

Abstract
The order Nidovirales is a diverse group of (+)RNA viruses, classified together based on their common genome organisation and conserved replicative enzymes, despite drastic differences in size and complexity. One such difference pertains to the mechanisms and enzymes responsible for generation of the proposed viral 5′ RNA cap. Within the Coronaviridae family, two separate methytransferases (MTase), nsp14 and nsp16, perform the RNA-cap N7-guanine and 2′-OH methylation respectively for generation of the proposed m7GpppNm type I cap structure. For the majority of other families within the Nidovirales order, the presence, structure and key enzymes involved in 5′ capping are far less clear. These viruses either lack completely an RNA MTase signature sequence, or lack an N7-guanine methyltransferase signature sequence, obscuring our understanding about how RNA-caps are N7-methylated for these families. Here, we report the discovery of a putative Rossmann fold RNA methyltransferase in 10 Tobaniviridae members in Orf1a, an unusual genome locus for this gene. Multiple sequence alignments and structural analyses lead us to propose this novel gene as a typical RNA-cap N7-guanine MTase with substrate specificity and active-site organization similar to the canonical eukaryotic RNA-cap N7-guanine MTase.
INTRODUCTION
The order Nidovirales is a large and diverse group of positive-stranded RNA viruses (Figure 1), capable of infecting a range of vertebrate and invertebrate hosts, causing significant global burden. The order has recently been reclassified into nine families ((1), Abyssoviridae, Arteriviridae, Coronaviridae (CoV), Medioniviridae, Mesoniviridae, Mononiviridae, Euroniviridae, Roniviridae and Tobaniviridae), several of which have attracted much attention in the past decades as the causative agents of serious diseases in humans and animals. This includes the human pathogens Severe Acute Respiratory Syndrome (SARS) and Middle-East Respiratory Syndrome (MERS) of the family CoV, which are associated with high case fatalities of ∼10% and ∼35%, respectively (reviewed in (2)). In addition, animal pathogens of the Arteriviridae family, including Equine Arteritis virus (EAV,) and Porcine Reproductive Respiratory Syndrome virus (PRSSV 1–2,) have caused substantial economic burden for the equine and swine industries.
Figure 1.
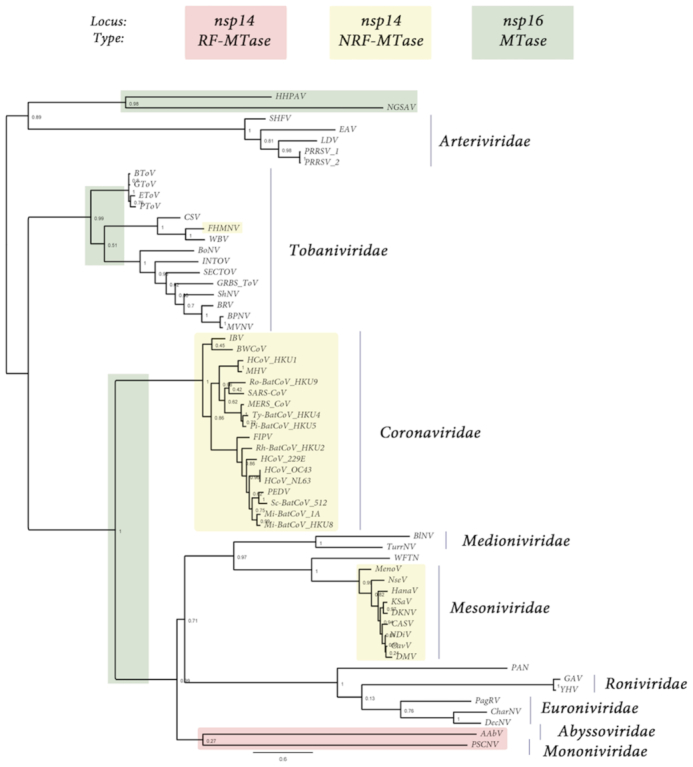
Presence, type and loci of signature-sequences of Nidovirales RNA MTases: RF-MTase of unknown specificity (light red), non-RF « nsp14-like » MT (yellow) and RF « nsp16-like » MTase (green). Both light red and yellow MTases map to the nsp14 C-ter locus, ie. immediately downstrean the ExoN domain, while « nsp16-like » MTase map to the nsp16 locus, at the end of Orf1b. The tree was made based on MAFFT v7.427 multiple sequence alignment with BLOSUM62 scoring matrix and G-INS-i iterative refinement method. The alignments were used as input for maximum likelihood trees generated with the FasTtree v2.1.5 software (best-fit model = JTT-Jones-Taylor-Thorton with single rate of evolution for each site = CAT). Local support values were computed using the Shimodaira-Hasegawa test (SH) with 1000 replicates. Numbers at the nodes represent FasTtree support values and scale var substitutions per site. The tree included two novel Ronivirus-like and Mesonivirus-like genome sequences: Western Flower Thrips Mesonivirus, and Palaemon Nidovirus, respectively (WFTV, PAN, unpublished, see the ‘Materials and Methods’ section and Supplementary Table S1). When present in the NCBI viral genomes database in the 92 Nidovirales complete genomes repository (https://www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi?taxid=76804), no accession number is indicated. When an accession number is given in parenthesis, it is referring to the GenBank accession number (https://www.ncbi.nlm.nih.gov/genbank/). The genome dataset used in this study is given in Supplementary Table S1. From top to bottom of the figure: Arteriviridae: HHPAV: Hainan hebius popei arterivirus (MG600021); NGSAV: Nanhai ghost shark arterivirus (MG600024); SHFV: simian hemorrhagic fever virus; EAV: Equine arteritis virus; LDV, Lactate elevating virus; PRRSV-1 and 2, porcine reproductive and respiratory syndrome virus. Tobaniviridae: BToV: Breda virus; GToV: Goat torovirus; EToV: Berne virus (CAA36747); PToV: Porcine torovirus; CSV: Chinook salmon bafinivirus; FHMNV: Fathead Minnow nidovirus 1; WBV: white bream virus; BoNV: Bovine nidovirus TCH5; INToV: Xinzhou nido-like virus 6; SECToV; Xinzhou toro-like virus 1; GRBS-ToV: Guangdong red banded snake torovirus (MG600030); ShNV: shingleback nidovirus 1 (KX184715); BPNV: Ball python nidovirus 1; MVNV: Morelia viridis nidovirus; BRV: Bellinger River virus (MF685025); Coronaviridae: IBV: Infectious Bronchitis Virus; BWCoV: Beluga Whale coronavirus SW1; HCoV_HKU1: Human coronavirus HK1; MHV: Mouse hepatitis virus; BatCoV_HKU9: Rousettus bat Coronavirus HKU9; SARS-CoV: Severe acute respiratory syndrome coronavirus; MERS_CoV: middle-east respiratory syndrome coronavirus; Ty-BatCoV_HKU4: Tylonycteris bat coronavirus HKU4; Pi-BatCoV_HKU5: pipistrellus bat coronavirus HKU5; FIPV: Feline infectious peritonitis virus; Rh-BatCoV-HKU2: Rhinolophus bat coronavirus HKU2; HCoV_229E: Human coronavirus 229E; HCoV_OC43: Human coronavirus OC43 (YP_003766); HCoV_NL63: Human coronavirus NL63; PEDV: Porcine epidemic diarrhea virus; Sc-BatCoV_512: Scotophilus bat coronavirus 512; Mi-BatCoV-1A: bat coronavirus 1A; Mi-BatCoV_HKU8: Miniopterus bat coronavirus HKU8. Medioniviridae: BlNV: Botrylloides leachii nidovirus; TurrNV: Turrinivirus 1; WFTV: Western Flower Thrips virus. Mesoniviridae: MenoV: Meno virus; NseV: Nse virus; HanaV: Hana virus; KSaV: Karang sari virus; DKNV: Dak Nong virus; CASV: Casuarina virus; NDiV: Nam Dinh virus; CavV: Cavally virus; DMV: Dianke mesonivirus. Roniviridae: PAN: Palaemon nidovirus; GAV: Gill-associated virus; YHV: Yellow head virus (EU487200). Euroniviridae: PagRV: Paguronivirus 1; CharNV: Charybnivirus 1; DecNV: Decronivirus 1. Abyssoviridae: AAbV: Aplysia abyssovirus 1. Mononiviridae: PSCNV: Planidovirus 1
The Nidovirus genome is predominantly comprised of two overlapping open reading frames (ORFs); ORF1a and ORF1b, which are translated directly from the viral genome to yield two polyproteins, pp1a and pp1ab, with the latter being the result of a −1 ribosomal frameshift. Cleavage of the polyproteins by viral proteases liberates between 12 and 17 non-structural proteins (nsp) that constitute the viral replicase/transcriptase complex (RTC). Template driven RNA synthesis by the RTC allows both genome amplification and the production of a set of subgenomic mRNAs (sg mRNAs) from 3′ end of the genome, which encode the structural and accessory proteins.
Two salient features of these viruses are, amongst others, their complex and still obscure replication/transcription mechanism along with their broad genome-size range. While these viruses are classed together based on a conserved genome organisation and a common ancestry of core replicative enzymes (3,4), their genomes vary considerably in size and complexity, from 12.7 to 15.7 kb for Arteriviridae (hereafter referred to as small-genome nidoviruses), to over 25 kb for the other families (hereafter referred to as large-genome nidoviruses) including Tobaniviridae and CoV (∼27–32 kb), and Mononiviridae (up to 41.1 kb)—among the largest (+)RNA viruses known (5). The expansion of the viral genome is believed to be due to the gradual acquisition of novel domains, which have allowed these viruses to develop into an unprecedented evolutionary space.
Of particular interest is the acquisition and adaptation of enzymes involved in the viral capping pathway. It is currently presumed that most Nidovirales genomes carry a 5′ cap structure which serves to both protect the viral genome from host degradation by 5′ exonucleases and to signal translation of the genome. The evidence for the presence of an RNA cap comes from immunological detection in Torovirus (family Tobaniviridae) genomic and sg mRNAs (6) and co-migration analysis using low-resolution chromatographic techniques for Mouse hepatitis virus (MHV, family CoV) (7) and Simian Hemorragic fever virus (family Arteriviridae) (8), however there is still no high-resolution structural analysis available for any Nidovirales RNA cap. Other families of the Nidovirales order are assumed to contain a canonical 5′ cap based on the presence of enzymes required in the capping pathway (reviewed in (9)), although direct demonstration of the presence of an RNA cap structure is still missing for many of these viruses.
In Eukaryotes, the RNA cap structure is thought to be synthesized post-transcriptionally through a well-described capping pathway (10). Most +RNA viruses supposedly follow this conventional RNA capping pathway in which nascent viral 5′-triphosphate genomic RNA (and sg mRNAs) is processed through three enzymatic reactions to yield an RNA cap whose structure is indistinguishable from that of cellular mRNAs. The capping pathway involves firstly the hydrolysis of the 5′-triphosphate RNA into a 5′-diphosphate by an RNA triphosphatase. A GMP residue (the « cap », originating from GTP) is subsequently covalently transferred to the 5′-diphosphate RNA in the 5′ to 5′ orientation by a guanlylyltransferase (GTase), releasing inorganic pyrophosphate. Both the cap and the first transcribed nucleotide are then methylated at the N7-guanine (mGpppN-RNA, so-called Cap0 structure) and the 2′-oxygen position (mGpppNm-RNA, Cap1 structure) respectively, by one or two S-Adenosyl-Methionine (SAM)-dependent RNA methyltransferases (MTases) (9).
Our understanding of Nidovirales RNA capping pathway, cap structures, and enzymes is still rather limited. In the case of CoVs, three out of four of the enzymes required in the presumed viral capping pathway have been identified, with the missing activity being that of the GTase. All proteins required for capping are located in Orf1b, including nsp13 which functions as the 5′-triphosphatase (along with RNA helicase) (11), nsp14 which contains an N7-guanine MTase domain fused to an N-terminal exoribonuclease (ExoN) (12,13) and nsp16 which performs the 2′-O MTase activity (14–16).
Remarkably, the CoV nsp14 N7-guanine MTase is the only example of a non-Rossman fold (NRF) viral MTase known so far. Until its discovery through elegant yeast-complementation assays (13), all known viral MTases (both N7 and 2′-O) belong to the Rossmann fold (RF) family, one of the five most ubiquitous and ancient super-secondary structures adopted throughout the superfamily of dinucleotide binding enzymes (17,18). Briefly, RF-MTases are characterized predominantly by two structural features; first, a βαβ architecture (formed by a seven-stranded β-sheet surrounded by 6 α-helices, with the seventh β-strand inserted in an anti-parallel orientation between the fifth and sixth strand) and secondly a glycine rich loop (G-x-G-xn-G) which interacts with the SAM cofactor. Viral 2′-O MTases can be further distinguished by the presence of a conserved K-D-K-E catalytic tetrad, and in general have been much better defined than N7-guanine MTases at the structural and functional level (reviewed in (9)). While various crystal structures of viral enzymes involved in N7-guanine methyltransferase activity have been resolved (19–23), the identification of a specific signature sequences is far less clear.
Structural analysis of the CoV nsp14 N7-guanine MTase revealed substantial structural deviations incompatible with classification into the RF family, including lack of both the βαβ structural motif and standard MTase sequence motifs (20). Rather than the alternating α – β architecture, SARS nsp14 is comprised of 12 β-strands and 5 α-helices, with the core of the structure formed by a five-stranded β-sheet. The SARS-CoV N7-MTase domain is unique to Nidoviruses so far, defining a new structural family of NRF N7-guanine MTases. Furthermore, it is currently the only N7-guanine MTase detected into the Nidovirales order.
Despite the presence of MTases amongst large-genome Nidoviruses, their presence, structure, specific activity and genomic distribution throughout the rest of the order varies considerably. For example, small-genome arteriviruses are not known to encode any evident MTase signature sequence, while only the 2′-O-MTase (but not the N7-MTase) has been identified for most other families (e.g Tobaniviridae, Medioniviridae, Roniviridae and Euroniviridae). Furthermore, for many viruses the presence of a specific MTase has not been specifically shown, but rather is extrapolated based on its presence in other members of the family. This raises several important questions as to how (and if) capping is performed in these families, and the evolution of the capping pathway in regards to the order as a whole.
In this paper, we performed large-scale genomic analysis of the order Nidovirales, in order to establish and clarify the presence and genomic location of MTase domains across different viral families. Sequence-based structural alignments were performed on newly identified MTase domains in order to predict activity and function. Several previously unidentified MTase signature-sequences have been identified, including the presence of a (presumably) 2′-O-MTase domain in two small-genome arteri-like viruses. Importantly, we also report the discovery of an RF-MTase sequence in the Orf1a of ten members of the Tobaniviridae family, an unusual and previously unseen location for this enzyme. Remarkably, sequence base structure alignments reveal that this enzyme is closely related to the canonical eukaryotic RF-N7-guanine MTase, suggesting it performs the currently missing N7-guanine RNA cap methylation for this family. If this is the case, this would represent the first RF-N7-guanine MTase identified for the Nidovirales order. Furthermore, this MTase was only identified in non-mammalian Tobaniviridae, and thus may represent a genetic marker distinguishing non-mammalian from mammalian Tobaniviridae.
MATERIALS AND METHODS
Virus genome sequences were retrieved from the NCBI database (https://www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi?taxid=439488) or Genbank (ncbi.nlm.nih.gov/genbank). The initial dataset was that of Lauber et al. (24), to which novel or recently described genome sequences (retrieved in NCBI of GenBank) were added. Additionally, the putative and unpublished Palaemon nidovirus and Western flower thrips nidovirus, and the proposed Botrylloides leachii nidovirus (25) were identified in the Sequence Read Archive (SRA) runs SRR5658389, SRR492945 and SRR2729873, respectively, and the virus sequences were assembled, curated and annotated as described elsewhere (26,27). The resulting sequences are available upon request to H.D. Accession numbers of the dataset are given in Supplementary Table S1.
The conserved Nidovirales C-terminus RdRp core was used to build phylogenetic trees based on MAFFT v7.427 multiple sequence alignment (MSA) with BLOSUM62 scoring matrix and G-INS-i iterative refinement method. The alignments were used as input for maximum likelihood trees generated with the FasTtree v2.1.5 software (best-fit model = JTT-Jones-Taylor-Thorton with single rate of evolution for each site = CAT). Local support values were computed using the Shimodaira-Hasegawa test (SH) with 1000 replicates.
Structural alignment of reference or retrieved MTases were done using EXPRESSO (28) and proofed with Chimera (29). N7-Mtases reference motifs were inferred and defined based on visual inspection and analysis of the structures. The subsequent motifs were used as orthogonal validation and not used for the search.
Sequences were analyzed using HHblits and HHpred tools of the BioInformatics tookit (30) searching against SCOPe70_2.07. Primary hits were selected by a two criteria cut-off: (i) a minimum domain length of 110 amino acids and (ii) an E-Value <10−5 (Original hit values are in Supplementary Table S2). Refined domain boundaries are based on alignments driven by secondary structure prediction generated with predict protein (31). When available, cleavage sites were used to predict protein gene products of the Orf1ab, Orf1a and Orf1b polyproteins. The boundaries were otherwise approximately (±10 aa) determined using structural homologies detected using HHPred, except for the N-term boundary of the Orf1b gene product: In Nidovirales, the absence of any structural data nor homology (outside the order) on the N-terminus of the RdRp gene (nsp9 Arteriviridae, nsp12 in CoV), which was used for phylogenic analysis, precludes precise sequence homology search in this limited area comprised between the nsp10 and nsp12 proteins (Coronavirus gene-product naming).
MSAs were generated using Muscle in SeaView (32). For each sequence of unknown structure, secondary structures were predicted using Predict Protein (31). The predicted secondary structures were used to validate the alignment with structural references. The MSA was rendered using ESPript 3.0 (33), together with appropriate structural models as indicated, to assign secondary structures. When possible, structural 3D models were generated using Phyre 2.0 (34). Conserved patches of amino-acids were generated using WebLogo (35) and mapped in the structural models rendered in Chimera (e.g. Figure 3).
Figure 3.

(A) Structural alignment of Tobaniviridae MTases together with various prokaryotic, eukaryotic and viral members; Pectobacterium Atrosepticum ECA1738 (PDB ID: 2P7I), Vaccinia virus D1:D12 heterodimer (PDB ID: 4CKB), Encephalitozooan cuniculi EcmI (PDB ID: 1Z3C), human RNA N7-guanine MTase (PDB ID: 3BGV). (B) Vaccinia Virus N7-guanine MTase structure (PDB: 4CKB) with highlighted motifs 1 to 5 and their corresponding amino-acid frequencies as determined by WebLogo analysis.
RESULTS
All non-arterivirus Nidovirales members carry at least one 2′-O-Mtase
We first wanted to establish and clarify the presence and genomic location of 2′-O-MTase domains located downstream of the RNA-dependent-RNA-polymerase (RdRp), as is the case for SARS-CoV nsp16. In order to do this, available genome sequences (Table 1 and Supplementary Table S1) were aligned based on the structurally conserved RdRp domain, and used to build a phylogenetic tree (Figure 1). From this alignment, we first determined that the majority of viruses within the Nidovirales order code for at least one RF-MTase protein, identified through the presence of the G-x-G-xn-G element of the SAM binding motif. These RF-MTase domains were additionally found to contain the canonical K-D-K E catalytic tetrad of 2′-O-MTases (Figure 1, green and Table 1). Conversely, this MTase was confirmed to be absent in members of the Arteriviridae family, including EAV, PRRSV and LDV. Interestingly however, the RF-MTase signature sequence was identified in two recently discovered arteri-like viruses: Hainan Hebius Popei Arterivirus (HHPAV, ∼12.5 kb) and Nanhai ghost shark arterivirus (NGSAV, ∼13.2 kb). Similar to the CoV 2′-O-MTase encoded on nsp16, all the identified RF- MTases, including the small-genome, arteri-like Nidoviruses, are located in a conserved genomic position at the 3′-end of Orf1b.
Table 1.
Distribution and putative substrate specificities of MTases in the four families of the Nidovirales Order
| Virus | Genome size | MTase | N7-MTase | 2′-O MTase |
|---|---|---|---|---|
| (nt) | Orf1a | nsp14-like | nsp16 | |
| Arteriviridae | ∼125–157 000 | − | − | (+/−)* |
| Tobaniviridae: | ||||
| Torovirus | ||||
| Porcine torovirus | 28 301 | − | − | − |
| Bovine torovirus | 28 479 | − | − | |
| Equine torovirus | 27 992 | − | − | − |
| Bostovirus | ||||
| Bovine nidovirus 1 | 20 261 | − | − | + |
| Bafinivirus | ||||
| White bream virus | 26 660 | + | − | + |
| Fathead minnow | ||||
| nidovirus 1 | 27 318 | + | + | + |
| Oncotshavirus | ||||
| Chinook salmon | ||||
| nidovirus 1 | 27 004 | + | − | + |
| Infratovirus | ||||
| Infratovirus 1 | 30 353 | + | − | + |
| Sectovirus | ||||
| Sectovirus 1 | 25 960 | + | − | + |
| Tiruvirus | ||||
| Shingleback | ||||
| nidovirus 1 | 23 832 | + | − | + |
| Pregotovirus | ||||
| Ball python | ||||
| nidovirus 1 | 33 452 | + | − | + |
| New/unnassigned genus | ||||
| Bellinger river virus | 30 742 | + | − | + |
| Guangdong | ||||
| red-banded snake | ||||
| torovirus | 30 859 | + | − | + |
| Python nidovirus | 32 399 | + | − | + |
| Goat torovirus | 28 487 | + | − | + |
| Coronaviridae | ∼27–32 000 | − | + | + |
| Medioni/ | ||||
| Mesoniviridae | ∼20–25 000 | − | −/+ | + |
| Roni/Euroniviridae | ∼26 000 | − | − | + |
| Abyssoviridae | 35 906 | − | +** | + |
| Mononiviridae | 41 178 | − | +** | + |
*: Arteriviruses do not usually carry any MTase, except the two newly identified Hainan Hebius Popei arterivirus and Nanhai gost shark arterivirus (see text).**: although in the nsp14 locus immediately downstream the ExoN gene, the MTase has a Rossmann fold. Virus species names are indicated and genome size corresponds to exemplar virus sequence used in this study.
Only the Tobaniviridae is expanded and genera indicated in italic plus unassigned viruses (see Figure 1).
We subsequently performed an MSA of nsp16 from the Roniviridae and Tobaniviridae families (Figure 1, in green), followed by modeling of a typical representative of these nsp16s (not shown). Consistent with phylogenetic analysis, all these enzymes are predicted to be canonical RNA 2′-O MTases, containing a typical K-D-K-E catalytic tetrad. As noted by others in the Ronivirus nsp16 model (36), minor structural differences are observed across the Tobaniviridae family, such as the absence of β3 strand and a shorter loop upstream helix αD, however overall the structure are consistent with a 2′-O-MTase function. Based on this, we conclude that non-arterivirus Nidovirales code for a RF 2′-O-MTase, containing a canonical K-D-K E catalytic tetrad which is located in a highly conserved position at the 3′-end of Orf1b.
The location and structure of N7-guanine MTases is not uniform along Nidovirales genomes
A distinguishing feature of large-genome Nidoviruses, is the possession of a unique NRF-MTase responsible for cap N7-guanine methylation. This has been most studied for the CoV family, where the N7-guanine MTase is fused to an N-terminal ExoN domain encoded on nsp14. It has been previously reported that the N7-guanine MTase domain is not uniformly present in nsp14-containing nidoviruses (24). Likewise, we could only identify NRF-N7-guanine MTase signature sequences (which line the SAM binding site of the nsp14 SARS MTase structure, PDB ID: 5C8U) for the CoVs and most mesoniviruses. In contrast, we were unable to detect any nsp14-like NRF-MTase for the majority of the other families within the Nidovirales order, including Arteriviridae, Medionivirdae, Roniviridae, Euroniviridae, Abyssoviridae, Mononiviridae and Tobaniviridae. Two notable exceptions are apparent. First, we were able to detect a nsp14-like NRF-MTase at the expected genomic Orf1b position in Fathead Minnow nidovirus 1 of the Tobaniviridae family. The conserved NRF folding, combined with the absence of the K-D-K-E catalytic tetrad of 2′-O MTases leads us to hypothesize that, like in CoVs, this enzyme is responsible for N7-guanine methylation of the RNA cap structure. Secondly, unique members of Abyssoviridae and Mononiviridae also possess an MTase signature-sequence at the C-terminus of their nsp14-like gene (i.e. fused to the ExoN domain). Curiously though, this MTase is readily detectable using HH-Pred as a RF-MTase, distinguishing it from the known Nidovirus NRF-N-7-guanine MTase. The identified nsp14-like MTase also lacks the characteristic K-D-K-E catalytic tetrad of the RF-2′-O MTases. We therefore cannot confirm the precise role of this identified MTase. We therefore deduce that the nidovirus enzyme responsible for N7-guanine methylation does not appear to be located in a conserved genomic location, nor contains a conserved structural architecture. The majority of viruses in the CoV and Mesoniviridae families appear to utilise a nidoviruses specific, NRF-MTase located directly downstream of the ExoN domain, as described for SARS-CoV. For other families, the enzyme responsible for N7-guanine methylation of the RNA-cap remains to be defined.
Selected Tobaniviridae members possess a RF-MTase signature-sequence in Orf1a lacking the canonical 2′-O catalytic K-D-K-E tetrad
The question still therefore remains as to how other members of the Nidovirales order methylate their RNA-cap at the N7-guanine position. One possibility would be that the RF-MTase identified at the 3′ end of Orf1b is bi-functional, carrying both 2′-O and N7-guanine methylation activity, as seen for the Flavivirus NS5 MTase. However, there is no obvious signature sequence to indicate bi-functionality, and furthermore no evidence to suggest that the activity for those virus families would deviate from the 2′-O methylation specificity shown for the same domain of large-genome nidoviruses (36,37). Another possibility would be that another gene would code for an enzyme performing this methylation, and had escaped detection by standard bioinformatic methods.
We thus performed a more extensive search for MTase signature-sequences along the whole Orf1ab in all Nidovirales. Surprisingly, a RF-MTase signature-sequence was detected in Orf1a of 10 members of the Tobaniviridae family (Figure 2). Strictly conserved amino-acids in these new viral MTases define three motifs (i) three glycines of the SAM binding site (Gly54, Gly56 and Gly58 in white bream virus (WBV)) located just downstream of a three amino acids hydrophobic segment in a β-strand motif, (ii) a histidine (His117 in WBV) immediately followed by either a phenylalanine or tyrosine and (iii) a glutamic acid (Glu175 in WBV) (Figure 3). Interestingly the catalytic K-D-K-E tetrad associated with 2′-O-MTases is lacking.
Figure 2.

Distribution of the Orf1a MTase along the (A) Tobaniviridae phylogenetic tree (see Figure 1). (B) approximate genome organisation and gene content along the family described in A). The genomes are not drawn to scale. Torovirinae: Bovine, Goat, Equine and Procine toroviruses; Bovine nidovirus 1 is the Bostovirus (Remotovirinae), whose genome is represented smaller than its fellow members to account for its ∼20 kb genome size; Piscavirinae and Serpentovirinae are the viruses boxed in green. See ref (1).
Remarkably, the novel Orf1a MTase gene was found to be predominantly associated with reptiles and fish, with the exception of two cases (Xinzhou Nematode virus 6 and Xinzhou toro-like virus 1, from the Sectovirus 1 and Infratovirus 1 species, respectively), where the viruses were isolated from snake-associated nematodes (although the host and life-cycle has not been clearly assessed). The mammalian toroviruses (EToV, BToV, Bovine TCH5 nidovirus, GToV and PToV) do not carry the MTase signature sequence.
We therefore conclude that certain members of the Tobaniviridae family possess a RF-MTase signature sequence in their Orf1a, and that this newly identified RF-MTase candidate may be a genetic marker distinguishing mammalian and non-mammalian members of this family.
What could define a N7-guanine MTase signature?
Unlike RF-2′-O-MTases (with their well-defined K-D-K-E tetrad) and the unique Nidovirus NRF-MTase discussed above, signature sequences of RNA cap N7-guanine MTases are much less evident. In order to expand our search criteria and establish potential activity of identified MTase domains in the Nidovirales order, we first attempted to discern a specific N7-guanine MTase signature sequence which could be used to aid in defining this family of enzymes. A wealth of structural and mechanistic data has been acquired from N7-guanine MTases from the microsporidian parasite Encephalitozoon cuniculi (Ecm1, PDB ID: 1Z3C) (38) and the D1:D12 heterodimer of the dsDNA Poxvirus (PDB ID: 4CKB) (37). Regarding RNA viruses, there are only three crystal structures of RF-MTases known to methylate N7-guanine caps. They are the rotavirus VP4 (PDB ID: 1KQR) the reovirus Lambda2 (PDB ID: 1EJ6) of the dsRNA Reoviridae family (21,22), and the West Nile Virus NS5 of the +RNA Flavivirus family (PDB ID: 2OYO) which performs bi-functional N7-guanine and 2′-O-MTase activities (39). Combining this data with structural information from the human RNA N7-guanine methyltransferase (PDB ID: 3BGV), we were unable to define specific N7-guanine MTase sequence motifs (Supplementary Figure S1), suggesting that structural conservation has likely prevailed over sequence conservation. We therefore narrowed the structural comparison to include only Encephalitozoon cuniculi, poxvirus and the human RNA N7-guanine Mtases. This allowed detection of five conserved amino acid motifs K/G/D/HY/E/Y (Supplementary Figure S2), spatially coherent in the structure to fulfil the binding and catalytic reaction.
The RF-MTase signature-sequence in Orf1a is a putative RNA cap N7-guanine MTase
We subsequently performed a structure-based alignment of the Orf1a MTase with several structurally-defined eukaryotic RNA-cap N7-guanine MTases (Figure 3A), of which Ecm1 can be considered as prototypic (38). The new alignment with these known N7-MTases reveals that although two deletions are observed relative to structurally characterized N7-guanine MTases (PDB IDs: 2P7I and 4CKB), they do not alter typical RF characteristic features. The conserved amino-acids remain essentially the same: two glycines within the SAM binding pocket (Gly54 and Gly56 in WBV, Gly74 and Gly76 in Ecm1) and a histidine (His117 in WBV, His144 in Ecm1) immediately followed by either phenylalanine or tyrosine. Glu175 is also remains highly conserved, and in the Ecm1 structure its homosteric counterpart (Glu225) is positioned close to His144 for interaction with the guanine base.
Taken together, these results suggest that this MTase is a N7-guanine MTase comprised of five conserved motifs, which are represented in Figure 3B structurally aligned onto the vaccinia N7-guanine MTase structure D12 (PDB ID: 4CKB).
DISCUSSION
It is currently assumed that (+)stranded viruses encode one or more MTases in their genomes to perform the necessary methylation leading to the RNA cap formation. RNA virus cap-MTases perform two types of reactions: the methylation of the N7-guanine of the RNA cap, and the methylation of the adenosine 2′-O ribose of the first transcribed nucleotide (9). Most of these viral MTases, with SARS-CoV nsp14 as a notable exception, belong to the RF family of enzymes (reviewed in (9)). The RF is an evolutionary ancient fold, which has been widely evolved to perform a variety of chemical reactions. Its structural plasticity is well illustrated by the Blue tongue virus VP4 MTase (family Reoviridae) which incorporates an entire functional domain within two additional secondary structure elements, in addition to the flavivirus NS5 MTase, which is able to perform both N7-guanine and 2′-O ribose methylation with the same ∼33 kDa domain fused to the N-terminus of the viral RdRp (23,39).
At a structural level there is a remarkable conservation of the RF for viral 2′-O MTases, combined with a conserved mechanism of action, reflected by the K-D-K-E catalytic tetrad. Conversely, N7-guanine methylation does not appear to obey any particular rule, neither at the structural nor at the biochemical level. Furthermore, the pathway and target molecule for N7-guanine methylation appears to be somewhat variable. For example in some families of viruses this reaction can be performed on a GTP molecule which is then used to cap the RNA (40). This is the case with the alphavirus nsp1 enzyme, whose structure is currently unknown. On the other hand, several crystal structures of viral enzymes involved in N7-guanine methylation of RNA caps have been reported, such as that of flaviviruses (NS5, (23,39)), rotaviruses and reoviruses (VP4 and lambda2, respectively (21,22)), and coronaviruses (nsp14, (19,20)). As stated above, the majority of these N7-MTases also adopt a RF (with the exception of SARS nsp14). However, at the sequence level no structurally conserved residues can be defined, thus complicating the detection of N7-MTases using bioinformatic analysis alone.
It is currently assumed that viruses within the order Nidovirales utilise a conventional capping pathway for synthesis of an RNA cap which is indistinguishable from that of the host RNA. However, in many cases the enzymes required in the capping pathway, including the N7- and 2′-O- MTase domains, have not been specifically identified, but rather are assumed to be present based on related genomes. Furthermore, the drastic variation in genome length suggests that the cap structure, enzymatic pathway and proteins may not be identical for all families, particularly in regards to the small-genome arteriviruses.
Here, we confirmed that with the exception of the Arteriviridae family, Nidovirales contain a RF-MTase located at a conserved position at the 3′ end of Orf1b. This enzyme is presumed to contain 2′-O-MTase activity, based on the presence of the conserved K-D-K-E catalytic tetrad and given the consistent genomic location with the well-characterized nsp16 2′-O-MTase of CoVs. The large-scale distribution of this domain, including in two small-genome arteri-like viruses (HHPA and NGSA) is somewhat surprising and may have a significant impact on our understanding of genome size evolution in Nidovirales. Phylogenetic branching of both HHPA and NGSA suggests that arteriviruses might not be primitive small version of larger Coronavirus genomes, but may rather originate from size-reduction of a large Nidovirus ancestor genome.
The presence of N7-guanine MTases in the Nidovirales order is more speculative. Until this point, the only confirmed N7-guanine MTase for the Nidovirales order was the unique NRF-MTase located in Orf1b just downstream of the ExoN domain in large-genome nidoviruses. While we could confirm the presence of nsp14-like NRF-MTases for viruses of the CoVs and mesoniviruses, we were unable to detect any nsp14-like NRF-MTase for the majority of other families, with the exception being a single member of the Tobaniviridae family, Fathead minnow virus 1. The question therefore remains as to how the other Nidovirus members methylate their RNA-cap at the N7-guanine position.
Interestingly, for unique members of the Abyssoviridae and Mononiviridae families, a predicted RF-MTase was identified in the analogous genomic location to the CoV nsp14 NRF-MTase, just downstream of the viral ExoN. The lack of the characteristic K-D-K-E catalytic tetrad suggests this protein does not contribute to 2′-O methylation, supported by the fact that both families already contain a conserved 2′-O MTase signature sequence at the end of Orf1b. Based on its conserved genomic location, we therefore suggest that this protein could function as the missing N7-guanine MTase, raising an interesting and curious question regarding the evolution and functional complementarity between the NRF-N7-guanine MTase of CoVs and the RF-MTases of these families.
Expansion of genomic search for potential MTase domains also revealed a RF-MTase signature-sequence in Orf1a of 10 members of the Tobaniviridae family. The presence of an MTase in Orf1a is unexpected, and has not been reported before, although signature sequences of an uncharacterized MTase had been detected in Ball Python nidovirus, Sectovirus 1 and Infratovirus 1 (25). Protein and enzyme functions in Nidovirales have been classified into a functional triangle (24), with each side of the triangle representing a carrier of the following roles: Orf1a- host defense; Orf1b—genome replication and maintenance; 3′ nested Orfs—structural and accessory proteins. In this triangle, the MTase activity involved in genome maintenance would usually map to Orf1b (24). The location in Orf1a may therefore suggest additional or auxiliary roles in tasks other than RNA capping, however this remains to be determined. Accordingly, it was recently reported that viral or cellular MTases can be recruited by viruses in order to induce internal methylation of their genome (41) and escape to the antiviral response mediated by MDA5 (42). Furthermore, −1 ribosomal frameshifting being a mechanism commonly used in viral and cellular proteins to regulate ratios and copy numbers of specific genes, it would suggest that the levels of Orf1a products is higher than those of Orf1b, raising the question as to why only Tobaniviridae members would need this additional MTase in higher quantities.
Taken together, this analysis clarifies the presence and location of MTase domains across the Nidovirales order, revealing a surprising variability. The identification of novel, putative RF-MTase domains may unveil the currently missing enzyme behind N7-guanine methylation for several viral families. If this is the case, it is a surprising structural deviation from the known CoV NRF-N7-guanine MTase nsp14. Dual N7 and 2′-O methylation activity by RF-MTases is also possible, as evidenced by the flavivirus NS5 MTase, particularly for families for which only a single (nsp16-like) MTase could be identified.
In any case, the type, diversity, and distribution of RNA MTases across the Nidovirales order reveals an enormous variability regarding genome organization, regulation and evolution, that need to be addressed both functionally and structurally. SAM-dependent MTases are ancient folds associated with RNA stability and evolution (43). Their presence and properties in a phylogenetic tree may well give interesting clues regarding RNA genome evolution and its associated issue of host defense mechanisms.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
CNRS (in part); Australian Endeavour Research Fellowship (to A.S.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Siddell S.G., Walker P.J., Lefkowitz E.J., Mushegian A.R., Adams M.J., Dutilh B.E., Gorbalenya A.E., Harrach B, Harrison R.L., Junglen S. et al.. Additional changes to taxonomy ratified in a special vote by the International Committee on Taxonomy of Viruses (October 2018). Arch. Virol. 2019; 164:943–946. [DOI] [PubMed] [Google Scholar]
- 2. Song Z., Xu Y., Bao L., Zhang L., Yu P., Qu Y., Zhu H., Zhao W., Han Y., Qin C.. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses. 2019; 11:E59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gorbalenya A.E., Enjuanes L., Ziebuhr J., Snijder E.J.. Nidovirales: evolving the largest RNA virus genome. Virus Res. 2006; 117:17–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon LLM., Guan Y., Rozanov M., Spaan WJM., Gorbalenya A.E.. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003; 331:991–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saberi A., Gulyaeva A.A., Brubacher J.L., Newmark P.A., Gorbalenya A.E.. A planarian nidovirus expands the limits of RNA genome size. PLoS Pathog. 2018; 14:e1007314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van Vliet ALW., Smits S.L., Rottier PJM., de Groot R.J.. Discontinuous and non-discontinuous subgenomic RNA transcription in a nidovirus. EMBO J. 2002; 21:6571–6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lai M.M., Patton C.D., Stohlman S.A.. Further characterization of mRNA’s of mouse hepatitis virus: presence of common 5′-end nucleotides. J. Virol. 1982; 41:557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sagripanti J.L., Zandomeni R.O., Weinmann R.. The cap structure of simian hemorrhagic fever virion RNA. Virology. 1986; 151:146–150. [DOI] [PubMed] [Google Scholar]
- 9. Decroly E., Ferron F., Lescar J., Canard B.. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 2011; 10:51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramanathan A., Robb G.B., Chan S-H.. mRNA capping: biological functions and applications. Nucleic Acids Res. 2016; 44:7511–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ivanov K.A., Ziebuhr J.. Human coronavirus 229E nonstructural protein 13: characterization of duplex-unwinding, nucleoside triphosphatase, and RNA 5′-triphosphatase activities. J. Virol. 2004; 78:7833–7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Minskaia E., Hertzig T., Gorbalenya A.E., Campanacci V., Cambillau C., Canard B., Ziebuhr J.. Discovery of an RNA virus 3′→5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:5108–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen Y., Cai H., ’an Pan J., Xiang N., Tien P., Ahola T., Guo D.. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:3484–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Decroly E., Imbert I., Coutard B., Bouvet M., Selisko B., Alvarez K., Gorbalenya A.E., Snijder E.J., Canard B.. Coronavirus nonstructural protein 16 is a cap-0 binding enzyme possessing (nucleoside-2′O)-methyltransferase activity. J. Virol. 2008; 82:8071–8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Decroly E., Debarnot C., Ferron F., Bouvet M., Coutard B., Imbert I., Gluais L., Papageorgiou N., Sharff A., Bricogne G. et al.. Crystal structure and functional analysis of the SARS-coronavirus RNA cap 2′-O-methyltransferase nsp10/nsp16 complex. PLoS Pathog. 2011; 7:e1002059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen Y., Su C., Ke M., Jin X., Xu L., Zhang Z., Wu A., Sun Y., Yang Z., Tien P. et al.. Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2′-O-methylation by nsp16/nsp10 protein complex. PLoS Pathog. 2011; 7:e1002294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rao S.T., Rossmann M.G.. Comparison of super-secondary structures in proteins. J. Mol. Biol. 1973; 76:241–256. [DOI] [PubMed] [Google Scholar]
- 18. Chouhan BPS., Maimaiti S., Gade M., Laurino P.. Rossmann-fold methyltransferases: taking a “β-Turn” around their cofactor, S-Adenosylmethionine. Biochemistry (Mosc). 2019; 58:166–170. [DOI] [PubMed] [Google Scholar]
- 19. Ma Y., Wu L., Shaw N., Gao Y., Wang J., Sun Y., Lou Z., Yan L., Zhang R., Rao Z.. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:9436–9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ferron F., Subissi L., Silveira De Morais A.T., Le NTT., Sevajol M., Gluais L., Decroly E., Vonrhein C., Bricogne G., Canard B. et al.. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E162–E171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sutton G., Grimes J.M., Stuart D.I., Roy P.. Bluetongue virus VP4 is an RNA-capping assembly line. Nat. Struct. Mol. Biol. 2007; 14:449–451. [DOI] [PubMed] [Google Scholar]
- 22. Tao Y., Farsetta D.L., Nibert M.L., Harrison S.C.. RNA synthesis in a cage–structural studies of reovirus polymerase lambda3. Cell. 2002; 111:733–745. [DOI] [PubMed] [Google Scholar]
- 23. Egloff M-P., Benarroch D., Selisko B., Romette J-L., Canard B.. An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 2002; 21:2757–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lauber C., Goeman J.J., Parquet M., del C., Nga P.T., Snijder E.J., Morita K., Gorbalenya A.E.. The footprint of genome architecture in the largest genome expansion in RNA viruses. PLoS Pathog. 2013; 9:e1003500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bukhari K., Mulley G., Gulyaeva A.A., Zhao L., Shu G., Jiang J., Neuman B.W.. Description and initial characterization of metatranscriptomic nidovirus-like genomes from the proposed new family Abyssoviridae, and from a sister group to the Coronavirinae, the proposed genus Alphaletovirus. Virology. 2018; 524:160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Debat H.J. Expanding the size limit of RNA viruses: Evidence of a novel divergent nidovirus in California sea hare, with a ∼35.9 kb virus genome. 2018; 24 April 2018, preprint: not peer reviewed 10.1101/307678. [DOI]
- 27. Debat H.J. An RNA virome associated to the Golden Orb-Weaver Spider Nephila clavipes. Front. Microbiol. 2017; 8:2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Armougom F., Moretti S., Poirot O., Audic S., Dumas P., Schaeli B., Keduas V., Notredame C.. Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 2006; 34:W604–W608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 30. Zimmermann L., Stephens A., Nam S-Z., Rau D., Kübler J., Lozajic M., Gabler F., Söding J., Lupas A.N., Alva V.. A completely reimplemented MPI bioinformatics toolkit with a New HHpred Server at its core. J. Mol. Biol. 2018; 430:2237–2243. [DOI] [PubMed] [Google Scholar]
- 31. Yachdav G., Kloppmann E., Kajan L., Hecht M., Goldberg T., Hamp T., Hönigschmid P., Schafferhans A., Roos M., Bernhofer M. et al.. PredictProtein–an open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 2014; 42:W337–W343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gouy M., Guindon S., Gascuel O.. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010; 27:221–224. [DOI] [PubMed] [Google Scholar]
- 33. Gouet P., Robert X., Courcelle E.. ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003; 31:3320–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg MJE.. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015; 10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Crooks G.E., Hon G., Chandonia J-M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zeng C., Wu A., Wang Y., Xu S., Tang Y., Jin X., Wang S., Qin L., Sun Y., Fan C. et al.. Identification and Characterization of a Ribose 2′-O-Methyltransferase Encoded by the Ronivirus Branch of Nidovirales. J. Virol. 2016; 90:6675–6685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Decroly E., Imbert I., Coutard B., Bouvet M., Selisko B., Alvarez K., Gorbalenya A.E., Snijder E.J., Canard B.. Coronavirus nonstructural protein 16 is a cap-0 binding enzyme possessing (nucleoside-2′O)-methyltransferase activity. J. Virol. 2008; 82:8071–8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fabrega C., Hausmann S., Shen V., Shuman S., Lima C.D.. Structure and mechanism of mRNA cap (guanine-N7) methyltransferase. Mol. Cell. 2004; 13:77–89. [DOI] [PubMed] [Google Scholar]
- 39. Ray D., Shah A., Tilgner M., Guo Y., Zhao Y., Dong H., Deas T.S., Zhou Y., Li H., Shi P-Y.. West Nile virus 5′-cap structure is formed by sequential guanine N-7 and ribose 2′-O methylations by nonstructural protein 5. J. Virol. 2006; 80:8362–8370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ahola T., Kääriäinen L.. Reaction in alphavirus mRNA capping: formation of a covalent complex of nonstructural protein nsP1 with 7-methyl-GMP. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martin B., Coutard B., Guez T., Paesen G.C., Canard B., Debart F., Vasseur J-J., Grimes J.M., Decroly E.. The methyltransferase domain of the Sudan ebolavirus L protein specifically targets internal adenosines of RNA substrates, in addition to the cap structure. Nucleic Acids Res. 2018; 46:7902–7912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ringeard M., Marchand V., Decroly E., Motorin Y., Bennasser Y.. FTSJ3 is an RNA 2′-O-Methyltransferase recruited by HIV to avoid innate immunity sensing. Nature. 2019; 565:500–504. [DOI] [PubMed] [Google Scholar]
- 43. Rana A.K., Ankri S.. Reviving the RNA World: An Insight into the Appearance of RNA Methyltransferases. Front. Genet. 2016; 7:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.