

Abstract
Porcine reproductive and respiratory syndrome viruses (PRRSV) are divided into North American and European types, which show about 40% difference in their amino acid sequences. The divergence time of these two types has been estimated to be about 1980 from epidemiological data. This suggested that PRRSV have evolved at a higher evolutionary rate (order of 10−2/site/year) compared with other RNA viruses of 10−3 to 10−5/site/year. Here, to test the evolutionary history of PRRSV speculated by the epidemiological background, we estimated the divergence time and evolutionary rate of PRRSV with molecular evolutionary analysis. Estimated divergence time (1972–1988) corresponded well to that estimated by the epidemiological data, and the evolutionary rate (4.71–9.8) × 10−2 of PRRSV was indeed the highest among RNA viruses so far reported. Furthermore, we inferred important sites for the adaptation in order to examine how PRRSV have adapted to swine since they emerged. The adaptive sites were located not only in the epitopes related to immunity but also in the transmembrane regions including a signal peptide. In particular, the adaptive sites in the transmembrane regions were considered to affect compatibility to the host cell membrane. We conclude that PRRSV were transmitted from another host species to swine in about 1980 and have adapted to swine by altering the transmembrane regions.
Keywords: PRRSV, evolutionary rate, divergence time, positive selection, emerging virus and transmembrane region
Introduction
Porcine reproductive and respiratory syndrome viruses (PRRSV), which belong to the family Arteriviridae in the order Nidovirales, are positive-sense single-stranded RNA viruses (Cavanagh 1997). PRRSV recently emerged in the domesticated swine and are recognized as the most important infectious agents causing reproductive failure in sows and severe pneumonia in piglets (Rossow et al. 1999). The first case of possible PRRSV infection with the above symptoms was reported in North America in the mid-1980s (Keffaber 1989; Ellis et al. 1999), and similar symptoms spread to other continents (Asia and Europe) by 1991 (Albina 1997; Ellis et al. 1999). In 1991, two strains of PRRSV were first isolated independently in the United States and the Netherlands (Wensvoort et al. 1991; Collins et al. 1992). The U.S. and Netherlands isolates are considered to be the reference strains of the American type (PRRSV-A) and European type (PRRSV-E), respectively. Currently, PRRSV-A is prevalent in the United States, Canada, and Asian countries, whereas PRRSV-E is prevalent in Europe.
Interestingly, when the genomic sequences of these reference sequences were determined, it was found that the amino acid identity between these two types is less than 60% (Wensvoort et al. 1992; Murtaugh et al. 1998). It follows that PRRSV diverged into two types varying by 40% of the amino acids within about 10 years and that PRRSV have evolved at a high evolutionary rate (order of 10−2/site/year) compared with those (orders of 10−3 to 10−5) of standard RNA viruses. However, the speculated evolutionary rate of PRRSV seemed too rapid compared with those of standard RNA viruses. Therefore, several researchers took the alternative view. The view is that the two types diverged well before the 1980s, evolved independently on American and European continents, and then emerged in swine on both continents at almost the same time (Nelsen, Murtaugh, and Faaberg 1999; Stadejek et al. 2002; Plagemann 2003). Thus, the divergence time and the evolutionary rate of PRRSV are unclear and deserving of molecular evolutionary analysis.
Furthermore, PRRSV provide an interesting opportunity to study the mechanisms of adaptation of PRRSV to swine because PRRSV may have adapted to swine over a short period of time. The important genes for viral adaptation to the host are usually considered to be those encoding the proteins located in the outside of the virion, where they interacted with the environment, including the host immune system. In PRRSV, there are nine open reading frames (ORFs). ORF1a–1b, ORF2a–2b, ORF3–5, ORF6, and ORF7 encode replication proteins, unknown structure proteins, envelope proteins, membrane proteins, and nucleocapsid proteins, respectively. Among these, ORFs 3, 4, 5, and 6 are known to be the genes encoding the outside components of the PRRSV virion. These ORFs are therefore considered to be important candidates for their adaptation to swine. Adaptive regions can be detected through inference of positively selected sites, which preferentially undergo amino acid replacements. Positively selected sites may provide important information not only for studying the adaptive evolution of PRRSV but also for developing vaccines.
The overall goal of our study is to understand when PRRSV emerged, how rapidly PRRSV evolved, and how PRRSV have adapted to swine. For these purposes, we estimated the rate of synonymous substitution and the divergence time of PRRSV by molecular evolutionary analysis. Furthermore, the positively selected sites in PRRSV genes (ORFs 3–6) were inferred to detect the adaptive sites for swine.
Materials and Methods
Constructing a Phylogenetic Tree of the Order Nidovirales
The order Nidovirales can be divided into two families, Arteriviridae and Coronaviridae. PRRSV belong to the Arteriviridae family (Cavanagh 1997). Despite the difference in genome sizes, the genome organizations of the order Nidovirales are remarkably similar to each other (De Vries et al. 1997). In the present study, in order to construct a phylogenetic tree of Nidovirales and to estimate the evolutionary position of PRRSV, we used the amino acid sequences of ORF1b because ORF1b was reported to be conserved among the order Nidovirales (Bredenbeek 1990; Snijder and Meulenberg 1998). The ORF1b sequences for transmissible gastroenteritis virus (TGEV), human corona virus (HcoV), murine hepatitis virus (MHV), bovine coronavirus (BCV), avian infectious bronchitis virus (AIBV), Berne virus (BEV), equine arteritis virus (EAV), lactate dehydrogenase–elevating virus (LDEV), simian hemorrhagic fever virus (SHFV), PRRSV-A, and PRRSV-E were collected from the International Nucleotide Sequence Database (INSD: DNA Data Bank of Japan/European Molecular Biology Laboratory/GenBank). The amino acid sequence accession numbers for TGEV, HcoV, MHV, BCV, AIBV, BEV, EAV, LDEV, SHFV, PRRSV-A, and PRRSV-E are AJ271965, AF304460, AF029248, AF391541, M95169, X52374, X53459, L13298, AF180391, AF046869, and M96262, respectively. The conserved regions in ORF1b were detected by the DotPlot program, DOTTER (Sonnhammer and Durbin 1995), and a multiple alignment of the conserved regions among the viruses was made by ClustalW (Thompson, Higgins, and Gibson 1994). The phylogenetic tree was constructed by the maximum likelihood (Felsenstein 1981) and Neighbor-Joining methods (Saitou and Nei 1987) using MOLPHY version 2.3 and PHYLIP program package, respectively. The Jones-Taylor-Thornton (JTT) model was used for constructing the maximum likelihood tree, whereas Kimura's equation was used to estimate the evolutionary distances for constructing the Neighbor-Joining tree. The robustness of the topology of each tree was examined by the bootstrap method.
The Synonymous Substitution Rate and Divergence Time of PRRSV
The nucleotide sequences of the whole envelope region (ORFs 3, 4, and 5) of PRRSV-A were collected from the INSD. PRRSV-A strains whose years of isolation were known were used in the present study. Wild strains isolated after 1995 were excluded from the analysis because they included vaccine-derived strains. The accession number and the isolation time of each sequence used are shown in Supplementary Materials. The synonymous substitution rate and divergence time of PRRSV were estimated by both the distance-based method and the maximum likelihood method.
In the distance-based method, the nucleotide sequences of PRRSV-A, PRRSV-E, and LDEV were aligned by ClustalW. A phylogenetic tree was constructed by the maximum likelihood method using the HKY (gamma) model (PAUP* version 4.0b) (Hasegawa, Kishino, and Yano 1985). To estimate the most recent ancestral sequence for PRRSV-A and PRRSV-E, LDEV was used as the out-group in the phylogenetic tree because LDEV is the closest virus to PRRSV among the known viruses of Arteriviridae. The ancestral sequence was estimated by the nucleotide model of the likelihood approach (PAML version 3.13) (Yang, Kumar, and Nei 1995). The number of synonymous substitutions between the ancestral sequence and each PRRSV-A sequence was estimated by the Nei-Gojobori model (Nei and Gojobori 1986). The year of isolation and the synonymous distance were plotted for each viral sequence in the two-dimensional space, and both the synonymous substitution rate of PRRSV-A and the divergence time between PRRSV-A and PRRSV-E were estimated by the least squares method (Suzuki, Yamaguchi-Kabata, and Gojobori 2000). The standard error of the divergence time was estimated by the bootstrap method under the assumption that the topology of the phylogenetic tree was correct. We constructed 500 sets of sequence alignments by randomly sampling each codon from the original alignment (Nei and Kumar 2000).
In the maximum likelihood method, the nucleotide sequences of PRRSV-A and PRRSV-E were aligned by ClustalW. Using PRRSV-E as the out-group, a phylogenetic tree was constructed by the Neighbor-Joining method. Then, we estimated both the synonymous substitution rate of PRRSV-A and the divergence time of PRRSV using the software PAML including TIPDATE algorithm (Rambaut 2000).
Nucleotide Sequence Data for Inferring Positively Selected Sites in the Genes Encoding the Outside Component of the Virion
To detect positively selected sites in the genes encoding the outer component of the PRRSV virion, ORFs 3, 4, 5, and 6 of PRRSV-A strains were collected from the INSD. Sequences including undetermined nucleotides and gaps were eliminated from the present analysis. Consequently, the numbers of sequences used for ORF3, ORF4, ORF5, and ORF6 were 31, 30, 141, and 41, respectively. A multiple alignment was made for each coding region using ClustalW. Positively selected amino acid sites were identified using the method of Suzuki and Gojobori (1999; Suzuki, Gojobori, and Nei 2001). In this method, a phylogenetic tree was constructed by the Neighbor-Joining method using the number of synonymous substitutions. The ancestral sequence was inferred at each node of the phylogenetic tree using the maximum parsimony method (Hartigan 1973). Then, the average numbers of synonymous (sS) and nonsynonymous (sN) sites and the total numbers of synonymous (cS) and nonsynonymous (cN) substitutions throughout the phylogenetic tree were estimated for each codon site. The probability (P) of obtaining the observed or more biased numbers of synonymous and nonsynonymous substitutions was computed for each codon site, assuming a binomial distribution. In the computation, sS/(sS + sN) and sN/(sS + sN) were used as the probabilities of the occurrence of synonymous and nonsynonymous substitutions, respectively. The number of synonymous substitutions per synonymous site (ds) and that of nonsynonymous substitutions per nonsynonymous site (dn) were estimated by cS/sS and cN/sN, respectively. The transmembrane and signal peptide regions of the envelope genes (ORFs 3, 4, 5, and 6) were estimated by TMPRED version 2.0 (Hofmann and Stoffel 1993).
Results and Discussion
The Phylogenetic Tree of Nidovirales
Three major conserved genomic regions (A, B, and C) among the order Nidovirales, including the families Arteriviridae and Coronaviridae, were identified for constructing the phylogenetic tree of the order Nidovirales, and amino acid alignments of the conserved regions were constructed (fig. 1). From the alignments, the phylogenetic tree of the order Nidovirales was constructed by the Neighbor-Joining method (fig. 2). The topology of the phylogenetic tree was the same as that constructed by the maximum likelihood method. The closest virus species to PRRSV was LDEV.
FIG. 1.—

Conserved regions among the order Nidovirales. Three conserved regions of ORF1b among the order Nidovirales were detected by the dot plot method. An amino acid alignment was constructed for each conserved region.
FIG. 2.—

The phylogenetic tree of the order Nidovirales. The numbers next to the branches are the bootstrap values by the Neighbor-Joining method. This phylogenetic tree indicates that the closest viral species to PRRSV is LDEV.
The Synonymous Substitution Rate and Divergence Time of PRRSV
Using LDEV as the out-group, the divergence position between PRRSV-A and PRRSV-E was identified using the whole envelope genes (fig. 3a). The rate of synonymous substitution and the divergence time of PRRSV were examined by both the distance-based and maximum likelihood methods (table 1 and fig. 4). The rate of PRRSV was estimated to be 7.7 ± 2.1 × 10−2 and 4.74 ± 0.3 × 10−2/synonymous site/year by the distance-based and maximum likelihood methods, respectively. Assuming that the rate estimated for three ORFs can be extrapolated to the whole genome, the divergence times are 1986 ± 1.8 and 1973 ± 1.3 according to the distance-based and maximum likelihood methods, respectively. Epidemiologically, there are two hypotheses for the divergence time. One is that PRRSV diverged in approximately 1980, the other is that PRRSV diverged well before 1980s as far back as approximately 1900. Our results strongly supported the former hypothesis because the estimated divergence times were 1972–1988. However, our results could not identify with confidence that the two major types of PRRSV diverged at approximately the same time as PRRSV appeared in the United States, as the maximum likelihood divergence time indicates an earlier divergence time. We conclude that PRRSV diverged approximately within 10 years before PRRSV spread in the United States.
FIG. 3.—
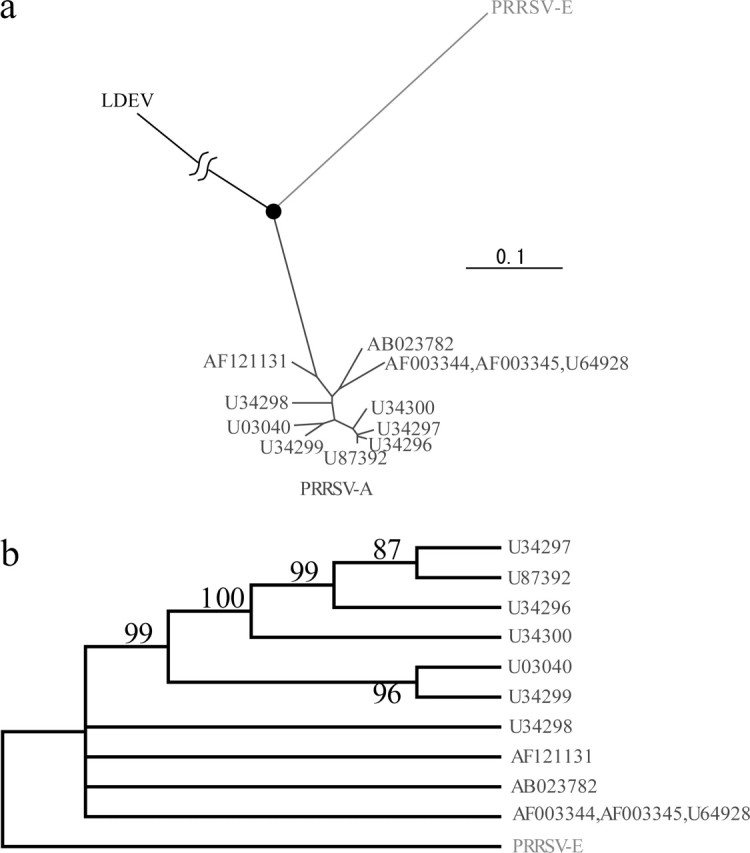
Divergence time of PRRSV. (a) Phylogenetic tree positioning the divergence node between PRRSV-A and PRRSV-E. The black solid circle indicates the divergence node. (b) Phylogenetic tree showing branches with >50% bootstrap support.
Table 1.
Rate of Synonymous Substitution and Divergence Time of PRRSV by Distance-Based Method and Maximum Likelihood Method
|
|
Distance-Based Method |
Maximum Likelihood Method |
|---|---|---|
| Rate of synonymous substitution | (7.7 ± 2.1) × 10−2 | (4.74 ± 0.3) × 10−2 |
| Divergence time of PRRSV |
1986 ± 1.8 |
1973 ± 1.3 |
FIG. 4.—

Divergence time was calculated by the least squares method. Values on the vertical axis indicate the synonymous distance from the ancestral node to each strain of PRRSV-A. Values on the horizontal axis indicate the year of isolation. A blue solid circle indicates each PRRSV-A strain. The red line was calculated from the original sequence data. Each blue line was obtained by the bootstrap method. A detailed explanation is presented in the Materials and Methods.
Moreover, we examined whether the data we used had recombination events or not. In the present study, we used three ORFs (ORF3, 4, and 5) for estimating the evolutionary rate. Phylogenetic trees for ORFs 3, 4, and 5 were independently constructed by the Neighbor-Joining method with 1,000 bootstrap replicates. Branches supported by more than 50% bootstrap values were all shared among three trees (fig. 3b). The high correlation of the phylogenetic trees among the three ORFs indicates a lack of recombination events in the data.
The rate of synonymous substitution estimated here was the highest among RNA viruses so far reported (Ellen et al. 1996; Suzuki and Gojobori 1998; Jenkins et al. 2002). We suggest two hypotheses for the extraordinarily rapid rate of PRRSV evolution, given the assumption that the rate of synonymous substitution is approximately equal to the mutation rate. The first is that the rate of replication error for PRRSV is extraordinarily high, and the second is that the number of replications per unit time (replication frequency) for PRRSV is extraordinarily high. To investigate support for these hypotheses, we compared the rate of replication error with the synonymous substitution rate among five RNA viruses, including PRRSV, as shown in table 2. In table 2, the replication error rate of PRRSV was estimated as follows. We first estimated the error rate per passage from the number of nucleotide substitutions occurring among passages (Yuan et al. 2001). Then, the replication error rate was estimated by the passage error rate divided by the time required for viral budding (Dea et al. 1995). The rates of replication error for other four RNA viruses were collected from literature (Holland et al. 1990; Schrag, Rota, and Bellini 1999; Stech et al. 1999; Escarmis et al. 2002). We also estimated the rates of synonymous substitution for these four RNA viruses (table 2). The results show that replication error rate is approximately equal between PRRSV and other RNA viruses. Even RNA viruses that have rates of synonymous substitution that are very different from PRRSV have replication error rates that are almost the same as that of PRRSV. It follows that the main source for the high synonymous substitution rate of PRRSV is the high replication frequency of PRRSV.
Table 2.
Comparison Between Rate of Replication Error and Rate of Synonymous Substitution
|
Virus Species |
Replication Error Rate (/site/replication) |
Synonymous Substitution Rate (/site/year) |
||
|---|---|---|---|---|
| Single-stranded (+) RNA viruses | ||||
| PRRSV-A | 3.7 × 10−5 | (4.71–9.8) × 10−2 | ||
| Foot-and-mouth disease virus | 3.7 × 10−5 | 8.2 × 10−3 | ||
| Single-stranded (−) RNA viruses | ||||
| Measles virus | (6.0–14) × 10−5 | 2.3 × 10−3 | ||
| Influenza A virus | (0.7–3.2) × 10−5 | 7.0 × 10−3 | ||
| Vesicular stomatitis virus |
10.0 × 10−5 |
7.0 × 10−5 |
||
There are some factors that can increase the replication frequency of PRRSV. PRRSV infect swine via the air, and swine continues to release the virus for half a year (Albina 1997). Viruses infected by air and inducing acute and persistent infection for long periods have been reported to have a rapid evolutionary rate (Hanada, Suzuki, and Gojobori 2004). PRRSV can also easily spread because of the dense population in swine facilities. Furthermore, as a reason why the speed of the spread could be accelerated, most swine do not have immunity against PRRSV because the virus only recently emerged in swine. Thus, it is considered that PRRSV not only repeated replications within each infected host but also quickly infected other individuals surrounding the infected host.
Positively Selected Sites of Envelope Genes
The positively and negatively selected amino acid sites in ORFs 3, 4, 5, and 6 of PRRSV-A are summarized in figure 5 and table 3. Figure 5 shows that ds exceeded dn at more than half the amino acid sites (470/810, 58.0%) and that negative selection was detected at more than 25% of all amino acid sites (216/810, 26.7%). Nevertheless, we detected sites in which dn exceeded ds. Among the sites where dn exceeded ds, we called a site where the value (1 − P) exceeded 0.95, a positively selected site (table 4). In figure 5, there are several sites where dn exceeded ds in the regions experimentally recognized as the B-cell epitopes of ORFs 3, 4, and 5 (Oleksiewicz et al. 2001; Ostrowski et al. 2002; Plagemann, Rowland, and Faaberg 2002), although (1 − P) did not exceed 0.95 at these sites. The ectodomain identified in ORF6 by TMPRED contained a positively selective site. This site may be susceptible to attack by the immune system. PRRSV may have escaped from the immune system of swine through replacements of such amino acid sites. These adaptive sites may also provide important information for developing vaccines for PRRSV because antibodies against the regions including adaptive sites could become ineffective after amino acid replacement at these sites. To develop an effective vaccine against PRRSV, it will be necessary to make a vaccine highly expressing the regions related to immunity but not those including adaptive sites (Suzuki 2004).
FIG. 5.—
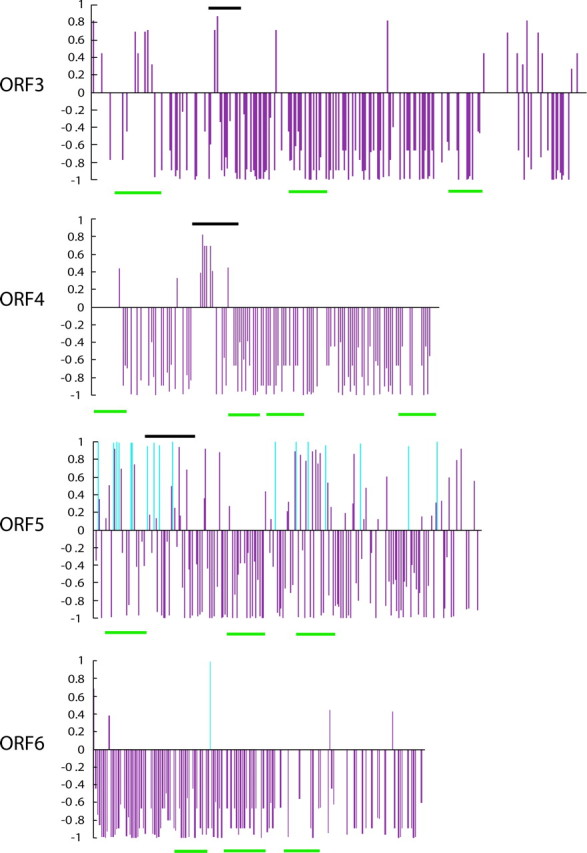
Distribution of the value of (1 − P) in ORFs 3, 4, 5, and 6 of PRRSV-A. The genes (ORFs 3, 4, 5, and 6) encode the envelope proteins (ORFs 3, 4, and 5) and the membrane protein (ORF6) of PRRSV-A. The figure indicates the distribution of the value (1 − P) for natural selection. When dn is larger than ds, the value is indicated above the abscissa, whereas in the opposite situation, below the abscissa. Light blue bars indicate positively selected sites (P < 0.05). The abscissa indicates the amino acid positions. The black rectangles indicate the experimentally recognized B-cell epitopes. The green rectangles indicate transmembrane regions.
Table 3.
Number of Positively and Negatively Selected Sites
|
ORF |
Average dn/ds Ratio |
Number of Positively Selected Sites |
Number of Negatively Selected Sites |
|---|---|---|---|
| ORF3 | 0.307 | 0 | 51 |
| ORF4 | 0.242 | 0 | 35 |
| ORF5 | 0.791 | 17 | 54 |
| ORF6 |
0.133 |
1 |
40 |
Table 4.
Positively Selected Sites
|
ORF |
Positions |
dn/ds Ratio |
Value (1 − P) |
|---|---|---|---|
| ORF5 | 3 | 3.59 | 0.99 |
| 11 | 1.88 | 0.99 | |
| 13 | 6.13 | 0.99 | |
| 14 | 3.39 | 0.99 | |
| 20 | 4.72 | 0.99 | |
| 21 | a | 0.99 | |
| 29 | 2.22 | 0.95 | |
| 32 | a | 0.99 | |
| 35 | 23.68 | 0.96 | |
| 42 | 10.48 | 0.99 | |
| 95 | 6.94 | 0.99 | |
| 106 | 6.47 | 0.99 | |
| 112 | 5.91 | 0.99 | |
| 121 | 2.86 | 0.96 | |
| 139 | 3.23 | 0.98 | |
| 164 | 1.89 | 0.95 | |
| 179 | 6.46 | 0.99 | |
| ORF6 |
63 |
2.58 |
0.99 |
dn/ds values could not be calculated, because of ds = 0.
Positively selected sites were concentrated in ORF5 (fig. 5). These sites are not only in the epitope regions often attacked by B cells but also in the transmembrane regions including the signal peptide (Fernandez et al. 2002; Ostrowski 2002). The replacement sites are positioned in the ectodomain regions in the ORF5 sequences isolated from the experimentally infected pigs (Rowland et al. 1999; Allende et al. 2000). However, these reports suggested that both the positively selected sites of the transmembrane regions including the signal peptide in ORF5 are not strongly related to escape from the host immune system.
The function of the transmembrane regions is to recognize the host cell membrane and attach to it. It has been reported that the transmembrane regions and the signal peptide are specific to a given membrane in terms of at least the expression level of a gene (Schatz and Dobberstein 1996). Therefore, the positive selection of the membrane regions in ORF5 is considered to be important for adaptation of PRRSV to the membrane of swine cells. In fact, PRRSV emerged so suddenly in swine only about 20 years ago and may have not yet adapted completely to swine cells. Given these circumstances, we speculate that PRRSV transferred from another host to swine in about 1980 and adapted to swine cells by altering the transmembrane regions of ORF5.
One of the potential problems of the Suzuki and Gojobori method (1999) for detecting positively selected amino acid sites was that the multiple substitutions were not taken into account in the estimation of the numbers of synonymous and nonsynonymous substitutions. However, as long as the nucleotide sequences compared are relatively closely related, the number of multiple substitutions would be sufficiently small so that the results obtained appear to be reliable (Saitou 1989). Indeed, in the present analysis, the branch lengths of the phylogenetic tree were very small (on average, 0.031, 0.029, 0.018, and 0.025 per synonymous site in ORFs 3, 4, 5, and 6, respectively).
It should also be noted that it is possible that the hitchhiking effect caused by strong positive selection as indicated above raised the rate of synonymous substitution for PRRSV. However, it appears that the increase in synonymous substitution is less affected by the hitchhiking effect than the effect of replication frequency because it has been reported that the hitchhiking effect minimally affects synonymous substitution (Birky and Walsh 1988).
Conclusions
In conclusion, PRRSV were transmitted from another host to swine in about 1980, and the virus explosively increased among domesticated swine. During this process, the synonymous substitution rate became the highest among RNA viruses so far reported. We identified positively selected sites in the transmembrane regions that do not affect adaptation to the host immune system but nevertheless affect adaptation to the host cell membrane for PRRSV.
Supplementary Material
Acknowledgments
We thank S. P. Prem, S. Compton, S. Dea, M. Morgan, N. Takikawa, P. Halbur, and Y. Murakami for providing us with the isolation years of PRRSV strains. We are grateful to Josh Rest, Aya Takahashi, Edward Holmes, two anonymous reviewers and all the members of the DNA Analysis Laboratory for valuable comments and discussion.
Footnotes
Edward Holmes, Associate Editor
References
- Albina, E. 1997. Epidemiology of porcine reproductive and respiratory syndrome (PRRS): an overview. Vet. Microbiol. 55:309–316. [DOI] [PubMed] [Google Scholar]
- Allende, R., W. W. Laegreid, G. F. Kutish, J. A. Galeota, R. W. Wills, and F. A. Osorio. 2000. Porcine reproductive and respiratory syndrome virus: description of persistence in individual pigs upon experimental infection. J. Virol. 74:10834–10837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birky, C. W., and J. B. Walsh. 1988. Effects of linkage on rates of molecular evolution. Proc. Natl. Acad. Sci. USA 85:6414–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredenbeek, P. J., E. J. Snijder, F. H. Noten, J. A. den Boon, W. M. Schaaper, M. C. Horzinek, and W. J. Spaan. 1990. The polymerase gene of corona- and toroviruses: evidence for an evolutionary relationship. Adv. Exp. Med. Biol. 276:307–316. [DOI] [PubMed] [Google Scholar]
- Cavanagh, D. 1997. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch. Virol. 142:629–633. [PubMed] [Google Scholar]
- Collins, J. E., D. A. Benfield, W. T. Christianson et al. (12 co-authors). 1992. Isolation of swine infertility and respiratory syndrome virus (isolate ATCC VR-2332) in North America and experimental reproduction of the disease in gnotobiotic pigs. J. Vet. Diagn. Invest. 4:117–126. [DOI] [PubMed] [Google Scholar]
- Dea, S., N. Sawyer, R. Alain, and R. Athanassious. 1995. Ultrastructural characteristics and morphogenesis of porcine reproductive and respiratory syndrome virus propagated in the highly permissive MARC-145 cell clone. Adv. Exp. Med. Biol. 380:95–98. [DOI] [PubMed] [Google Scholar]
- De Vries, A. A. F., M. C. Horzinek, P. J. M. Rottier, and R. J. de Groot. 1997. The genome organization of Nidovirales: similarities and differences between arteri-, toro- and coronaviruses. Semin. Virol. 8:33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis, J. A., S. Krakowa, G. Allan, E. Clark, and S. Kennedy. 1999. The clinical scope of porcine reproductive and respiratory syndrome virus infection has expanded since 1987: an alternative perspective. Vet. Pathol. 36:262–265. [DOI] [PubMed] [Google Scholar]
- Escarmis, C., G. Gomez-Mariano, M. Davila, E. Lazaro, and E. Domingo. 2002. Resistance to extinction of low fitness virus subjected to plaque-to-plaque transfers: diversification by mutation clustering. J. Mol. Biol. 315:647–661. [DOI] [PubMed] [Google Scholar]
- Felsenstein, J. 1981. Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17:368–376. [DOI] [PubMed] [Google Scholar]
- Fernandez, A., P. Suarez, J. M. Castro, E. Tabares, and M. Diaz-Guerra. 2002. Characterization of regions in the GP5 protein of porcine reproductive and respiratory syndrome virus required to induce apoptotic cell death. Virus Res. 83:103–118. [DOI] [PubMed] [Google Scholar]
- Hanada, K., Y. Suzuki, and T. Gojobori. 2004. A large variation in the rates of synonymous substitution for RNA viruses and its relationship to a diversity of viral infection and transmission modes. Mol. Biol. Evol. 21:1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartigan, J. A. 1973. Minimum evolution fits to a given tree. Biometrics 29:53–65. [Google Scholar]
- Hasegawa, M., H. Kishino, and T. Yano. 1985. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22:160–174. [DOI] [PubMed] [Google Scholar]
- Hofmann, K., and Wf. Stoffel. 1993. Tmbase—a database of membrane spanning proteins segments. Biol. Chem. Hoppe-Seyler 347:166. [Google Scholar]
- Holland, J. J., E. Domingo, J. C. de la Torre, and D. A. Steinhauer. 1990. Mutation frequencies at defined single codon sites in vesicular stomatitis virus and poliovirus can be increased only slightly by chemical mutagenesis. J. Virol. 64:3960–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins, G. M., A. Rambaut, O. G. Pybus, and E. C. Holmes. 2002. Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J. Mol. Evol. 54:156–165. [DOI] [PubMed] [Google Scholar]
- Keffaber, K. K. 1989. Reproductive failure of unknown etiology. Am. Assoc. Swine Pract. Newslett. 1–9.
- Murtaugh, M. P., K. S. Faaberg, J. Laber, M. Elam, and V. Kapur. 1998. Genetic variation in the PRRS virus. Adv. Exp. Med. Biol. 440:787–794. [DOI] [PubMed] [Google Scholar]
- Nei, M., and T. Gojobori. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3:418–426. [DOI] [PubMed] [Google Scholar]
- Nei, M., and S. Kumar. 2000. Evolutionary changes of amino acid sequences. Pp. 17–50 in molecular evolution and phylogenetics. Oxford: Oxford University Press.
- Nelsen, C. J., M. P. Murtaugh, and K. S. Faaberg. 1999. Porcine reproductive and respiratory syndrome virus comparison: divergent evolution on two continents. J. Virol. 73:270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleksiewicz, M. B., A. Botner, P. Toft, P. Normann, and T. Storgaard. 2001. Epitope mapping porcine reproductive and respiratory syndrome virus by phage display: the nsp2 fragment of the replicase polyprotein contains a cluster of B-cell epitopes. J. Virol. 75:3277–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski, M., J. A. Galeota, A. M. Jar, K. B. Platt, F. A. Osorio, and O. J. Lopez. 2002. Identification of neutralizing and nonneutralizing epitopes in the porcine reproductive and respiratory syndrome virus GP5 ectodomain. J. Virol. 76:4241–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagemann, P. G. 2003. Porcine reproductive and respiratory syndrome virus: origin hypothesis. Emerg. Infect. Dis. 9:903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagemann, P. G., R. R. Rowland, and K. S. Faaberg. 2002. The primary neutralization epitope of porcine respiratory and reproductive syndrome virus strain VR-2332 is located in the middle of the GP5 ectodomain. Arch. Virol. 147:2327–2347. [DOI] [PubMed] [Google Scholar]
- Rambaut, A. 2000. Estimating the rate of molecular evolution: incorporating non-contemporaneous sequences into maximum likelihood phylogenies. Bioinformatics 16:395–399. [DOI] [PubMed] [Google Scholar]
- Rossow, K. D., J. L. Shivers, P. E. Yeske, D. D. Polson, R. R. Rowland, S. R. Lawson, M. P. Murtaugh, E. A. Nelson, and J. E. Collins. 1999. Porcine reproductive and respiratory syndrome virus infection in neonatal pigs characterised by marked neurovirulence. Vet. Rec. 144:444–448. [DOI] [PubMed] [Google Scholar]
- Rowland, R. R., M. Steffen, T. Ackerman, and D. A. Benfield. 1999. The evolution of porcine reproductive and respiratory syndrome virus: quasispecies and emergence of a virus subpopulation during infection of pigs with VR-2332. Virology 259:262–266. [DOI] [PubMed] [Google Scholar]
- Saitou, N. 1989. A theoretical study of the underestimation of branch lengths by the maximum parsimony principle. Syst. Zool. 38:1–6. [Google Scholar]
- Saitou, N., and M. Nei. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4:406–425. [DOI] [PubMed] [Google Scholar]
- Schatz, G., and B. Dobberstein. 1996. Common principles of protein translocation across membranes. Science 271:1519–1526. [DOI] [PubMed] [Google Scholar]
- Schrag, S. J., P. A. Rota, and W. J. Bellini. 1999. Spontaneous mutation rate of measles virus: direct estimation based on mutations conferring monoclonal antibody resistance. J. Virol. 73:51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder, E. J., and J. J. Meulenberg. 1998. The molecular biology of arteriviruses. J. Gen. Virol. 79:961–979. [DOI] [PubMed] [Google Scholar]
- Sonnhammer, E. L., and R. Durbin. 1995. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene 167:1–10. [DOI] [PubMed] [Google Scholar]
- Stadejek, T., A. Stankevicius, T. Storgaard, M. B. Oleksiewicz, S. Belak, T. W. Drew, and Z. Pejsak. 2002. Identification of radically different variants of porcine reproductive and respiratory syndrome virus in Eastern Europe: towards a common ancestor for European and American viruses. J. Gen. Virol. 83:1861–1873. [DOI] [PubMed] [Google Scholar]
- Stech, J., X. Xiong, C. Scholtissek, and R. G. Webster. 1999. Independence of evolutionary and mutational rates after transmission of avian influenza viruses to swine. J. Virol. 73:1878–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss, E.G., J.H. Strauss, and A.J. Levine. 1996. Virus Evolution. Pp 153–172 in B.N. Fields et al. (eds.) Fields Virology, 2nd ed. New York: Lippincott-Raven.
- Suzuki, Y. 2004. Negative selection on neutralization epitopes of poliovirus surface proteins: implications for prediction of candidate epitopes for immunization. Gene 328:127–133. [DOI] [PubMed] [Google Scholar]
- Suzuki, Y., and T. Gojobori. 1998. The origin and evolution of human T-cell lymphotropic virus types I and II. Virus Genes. 16:69–84. [DOI] [PubMed] [Google Scholar]
- ——— 1999. A method for detecting positive selection at single amino acid sites. Mol. Biol. Evol. 16:1315–1328. [DOI] [PubMed] [Google Scholar]
- Suzuki, Y., T. Gojobori, and M. Nei. 2001. ADAPTSITE: detecting natural selection at single amino acid sites. Bioinformatics 17:660–661. [DOI] [PubMed] [Google Scholar]
- Suzuki, Y., Y. Yamaguchi-Kabata, and T. Gojobori. 2000. Nucleotide substitution rates of HIV-1. AIDS Rev. 2:39–47. [Google Scholar]
- Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensvoort, G., E. P. de Kluyver, E. A. Luijtze, A. den Besten, L. Harris, J. E. Collins, W. T. Christianson, and D. Chladek. 1992. Antigenic comparison of Lelystad virus and swine infertility and respiratory syndrome (SIRS) virus. J. Vet. Diagn. Invest. 4:134–138. [DOI] [PubMed] [Google Scholar]
- Wensvoort, G., C. Terpstra, J. M. Pol et al. (22 co-authors). 1991. Mystery swine disease in The Netherlands: the isolation of Lelystad virus. Vet. Q. 13:121–130. [DOI] [PubMed] [Google Scholar]
- Yang, Z., S. Kumar, and M. Nei. 1995. A new method of inference of ancestral nucleotide and amino acid sequences. Genetics 141:1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, S., D. Mickelson, M. P. Murtaugh, and K. S. Faaberg. 2001. Complete genome comparison of porcine reproductive and respiratory syndrome virus parental and attenuated strains. Virus Res. 79:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.