0002985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan MC, Cheung CY, Chui WH, et al. Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir Res. 2005;6:135. doi: 10.1186/1465-9921-6-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheung CY, Poon LL, Lau AS, et al. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet. 2002;360:1831–7. doi: 10.1016/s0140-6736(02)11772-7. [DOI] [PubMed] [Google Scholar]
- 16.de Jong MD, Simmons CP, Thanh TT, et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med. 2006;12:1203–7. doi: 10.1038/nm1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peiris JS, Yu WC, Leung CW, et al. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet. 2004;363:617–9. doi: 10.1016/S0140-6736(04)15595-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 2008;4:e1000115. doi: 10.1371/journal.ppat.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng R, Lu M, Korteweg C, et al. Distinctly different expression of cytokines and chemokines in the lungs of two H5N1 avian influenza patients. J Pathol. 2008;216:328–36. doi: 10.1002/path.2417. [DOI] [PubMed] [Google Scholar]
- 20.Droebner K, Reiling SJ, Planz O. Role of hypercytokinemia in NF-kappaB p50-deficient mice after H5N1 influenza A virus infection. J Virol. 2008;82:11461–6. doi: 10.1128/JVI.01071-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salomon R, Hoffmann E, Webster RG. Inhibition of the cytokine response does not protect against lethal H5N1 influenza infection. Proc Natl Acad Sci U S A. 2007;104:12479–81. doi: 10.1073/pnas.0705289104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szretter KJ, Gangappa S, Lu X, et al. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J Virol. 2007;81:2736–44. doi: 10.1128/JVI.02336-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baskin CR, Bielefeldt-Ohmann H, Tumpey TM, et al. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc Natl Acad Sci U S A. 2009;106:3455–60. doi: 10.1073/pnas.0813234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herzog EL, Brody AR, Colby TV, Mason R, Williams MC. Knowns and unknowns of the alveolus. Proc Am Thorac Soc. 2008;5:778–82. doi: 10.1513/pats.200803-028HR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuchs S, Hollins AJ, Laue M, et al. Differentiation of human alveolar epithelial cells in primary culture: morphological characterization and synthesis of caveolin-1 and surfactant protein-C. Cell and Tissue Res. 2003;311:31–45. doi: 10.1007/s00441-002-0653-5. [DOI] [PubMed] [Google Scholar]
- 26.Stevens A, Lowe JS. Human histology. 2nd ed. London: Chapman & Hall; 1996. [Google Scholar]
- 27.Gu J, Xie Z, Gao Z, et al. H5N1 infection of the respiratory tract and beyond: a molecular pathology study. Lancet. 2007;370:1137–45. doi: 10.1016/S0140-6736(07)61515-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guarner J, Shieh WJ, Dawson J, et al. Immunohistochemical and in situ hybridization studies of influenza A virus infection in human lungs. Am J Clin Pathol. 2000;114:227–33. doi: 10.1309/HV74-N24T-2K2C-3E8Q. [DOI] [PubMed] [Google Scholar]
- 29.Kuiken T, Taubenberger JK. Pathology of human influenza revisited. Vaccine. 2008;26(Suppl 4):D59–66. doi: 10.1016/j.vaccine.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zielecki F, Semmler I, Kalthoff D, et al. Virulence determinants of avian H5N1 influenza A virus in mammalian and avian hosts: role of the C-terminal ESEV motif in the viral NS1 protein. J Virol. 2010;84:10708–18. doi: 10.1128/JVI.00610-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolfe ND, Dunavan CP, Diamond J. Origins of major human infectious diseases. Nature. 2007;447:279–83. doi: 10.1038/nature05775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe T, Watanabe S, Kawaoka Y. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe. 2010;7:427–39. doi: 10.1016/j.chom.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Myers KP, Olsen CW, Gray GC. Cases of swine influenza in humans: a review of the literature. Clin Infect Dis. 2007;44:1084–8. doi: 10.1086/512813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobrovolny HM, Baron MJ, Gieschke R, Davies BE, Jumbe NL, Beauchemin CA. Exploring cell tropism as a possible contributor to influenza infection severity. PLoS One. 2010;5:e13811. doi: 10.1371/journal.pone.0013811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ibricevic A, Pekosz A, Walter MJ, et al. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J Virol. 2006;80:7469–80. doi: 10.1128/JVI.02677-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk HD. Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc Natl Acad Sci U S A. 2004;101:4620–4. doi: 10.1073/pnas.0308001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mason RJ. Biology of alveolar type II cells. Respirology. 2006;11(Suppl):S12–5. doi: 10.1111/j.1440-1843.2006.00800.x. [DOI] [PubMed] [Google Scholar]
- 38.Shieh WJ, Blau DM, Denison AM, et al. 2009 pandemic influenza A (H1N1): pathology and pathogenesis of 100 fatal cases in the United States. Am J Pathol. 2010;177:166–75. doi: 10.2353/ajpath.2010.100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Zhang Z, Fan X, et al. 2009 pandemic H1N1 influenza virus replicates in human lung tissues. J Infect Dis. 2010;201:1522–6. doi: 10.1086/650544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chow KC, Hsiao CH, Lin TY, Chen CL, Chiou SH. Detection of severe acute respiratory syndrome-associated coronavirus in pneumocytes of the lung. Am J Clin Pathol. 2004;121:574–80. doi: 10.1309/C0EDU0RAQBTXBHCE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shieh WJ, Hsiao CH, Paddock CD, et al. Immunohistochemical, in situ hybridization, and ultrastructural localization of SARS-associated coronavirus in lung of a fatal case of severe acute respiratory syndrome in Taiwan. Hum Pathol. 2005;36:303–9. doi: 10.1016/j.humpath.2004.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ye J, Zhang B, Xu J, et al. Molecular pathology in the lungs of severe acute respiratory syndrome patients. Am J Pathol. 2007;170:538–45. doi: 10.2353/ajpath.2007.060469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee SM, Chan RW, Gardy JL, et al. Systems-level comparison of host responses induced by pandemic and seasonal influenza A H1N1 viruses in primary human type I-like alveolar epithelial cells in vitro. Respir Res. 2010;11:147. doi: 10.1186/1465-9921-11-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu WC, Chan RW, Wang J, et al. Viral replication and innate host responses in primary human alveolar epithelial cells and alveolar macrophages infected with influenza H5N1 and H1N1 viruses. J Virol. 2011;85:6844–55. doi: 10.1128/JVI.02200-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Riel D, Leijten LM, van der Eerden M, et al. Highly pathogenic avian influenza virus H5N1 infects alveolar macrophages without virus production or excessive TNF-alpha induction. PLoS Pathog. 2011;7:e1002099. doi: 10.1371/journal.ppat.1002099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolff T, Ludwig S. Influenza viruses control the vertebrate type I interferon system: factors, mechanisms, and consequences. J Interferon Cytokine Res. 2009;29:549–57. doi: 10.1089/jir.2009.0066. [DOI] [PubMed] [Google Scholar]
- 47.Chan MC, Chan RW, Yu WC, et al. Tropism and innate host responses of the 2009 pandemic H1N1 influenza virus in ex vivo and in vitro cultures of human conjunctiva and respiratory tract. Am J Pathol. 2010;176:1828–40. doi: 10.2353/ajpath.2010.091087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Osterlund P, Pirhonen J, Ikonen N, et al. Pandemic H1N1 2009 influenza A virus induces weak cytokine responses in human macrophages and dendritic cells and is highly sensitive to the antiviral actions of interferons. J Virol. 2010;84:1414–22. doi: 10.1128/JVI.01619-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramos I, Bernal-Rubio D, Durham N, et al. Effects of receptor binding specificity of avian influenza virus on the human innate immune response. J Virol. 2011;85:4421–31. doi: 10.1128/JVI.02356-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang J, Nikrad MP, Phang T, et al. Innate immune response to influenza A virus in differentiated human alveolar type II cells. Am J Respir Cell Mol Biol. 2011;45:582–91. doi: 10.1165/rcmb.2010-0108OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.