

Abstract
Background
Human infection with Middle East respiratory syndrome coronavirus (MERS-CoV) poses an ongoing threat to public health worldwide. The studies of MERS patients with severe disease and experimentally infected animals showed that robust viral replication and intensive proinflammatory response in lung tissues contribute to high pathogenicity of MERS-CoV. We sought to identify pattern recognition receptor (PRR) signaling pathway(s) that mediates the inflammatory cascade in human macrophages upon MERS-CoV infection.
Methods
The potential signaling pathways were manipulated individually by pharmacological inhibition, small interfering ribonucleic acid (siRNA) depletion, and antibody blocking. The MERS-CoV-induced proinflammatory response was evaluated by measuring the expression levels of key cytokines and/or chemokines. Reverse transcription-quantitative polymerase chain reaction assay, flow cytometry analysis, and Western blotting were applied to evaluate the activation of related PRRs and engagement of adaptors.
Results
MERS-CoV replication significantly upregulated C-type lectin receptor (CLR) macrophage-inducible Ca2+-dependent lectin receptor (Mincle). The role of Mincle for MERS-CoV-triggered cytokine/chemokine induction was established based on the results of antibody blockage, siRNA depletion of Mincle and its adaptor spleen tyrosine kinase (Syk), and Syk pharmacological inhibition. The cytokine and/or chemokine induction was significantly attenuated by siRNA depletion of retinoic acid-inducible-I-like receptors (RLR) or adaptor, indicating that RLR signaling also contributed to MERS-CoV-induced proinflammatory response.
Conclusions
The CLR and RLR pathways are activated and contribute to the proinflammatory response in MERS-CoV-infected macrophages.
Keywords: CLR, MERS-CoV, Mincle, proinflammatory response, RLR
Middle East respiratory syndrome coronavirus (MERS-CoV) has been identified as a novel pathogen causing human respiratory infections since 2012 [1]. Most MERS patients presented with viral pneumonia, and some of them developed acute respiratory distress syndrome and multiorgan failure with a case-fatality rate over 30% [2]. Cytological examination of bronchoalveolar lavage fluids from MERS patients exhibited large numbers of neutrophils and macrophages, indicating a massive pulmonary inflammation [3]. The postmortem study of a MERS patient revealed compatible pathological changes, including edematous alveolar septa with infiltration of lymphocytes, neutrophils, and macrophages, as well as diffuse alveolar damage [4]. A detailed examination of lung tissues of MERS-CoV-infected nonhuman primates including common marmosets and Rhesus macaques revealed similar pathological changes, reminiscent of prominent lung disease in severe MERS patients. Thus, the authors reached a central conclusion that robust viral replication, together with an intense local immune response to MERS-CoV infection, may result in severe respiratory disease in these experimental animals [5].
Macrophages are important sentinel cells of the innate immune system. Macrophages sense and recognize invading pathogens or endogenous ligands through a broad range of sensors, and they generally elicit effective clearance via phagocytosis. On the other hand, macrophages may fail to eliminate invading microbes. Instead, they become inappropriately activated and initiate dysregulated inflammatory responses in some cases [6, 7]. We have previously reported that MERS-CoV productively infected human monocyte-derived macrophages (MDMs) and triggered aberrant proinflammatory response [8], providing direct evidence to explain the massive inflammation observed in severe MERS patients and experimentally infected nonhuman primates.
Upon viral infection, host germline-encoded pattern recognition receptors (PRRs) including retinoic acid-inducible gene (RIG)-I-like receptors (RLRs), Toll-like receptors (TLRs), nucleotide-binding oligomerization domains-like receptors (NLRs), and C-type lectin receptors (CLRs) detect the presence of foreign motifs or ligands known as pathogen-associated molecular patterns [9–12]. The intracellular signaling cascades triggered by these PRRs lead to transcriptional activation of type I interferons (IFNs) and inflammatory mediators that coordinate the elimination of pathogens and infected cells and, meanwhile, contribute to the inflammation and clinical symptoms of viral infections.
The RLRs, such as RIG-I and melanoma differentiation-associated gene 5 (MDA5), have been extensively characterized as essential cellular sensors to recognize RNA viruses [11, 13]. Ding et al [14] demonstrated the RLR-mediated signaling for nuclear factor (NF)-κB activation in transmissible gastroenteritis virus infection. Zalinger et al [15] elucidated the importance of MDA5 to host defense during murine CoV infection. Meanwhile, CLR signaling has shown the increasing importance for triggering inflammatory response upon viral infections [16, 17]. The transcription factor NF-κB is an essential mediator of inducible gene expression of cytokines and chemokines. CLR induced signal transduction seems to mainly activate and modulate NF-κB functions [17, 18]. The mechanisms by which CoVs evade host innate antiviral response, including MERS-CoV and severe acute respiratory syndrome (SARS)-CoV, have been extensively investigated [19–21]. However, it is poorly understood how the highly pathogenic MERS-CoV triggers the aberrant proinflammatory response, one of the pathological bases of severe respiratory diseases in MERS patients and experimentally infected animals. In this study, we sought to elucidate the contribution of RLR and CLR signaling pathway for mediating the exuberant inflammatory responses in macrophages upon MERS-CoV infection.
MATERIALS AND METHODS
Virus Culture
The MERS-CoV of strain EMC/2012 [1] was propagated in Vero E6 cells. Three days after virus inoculation, the cell-free media were collected and stored at −80°C in aliquots. Ultraviolet (UV) inactivation of MERS-CoV was performed by exposing the virus to UV cross-linker for 10 minutes as described previously [22].
Preparation of Human Monocyte-Derived Macrophages, Virus Infection, and Manipulation of the Macrophages
Peripheral blood was obtained from healthy blood donors at the Hong Kong Red Cross Blood Transfusion Center according to a protocol approved by the Institutional Review Board of The University of Hong Kong/Hospital Authority Hong Kong West Cluster. Monocyte preparation and differentiation were performed according to a well established protocol described previously [23]. For viral infection, treated or mock-treated MDMs were inoculated with MERS-CoV at a multiplicity of infection (MOI) of 2 or were mock inoculated for 1 hour at 37°C. Cell lysates were harvested at 24 hours postinfection (hpi) for quantification of messenger RNA (mRNA) expression levels of cellular genes and detection of viral load.
TANK-binding kinase 1 (TBK1) and inhibitor of NF-κB kinase epsilon (IKKε) inhibitor Amlexanox, spleen tyrosine kinase (Syk) inhibitor R406, and caspase-1 inhibitor VX-765 were purchased from InvivoGen. The individual inhibitors or a neutralization antibody against human Mincle (InvivoGen, mabg-hmcl) or mouse monoclonal immunoglobulin (Ig)G2B (MAB004; R&D) were administered to MDMs 1 hour before virus inoculation at the indicated concentrations and supplemented in the culture media throughout after infection. Silencer Select small interfering RNA (siRNA) targeting human mitochondrial antiviral-signaling proteins (MAVS) (s33179; Thermo Fisher Scientific), Syk (s13681), RIG-I (s223615), MDA5 (s34499), Mincle (s25297), and scrambled siRNA were transfected to MDMs using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s instruction. In brief, MDMs were transfected with 200 nM siRNA for 2 consecutive days. At day 3 after siRNA transfection, the cells were inoculated with MERS-CoV.
Quantification of Cellular Messenger Ribonucleic Acid Transcript and Viral Load By Reverse Transcription-Quantitative Polymerase Chain Reaction Assay
Detection of cellular mRNA expression and viral load was performed as described previously [24]. In brief, cell lysates were applied to RNA extraction, followed by reverse transcription (RT) using oligo(dT) or the virus-specific primer. The resultant complementary deoxyribonucleic acids (cDNAs) were used for quantitative polymerase chain reaction (qPCR) assay to measure mRNA expression level of cellular gene and viral load. The primer sequences used in qPCR assay are shown in the Supplementary Table.
Flow Cytometry Analysis, Immunofluorescence Staining, and Western Blot Analysis
Immunostaining and flow cytometry analysis were performed according to the standard protocol as described elsewhere [25, 26]. In brief, after detachment, fixation, and permeabilization, MDMs were labeled with a mouse antibody against human Mincle (ab100846; Abcam) and in-house made antibody against MERS-CoV NP or antibody of isotype control, followed by the corresponding secondary antibodies. Flow cytometry analysis was performed using a BD FACSCanto II flow cytometer (BD Biosciences), and the data were analyzed using FlowJo vX (Tree Star).
For immunofluorescence staining, MDMs seeded on glass coverslips were inoculated with MERS-CoV at 5 MOI. At 24 hpi, the cells were fixed with 4% paraformaldehyde and labeled with anti-NP and anti-Mincle, followed by corresponding secondary antibodies. Slides were mounted with ProLong Gold antifade reagent with 4’,6-diamidino-2-phenylindole (Themo Fisher Scientific) and imaged with a Carl Zeiss LSM 800 confocal microscope.
The whole-cell extracts of infected and mock-infected macrophages were separated in sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane. After blocking, the membranes were probed with antibodies against MAVS (ab31334; Abcam), Syk (ab40781), RIG-I (ab45428), MDA5 (ab126630), Mincle (ab100846), and CARD9 (ab124922), NP antibody or mouse β-Actin antibody (A5441; Sigma), followed by secondary staining. The blots were visualized by Luminata Classico Western HRP Substrate (WBLUC0500; Millipore). Quantification was performed using ImageJ software. A human IP-10 ELISA Kit (ab173194; Abcam) was used for quantification of IP-10 secretion in the culture media.
Statistical Analysis
An unpaired t test was performed for data analysis using GraphPad Prism 6. P < .05 was considered to be statistically significant. Data are presented as mean and standard deviation of representative experiments.
RESULTS
Inhibition Assay Hinted the Potential Involvement of RLR and CLR Pathways
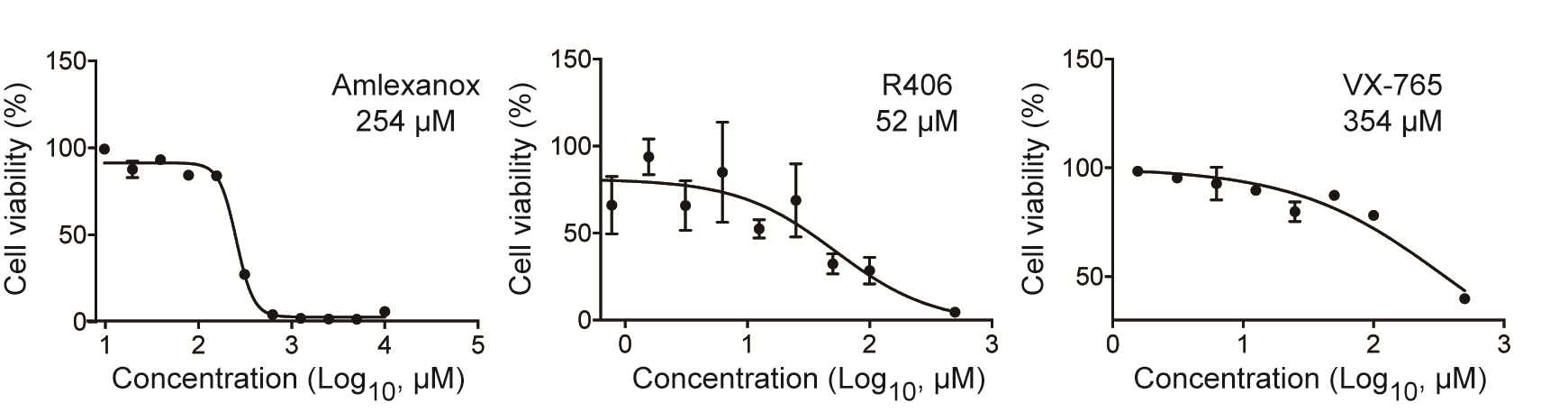
We first used the inhibitors of RLR and CLR signaling pathway to evaluate their potential contribution for mediating the proinflammatory response in MERS-CoV-infected macrophages. Amlexanox, an inhibitor of RLR signaling pathway, specifically supresses the noncanonical IkB kinases IKKε and TBK1, and both are essential players for the coordination of IFN regulatory factor 3 (IRF3)- and NF-κB-mediated innate immune response [27, 28]. R406 is a specific, ATP-competitive inhibitor of Syk, the essential adaptor of CLR pathway [29]. Caspase-1 inhibitor VX-765, a commonly used inhibitor for NLR signaling, is used for comparison [30]. MTT assay was used to detect the 50% cytotoxic concentration (CC50) of each inhibitor in MDMs (Supplementary Figure 1), to ensure that the concentrations used for inhibition of PRRs have no adverse effect on macrophage viability. Based on the effective concentration of each inhibitor and its CC50, the indicated working concentrations (Figure 1A) were used throughout the study.
Figure 1.

Inhibition of individual pattern recognition receptor (PRR) signaling pathways implicates the involvement of retinoic acid-inducible gene-I-like receptor (RLR) and C-type lectin receptor (CLR) for the immune activation in Middle East respiratory syndrome coronavirus (MERS-CoV)-infected macrophages. (A) The inhibitors for various PRR signal pathways, the 50% cytotoxic concentration (CC50), and the working concentration used in the study are listed. (B) The MERS-CoV-infected monocyte-derived macrophages (MDMs) were treated with the inhibitors or mock-treated in triplicate. At 24 hours postinfection, cells were lysed for detecting messenger ribonucleic acid expression levels of proinflammatory cytokines and chemokines. Results show the fold change of glyceraldehyde 3-phosphate dehydrogenase-normalized expression level in the treated or mock-treated cells relative to that in the mock-infected cells. Data are presented as mean ± standard deviation of triplicate wells of MDMs from 3 different donors. Unpaired t test was used for data analysis. *, P < .05; **, P < .01; ***, P < .001. IFN, interferon; IKKε, inhibitor of NF-κB kinase epsilon; IL-6, interleukin 6; MIP, macrophage-inflammatory protein; NLR, nucleotide-binding oligomerization domains-like receptor; Syk, spleen tyrosine kinase; TBK1, TANK-binding kinase 1; TNF, tumor necrosis factor.
To assess the contribution of RLR or CLR pathway to induce proinflammatory response, we measured the expression levels of a series of key proinflammatory cytokines and/or chemokines in MERS-CoV-infected MDMs in the presence of these inhibitors (Figure 1B). Consistent with our previous observation, MERS-CoV infection globally stimulated an array of proinflammatory cytokines and chemokines including interleukin (IL)-6, tumor necrosis factor (TNF)-α, macrophage-inflammatory protein (MIP)-1α, RANTES, IFN-γ, and IP-10 [8]. The induction of cytokine and/or chemokine was dependent on viral replication because the inoculation of the UV-inactivated viruses was unable to trigger these inflammatory mediators, except RANTES (Supplementary Figure 2). It is notable that MERS-CoV-mediated induction of TNF-α, MIP-1α, IFN-γ, and IP-10 was generally diminished in the presence of Amlexanox and R406, although Amlexanox treatment only marginally reduced TNF-α production. In addition, the treatment of Amlexanox, but not R406, significantly attenuated the induction of IL-6 and RANTES. However, NLR signaling inhibitor VX-765 seemed to have minimal effect on these inflammatory mediators. Because the replication of MERS-CoV is the driving force for the induction of most cytokines and/or chemokines, we evaluated the viral replication in the presence of the inhibitors. It was shown that the addition of these inhibitors did not affect viral replication (Supplementary Figure 3).
Accumulating evidence suggested that RLRs and CLRs are inducible upon stimulation or microbial infection [14, 31–33]. Among CLRs, Dectin-1, Dectin-2, and Mincle are highly expressed in myeloid cells such as monocytes, macrophages, and dendritic cells [17]. Thus, we measured the mRNA profiles of RLRs and myeloid CLRs in MERS-CoV-infected macrophages. As shown in Figure 2A, the mRNA expression levels of RLRs including RIG-I, MDA5, and LPG2, as well as CLRs Mincle and Dectin-2 were significantly upregulated at 24 hpi. The positive modulation pattern of RLR and CLR, especially the increased expression of RIG-I and Mincle, was manifested as early as 6 hpi (Figure 2B), suggesting an accelerating activation of these PRRs during MERS-CoV infection. Overall, the above results suggested that RLR and CLR signaling might be involved in viral recognition and trigger the proinflammatory response upon MERS-CoV infection in macrophages.
Figure 2.

Retinoic acid-inducible gene (RIG)-I-like receptors (RLRs) and some C-type lectin receptors (CLRs) are inducible upon Middle East respiratory syndrome coronavirus (MERS-CoV) infection. At 24 hours postinfection (hpi) (A) and 6 hpi (B), the MERS-CoV-infected monocyte-derived macrophages (MDMs) and mock-infected cells were harvested for detection of messenger ribonucleic acid expression levels of RLRs and myeloid CLRs. Results show the fold change of glyceraldehyde 3-phosphate dehydrogenase-normalized expression level in the infected cells relative to that in the mock-infected cells. Data are presented as mean ± standard deviation of triplicate wells of MDMs from 3 different donors. Unpaired t test was used for data analysis. *, P < .05. MDA5, melanoma differentiation-associated gene 5; Mincle, macrophage-inducible Ca2+-dependent lectin receptor.
Depletion of CLR or RLR Adaptors and Receptors Dampened the Induction of Cytokines and Chemokines
To dissect the role of RLR and CLR signaling in inducing proinflammatory response, we depleted the RLR adaptor, MAVS, or the CLR adaptor, Syk, by siRNA knockdown and examined MERS-CoV-elicited cytokine and/or chemokine response. The effective depletion of both adaptors was shown by RT-qPCR assay at 48 hours post-siRNA transfection, to approximately 20% for Syk and 40% for MAVS relative to the control cells (Figure 3A). The reduced expression of the adaptors was also verified by Western blot. Subsequently, we infected the transfected cells with MERS-CoV and measured the expression profiles of the cytokines and/or chemokines. As shown in Figure 3B, siRNA depletion of either MAVS or Syk significantly diminished the induction of all tested inflammatory molecules including IL-6, TNF-α, MIP-1α, RANTES, IFN-γ, and IP-10.
Figure 3.

Genetic depletion of retinoic acid-inducible gene-I-like receptor (RLR) or C-type lectin receptor (CLR) adaptor significantly attenuated the induction of proinflammatory cytokines and chemokines. (A) Small interfering ribonucleic acid (siRNA) targeting RLR adaptor mitochondrial antiviral-signaling proteins (MAVS) or CLR adaptor spleen tyrosine kinase (Syk) or scrambled siRNA (siCtrl) were transfected into monocyte-derived macrophages (MDMs) in 2 consecutive days. At day 3 after siRNA transfection, cells were harvested to assess the knockdown effect of MAVS and Syk by reverse transcription-quantitative polymerase chain reaction assay and Western blotting. (B) At day 3 after siRNA transfection, the depleted cells were infected with Middle East respiratory syndrome coronavirus (MERS-CoV). At 24 hours postinfection, cells were lysed for detecting messenger RNA expression levels of proinflammatory cytokines and chemokines. Results show the fold change of glyceraldehyde 3-phosphate dehydrogenase-normalized expression level in the infected cells relative to that in the mock-infected cells. Data are presented as mean ± standard deviation of triplicate wells of MDMs from 3 different donors. Unpaired t test was used for data analysis. *, P < .05; **, P <. 01; ***, P < .001. IFN, interferon; IL-6, interleukin 6; MIP, macrophage-inflammatory protein; TNF, tumor necrosis factor.
RIG-I, MDA5, and Mincle were upregulated the most among RLR and CLR receptors (Figure 2). We then tested whether siRNA depletion of RIG-I, MDA5, and Mincle have any effect on MERS-CoV-triggered immune activation. As shown in Figure 4A, the receptors were depleted to approximately 30% at transcriptional level, which were verified by Western blot analysis. In RIG-I-, MDA5-, and Mincle-depleted cells, the induction of IFN-γ and IP-10 was generally abolished; and MIP-1α and RANTES expression were significantly reduced (Figure 4B). A significantly attenuated IL-6 induction was observed in RIG-I-depleted cells, but not in MDA5- or Mincle-depleted cells; whereas TNF-α production was basically unaffected after depletion of these receptors individually.
Figure 4.

Genetic depletion of retinoic acid-inducible gene (RIG)-I-like receptor (RLR) or C-type lectin receptor (CLR) dampened the induction of proinflammatory cytokines and chemokines. (A) Small interfering ribonucleic acid (siRNA) targeting RLR (RIG-I and melanoma differentiation-associated gene 5 [MDA5]) or macrophage-inducible Ca2+-dependent lectin receptor (Mincle) or scrambled siRNA (siCtrl) were transfected into monocyte-derived macrophages (MDMs) in 2 consecutive days. At day 3 after siRNA transfection, cells were harvested to measure the knockdown effect of receptors by reverse transcription-quantitative polymerase chain reaction assay and Western blotting. (B) At day 3 after siRNA transfection, the depleted cells were infected with Middle East respiratory syndrome coronavirus (MERS-CoV). At 24 hours postinfection (hpi), cells were lysed for detecting messenger RNA expression levels of proinflammatory cytokines and chemokines. Results show the fold change of glyceraldehyde 3-phosphate dehydrogenase-normalized expression level in the infected cells relative to that in the mock-infected cells. Data are presented as mean ± standard deviation of triplicate wells of MDM from 3 different donors. (C) At day 3 after the siRNA transfection, the depleted cells were infected with MERS-CoV. At 24 hpi, cell-free media were harvested for detection of IP-10 secretion by enzyme-linked immunosorbent assay. Unpaired t test was used for data analysis. *, P < .05; **, P < .01; ***, P < .001.
The induction of IFN-γ and IP-10 was constantly and significantly reduced in various assays, including the inhibitor treatment and siRNA depletion. In addition, these 2 proinflammatory mediators are highly stimulated in severe SARS and MERS patients [34–36]. Thus, we examined MERS-CoV-induced secretion of IFN-γ and IP-10 after genetic depletion of the above-mentioned RLR and CLR receptors and adaptors. IP-10 secretion was readily detectable from both MERS-CoV-infected and mock-infected MDMs; whereas IFN-γ secretion was below the detection limit, even in the virus-infected cells. Thus, we examined MERS-CoV-induced IP-10 secretion in MDMs upon siRNA transfection. As shown in Figure 4C, genetic depletion of RIG-I, or Syk and Mincle resulted in a signification reduction of IP-10 secretion. Knockdown of RIG-I adaptor MAVS also marginally attenuated IP-10 production. Taken together, we found that disrupting RLR or CLR signaling, especially at the adaptor level, largely dampened the production of MERS-CoV-induced proinflammatory cytokines and chemokines.
Mincle Blockage by Antibody Reduced Proinflammatory Response
The interplay of CLR signaling and human CoVs has not been appreciated yet. To further characterize the role of Mincle for immune activation in MERS-CoV-infected macrophages, we measured the MERS-CoV-related IFN-γ induction in the presence of increasing concentrations of a neutralization antibody against Mincle. The results showed that the addition of α-Mincle significantly suppressed MERS-CoV-triggered IFN-γ induction in a dose-dependent manner (Figure 5A). In addition, treatment of α-Mincle significantly diminished the induction of IL-6, RANTES, IFN-γ, and IP-10 (Figure 5B). However, the inhibitory effect was negligible for TNF-α and MIP-1α. Taken together, in line with the observations from Mincle siRNA depletion, CLR receptor Mincle contributed to the immune activation in MERS-CoV-infected macrophages.
Figure 5.

Antibody blockage of macrophage-inducible Ca2+-dependent lectin receptor (Mincle) diminished the proinflammatory response. (A) Middle East respiratory syndrome coronavirus (MERS-CoV)-infected monocyte-derived macrophages (MDMs) were treated with increasing concentrations of Mincle antibody or mouse immunoglobulin (Ig)G2B. At 24 hours postinfection (hpi), cell lysates were used for detecting interferon (IFN)-γ expression. Results show the glyceraldehyde 3-phosphate dehydrogenase (GAPDH)-normalized IFN-γ expression level in the treated or mock-treated cells relative to that in the mock-infected cells. Data are presented as mean ± standard deviation (SD) of triplicate wells of MDM from 2 different donors. (B) The MERS-CoV-infected MDMs were treated with 5 µg/mL anti-Mincle or IgG2B with same concentration. At 24 hpi, cell lysates were used for detecting the induction of proinflammatory response. Results show the fold change of GAPDH-normalized expression level in the treated or mock-treated infected cells relative to that in the mock-infected cells. Data are presented as mean ± SD of triplicate wells of MDMs from 4 different donors. Unpaired t test was used for data analysis. *, P < .05. IL-6, interleukin 6; MIP, macrophage-inflammatory protein; TNF, tumor necrosis factor.
Mincle Was Activated in MERS-CoV-Infected Cells
To characterize the engagement of Mincle in MERS-CoV-infected cells, we assessed Mincle protein expression by flow cytometry analysis and immunofluorescence staining. As shown in Figure 6A, in the mock-infected cells, approximately 50% of macrophages were Mincle+, indicating that Mincle is indeed highly expressed in macrophages as reported previously [17]. The percentage of NP+ (infected) cells increased from 2.9% at 3 hpi to 37.8% at 24 hpi, indicating a productive viral replication. We then compared Mincle expression in NP+ cells versus that in NP− cells at 24 hpi. In NP− cells, Mincle protein expression, ie, the percentage of Mincle-expressing cells and the abundance of Mincle protein (mean fluorescence intensity), remained similar in the NP− cells and the mock-infected cells. It is notable that Mincle protein expression was significantly elevated in the NP+ cells (Figure 6B). Mincle upregulation in the virus-infected cells was verified by costaining of Mincle and NP. The NP+ macrophages displayed the substantially elevated Mincle expression (Figure 6C). Therefore, the results suggested that MERS-CoV replication significantly enhanced Mincle expression in macrophages.
Figure 6.

Middle East respiratory syndrome coronavirus (MERS-CoV) replication in macrophages enhanced macrophage-inducible Ca2+-dependent lectin receptor (Mincle) protein expression. At the indicated hours postinfection (hpi), MERS-CoV-infected or mock-infected monocyte-derived macrophages (MDMs) were fixed, immunolabeled with an antibody against MERS-CoV NP, and α-Mincle or mouse immunoglobulin (Ig)G2B, followed by flow cytometry analysis. (A) A representative histogram of Mincle and NP expression after virus inoculation. The bar charts show the percentage of NP+ cells and the abundance (mean fluorescence intensity [MFI]) of NP at the indicated hpi. Data are presented as mean ± standard deviation (SD) of MDMs from 3 different donors. (B) The percentage of Mincle+ cells (left) and relative abundance of Mincle (right) in the virus-inoculated cells (including NP+ and NP− cells) versus that in the mock-infected cells at 24 hpi. Data are presented as mean ± SD of MDMs from 3 different donors. Unpaired t test was used for data analysis. *, P < .05. (C) Representative images of Mincle expression in MERS-CoV-infected MDMs. The MDMs harvested at 24 hours postinoculation are applied to immunofluorescence staining using the antibody against MERS-CoV NP (red) and Mincle (green). Cellular F-actin and nuclei are counterstained with Phalloidin (white) and 4’,6-diamidino-2-phenylindole (blue), respectively.
The Upregulated CARD9 Transduced CLR Signaling
Engagement of adaptor Syk with CLRs, via CARD9, activates mitogen-activated protein kinases and transcription factor NF-kB and results in the induction of proinflammatory cytokines [9]. CARD9 activation is not only mediated by phosphorylation and ubiquitination processes, but it is also enabled by upregulation [37]. To dissect the role of CARD9 in CLR signaling upon MERS-CoV infection, we examined CARD9 activation upon MERS-CoV infection or in the presence of the CLR inhibitor R406. As shown in Supplementary Figure 4, R406 had a negligible effect on CARD9 expression in the naive macrophages. However, CARD9 was significantly upregulated in MERS-CoV-infected macrophages. In contrast to the mock-infected cells, inhibition of CLR signaling by R406 significantly suppressed the degree of CARD9 activation. Thus, the results revealed the importance of CARD9 as an essential adaptor to relay CLR signaling in MERS-CoV-infected macrophages.
DISCUSSION
MERS-CoV, the most virulent human CoV identified so far, has posed an ongoing threat to public health worldwide. It has been recognized that the rapid viral replication in lung tissues, massive inflammation, and elevated proinflammatory cytokine and chemokine response collectively contribute to acute lung injury and underlie the high pathogenicity of this life-threatening human CoV [2, 5, 34, 38]. The postmortem examination of MERS-CoV patient’s lung tissues, which were obtained by needle sampling shortly after death, revealed severe pneumonia with diffuse alveolar damage and mixed inflammatory cell infiltration. Immunostaining revealed numerous intra-alveolar macrophages and infiltrating T lymphocytes in the parenchyma [4], which are consistent with the histopathological changes in lung tissues of MERS-CoV-infected marmosets. It is notable that profound macrophage proliferation was the most prominent pathological change in the postmortem examination of lung tissues of fatal SARS patients [39]. It was postulated that the proinflammatory cytokines and chemokines released from the proliferative alveolar macrophages play a prominent role in the pathogenesis of SARS [40–42]. We have reported that MERS-CoV can productively infect human macrophages and trigger exuberant proinflammatory response [8]. It is conceivable that the proliferating macrophages in lung tissue of a MERS patient and the dysregulated immune response in these cells may contribute significantly to the high pathogenicity of MERS-CoV.
In this study, we sought to identify the cellular signaling pathway(s) that mediate the proinflammatory response in MERS-CoV- infected human macrophages. The initial observations from the effect of the pathway inhibitors and profiling of receptor activation hinted at the possible involvement of RLR and CLR signaling in MERS-CoV-induced immune activation in macrophages. We then performed a series of experiments to characterize the role of RLR and CLR, especially CLR, in MERS-CoV-infected macrophages, with the intention to better understand the pathogenesis of human MERS.
The role of RLR and CLR for MERS-CoV for eliciting immune activation was noted upon genetic depletion of the RLR adaptor MAVS or the CLR adaptor Syk. The genetic depletion generally compromised or abolished the induction of cytokines and/or chemokines. The attenuated production of most proinflammatory mediators including MIP-1α, RANTES, IFN-γ, and IP-10 was replicated when RLR receptors (RIG-I or MDA5) or CLR receptor (Mincle) were depleted individually. However, the dampened inflammatory response was more prominent in adaptor depletion than receptor depletion, which could be ascribed to the fact that the activation signals from multiple receptors (eg, RIG-I and MDA5) converge at 1 adaptor (eg, MAVS). Upon MERS-CoV infection, multiple PRRs were activated in the macrophages (Figure 2). Therefore, depleting individual receptors was unable to entirely block the activation signals.
The RLRs are ubiquitously expressed in many cell types and are among the first sensors to detect many viral infections. They are localized in the cytoplasm and recognize the genomic RNA of double-stranded RNA (dsRNA) viruses and dsRNA generated as the replication intermediate of single-stranded RNA viruses. The RIG-I and MDA5 are inducible upon IFN treatment or virus infection. The activated RIG-I or MDA5 interacts with an essential RLR adaptor, MAVS, initiating a signaling cascade that includes the activation of TBK1 and IKKε kinases, which subsequently phosphorylate and activate the transcription factor IRF3 and NF-κB. In this study, we demonstrate that RIG-I or MDA5 were both induced in macrophages upon MERS-CoV infection. The engagement of the receptors and subsequent signaling transduction were manifested in siRNA depletion of both receptors and their adaptor MAVS, as well as TBK1/IKKε inhibition. Thus, RLRs indeed recognize replicating MERS-CoV and mediate the proinflammation response in macrophages.
Recent studies have identified CLRs as an important family of PRRs that are involved in the induction of inflammation response to specific pathogens. Dectin-1, Dectin-2, and Mincle are members of a family of C-type lectins that are typically expressed by dendritic cells and macrophages [17]. Mincle is known to associate noncovalently with the adaptor molecule FcRγ. The phosphorylation of the adaptor serves to recruit Syk and induces downstream NF-κB activation through the Syk-CARD9-BCL10-MALT1 complex. The in vivo genetic studies have revealed an essential role of CLR-triggered CARD9 signaling in host protection. CARD9-deficient mice exhibited deregulated innate responses and failed to control infection of Mycobacterium tuberculosis [43]. In addition, Mincle was identified as a lipopolysaccharide-inducible protein in mouse macrophages and was transcriptionally upregulated after exposed to various stimuli and cellular stress [44]. In human monocytes, fungus infection elevated Mincle expression [31]. In this study, we demonstrated that MERS-CoV replication upregulated Mincle expression. The antibody blockage of Mincle, siRNA depletion of Mincle, Syk pharmacological inhibition, and CARD9 activation invariably highlighted the role of Mincle for mediating the proinflammatory cascade in MERS-CoV-infected macrophages.
In terms of cellular inflammatory response, 4 major family of PRRs, including TLRs, RLRs, CLRs, and NLRs, share overlapping ligand specificities and converge on common downstream pathways. The cooperative detection by different PRRs has been reported in other DNA and RNA viruses [33, 45] and bacteria [46]. In rhinovirus-infected bronchial epithelial cells, the innate immune response requires a coordinated recognition, initially via TLR3 or TRIF and followed by RNA helicases RIG-I and MDA5 [33]. Our results indicate that CLR and RLR signaling may be involved in mediating the immune activation in MERS-CoV-infected human macrophages. However, the possible crosstalk or coordination of different pathways has yet to be investigated.
CONCLUSIONS
Taken together, our study has advanced current knowledge of the recognition of human CoVs by cellular PRRs. In agreement with previous literature on other human CoVs, our data revealed the important role of RLRs in mediating the proinflammatory response upon MERS-CoV infection. More important, our study further investigated the involvement of CLRs in sensing MERS-CoV and provided the first evidence that CLRs contributed to the recognition of MERS-CoV. In addition, we demonstrated that a significant proportion of MERS-CoV-triggered proinflammatory response was governed by Mincle. In this regard, CLRs may play an important role in modulating the pathogenesis of MERS-CoV.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Supplementary Figure 1. Determination of 50% cytotoxic concentration (CC50) of 3 pattern recognition receptor (PRR) pathway inhibitors. Monocyte-derived macrophages (MDMs) were treated with the indicated concentrations of inhibitors and incubated at 37°C for 24 hours. The cells were then applied to MTT assay. The results are used for calculation of the CC50 of each inhibitor. Data are presented as mean ± standard deviation (SD) of triplicate wells of MDMs from 3 different donors.
Supplementary Figure 2. Inactivated Middle East respiratory syndrome coronavirus (MERS-CoV) was unable to induce proinflammatory cytokines and chemokines. Ultraviolet (UV) inactivation was performed by exposing the viruses to UV cross-linker for 10 minutes at 37°C. The inactivation was confirmed after inoculation in VERO E6 cells. Monocyte-derived macrophages (MDMs) were infected with MERS-CoV or the UV-inactivated MERS-CoV at a multiplicity of infection (MOI) of 2 or were mock infected. At 24 hours postinoculation, cells were harvested for detecting the expression levels of cytokines and chemokines by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) assay. Data are presented as mean ± standard deviation (SD) of MDMs from 3 different donors.
Supplementary Figure 3. Treatment of pattern recognition receptor (PRR) inhibitors and blocking antibody did not affect Middle East respiratory syndrome coronavirus (MERS-CoV) replication. (A) The MERS-CoV-infected MDMs were treated with Amlexanox or R406 or VX-765. At 2 and 24 hours postinfection (hpi), cells were harvested for detection of viral load in the cell lysates. (B) The expression of MERS-NP protein was verified at 6 and 24 hpi by Western blot. (C) At 2 and 24 hpi, the infected cells were harvested to detect the effect of α-Mincle antibody on viral replication by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) assay.
Supplementary Figure 4. CARD9 was activated upon Middle East respiratory syndrome coronavirus (MERS-CoV) infection. Monocyte-derived macrophages (MDMs) were treated with 1 µM R406 or mock-treated and inoculated with MERS-CoV or mock-infected. At 6 hours postinfection (hpi), cells were harvested to detect CARD9 by Western Blotting. The right panel shows intensities of CARD9 normalized with those of β-actin. Data are presented as mean ± standard deviation (SD) of MDMs from 3 different donors. Unpaired t test was used for data analysis. *, P < .05.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Notes
Acknowledgments. We thank the Faculty Core Facility, Li Ka Shing Faculty of Medicine, University of Hong Kong for assistance in confocal imaging and flow cytometry analysis
Financial support. This work was partly supported by funding from Health and Medical Research Fund (Project Nos. 14131392 and 17161272) of the Food and Health Bureau of the Hong Kong Special Administrative Region (HKSAR) Government; Theme-based Research Scheme (T11-707/15-R) of the Research Grants Council, the HKSAR Government; the High Level Hospital-Summit Program in Guangdong, The University of Hong Kong-Shenzhen Hospital; and the donations by the Shaw Foundation Hong Kong, Richard Yu and Carol Yu, Michael Seak-Kan Tong, Respiratory Viral Research Foundation Limited, Hui Ming, Hui Hoy & Chow Sin Lan Charity Fund Limited, and Chan Yin Chuen Memorial Charitable Foundation.
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest.
References
- 1. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 2012; 367:1814–20. [DOI] [PubMed] [Google Scholar]
- 2. Assiri A, Al-Tawfiq JA, Al-Rabeeah AA, et al. Epidemiological, demographic, and clinical characteristics of 47 cases of Middle East respiratory syndrome coronavirus disease from Saudi Arabia: a descriptive study. Lancet Infect Dis 2013; 13:752–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guery B, Poissy J, el Mansouf L, et al. Clinical features and viral diagnosis of two cases of infection with Middle East respiratory syndrome coronavirus: a report of nosocomial transmission. Lancet 2013; 381:2265–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alsaad KO, Hajeer AH, Al Balwi M, et al. Histopathology of Middle East respiratory syndrome coronovirus (MERS-CoV) infection - clinicopathological and ultrastructural study. Histopathology 2018; 72:516–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baseler LJ, Falzarano D, Scott DP, et al. An acute immune response to Middle East respiratory syndrome coronavirus replication contributes to viral pathogenicity. Am J Pathol 2016; 186:630–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 2011; 11:723–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smith AM, Rahman FZ, Hayee B, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J Exp Med 2009; 206:1883–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou J, Chu H, Li C, et al. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J Infect Dis 2014; 209:1331–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol 2012; 4:a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010; 140:805–20. [DOI] [PubMed] [Google Scholar]
- 11. Kato H, Takeuchi O, Sato S, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006; 441:101–5. [DOI] [PubMed] [Google Scholar]
- 12. Wang X, Jiang W, Yan Y, et al. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat Immunol 2014; 15:1126–33. [DOI] [PubMed] [Google Scholar]
- 13. Lian H, Zang R, Wei J, et al. The zinc-finger protein ZCCHC3 binds RNA and facilitates viral RNA sensing and activation of the RIG-I-like receptors. Immunity 2018; 49:438–48.e5. [DOI] [PubMed] [Google Scholar]
- 14. Ding Z, An K, Xie L, et al. Transmissible gastroenteritis virus infection induces NF-κB activation through RLR-mediated signaling. Virology 2017; 507:170–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zalinger ZB, Elliott R, Rose KM, Weiss SR. MDA5 is critical to host defense during infection with murine coronavirus. J Virol 2015; 89:12330–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen ST, Lin YL, Huang MT, et al. CLEC5A is critical for dengue-virus-induced lethal disease. Nature 2008; 453:672–6. [DOI] [PubMed] [Google Scholar]
- 17. Monteiro JT, Lepenies B. Myeloid C-type lectin receptors in viral recognition and antiviral immunity. Viruses 2017; 9. doi:10.3390/v9030059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yamasaki S, Matsumoto M, Takeuchi O, et al. C-type lectin Mincle is an activating receptor for pathogenic fungus, Malassezia. Proc Natl Acad Sci U S A 2009; 106:1897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Siu KL, Kok KH, Ng MH, et al. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J Biol Chem 2009; 284:16202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Siu KL, Yeung ML, Kok KH, et al. Middle east respiratory syndrome coronavirus 4a protein is a double-stranded RNA-binding protein that suppresses PACT-induced activation of RIG-I and MDA5 in the innate antiviral response. J Virol 2014; 88:4866–76. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21. Zhao L, Jha BK, Wu A, et al. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe 2012; 11:607–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chu H, Zhou J, Wong BH, et al. Middle East respiratory syndrome coronavirus efficiently infects human primary T lymphocytes and activates the extrinsic and intrinsic apoptosis pathways. J Infect Dis 2016; 213:904–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chu H, Wang JJ, Qi M, et al. Tetherin/BST-2 is essential for the formation of the intracellular virus-containing compartment in HIV-infected macrophages. Cell Host Microbe 2012; 12:360–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou J, Li C, Sachs N, et al. Differentiated human airway organoids to assess infectivity of emerging influenza virus. Proc Natl Acad Sci U S A 2018; 115:6822–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou J, To KK, Dong H, et al. A functional variation in CD55 increases the severity of 2009 pandemic H1N1 influenza A virus infection. J Infect Dis 2012; 206:495–503. [DOI] [PubMed] [Google Scholar]
- 26. Zhou J, Wang D, Wong BH, et al. et al. Identification and characterization of GLDC as host susceptibility gene to severe influenza. EMBO Mol Med 2019; 11:e9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reilly SM, Chiang SH, Decker SJ, et al. An inhibitor of the protein kinases TBK1 and IKK-ɛ improves obesity-related metabolic dysfunctions in mice. Nat Med 2013; 19:313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Niederberger E, V Moser C, L Kynast K, Geisslinger G. The non-canonical IκB kinases IKKε and TBK1 as potential targets for the development of novel therapeutic drugs. Curr Mol Med 2013; 13:1089–97. [DOI] [PubMed] [Google Scholar]
- 29. Gross O, Poeck H, Bscheider M, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 2009; 459:433–6. [DOI] [PubMed] [Google Scholar]
- 30. Doitsh G, Galloway NLK, Geng X, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2013; 505:509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vijayan D, Radford KJ, Beckhouse AG, Ashman RB, Wells CA. Mincle polarizes human monocyte and neutrophil responses to Candida albicans. Immunol Cell Biol 2012; 90:889–95. [DOI] [PubMed] [Google Scholar]
- 32. Li J, Liu Y, Zhang X. Murine coronavirus induces type I interferon in oligodendrocytes through recognition by RIG-I and MDA5. J Virol 2010; 84:6472–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Slater L, Bartlett NW, Haas JJ, et al. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog 2010; 6:e1001178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol 2017; 39:529–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang KJ, Su IJ, Theron M, et al. An interferon-gamma-related cytokine storm in SARS patients. J Med Virol 2005; 75:185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Min CK, Cheon S, Ha NY, et al. Comparative and kinetic analysis of viral shedding and immunological responses in MERS patients representing a broad spectrum of disease severity. Sci Rep 2016; 6:25359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kock G, Bringmann A, Held SA, Daecke S, Heine A, Brossart P. Regulation of dectin-1-mediated dendritic cell activation by peroxisome proliferator-activated receptor-gamma ligand troglitazone. Blood 2011; 117:3569–74. [DOI] [PubMed] [Google Scholar]
- 38. Zhou J, Chu H, Chan JF, Yuen KY. Middle East respiratory syndrome coronavirus infection: virus-host cell interactions and implications on pathogenesis. Virol J 2015; 12:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nicholls JM, Poon LL, Lee KC, et al. Lung pathology of fatal severe acute respiratory syndrome. Lancet 2003; 361:1773–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Perlman S, Dandekar AA. Immunopathogenesis of coronavirus infections: implications for SARS. Nat Rev Immunol 2005; 5:917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gu J, Gong E, Zhang B, et al. Multiple organ infection and the pathogenesis of SARS. J Exp Med 2005; 202:415–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Channappanavar R, Fehr AR, Vijay R, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe 2016; 19:181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dorhoi A, Desel C, Yeremeev V, et al. The adaptor molecule CARD9 is essential for tuberculosis control. J Exp Med 2010; 207:777–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Matsumoto M, Tanaka T, Kaisho T, et al. A novel LPS-inducible C-type lectin is a transcriptional target of NF-IL6 in macrophages. J Immunol 1999; 163:5039–48. [PubMed] [Google Scholar]
- 45. Delaloye J, Roger T, Steiner-Tardivel QG, et al. Innate immune sensing of modified vaccinia virus Ankara (MVA) is mediated by TLR2-TLR6, MDA-5 and the NALP3 inflammasome. PLoS Pathog 2009; 5:e1000480. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46. Ferwerda G, Kullberg BJ, de Jong DJ, et al. Mycobacterium paratuberculosis is recognized by Toll-like receptors and NOD2. J Leukoc Biol 2007; 82:1011–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.