

Abstract
For antiviral signaling mediated by retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), the recruitment of cytosolic RLRs and downstream molecules (such as TBK1 and IKKε) to mitochondrial platform is a central event that facilitates the establishment of host antiviral state. Here, we present an example of viral targeting for immune evasion through spatial isolation of TBK1/IKKε from mitochondrial antiviral platform, which was employed by severe fever with thrombocytopenia syndrome virus (SFTSV), a deadly bunyavirus emerging recently. We showed that SFTSV nonstructural protein NSs functions as the interferon (IFN) antagonist, mainly via suppressing TBK1/IKKε–IRF3 signaling. NSs mediates the formation of cytoplasmic inclusion bodies (IBs), and the blockage of IB formation impairs IFN-inhibiting activity of NSs. We next demonstrate that IBs are utilized to compartmentalize TBK1/IKKε. The compartmentalization results in spatial isolation of the kinases from mitochondria, and deprived TBK1/IKKε may participate in antiviral complex assembly, leading to the blockage of IFN induction. This study proposes a new role of viral IBs as virus-built ‘jail’ for imprisoning cellular factors and presents a novel and likely common mechanism of viral immune evasion through spatial isolation of critical signaling molecules from the mitochondrial antiviral platform.
Keywords: innate immunity, immune evasion, severe fever with thrombocytopenia syndrome virus, inclusion bodies, TBK1/IKKε, spatial isolation
Introduction
Innate immunity is the first line of host defense against pathogen invasion. For RNA virus infection, cytosolic viral RNAs are initially recognized by RIG-I-like receptors (RLRs) RIG-I and MDA5 (Yoneyama and Fujita, 2009). Recognizing RNA by RLRs triggers their activation and subsequent translocation to mitochondria where they interact with the common adaptor MAVS (also known as IPS-1, CARDIF, and VISA) (Kawai et al., 2005; Meylan et al., 2005; Seth et al., 2005; Xu et al., 2005), resulting in the formation of active MAVS polymers (Hou et al., 2011). MAVS then recruits various signaling proteins to form antiviral complexes, such as TRAF6 and TRAF3. The recruitment of TRAF6 to MAVS activates NF-κB signaling, while TRAF3 binds to MAVS to promote TBK1/IKKε-mediated IRF3 activation (Belgnaoui et al., 2011). MAVS also interacts with another mitochondrial protein TOM70 for recruiting TBK1/IRF3 via chaperone Hsp90 to enhance the activation of IRF3 (Liu et al., 2010). These activation events of NF-κB and IRF3 lead to the production of cytokines including type I interferons (IFNs), while the optimum activation also involves organelle antiviral platforms as well as many other molecules (such as RAVER1 and GSK3β) for association and cooperation in signaling (Lei et al., 2010; Chen et al., 2013). As the antiviral signaling hubs, mitochondria provide an appropriate environment for antiviral complex assembly, and mitochondrial location of MAVS is required for RLR-mediated IFN induction (Seth et al., 2005); while, in turn, MAVS functions as a sorting adaptor and further defines mitochondria as the central signaling sites (Kagan, 2012). Thus the signaling transduction spatiotemporally involves translocation of multiple cytosolic signaling proteins to mitochondria.
To circumvent host antiviral responses, viruses have evolved multifarious strategies at different levels of RLR signaling. Since mitochondrial relocation is a notable event for many signaling molecules, it is likely to be targeted by viruses for immune evasion. In the current study, we present an example of spatial isolation of TBK1/IKKε from mitochondrial antiviral platform by severe fever with thrombocytopenia syndrome virus (SFTSV). SFTSV is a novel phlebovirus of Bunyaviridae family, recently emerging in China (Yu et al., 2011). Human infection by SFTSV can cause severe fever with thrombocytopenia syndrome (SFTS) with a high fatality rate up to 30% (Yu et al., 2011). As an emerging deadly virus, SFTSV has posed a serious health threat. However, no antiviral drugs or vaccines are available at present, and little is known about the viral pathogenesis and virus–host interactions in humans infected with SFTSV. Clinically, the production of IFNs was almost undetectable in the blood of patients (Li, 2011), suggesting that innate immune responses are suppressed effectively, which may contribute to viral systematic infection and pathogenesis. Like other phleboviruses, SFTSV is a negative, single-stranded RNA virus with a three-segmented genome. The large (L) and medium (M) segments encode RNA-dependent RNA polymerase (RdRP) and glycoproteins, respectively, while the small (S) segment uses an ambisense strategy to encode the nucleoprotein (NP) and the nonstructural protein (NSs). Bunyavirus NSs proteins are hypervariable in sequences, sizes, and encoding strategies, but they appear to have some conservative functions, such as regulation of viral replication and IFN antagonism (Schmaljohn and Nichol, 2007). SFTSV NSs might be involved in the suppression of IFN induction as well, but the mechanism was not fully elucidated (Qu et al., 2012).
In this study, we show that the SFTSV NSs functions as type I IFN antagonist mainly by interfering with the TBK1/IKKε–IRF3 signaling. NSs mediates the formation of cytoplasmic inclusion bodies (IBs) and efficiently captures TBK1/IKKε to the IBs. The capture leads to a spatial isolation of TBK1/IKKε from the mitochondrial antiviral platform, thus suppressing antiviral signaling and IFN induction.
Results
SFTSV NSs inhibits Sendai virus-triggered activation of IFN-β promoter mainly by targeting the IRF-responsive element activation
Proteins encoded by bunyaviruses, either nonstructural or structural, can act as potential IFN antagonists (Elliott and Weber, 2009). To identify the type I IFN antagonist(s) of SFTSV, a functional screening of the proteins from SFTSV (WCH-2011/HN/China/isolate97) was performed. Plasmids encoding these proteins and influenza PR8 virus NS1 (as a positive control) were transfected individually into HEK293T cells, and their effects on the activation of IFN-β promoter following Sendai virus (SeV) infection were examined by dual luciferase reporter (DLR) assays. NSs encoded by the S segment was identified as the inhibitor of IFN-β promoter activation (Figure 1A).
Figure 1.

SFTSV NSs-induced suppression of IFN-β, ISRE, and NF-κB promoters. HEK293T cells were co-transfected with 600 ng empty plasmid (vector) or plasmids encoding the indicated viral proteins, along with the reporter plasmids for IFN-β (A), ISRE (B), or NF-κB (C and D) promoter and the Renilla luciferase plasmid pRL-TK. Twenty-four hours after transfection, cells were infected with SeV or left uninfected for 16 h (A–C), or treated with TNF-α (20 ng/ml) or left untreated for 12 h (D) before luciferase activities were measured. Graphs show mean ± SD, n = 3.
The activation of IFN-β promoter depends on synergistic interactions among NF-κB, IRF3, and other transcription factors that bind to distinct regulatory domains in the promoter. To identify which transcription factor-mediated signaling was targeted by NSs, we tested the ability of NSs to suppress transcription from individual regulatory elements bound by NF-κB or IRF3. As shown in Figure 1B, NSs overexpression almost completely blocked SeV-triggered activation of ISRE, and the inhibition effect was even stronger than that mediated by the positive control, NS1 protein (Figure 1B). In contrast, the suppression of NF-κB promoter induced by NSs appeared to be slight, compared with that caused by NS1 (Figure 1C). Similar results were obtained when TNF-α was used to activate NF-κB promoter (Figure 1D). We did not observe significant suppression of ISRE or NF-κB promoter by other viral proteins. Thus, these data indicate that NSs functions as the IFN antagonist of SFTSV and likely mainly targets IRF signaling for the inhibition of IFN-β induction.
NSs-induced formation and characterization of SFTSV IBs
In order to further elucidate the function of NSs, we investigated its subcellular localization. As revealed by immunofluorescence staining of cells transfected with NSs expression plasmids under the confocal microscope, NSs seemed to be localized in cytoplasmic structures that were heterogeneous in size and shape (Figure 2A). Under light microscope, the structures looked like compact granules. Further analyses of cells expressing EGFP-tagged NSs by confocal microscopy showed that they were solid and the EGFP-NSs fused protein was not limited on the surface of the structures (Figure 2B and C). Various types of mammalian cells were used for examining SFTSV infection with rabbit anti-NSs antiserum, and similar structures could be observed in SFTSV-infected cells, including Vero (green monkey kidney cells), HEK293 (human kidney cells), HepG2 (human hepatocytes), H9c2 (rat cardiac myoblasts), RAW264.7 (mouse macrophages), and DH82 (canine macrophage-monocyte cells) (Figure 2D and Supplementary Data). For the staining characteristics, subcellular distribution, size, and shape, the structures were quite similar to IBs observed in cells infected with some non-segmented negative-strand RNA viruses such as rabies virus (Lahaye et al., 2009) and Ebola virus (Hoenen et al., 2012).
Figure 2.
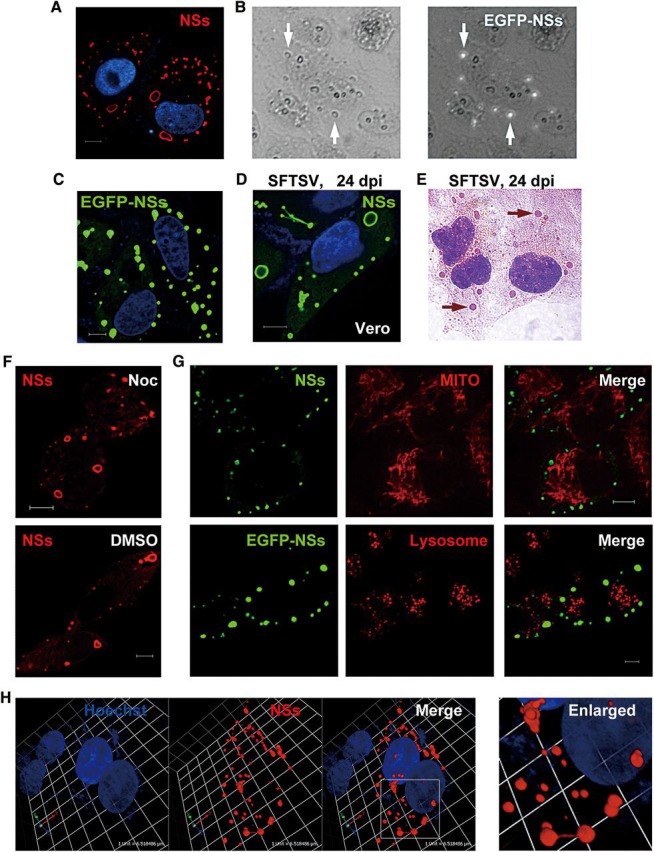
Formation and characterization of SFTSV NSs IBs. (A) Vero cells were transfected with the plasmid encoding NSs fused with S.tag (NSs-S.tag). Twenty-four hours later, cells were fixed for immunofluorescence assay (IFA). NSs (red) was visualized with anti-S.tag antibody under confocal microscope. (B) Vero cells were transfected with the EGFP-NSs expression plasmid for 24 h and then fixed with 95% ethanol. The structures harboring NSs are indicated by white arrows. Images show the bright field (left) and the same field with EGFP fluorescence (right). (C) Cells expressing EGFP-NSs were visualized under confocal microscopy. (D) Vero cells were infected with SFTSV for 24 h and stained with anti-NSs antiserum. (E) Cells were infected with SFTSV for 24 h, and then fixed with 95% ethanol for H&E staining. Arrows indicate eosinophilic IB structures. (F) After transfection with NSs-S.tag expression plasmids, cells were immediately treated with 40 μM nocodazole (Noc, upper) or the solvent as mock (DMSO, lower) for 20 h. NSs IBs were stained with anti-S.tag and visualized under confocal microscope. (G) Cells expressing NSs-S.tag (upper panels) or EGFP-NSs (lower panels) were stained with organel markers to visualize mitochondria (upper panels) or lysosomes (lower panels). (H) A 3D view of NSs IBs in SFTSV-infected cells. Viral IBs were stained with anti-NSs antiserum. Scale bars, 6 μm (A, C, D, F, and G).
We further characterized these IBs induced by SFTSV NSs. Hematoxylin-and-eosin (H&E) staining was performed and showed that the SFTSV IBs appeared to be eosinophilic in SFTSV-infected cells (Figure 2E).
There are some cytoplasmic IBs called aggresomes that represent misfolded protein aggregates. The aggregated proteins are driven along microtubule tracks to juxtanuclear microtubule-organizing center to form aggresomes (Kopito, 2000). Aggresomes may recruit mitochondria and they can be further delivered to the autophagy–lysosome system for degradation (Garcia-Mata et al., 1999; Kopito, 2000). However, the distribution of NSs IBs was not limited in perinuclear zone and the treatment with nocodazole, which induces microtubule depolymerization, did not inhibit the formation of NSs IBs (Figure 2F). Furthermore, no significant recruitment of mitochondria to the NSs IBs was observed (Figure 2G, upper panels). In addition, these NSs IBs were likely not delivered to lysosomes neither, as revealed by EGFP-NSs expression and lysosome staining (Figure 2G, lower panels). These results suggest that NSs-induced IBs are distinct from aggresomes.
For some other cytoplasmic IBs induced by viruses like rabies virus and Ebola virus, it has been demonstrated that they are dynamic and functional structures compartmentalizing and hence implementing some essential events of the viral life cycle (Lahaye et al., 2009; Hoenen et al., 2012). Employing a finer temporal resolution for live-cell analysis, Hoenen et al. (2012) reported that Ebola virus IBs were highly dynamic and they could be fused together to form larger ones or dispersed into smaller ones. Interestingly, we indeed observed that some of SFTSV NSs IBs were likely fusing, dispersing, or being linked with each other by a ‘fiber’, implying that NSs IBs might also undergo dynamic fusion or fission during their developments (Figure 2H). These findings suggest that SFTSV NSs IBs are distinct from aggresomes, but share some characteristics with the active IBs induced by some other viruses, in which viral or cellular proteins are compartmentalized but not damaged (Lahaye et al., 2009; Hoenen et al., 2012; Fricke et al., 2013).
The PXXP motif is required for IB formation and NSs IFN-inhibiting activity
Two PXXP motifs (P refers to proline and X refers to any amino acid) at N-terminus of the NSs protein encoded by Rift Valley fever virus (RVFV, also a phlebovirus) were suggested to play important roles in both the localization of NSs and its function as an IFN antagonist (Billecocq et al., 2004). Sequence analysis revealed that SFTSV NSs also contains a conserved PXXP motif at its N-terminus (aa residues 66–69). Thus, we analyzed the role of this proline-rich motif in NSs location through replacing the proline(s) by alanine(s). Vero cells were transfected with plasmids expressing EGFP, EGFP-tagged NSs, or NSs mutants, respectively. Any single-point mutation at P66 (NSs P66A) or P69 (NSs P69A) did not prevent the formation of IBs, whereas simultaneous substitution of P66 and P69 (NSs PP) disrupted the formation of NSs IBs and rendered even cytoplasmic localization of the protein (Figure 3A). Next, we evaluated their capacities for suppressing ISRE and IFN-β promoters. Intriguingly, NSs PP that was no longer able to induce IB formation lost most of the inhibitory activities as well, while single-point mutations were only slightly impacted and still strongly interfered with the activation of IFN-β and ISRE promoters triggered by viral infection (Figure 3B). These results suggest that, similar to RVFV NSs, the N-terminal PXXP motif of SFTSV NSs is also required for IB formation, and localization and function of the protein. It is well known that the localization of NSs is required for NSs activity for RVFV. Here, the formation of SFTSV NSs IBs appears to be also required for NSs inhibitory activities against IRF signaling and IFN induction, as suggested by the clear correlation between NSs IB formation and NSs capacity of inhibition on reporter gene transcription.
Figure 3.

A N-terminal PXXP motif plays roles in IB formation and NSs function. (A) Double mutation of PXXP affects the NSs location and IB formation. Cells were transfected with plasmids expressing EGFP, or EGFP-tagged wild type or mutant NSs. Twenty-four hours later, the localization of EGFP or EGFP-tagged proteins were visualized under confocal microscope. (B) IFN-inhibiting activity of NSs and its mutants. HEK293T cells were co-transfected with 600 ng plasmids expressing the indicated proteins, along with the reporter plasmids for IFN-β (left) or ISRE (right) promoter and the Renilla luciferase plasmid pRL-TK. Twenty-four hours after transfection, cells were infected with SeV or left uninfected for 16 h before luciferase activities were measured. Graphs show mean ± SD, n = 3.
NSs prevents the activation and nuclear translocation of endogenous IRF3
IRF3 plays as a key regulator of type I IFN gene expression elicited by viruses. During virus infection, IRF3 is phosphorylated by TBK1 and IKKε, resulting in the dimerization and subsequent nuclear translocation of IRF3 as activated transcription factor. Since NSs appears to target IRF signaling, the activation of IRF3 was also investigated. Firstly, the level of IRF3 phosphorylation was evaluated by western blot (WB) analysis using a phosphorylation-specific antibody, anti-IRF3 (S396). SeV infection noticeably triggered IRF3 phosphorylation, which, however, was inhibited significantly by NSs expression (Figure 4A). Complementing the phosphorylation status, the effect of NSs on IRF3 dimerization was analyzed by Native-PAGE using an anti-IRF3 antibody. As shown in Figure 4B, SeV-induced dimerization of IRF3 was reduced by the expression of NSs. Virus-triggered nuclear translocation of IRF3 was also investigated by immunofluorescence. As indicated in Figure 4C, SeV infection resulted in the translocation of IRF3 from the cytoplasm to the nuclei, which was blocked in the cells expressing NSs. In contrast, NSs PP almost lost the capacity for blocking IRF3 nuclear translocation (Figure 4D and Supplementary Data). These results demonstrate that NSs indeed interferes with IRF3 activities.
Figure 4.
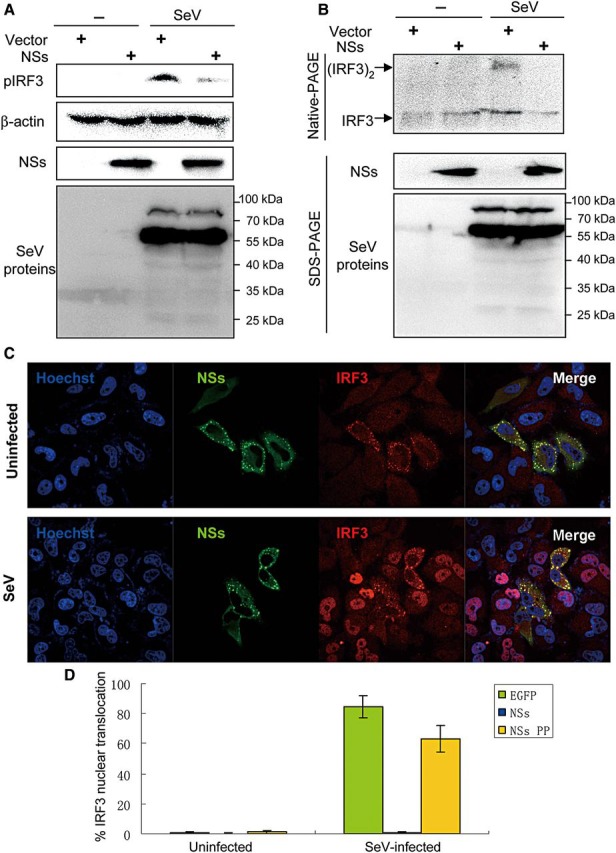
NSs inhibits the activation and nuclear translocation of IRF3 induced by SeV infection. (A and B) HEK293T were transfected with the vector plasmid or the plasmid expressing NSs-S.tag. Twenty-four hours post-transfection, cells were infected with SeV or left uninfected (−) for 6 h (A) or 8 h (B). Cell lysates were separated by SDS- and native-PAGE and analyzed for the phosphorylation (A) or dimerization (B) of IRF3 by WB with antibodies against the indicated proteins except that NSs expression was detected with the anti-S.tag antibody. (C) NSs blocks the nuclear translocation of IRF3 triggered by SeV. Cells were transfected with the plasmid encoding HA-tagged NSs. Twenty-four hours after transfection, cells were infected with SeV or left uninfected for 12 h. The localization of proteins was visualized by confocal microscopy after IFA. (D) Experiments were performed similarly as in C. Cells expressing NSs, NSs PP, or EGFP (as control) were scored for SeV-induced nuclear translocation of IRF3. At least 200 cells were counted for each sample. Data shown are presented as mean ± SD, n = 3.
NSs inhibits RLR-mediated signaling at the level of TBK1/IKKɛ
SeV is a model virus that can trigger RLR signaling and hence IRF3 activation. In our reporter assays, knockdown of RIG-I by RNAi inhibited SFTSV-induced IFN-β promoter activation, suggesting that RIG-I is also involved in the recognition of SFTSV infection (Supplementary Data). Since SFTSV NSs inhibited SeV-induced activation of IRF3, we hypothesized that NSs may interfere with RLR-mediated signaling pathway. To certify this, the NSs-expressing plasmid and plasmids expressing MDA5 or a constitutively active form of RIG-I (RIG-IN) were, respectively, transfected into HEK293T cells for reporter gene assays. The overexpression of RIG-IN or MDA5 could strongly activate the IFN-β promoter, while co-expression of NSs inhibited the activation of IFN-β promoter in a dose-dependent manner (Figure 5A and B), indicating that NSs indeed interferes with RLR signaling pathway.
Figure 5.
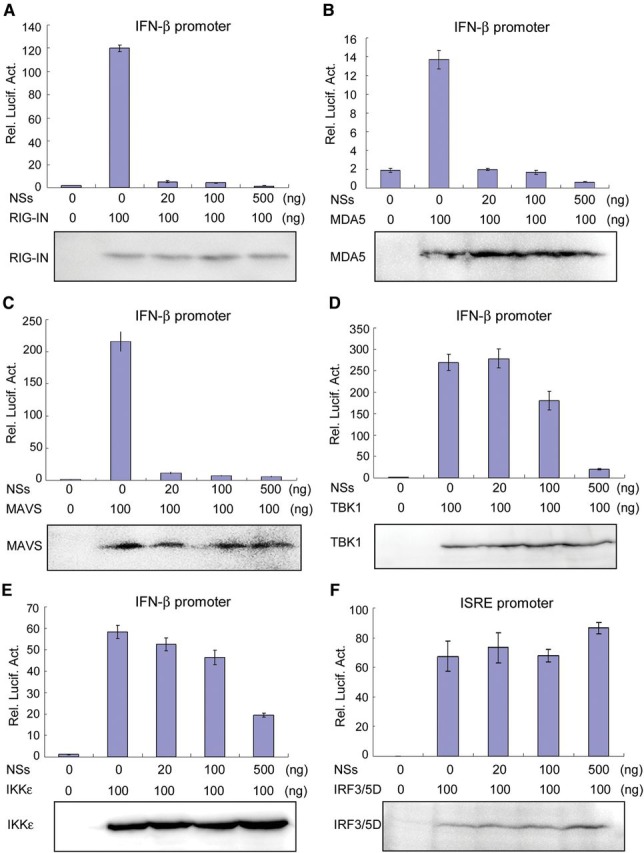
NSs suppresses RLR signaling at the level of TBK1/IKKε. HEK293T cells were co-transfected with the indicated amount of NSs expression plasmids and plasmids encoding RIG-IN (A), MDA5 (B), MAVS (C), TBK1 (D), IKKε (E), or IRF3/5D (F), along with IFN-β (A–E) or ISRE (F) promoter reporter plasmids and pRL-TK. Thirty-six hours after transfection, luciferase activities were measured. Expression of signaling proteins was also analyzed by WB. Graphs show mean ± SD, n = 3.
Furthermore, we addressed which step of the RLR signaling cascade is targeted by NSs. Signaling proteins downstream of RLRs including the adaptor MAVS and the kinases TBK1 and IKKε were used to activate the IFN-β promoter. Overexpression of these proteins resulted in significant activation of the IFN-β promoter, which was inhibited by NSs (Figure 5C and E), suggesting that NSs acts at the level of TBK1/IKKε or further downstream. To gain further insight, a constitutively active form of IRF3 (IRF3/5D) was constructed and used to induce the activation of ISRE promoter. As shown in Figure 5F, NSs was not able to suppress IRF3/5D-mediated activation of ISRE promoter, indicating that NSs functions at the upstream of IRF3. Taken together, these data suggest that NSs targets at the level of TBK1/IKKε to inhibit RLR signaling and hence IRF3 activity.
NSs interacts with TBK1/IKKɛ and relocates the kinases to NSs IBs
To investigate how NSs antagonizes the RLR signaling pathway at the level of TBK1/IKKε, the interactions between NSs and RLR signaling molecules were examined by S-protein pull-down assays. HEK293 cells were co-transfected with the plasmids encoding Flag-tagged signaling proteins and NSs fused with S.tag (NSs-S.tag), and lysed for S.tag precipitation. As shown in Figure 6A, TBK1 and IKKε, but not RIG-I, MDA5, MAVS, or IRF3, were co-precipitated strongly with NSs-S.tag. Then, the interaction of NSs with endogenous TBK1 was also confirmed by the endogenous Co-IP assay using SFTSV-infected cells with NSs-specific rabbit antiserum (Figure 6B).
Figure 6.

NSs interacts with host kinases TBK1 and IKKε. (A) Screening of host proteins interacting with NSs. HEK293 cells were co-transfected with the indicated plasmids. Thirty-six hours after transfection, cells were lysed and pull-down assays were performed with the slurry of S-protein agarose. The precipitates were analyzed by WB with anti-Flag and anti-S.tag for revealing the signaling proteins interacting with NSs and the pull-down of NSs-S.tag, respectively. The expression of transfected Flag-tagged signaling proteins (lysate input) was detected by WB with anti-Flag. Vectors were the corresponding empty plasmids. (B) Co-IP of endogenous TBK1 with viral NSs. Mock- or SFTSV-infected cells were lysed and the cell supernatants pretreated with pre-immune serum and protein A/G agarose were immunoprecipitated with NSs-specific antiserum and protein A/G agarose. The immunoprecipitates were then analyzed by WB with anti-TBK1 and anti-NSs antibodies, respectively (left panels). Cell lysates were also subjected to WB analysis for detecting the expression of endogenous TBK and viral NSs (lysate input, right panels).
Furthermore, immunofluorescence staining of NSs and TBK1/IKKε was performed to determine their co-localization in cells. TBK1 and IKKε distributed diffusely cytosolic in absence of NSs, whereas they were abundantly localized to the IBs during co-expression with NSs (Supplementary Data), indicating that NSs interacts with the kinases and relocates them to the IB compartments. In contrast, NSs did not noticeably affect the localization of MAVS and they were separately located (Supplementary Data).
NSs-mediated TBK1/IKKɛ relocation to IBs causes irreversible spatial isolation of kinases from mitochondria and blocks antiviral signaling
Upon viral infection, signaling molecules including TBK1/IKKε are recruited to mitochondrial platforms harboring MAVS for antiviral complex assembly. Since NSs relocates TBK/IKKε to IBs, we examined whether the relocated TBK1/IKKε can still be responsive to viral infection, i.e. released from IBs and translocated to other subcellular sites. As shown in Figure 7A, NSs expression resulted in the relocation of IKKε to NSs IBs, but viral infection could not trigger any release or translocation of IKKε from IBs, indicating that the imprisonment of the kinase in IBs is not reversible in response to viral infection.
Figure 7.
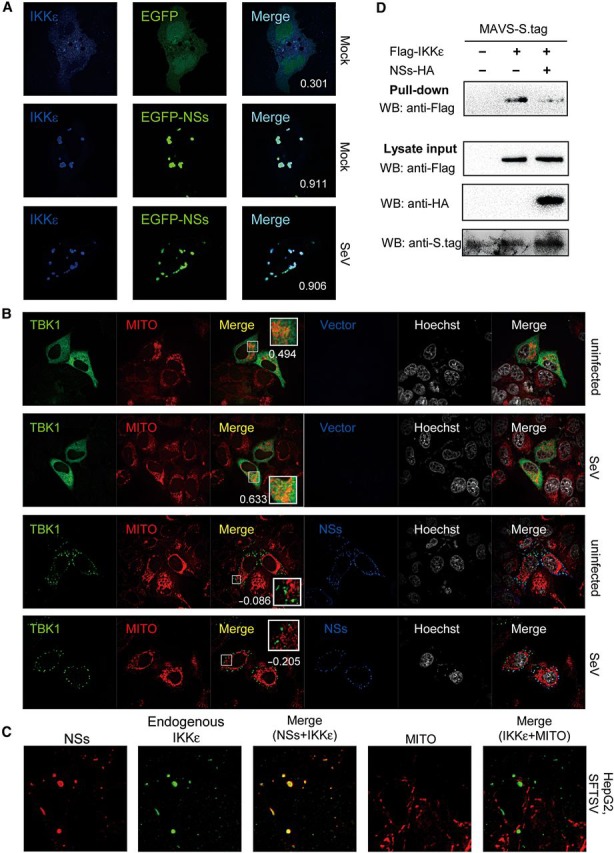
NSs-mediated relocation of TBK1/IKKε leads to irreversible spatial isolation from mitochondria and blocks antiviral signaling. (A) Relocation of kinases to IBs was irreversible upon viral infection. HeLa cells were co-transfected with Flag-IKKε expression plasmids, together with the EGFP or EGFP-NSs expression plasmid. Twenty-four hours later, cells were infected with SeV or left uninfected for 6 h and fixed for IFA. IKKε was stained with anti-Flag. To quantify the co-localization of IKKε and NSs, Pearson's correlation coefficients (PCC) are calculated and shown in the merge column (white numerals). (B) Relocation to IBs leads to irreversible spatial isolation of kinases from mitochondria. Cells were co-transfected with the Flag-TBK1 expression plasmid, and the NSs-S.tag expression plasmid or the empty vector. Twenty-four hours later, cells were infected with SeV or left uninfected for 2 h and stained with mitochondrial marker before being fixed for IFA. TBK1 and NSs were stained with anti-Flag and anti-S.tag, respectively. Nuclei stained with Hoechst are shown in grayscale. To evaluate the localization of TBK1 at mitochondria (MITO), Pearson's correlation coefficients of the signals from green (TBK1) and red (MITO) channels were shown for the representative images (white numerals). See also Supplementary DataA. (C) HepG2 cells were infected with SFTSV for 24 h and stained with mitochondrial marker before IFA. Endogenous IKKε and NSs were revealed with mouse anti-IKKε antibody and rabbit anti-NSs serum, respectively. See also Supplementary DataB. (D) Suppression of IKKε–MAVS association. HEK293 cells were co-transfected with the plasmid encoding MAVS fused with S.tag, along with plasmids expressing IKKε and NSs or their control empty plasmids as indicated. Thirty-six hours post-transfection, cells were treated with SeV for 2 h, and the interaction between IKKε and MAVS was detected by S-protein pull-down assays.
Furthermore, the effects of NSs on virus-induced recruitment of TBK1/IKKε to mitochondria were investigated. As indicated in Figure 7B, overexpressed TBK1 appeared to be partially co-localized with the mitochondrial marker even in absence of viral infection, which seemed to be enhanced upon SeV infection. In NSs-expressing cells, however, TBK1 was efficiently trapped to IBs by NSs, leading to spatial isolation of the kinase from mitochondria, which could not be reversed by viral infection (Figure 7B). Similar results were obtained for overexpressed IKKε (Supplementary DataA).
Moreover, we investigated the subcellular localization of endogenous IKKε in SFTSV-infected human cells using a mouse anti-IKKε antibody. We found that SFTSV could efficiently infect HepG2 and HEK293 cells, and the expression of endogenous IKKε seemed too low to be visualized in uninfected cells or at early phase in infected cells (data not shown). Twenty-four hours post-infection, endogenous IKKε were visualized to be compartmentalized in NSs IBs and spatially isolated from the mitochondrial network (Figure 7C and Supplementary DataB).
Since TBK1/IKKε are isolated from mitochondria, there should be no chance for them to interact with their upstream signaling molecules like MAVS or form TBK/IKKε-containing complex. To certify it, we further investigated the influence of NSs on the association between IKKε and MAVS. As shown by the S-protein pull-down assay, NSs expression significantly inhibited the association of IKKε and MAVS (Figure 7D).
Taken together, these results suggest that by trapping TBK1/IKKε to IBs, NSs mediates an effective spatial isolation of TBK1/IKKε from the mitochondrial platform, which interrupts the linkup of the kinases with their upstream molecules and results in the blockage of antiviral signaling cascade.
Discussion
Subcellular localization is important for functional regulation of cellular proteins. In response to viral infection, mitochondrial relocation of upstream RLRs and subsequently many downstream signaling molecules, such as WDR5 (Wang et al., 2010), TRAF3 (van Zuylen et al., 2012), and TBK1/IKKε (Hiscott et al., 2006), are required for MAVS antiviral complex assembly and transcription factor activation. This study provides an example of viral immune evasion through targeting this event where SFTSV NSs functions as the type I IFN antagonist through trapping TBK1/IKKε to the virus-induced IB compartments and hence efficiently mediating the irreversible spatial isolation of the kinases from mitochondria to terminate the signaling cascade and IFN induction (Figure 8).
Figure 8.

A model for viral suppression of innate immunity via spatial isolation of signaling proteins. (A) RLR signaling triggered by viral infection. (B) SFTSV-mediated interruption of RLR signaling. NSs expression mediates formation of IBs that are spatially isolated from mitochondria. TBK1/IKKε are arrested by NSs and efficiently relocalized to the IB compartments, resulting in spatial isolation of the kinases from the mitochondrial platform and hence interruption of the signaling. (C) Spatial isolation of signaling molecules from antiviral platforms is likely a common viral strategy for immune evasion. This involves many viral proteins, such as Ebola virus (EBOV) VP35, hepatitis C virus (HCV) NS2, NY-1 hantavirus Gn, and Severe acute respiratory syndrome coronavirus (SARS–CoV) M, that interact with signaling proteins and locate at various cellular or virus-induced structures spatially isolated from antiviral platforms.
Recent structural and interaction proteomic studies support a model that cellular localization of TBK1 determines specific TBK1 signaling (Helgason et al., 2013). Our study suggests that SFTSV NSs blocks the relocation of TBK1/IKKε to mitochondria via the spatial isolation and thus suppresses RLR-mediated IRF3 activation and type I IFN induction upon SeV infection, therefore highlighting the importance of the kinase location. Additionally, since TBK1/IKKε are crucial components at the convergence point of antiviral signalings mediated by several pattern recognition receptors, including RLRs, toll-like receptors (TLRs), and DNA sensors, it remains to be determined whether TBK1/IKKε signalings at other subcellular sites are prevented by the spatial isolation in IBs as well.
For circumventing IFN responses, viral proteins can target multiple molecules of RLR signaling. Coincidently, many of these viral proteins share two biological features: (i) interacting with certain critical signaling molecule(s) for which translocation to specific antiviral platforms is required for optimal antiviral signaling; and (ii) locating in specific subcellular sites (virus-induced or cellular structures) discrete from the proper antiviral platforms. For instance, NY-1 hantavirus Gn is associated with TRAF3 through its cytoplasmic tail (Alff et al., 2008) and locates in Golgi apparatus (Schmaljohn and Nichol, 2007); HCV NS2 that can interact with TBK1/IKKε (Kaukinen et al., 2013) is resident in endoplasmic reticulum (Popescu et al., 2011); New World arenaviruse Z proteins binding RIG-I target to cellular membranes (Fan et al., 2010); and intriguingly similar to SFTSV NSs, Ebola virus VP35 that interacts with TBK1/IKKε (Prins et al., 2009) is localized in viral IBs as well (Groseth et al., 2009). Previously, the physical mechanisms for antiviral signaling impairment were often proposed to be the inhibition of certain interactions or antiviral complex assembly via steric hindrance, competitive binding, or unknown means. In this study, we also detected the suppression of the interaction between the kinase IKKε and upstream MAVS, but more importantly, spatial isolation was shown to be the further mechanism. It hence presents the first clear instance of spatial isolation from antiviral platforms and prompts the possibility that this may be the direct reason for the interruption of antiviral signaling at least in some cases, in consideration that the spatial isolation of the essential component(s) likely hinders interactions and complex assembly on antiviral platforms. Thus, spatial isolation from antiviral platforms may be a common and radical strategy employed by many viruses to interrupt host antiviral IFN signaling (Figure 8C).
Intracytoplasmic IBs of RNA viruses have been characterized for some members of the Mononegavirales order, such as rabies virus (Rhabdoviridae) (Lahaye et al., 2009), respiratory syncytial virus (RSV, Paramyxoviridae) (Lindquist et al., 2010), and Ebola virus (Filoviridae) (Hoenen et al., 2012), while little was reported on IBs induced by segmented negative-stranded RNA viruses. Here, we show IB formation in SFTSV-infected cells. We demonstrated that both SFTSV infection and NSs transient expression caused the production of intracytoplasmic IBs, which were eosinophilic and could be easily visualized using H&E staining (Figure 2). Although SFTSV could infect many cell lines, common cytopathic effects were not evident in most of these cells except for DH82 cells (Yu et al., 2011). Nevertheless, NSs-induced IBs could be observed under light microscopy in all types of mammalian cells detected in this study, including HeLa, HEK293, HEK293T, HepG2, Vero, Raw 264.7, H9c2, and DH82 cells (Figure 2 and Supplementary Data), indicating the likely general roles of NSs and IBs in multiple tissues and organs. Thus, the intracytoplasmic eosinophilic IBs may be conveniently used as a pathological marker for the recognition of SFTSV infection in cultured cells or for the clinical diagnosis. Additionally, the primary target organ(s) and cell(s) of SFTSV infection in humans are unknown, though functions of multiple organs, such as liver, kidney, heart, and gastrointestinal tract, were likely impaired (Yu et al., 2011).
Our results show that SFTSV IBs are not aggresomes but resemble the active IBs of some other viruses, which are functionally involved in the viral life cycle. In cells infected with rabies virus, viral N, P, and L proteins implicated in viral transcription and replication were shown to accumulate in IBs, defining IBs as the viral factory for RNA synthesis (Lahaye et al., 2009). Although SFTSV NSs alone is sufficient to induce the formation of IBs, it is currently unknown whether SFTSV IBs contain other viral proteins like RdRP or/and NP during viral infection. Further investigations on viral protein composition of SFTSV IBs will be required to dissect the possible roles of NSs and NSs-induced IBs in viral life cycles.
This study shows that TBK1/IKKε is trapped to SFTSV IBs, almost completely blocking the IFN signaling. Similarly, TLR3 was found to be relocated to IBs in rabies virus-infected neuronal cells, and the hijack of TLR3 might interfere with TLR3 signaling (Menager et al., 2009). Apart from being involved in IFN-inducing signaling, IBs of several viruses also function in suppression of other cellular pathways. For instance, phosphorylated p38 (p38-P) and O-linked N-acetylglucosamine transferase (OGT) were sequestered into IBs of human RSV, resulting in suppression of cellular responses to viral infection including MAPK-activated protein kinase 2 (MK2) pathway and stress granule assembly (Fricke et al., 2013). Combined with previous studies (Menager et al., 2009; Heinrich et al., 2010; Radhakrishnan et al., 2010; Lahaye et al., 2012; Lifland et al., 2012; Fricke et al., 2013), here we propose that viral IBs likely represent the ‘hot spot’ or ‘interface’ of virus–cell interactions, and especially function as the virus-built ‘jail’ for imprisoning some host factors and hence interfering with corresponding cellular processes. The identification of additional cellular proteins in viral IBs will help us to further uncover the mechanism of IB formation and roles of IBs in virus–host interactions.
In parallel with our study, Qu et al. (2012) reported that both SFTSV NSs and NP might suppress the activation of NF-κB and IFN-β promoters. However, we did not detect significant suppressive activity of NP using our experimental system (Figure 1) and the reason for the discrepancy remains unclear. Additionally, we showed that NSs-mediated suppression of the NF-κB element activation triggered by both SeV infection and TNF-α treatment appeared to be slight, in comparison with the strong inhibition of the IRF-responsive element (ISRE) activation (Figure 1). Hence, NSs strongly inhibits IFN-β induction mainly through ISRE activation but with slight influence on NF-κB signaling, which we suppose is beneficial to virus survival, given that NF-κB is required for efficient replication of SFTSV (Qu et al., 2012).
Emerging viral diseases are posing the serious threat to global economy and public health. As the largest RNA virus family, Bunyaviridae contains various notable viruses pathogenic in plants and animals including human, and many of them are emerging. Recently, another novel bunyavirus named Heartland virus that caused the severe disease with clinical manifestations similar to those of SFTS was discovered in Missouri, USA (McMullan et al., 2012). The phylogenetic analysis demonstrated that Heartland virus is most closely related to SFTSV, and the sequences of their NSs proteins share >60% identity. Thus, it will be interesting to examine whether the NSs of Heartland virus functions similar to that of SFTSV showed in this study.
Materials and methods
Cells and viruses
HEK293T, DH82, and H9c2 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM, GIBCO) supplemented with 10% fetal bovine serum (FBS, GIBICO). Raw 264.7 cells were maintained in RPMI 1640 medium supplemented with 10% FBS. HEK293, Vero, HepG2, and HeLa cells were grown in DMEM supplemented with 10% newborn calf serum. SeV was propagated in 10-day-old embryonated eggs, and the virus titer was measured by hemagglutination assay using chicken erythrocytes. The HA titer of the virus stock was 1:256, and 1:20 was used for infection. SFTSV WCH-2011/HN/China/isolate97 (Lam et al., 2013) was expanded in Vero, HEK293, or DH82 cells in a biosafety level 3 laboratory.
Plasmids
Firefly luciferase reporter plasmids for NF-κB, ISRE, and IFN-β promoters, Renilla luciferase control plasmid (pRL-TK), and mammalian cell expression plasmids for Flag-RIG-I, Flag-MDA5, Flag-VISA (i.e. MAVS), HA-TBK1, Flag-IKKε, and Flag-IRF3 were kindly provided by Dr Hong-Bing Shu (Wuhan University, China) and were described previously (Lei et al., 2010; Chen et al., 2013; He et al., 2013). The RIG-I RNAi plasmid that can significantly inhibit RIG-I expression and the control RNAi plasmid for the RNAi experiment were described previously (Diao et al., 2007; Ran et al., 2011). ORFs encoding the L protein (i.e. RdRp), glycoprotein polyprotein (GP), nucleocapsid protein (NP), and nonstructural protein (NSs) were amplified by RT–PCR from SFTSV genomic RNA and cloned into expression vector pcDNA3.1 or/and pCAGGSP7 with or without S.tag or HA tag. The plasmid encoding influenza A virus NS1 was generated by cloning PR8 virus NS1 ORF into pcDNA3.1. Expression plasmids for other tagged proteins, NSs mutants, and constitutively active forms of RIG-I (RIG-IN) and IRF3 (IRF3/5D) were constructed by standard molecular biology techniques.
Antibodies and reagents
Primary antibodies were purchased from the indicated manufacturers as follows: mouse monoclonal antibodies (mAbs) against Flag (Sigma–Aldrich), HA tag (Beyotime), β-actin (Beyotime), human IKK-i (i.e. IKKε, Santa Cruz Biotechnology), TBK1 (Santa Cruz Biotechnology), Mannose 6 Phosphate Receptor (M6PR, Abcam), and EEA1 (Abcam); rabbit mAbs against phospho-IRF3(Ser396) and IRF3 (Cell Signaling Technology); and rabbit polyclonal antibodies anti-S.tag (Abcam) and anti-SeV (MBL). Rabbit anti-NSs antiserum was raised against NSs protein purified from Escherichia coli. Secondary antibodies used were Alexa Fluor 350 goat anti-mouse IgG (Beyotime), FITC goat anti-rabbit IgG (Rockland), FITC goat anti-mouse IgG (Proteintech), Rhodamine goat anti-rabbit IgG (Chemicon), and DyLight 549 goat anti-mouse IgG (Beyotime). MitoTracker Deep Red (Invitrogen), LysoTracker Red (Beyotime), and Hoechst 33258 (Beyotime) dyes were purchased from the indicated manufacturers. Nocodazole was purchased from Sigma–Aldrich, and human TNF-α from PeproTech, Inc.
Reporter gene assays
HEK293T cells (∼1.5 × 105) were plated onto 24-well dishes and transfected the following day using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. A hundred nanograms reporter plasmid, 20 ng pRL-TK plasmid, and indicated amount of expression plasmids were used per well. In the same experiment, empty vector plasmids were added to ensure that the same amount of total DNA was used for each transfection. Twenty-four hours post-transfection, cells were infected with SeV or left uninfected for 16 h. Luciferase activities were then measured with a DLR assay kit (Promega), and firefly luciferase activity was normalized to Renilla luciferase activity. Thus, the relative luciferase activities (Rel.Lucif.Act.) are shown. For some experiments, cells were co-transfected with plasmids encoding RLR signaling components including RIG-IN, MDA5, MAVS, TBK1, IKKε, IRF3, and IRF3/5D to activate IFN-β or ISRE promoter, and 36 h post-transfection, the luciferase activities were measured.
H&E staining, immunofluorescence, and confocal microscopy
Transfected or infected cells were fixed with 95% ethanol for 20 min before staining with hematoxylin and eosin, and observed under an Olympus IX51 microscope with 100× objective lens. For immunofluorescence, transfected or infected cells were fixed with 4% paraformaldehyde in PBS for 10 min and then permeabilized in 0.5% Triton X-100. After blocking with 2% BSA (Biosharp) and 2% normal goat serum (NGS, Jackson ImmunoResearch) in PBS, cells were stained with primary antibodies for 1 h or overnight, and secondary antibodies for 1 h at 4°C. Cells were then incubated with Hoechst 33258 for 5 min at room temperature for nucleus staining. After each incubation step, cells were washed extensively with PBS. For marking lysosomes and mitochondria, transfected or infected living cells were directly stained with LysoTracker Red and MitoTracker Deep Red, respectively, according to the manufacturers' specifications. Images were taken on Nikon Eclipse TE2000-S inverted fluorescence microscope, or Nikon Ti confocal microscope with the Volocity software (PerkinElmer).
Protein–protein interaction analysis
S-protein pull-down assays were used for identification of overexpressed protein interactions. Forty-eight hours after transfection, 2 × 106 cells were lysed in 400 μl lysis buffer (25 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100) supplemented with protease inhibitor cocktail (Roche) on ice for 15 min. The lysates were then cleared by centrifugation and mixed with 30 μl slurry of S-protein agarose (Merck Novagen), rotating at 4°C for 4 h. After extensive washing with the lysis buffer, the beads were resuspended in 30 μl 1× SDS sample buffer and boiled for 5 min, followed by SDS–PAGE and WB analysis.
For endogenous co-immunoprecipitation (Co-IP) experiments, mock- or SFTSV-infected HEK293 cells (∼5 × 107) were lysed with the lysis buffer as described above. Cell supernatants pretreated with pre-immune serum and protein A/G PLUS-agarose (Santa Cruz Biotechnology) were incubated with anti-NSs antiserum at 4°C for 1 h and then with protein A/G PLUS-agarose at 4°C overnight. After washes with the lysis buffer, immunoprecipitates were subjected to SDS–PAGE and WB analysis.
Native-PAGE and WB analysis
IRF3 dimerization was analyzed with Native-PAGE as described previously (Iwamura et al., 2001) with some modifications. Briefly, whole cell extracts were prepared in lysis buffer and then subjected to electrophoresis in a 8% native acrylamide gel already pre-run for 30 min at 4°C. The electrophoresis buffers were composed of a lower chamber buffer (25 mM Tris, pH 8.4, 192 mM glycine) and an upper chamber buffer (the lower chamber buffer supplemented with 1% sodium deoxycholate). The gel was soaked in SDS running buffer (25 mM Tris, pH 8.4, 250 mM glycine, 0.1% SDS) for 30 min at 25°C and then ready for WB analysis.
For WB, protein samples were firstly separated by SDS–PAGE or Native-PAGE, and then transferred to a PVDF membrane (Millipore). After blocking with 5% BSA in TBS-T, the membrane was probed with primary antibodies and then corresponding horseradish peroxidase-conjugated secondary antibodies (1:2500, Proteintech). Protein signals were detected by an enhanced chemiluminescence (ECL) kit (Thermo Fisher Scientific).
Supplementary material
Funding
This work was supported by the National Science Foundation of China (grant numbers 31125003 and 31321001), the Science and Technology Basic Work Program (grant number 2013FY113500), and the National Basic Research Program (973 Program) of China (grant numbers 2010CB530100 and 2013CB911101).
Conflict of interest: none declared.
Supplementary Material
Acknowledgements
We thank Dr Hong-Bing Shu (Wuhan University, China) for supplying reporter and expression plasmids.
References
- Alff P.J., Sen N., Gorbunova E., et al. The NY-1 hantavirus Gn cytoplasmic tail coprecipitates TRAF3 and inhibits cellular interferon responses by disrupting TBK1-TRAF3 complex formation. J. Virol. 2008;82:9115–9122. doi: 10.1128/JVI.00290-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgnaoui S.M., Paz S., Hiscott J. Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr. Opin. Immunol. 2011;23:564–572. doi: 10.1016/j.coi.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Billecocq A., Spiegel M., Vialat P., et al. NSs protein of Rift Valley fever virus blocks interferon production by inhibiting host gene transcription. J. Virol. 2004;78:9798–9806. doi: 10.1128/JVI.78.18.9798-9806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Li Y., Zhang J., et al. RAVER1 is a coactivator of MDA5-mediated cellular antiviral response. J. Mol. Cell Biol. 2013;5:111–119. doi: 10.1093/jmcb/mjt006. [DOI] [PubMed] [Google Scholar]
- Diao F., Li S., Tian Y., et al. Negative regulation of MDA5- but not RIG-I-mediated innate antiviral signaling by the dihydroxyacetone kinase. Proc. Natl Acad. Sci. USA. 2007;104:11706–11711. doi: 10.1073/pnas.0700544104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott R.M., Weber F. Bunyaviruses and the type I interferon system. Viruses. 2009;1:1003–1021. doi: 10.3390/v1031003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L., Briese T., Lipkin W.I. Z proteins of New World arenaviruses bind RIG-I and interfere with type I interferon induction. J. Virol. 2010;84:1785–1791. doi: 10.1128/JVI.01362-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke J., Koo L.Y., Brown C.R., et al. p38 and OGT sequestration into viral inclusion bodies in cells infected with human respiratory syncytial virus suppresses MK2 activities and stress granule assembly. J. Virol. 2013;87:1333–1347. doi: 10.1128/JVI.02263-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mata R., Bebok Z., Sorscher E.J., et al. Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J. Cell Biol. 1999;146:1239–1254. doi: 10.1083/jcb.146.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groseth A., Charton J.E., Sauerborn M., et al. The Ebola virus ribonucleoprotein complex: a novel VP30-L interaction identified. Virus Res. 2009;140:8–14. doi: 10.1016/j.virusres.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X., Li Y., Li C., et al. USP2a negatively regulates IL-1beta- and virus-induced NF-kappaB activation by deubiquitinating TRAF6. J. Mol. Cell Biol. 2013;5:39–47. doi: 10.1093/jmcb/mjs024. [DOI] [PubMed] [Google Scholar]
- Heinrich B.S., Cureton D.K., Rahmeh A.A., et al. Protein expression redirects vesicular stomatitis virus RNA synthesis to cytoplasmic inclusions. PLoS Pathog. 2010;6:e1000958. doi: 10.1371/journal.ppat.1000958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgason E., Phung Q.T., Dueber E.C. Recent insights into the complexity of Tank-binding kinase 1 signaling networks: the emerging role of cellular localization in the activation and substrate specificity of TBK1. FEBS Lett. 2013;587:1230–1237. doi: 10.1016/j.febslet.2013.01.059. [DOI] [PubMed] [Google Scholar]
- Hiscott J., Lacoste J., Lin R. Recruitment of an interferon molecular signaling complex to the mitochondrial membrane: disruption by hepatitis C virus NS3-4A protease. Biochem. Pharmacol. 2006;72:1477–1484. doi: 10.1016/j.bcp.2006.06.030. [DOI] [PubMed] [Google Scholar]
- Hoenen T., Shabman R.S., Groseth A., et al. Inclusion bodies are a site of ebolavirus replication. J. Virol. 2012;86:11779–11788. doi: 10.1128/JVI.01525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou F., Sun L., Zheng H., et al. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamura T., Yoneyama M., Yamaguchi K., et al. Induction of IRF-3/-7 kinase and NF-kappaB in response to double-stranded RNA and virus infection: common and unique pathways. Genes Cells. 2001;6:375–388. doi: 10.1046/j.1365-2443.2001.00426.x. [DOI] [PubMed] [Google Scholar]
- Kagan J.C. Defining the subcellular sites of innate immune signal transduction. Trends Immunol. 2012;33:442–448. doi: 10.1016/j.it.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaukinen P., Sillanpaa M., Nousiainen L., et al. Hepatitis C virus NS2 protease inhibits host cell antiviral response by inhibiting IKKepsilon and TBK1 functions. J. Med. Virol. 2013;85:71–82. doi: 10.1002/jmv.23442. [DOI] [PubMed] [Google Scholar]
- Kawai T., Takahashi K., Sato S., et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- Kopito R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- Lahaye X., Vidy A., Pomier C., et al. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: evidence that NBs are sites of viral transcription and replication. J. Virol. 2009;83:7948–7958. doi: 10.1128/JVI.00554-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahaye X., Vidy A., Fouquet B., et al. Hsp70 protein positively regulates rabies virus infection. J. Virol. 2012;86:4743–4751. doi: 10.1128/JVI.06501-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam T.T., Liu W., Bowden T.A., et al. Evolutionary and molecular analysis of the emergent severe fever with thrombocytopenia syndrome virus. Epidemics. 2013;5:1–10. doi: 10.1016/j.epidem.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei C.Q., Zhong B., Zhang Y., et al. Glycogen synthase kinase 3beta regulates IRF3 transcription factor-mediated antiviral response via activation of the kinase TBK1. Immunity. 2010;33:878–889. doi: 10.1016/j.immuni.2010.11.021. [DOI] [PubMed] [Google Scholar]
- Li D.X. Fever with thrombocytopenia associated with a novel bunyavirus in China. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi. 2011;25:81–84. [PubMed] [Google Scholar]
- Lifland A.W., Jung J., Alonas E., et al. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J. Virol. 2012;86:8245–8258. doi: 10.1128/JVI.00215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist M.E., Lifland A.W., Utley T.J., et al. Respiratory syncytial virus induces host RNA stress granules to facilitate viral replication. J. Virol. 2010;84:12274–12284. doi: 10.1128/JVI.00260-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.Y., Wei B., Shi H.X., et al. Tom70 mediates activation of interferon regulatory factor 3 on mitochondria. Cell Res. 2010;20:994–1011. doi: 10.1038/cr.2010.103. [DOI] [PubMed] [Google Scholar]
- McMullan L.K., Folk S.M., Kelly A.J., et al. A new phlebovirus associated with severe febrile illness in Missouri. N. Engl. J. Med. 2012;367:834–841. doi: 10.1056/NEJMoa1203378. [DOI] [PubMed] [Google Scholar]
- Menager P., Roux P., Megret F., et al. Toll-like receptor 3 (TLR3) plays a major role in the formation of rabies virus Negri Bodies. PLoS Pathog. 2009;5:e1000315. doi: 10.1371/journal.ppat.1000315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan E., Curran J., Hofmann K., et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- Popescu C.I., Callens N., Trinel D., et al. NS2 protein of hepatitis C virus interacts with structural and non-structural proteins towards virus assembly. PLoS Pathog. 2011;7:e1001278. doi: 10.1371/journal.ppat.1001278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins K.C., Cardenas W.B., Basler C.F. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J. Virol. 2009;83:3069–3077. doi: 10.1128/JVI.01875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu B., Qi X., Wu X., et al. Suppression of the interferon and NF-kappaB responses by severe fever with thrombocytopenia syndrome virus. J. Virol. 2012;86:8388–8401. doi: 10.1128/JVI.00612-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A., Yeo D., Brown G., et al. Protein analysis of purified respiratory syncytial virus particles reveals an important role for heat shock protein 90 in virus particle assembly. Mol. Cell. Proteomics. 2010;9:1829–1848. doi: 10.1074/mcp.M110.001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran Y., Liu T.T., Zhou Q., et al. SENP2 negatively regulates cellular antiviral response by deSUMOylating IRF3 and conditioning it for ubiquitination and degradation. J. Mol. Cell Biol. 2011;3:283–292. doi: 10.1093/jmcb/mjr020. [DOI] [PubMed] [Google Scholar]
- Schmaljohn C.S., Nichol S.T. Fields Virology. Philadelphia: Lippincott, Williams & Wilkins; 2007. Bunyaviridae; pp. 1741–1789. Knipe, D.M., and Howley, P.M. (eds) [Google Scholar]
- Seth R.B., Sun L., Ea C.K., et al. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- van Zuylen W.J., Doyon P., Clement J.F., et al. Proteomic profiling of the TRAF3 interactome network reveals a new role for the ER-to-Golgi transport compartments in innate immunity. PLoS Pathog. 2012;8:e1002747. doi: 10.1371/journal.ppat.1002747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.Y., Liu L.J., Zhong B., et al. WDR5 is essential for assembly of the VISA-associated signaling complex and virus-triggered IRF3 and NF-kappaB activation. Proc. Natl Acad. Sci. USA. 2010;107:815–820. doi: 10.1073/pnas.0908967107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L.G., Wang Y.Y., Han K.J., et al. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Yoneyama M., Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 2009;227:54–65. doi: 10.1111/j.1600-065X.2008.00727.x. [DOI] [PubMed] [Google Scholar]
- Yu X.J., Liang M.F., Zhang S.Y., et al. Fever with thrombocytopenia associated with a novel bunyavirus in China. N. Engl. J. Med. 2011;364:1523–1532. doi: 10.1056/NEJMoa1010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.