

Abstract
Two pathways are described for submission to FDA for clearance of a diagnostic device: a Premarket Application (PMA), which can lead to approval of a diagnostic device, and a Premarket Notification, which can lead to clearance. The latter is often called a 510(k), named for the statute providing for this path. Recent FDA clearance of molecular-based multiplex panels represents the beginning of a new era for the diagnosis of respiratory infections. The ability to test for multiple pathogens simultaneously, accompanied by the increasing availability of molecular-based assays for newly recognized respiratory pathogens will likely have a major impact on patient care, drug development, and public health epidemiology. We provide a general overview of how FDA evaluates new diagnostics for respiratory tract infections and the agency’s expectations for sponsors developing new tests in this area.
DEFINITION OF IN VITRO DIAGNOSTIC DEVICES
The Food and Drug Administration (FDA) has regulatory responsibility in the United States for review and oversight of the products that form the backbone of modern medical practice. FDA premarket and postmarket activities are administratively organized around centers, with the Center for Drug Evaluation and Research (CDER), Center for Biologics, Evaluation, and Research (CBER), and Center for Devices and Radiological Health (CDRH) bearing most of the responsibility for patient diagnosis and treatment. CDRH, through the Division of Microbiology Devices, in the Office of In Vitro Diagnostic Device Evaluation and Safety has regulatory responsibility for in vitro diagnostic devices (IVDs) intended for use in the diagnosis of almost all infectious diseases, including respiratory tract infections. The specific regulations that guide FDA are defined under Title 21 of the Code of Federal Regulations (CFR); in this overview, we highlight both the elements of the CFR relevant to in vitro microbiology diagnostic devices as well as how FDA approaches the review of microbiology device applications for respiratory infections. (Specific regulations cited are included in brackets when referenced.)
IVDs are defined as “reagents, instruments, and systems intended for use in the diagnosis of disease or other conditions, including a determination of the state of health, in order to cure, mitigate, treat, or prevent disease or its sequelae…for use in the collection, preparation, and examination of specimens from the human body” [21 CFR 809.3]. Similar to drug and biological products, new in vitro devices must undergo FDA review prior to marketing. For a new IVD, both “safety” and “effectiveness” of a device must be demonstrated. The broad criteria for evaluating the safety and effectiveness of new IVD are also defined by regulation:
Safety: Are there reasonable assurances based on valid scientific evidence that probable benefits to health from use of the device outweigh any probable risks? [21 CFR 860.7 (d)(1)]
Effectiveness: Is there reasonable assurance based on valid scientific evidence that the use of the device in the target population will provide clinically significant results? [21 CFR 860.7(e)(1)]
Requirements for marketing also include the need for labeling that includes, among other requirements, product performance characteristics “as appropriate… describing such things as accuracy, precision, specificity, and sensitivity” [21 CFR 809.10]. The following discussion provides an overview of how FDA approaches these requirements during the development and review of a new microbiology diagnostic device.
FDA regularly issues guidances in an effort to make the FDA submission and review process more consistent. Guidances are not rules but provide FDA’s current thinking on a topic. Guidances may be applicable to all of FDA (eg, see [1]), to a center within FDA, to a class of products, or tied to a novel product (often called Special Controls Guidance; eg, see [2, 3]). Some are initially issued as draft guidances where the public has an opportunity to comment for a prespecified period. In this article, we provide references to several FDA guidances.
THERAPEUTICS VERSUS DIAGNOSTICS
The FDA drug approval process is more familiar to physicians—adequate and well-controlled trials to establish safety and efficacy [21 CFR 314.50] are required as the standard for regulatory approval, usually conducted as randomized, comparative trials. In contrast, study designs used to support device applications are typically not randomized, controlled trials; most often, clinical performance is documented by a single prospective multicenter study in the intended use population, supported by extensive nonclinical analytical studies. For diseases of low prevalence, prospective studies may not be feasible.
One major difference between drugs and devices is the existence of two distinct pathways for the marketing of devices: devices may be “cleared” through the 510(k) process when a new device is determined to be substantially equivalent to a previously marketed product. Devices for entirely new indications or higher-risk intended use are “approved” by the Premarket Application (PMA) process. It is important that clinical and analytical requirements for submissions are more aligned with the technology and the clinical settings for use of the device rather than the specific pathway used to go to market. Some devices are novel, but may be considered to be moderate risk in the context of how they are used. These devices may still may be cleared via the 510(k) process using what is called a de novo 510(k). However, the potential for a de novo pathway should be discussed with FDA well in advance of a formal submission.
Essential elements considered in the review of a new in vitro diagnostic device for a respiratory pathogen are outlined in Table 1 and discussed in more detail below.
Table 1.
Major Elements in the Evaluation of a New In Vitro Diagnostic Device for a Respiratory Pathogen
| 1 | Intended Use |
| 2 | Reference Methods and Clinical Studies |
| 3 | Assay Interpretation |
| 4 | Assay Performance Characteristics |
| 5 | Evaluating Multiplex Assays |
| 6 | CLIA Waivers |
Intended Use
How diagnostic devices are evaluated is strongly influenced by the intended use (IU) and the risks associated with that use. The proposed IU for a device is an explicit statement of the analyte that the device is measuring or detecting (eg, a specific organism or a biomarker for that organism, and the clinical disease resulting from infection). The IU also describes how the results are reported: qualitative assays most often report whether a specific pathogen is detected or not (eg, culture or direct antigen tests); less commonly (for respiratory pathogens), the results may be quantitative (eg, viral load assays or antibody titers).
The specimen type (also referred to as the sample matrix) that can be tested is also an important component of the IU of the device. Typical specimens for an upper respiratory tract infection diagnostic device are nasal swabs, nasopharyngeal swabs, nasal washes; or for lower respiratory tract specimens, bronchoalveolar lavage or sputum. Less obvious matrices for respiratory pathogens include urine or gastric lavage specimens. Evaluation of an analyte in a specific matrix may be critical for a proposed IU as test performance may significantly vary across different matrices.
The setting (and/or timing) where test specimens should be collected may also be part of the IU; for example, some devices (including several rapid influenza tests) are cleared for point-of-care settings (ie, used near the patients while under the auspices of a laboratory with professional laboratorians). Timing may be equally critical to test performance; for example, sensitivity of certain rapid antigen tests may drop dramatically as an illness evolves.
Reference Methods and Clinical Studies
New devices must be compared with a benchmark for establishing whether or not a specific target condition is present. FDA recognizes two major situations for assessing the diagnostic performance of new qualitative diagnostic tests: when a clinical reference method is available or when a comparator other than a reference method is used. In the former circumstance, a clinical reference method is considered to be the best available method for establishing the presence or absence of the target organism [1] but the reference method cannot incorporate results reported by the new device; for example, a new enzyme-linked immunosorbent assay (ELISA) could not be used as one of the diagnostic criteria for the reference method in a study used to evaluate the performance of the assay.
Confusion may sometimes arise when distinguishing between analytical and clinical reference method since these may be identical or separate in different contexts; for example, the performance characteristics for a new assay for group A streptococcus against an analytical reference method such as culture on appropriate media may only be valid in the clinical setting of pharyngitis, since group A streptococcus may be a normal colonizer and performance may be different in the setting of colonization versus infection. In contrast, the analytical and clinical reference methods for influenza A are identical since the presence of influenza A is always considered abnormal. Determining the appropriate clinical reference methods may be particularly complex in the setting of multiplex assays for diseases with multiple etiologies such as community-acquired pneumonia, and where two or more possible pathogens may be isolated.
Newly identified organisms introduce different challenges in selecting the appropriate reference method; for example, more recently identified pathogens such as metapneumovirus may be difficult to isolate or confirm by traditional methods such as culture. Because a clinical reference method cannot include the new device in its ascertainment, methods such as polymerase chain reaction (PCR) followed by bidirectional DNA sequencing may be necessary to confirm test performance. Similar concerns exist for assays that differentiate subtypes of novel influenza, a disease where diagnostics are essential but where relatively few clinical specimens are available for confirming performance. There are recent FDA guidances that are useful in aiding companies in developing valid comparators. One is a draft guidance on establishing the performance of an influenza IVD [4], whereas the other two guidances are special controls guidances: one for multiplex devices for respiratory viral pathogens [2] and the other for multiplex devices for influenza A subtyping [3].
Another common issue that arises in clinical studies for new devices is result discordance; that is, when discrepancies occur between the result with the new device and the reference method. The objective of a pivotal study is to ascertain performance of the new device. Retesting discrepant samples tends to result in an overly optimistic view of performance of the new device. If the new device is truly better than the current clinical reference method, companies should consider contacting FDA to discuss an alternative comparator prior to beginning their pivotal trial.
Clinical studies for evaluating new devices should support the intended use of the device and be conducted in a manner that will yield results consistent with how the device would be integrated into US clinical practice. New devices under study may include foreign sites depending on the specific analyte and the specific intended use for the device; however, clinical studies with foreign sites must include a justification for why similar performance is expected in the United States. Study protocols should include clear inclusion and exclusion criteria, case report forms, and testing procedures. Sample sizes should be sufficient to ensure that the clinical performance is acceptable in its IU setting. See section on Assay performance characteristics: Statistics 101 for Diagnostic Devices for further discussion.
Assay Interpretation
The interpretation of results from a new investigational device should be determined prior to conducting ‘pivotal’ clinical studies. For a qualitative assay, this includes the criteria for scoring device results (eg, as either analyte detected, analyte not detected, equivocal, or invalid). Invalid results may occur when one or more test parameters fail to meet the expected criteria. For equivocal results, rules for retesting specimens should be specified.
Assay Performance Characteristics: Statistics 101 for Diagnostic Devices
Sensitivity/specificity. The simplest approach to describing diagnostic performance characteristics is when every test result can be described qualitatively (eg, analyte detected or not detected), and where a reference method exists. For a prospectively enrolled study where patients meet the inclusion/exclusion criteria for the IU of the device, agreement of the test results with the reference method establishes sensitivity and specificity of the test device.
Sensitivity is the probability that the new device will have a positive test result given that the clinical reference method is positive; it is estimated in a clinical study as the fraction of patients that are positive by the new test among those that are positive by the clinical reference method. For example, in a study of novel H1N1 influenza, it is the percent of patients that both test positive by the new device and are positive via the reference assay divided by the number of patients positive by the reference assay. Low test sensitivity reflects less security with the results of a negative test; that is, patients with the disease may be falsely labeled as negative by the new assay. (This fundamental consideration, i.e., —equating “analyte not detected” with the absence of disease [or the analyte]—is a common misinterpretation that bedevils the clinical use of devices with imperfect sensitivity.). Specificity is the converse; that is, the probability that the new device will have a negative test result given that the clinical reference method is negative. Specificity is estimated in the pivotal study by the fraction of subjects that are negative by the new test among those that are negative by the clinical reference method. Higher specificity yields greater confidence in a positive result. It is important to recognize that although patients included in specificity calculations do not have the disease of interest, in most cases they are still symptomatic (depending on the specific IU for the test); asymptomatic subjects would not be considered part of the IU population for such a device and therefore would not be appropriate for use in a specificity calculation.
There are examples where a new device is not compared with a reference but to a previously cleared/approved device. In this setting, sensitivity and specificity cannot be estimated due to the lack of known infected status. (For example, a benchmark reference standard for aspergillosis infection may include galactomannan as part of the definition of aspergillosis infection in certain populations, even in the absence of invasive procedures to document infection.). Identical statistical calculations can be used in this setting but are interpreted as being positive and negative percent agreement rather than sensitivity and specificity; these considerations are also described in FDA guidance [1]. Percent agreement estimates may be misleading as similar devices can both be in error; for example, 100% agreement between two similar devices would be unlikely to indicate the new device has perfect clinical sensitivity/specificity were the clinical truth available.
Prevalence, Negative Predictive Values, and Positive Predictive Values
There may be significant challenges with estimating the sensitivity of a new assay if the prevalence of the disease being tested for is low. As Table 2 illustrates, estimates of sensitivity are dependent both on the true underlying sensitivity and sample size. In a clinical study with only 5 specimens positive for a specific pathogen (via the reference method) and an observed sensitivity of 5/5 (100%) results in a lower confidence bound of only 55.6%, meaning diagnostic performance is very uncertain. Even for a test with perfect sensitivity, at least 35 patients with the condition of interest would be needed to yield a 95% lower bound greater than 90%. In a pivotal study for devices to detect more common conditions, it is not uncommon to see a sample size of 50 or more patients that are positive via the reference method. FDA considers the observed sensitivity and specificity and their respective 95% lower confidence bounds as part of device performance.
Table 2.
Estimates of Sensitivity
| Number of samples | Observed performance | 95% lowerconfidence bounda |
| 5 | 5/5 = 100% | 56.6% |
| 30 | 30/30 = 100% | 88.6% |
| 35 | 35/35 = 100% | 90.1% |
| 50 | 50/50 = 100% | 92.9% |
| 30 | 24/30 = 80% | 62.7% |
| 50 | 40/50 = 80% | 67.0% |
Lower limit of a two-sided 95% score confidence interval (see [1]). The lower confidence bound is influenced by the number of specimens with pathogen of interest and how good the device is.
When sensitivity, specificity, and prevalence are known, one can calculate positive and negative predictive values (PPV and NPV, respectively). PPV and NPV ask an alternative question: given a positive or negative test result, what is the probability that the patient has (or does not have) the condition being tested for? In a prospective study, this would be estimated as the percentage of patients with the condition of interest from all patients that test positive; similarly, the negative predictive value is estimated as the percentage of patients without the condition of interest among those that test negative.
The relationship between sensitivity, specificity, prevalence, PPV, and NPV is illustrated in Table 3. As can be seen, PPV and NPV are expressed as percentages; ideally, both will be close to 100%. The importance of test prevalence is illustrated in Table 4 where, despite high sensitivity and specificity, test utility is extremely limited in the context of ruling in a diagnosis as the PPV of the test result is still only 8%, underscoring the value of tests as an adjunct to diagnosis rather than the sole means for diagnosis.
Table 3.
Sensitivity, Specificity, and Predictive Values
| Clinical reference standard |
|||
| Condition present | Condition absent | Total | |
| Test positive | True positive (TP) | False positive (FP) | |
| Test negative | False negative (FN) | True negative (TN) | |
| Total | N+ | N− | N |
NOTE. For a prospective study: prevalence is estimated as 100% x (TP + FN)/(TP + FN + TN + FP) = 100% x (N+/N), positive predictive value (PPV) is 100% x TP/(TP + FP), negative predictive value (NPV) is 100% x TN/(TN + FN).
Table 4.
Hypothetical Example for Diagnostic: Low Prevalence of Condition
| Clinical reference standard |
|||
| Condition present | Condition absent | Total | |
| Test positive | 9 | 99 | 108 |
| Test negative | 1 | 891 | 892 |
| Total | 10 | 900 | 1000 |
NOTE. Sensitivity = 90%; specificity = 90%; positive predictive value (PPV) = 9/108 or 8.33%; negative predictive value (NPV) = 891/892 or 99.9%.
However, tests may still have substantial clinical value even when only the NPV is very high. A high NPV indicates that if the patient tests negative, it is very unlikely that the disease is present and the physician may want to pursue further work up to obtain a diagnosis.
However, the formulas for NPV and PPV in Table 3 are inappropriate when prevalence is unknown (e.g., when representative cases and controls are selected for study). Instead, a more complex set of formulas found in Pepe [5] provides a means of estimating NPV and PPV under various scenarios for prevalence.
Assuming one believes reasonable estimates of sensitivity and specificity are available, these equations can be used to illustrate what impact prevalence has on PPV and NPV:
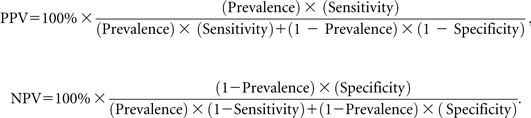 |
In these equations, prevalence is assumed known (ie, measured without error). So if we had a device with a sensitivity of 90% and a specificity of 90% as in Table 4, but the prevalence of the target condition of interest was now 10% rather than the 1% in Table 4, the PPV would be 0.50 or 50% rather than 8.33% as in Table 3, and NPV would be 0.99 or 99%. NPV is negligibly lower than in Table 3.
When a clinical study only assesses agreement to another previously cleared device rather than to a reference method, reporting NPV and PPV would be inappropriate.
Retrospective Versus Prospective Studies
Prospective studies in the IU population provide the best estimate of real-world performance for a diagnostic device. However, clinical performance of devices for detecting rare conditions, such as potential bioterrorism agents (eg, anthrax) may be impossible to ascertain.
Low prevalence often mandates the use of retrospective or banked specimens as clinical specimens for new devices. With banked specimens and/or enriched studies, prevalence usually cannot be estimated. (Retrospective samples that were prospectively archived [e.g., all patients meeting a prespecified inclusion/exclusion criteria are included] can still be representative provided storage conditions do not impact the assay results. However, banked [repository] specimens may not be representative and should be considered separately.). As an alternative, the PPV and NPV can be calculated for a set of plausible prevalence estimates so that the clinical impact of a different disease prevalence can be understood. It is also important to appreciate that including a nontrivial proportion of banked specimens or substantial enrichment of specimens can change the disease spectrum in the study, which in turn changes the estimates of sensitivity and specificity and not just prevalence; in this situation, sensitivity, specificity, PPV, and NPV could all be biased and misleading.
Given the critical role of study design in determining performance characteristics for a new test, companies are strongly encouraged to discuss their study proposals with FDA prior to initiation of studies essential for device clearance or approval.
Reproducibility/Precision and Other Analytical Studies
To evaluate the performance of the assay in multiple settings, precision studies must be conducted to confirm the reproducibility of assay results. These studies usually include multiple days or multiple runs across clinical sites. Studies should be designed to capture all major sources of variability, including site-to-site variation, the effect of different operators, and instrument-specific variability. Detailed guidance regarding precision studies for in vitro diagnostic devices is available in the Clinical and Laboratory Standards Institute (CLSI) guideline EP5-2A [6].
For qualitative assays, test panels used for precision/reproducibility studies should not only include both positive and negative samples, but particularly samples close to assay cutoffs; that is, thresholds for distinguishing positive from negative or equivocal. For quantitative assays, panels should include samples that are at the extremes of the analytical measurement range as well as values near key clinical decision points.
Analytical sensitivity (limit of detection) for each detected pathogen (analyte) and each specimen type (ie, matrix) need to be assessed [2–4, 7, 8]. Analytical specificity, including cross-reactivity with other possible respiratory pathogens, should be assessed, as well as interference by common substances such as blood, medications, and so forth.
For some quantitative assays, it may be necessary to rule out a possible hook effect with high positive samples. For studies utilizing banked specimens to establish clinical or analytical performance, it would be necessary to provide evidence that storage conditions do not impact assay results. Other analytical studies may be requested depending on the type of technology used.
Evaluating Multiplex Assays
A recent major advance for respiratory in vitro diagnostic testing has been FDA clearance of multiplex devices that can test for multiple pathogens simultaneously. The technical definition of a multiplex assay is “two or more targets simultaneously detected via a common process of sample preparation, target or signal amplification, allele discrimination, and collective interpretation” [7]. Essentially, a multiplex assay takes a single sample and then provides more than one output (eg, adenovirus present or absent and influenza A present or absent).
There are several known hurdles to developing a multiplex assay with many pathogens, as illustrated by the multiplex respiratory viral infection panel example [2]. Adequate performance must be demonstrated for each viral pathogen included in the multiplex panel; furthermore, individual viral analytes must still remain detectable in the setting of coinfections with bacterial pathogens or other viral pathogens. As the number of targets in the panel expand, concerns with interference from other analytes grow as does the probability of “overall false positive results.” An example would be the inclusion of Mycoplasma pneumoniae as an analyte in a general respiratory panel; the occurrence of a false positive result would likely override true positives. Physicians could be misled by device outputs in this setting because of having a test with a low PPV.
Adding a high-risk pathogen such as severe acute respiratory syndrome (SARS) coronavirus to a multiplex assay would mean submitting a PMA for approval. Thus, the regulatory path may be a consideration when designing a multiplex device.
An FDA special controls guidance for respiratory viral panel multiplex devices has been recently published [2] and addresses some of these issues. FDA also publishes special control guidance on devices detecting new pathogens, which can be either standalone tests or part of multiplex assays.
Clinical Laboratory Improvement Amendments Waivers
The Clinical Laboratory Improvement Amendments of 1988 (CLIA) defines categorization for a subset of devices sufficiently simple to use in a CLIA-waived settings, thus in essence permitting the use of these diagnostic tests outside of a clinical laboratory [9]. (A CLIA-waived test is different from “home use devices” [eg, pregnancy tests] which can be used with no medical oversight, or “point-of-care” tests [eg, tests performed in an emergency room, which remains under the auspices of the hospital laboratory]. “Point-of-care” tests may or may not be CLIA-waived depending on the specific device.) Upon application by a sponsor, FDA has the authority to designate a specific in vitro diagnostic test as a waived test. Waived tests such as urine dipsticks or group A streptococcal throat swab tests have widespread clinical use due to their availability at sites such as physician offices or commercial pharmacies.
Devices being considered for a CLIA waiver are first subject to the same regulatory process as other diagnostic devices; that is, the device must demonstrate it has adequate performance characteristics in the hands of a trained laboratorian. However, beyond the demonstration that the device has adequate performance characteristics, the device must perform well in a CLIA-waived setting. This can be done either after a device is cleared or approved, or the initial study of the device can be designed to support a CLIA waiver application, which can be submitted after clearance/approval is granted.
As part of an application for a CLIA waiver, sponsors are required to include studies to show the outcomes of not following the “instructions for use” provided with the device (e.g., prolonging the incubation of reagents, adding reagents in the wrong order, etc). These are studies designed to ensure that the device is robust and simple enough to operate without a trained laboratorian. By this definition, a sample that required centrifugation for processing, for example, would not be eligible for a waiver.
The study participants in a CLIA-waiver study represent end users typical of those who would perform the test in a waived setting (eg, nurses or aides in a doctor’s office or nursing home). Three or more sites should be included in the study, with 1–3 users per site for a minimum of 9 intended users. Testing must also include specimens with results close to the test cutoff values (ie, specimens that may be the most challenging to nonlaboratorians). Detailed information regarding the design of studies for a CLIA waiver application, including the minimal performance criteria acceptable for a CLIA-waived device, is described in “FDA guidance for industry and FDA staff: recommendations for Clinical Laboratory Improvement Amendments of 1988 (CLIA) waiver applications for manufacturers of in vitro diagnostic devices” [9].
It is highly recommended that sponsors seeking to market a device for use in a waived setting consult with FDA early in the regulatory process, especially for new products under development. It is important to note that CLIA-waiver requirements are very stringent; sponsors should consider the value of device use in CLIA-waived settings before pursuing the waiver process.
PRE–INVESTIGATIONAL DEVICE EXEMPTION CONSULTATION (PRESUBMISSION ADVICE)
Most pivotal studies for IVDs do not involve managing the patient based on the result of the investigational device; thus, IVD companies are rarely required to have an investigational device exemption (IDE) to start their pivotal study. However, FDA Center for Devices and Radiological Health, including the Division of Microbiology Devices, provides advice to sponsors on whether an IDE is required and other issues regarding the pre-IDE submission consultative process. Given the extensive number of diagnostic indications, the evolving technologies, and potential regulatory pathways, FDA strongly recommends that companies consider participating in the pre-IDE submission process early in the device development cycle and especially prior to conducting pivotal studies. The pre-IDE process allows FDA to evaluate if the proposed study designs are appropriate to support the IU of a device. For new instrumentation that have not been reviewed by the FDA, documentation of hardware, software, and manufacturing processes will need to be assessed, and could be topics for discussion in a pre-IDE submission [10, 11].
As an aid to sponsors developing new diagnostic devices, FDA posts summaries of the review decisions for cleared devices on the FDA Web site (http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm). Companies may want to examine these summaries for recently cleared devices that are similar to the ones they are proposing to submit. In addition, substantial advice concerning the device development process is available through published FDA guidance [12].
CONCLUSIONS
The previous discussion has only briefly reviewed some of the concerns manufacturers of new in vitro diagnostic tests should consider during their development. The extensive number of diagnostic indications, evolving technologies, increasing use of multiplex instruments in microbiology, and potential regulatory pathways mandate careful planning by sponsors when developing new diagnostic assays. It is essential that sponsors plan for clinical and analytical studies that consider the device performance needed to support the proposed intended use. The use of appropriate statistical methods in both planning and evaluating these studies is also highly recommended. Interactive dialogue with FDA during the device development process is strongly encouraged.
Acknowledgments
Supplement sponsorship. This article was published as part of a supplement entitled “Workshop on Molecular Diagnostics for Respiratory Tract Infections.” The Food and Drug Administration and the Infectious Diseases Society of America sponsored the workshop. AstraZeneca Pharmaceuticals, Bio Merieux, Inc., Cepheid, Gilead Sciences, Intelligent MDX, Inc., Inverness Medical Innovations, and Roche Molecular Systems provided financial support solely for the purpose of publishing the supplement.
Potential conflicts of interest: All authors: no conflicts.
References
- 1.FDA guidance for industry and FDA staff: statistical guidance on reporting results from studies evaluating diagnostic tests. Available at: http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm071287.pdf. Accessed March 2007. [Google Scholar]
- 2.FDA guidance for industry and FDA staff: class II special controls guidance documents: respiratory virus panel multiplex nucleic acid assay. Available at: http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm180324.pdf. Accessed October 2009. [Google Scholar]
- 3.FDA guidance for industry and FDA staff: class II special controls guidance document: testing for detection and differentiation of influenza A virus subtypes using multiplex nucleic acid tests. Available at: http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm180310.pdf. Accessed October 2009. [Google Scholar]
- 4.FDA draft guidance: establishing performance characteristics of in vitro diagnostic devices for the detection or detection and differentiation of influenza viruses. Available at: http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm071458.pdf. Accessed February 2008. [Google Scholar]
- 5.Pepe MS. The statistical evaluation of medical tests for classification and prediction. Oxford: Oxford University Press; 2003. p. 302. [Google Scholar]
- 6.CLSI EP5-2A: establishment of precision of quantitative measurement procedures. Wayne, PA: Clinical Laboratory and Standards Institute; 2004. [Google Scholar]
- 7.CLSI MM17-A: verification and validation of multiplex nucleic acid assays. Wayne, PA: Clinical Laboratory and Standards Institute; 2008. [Google Scholar]
- 8.CLSI EP17-A: protocols for determination of limits of detection and limits of quantitation. Wayne, PA: Clinical Laboratory and Standards Institute; 2004. [Google Scholar]
- 9.FDA guidance for industry and FDA staff: recommendations for Clinical Laboratory Improvement Amendments of 1988 (CLIA) waiver applications for manufacturers of in vitro diagnostic devices. Available at: http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm070890.pdf. Accessed January 2008. [Google Scholar]
- 10.FDA guidance for industry and FDA staff: guidance for the content of premarket submissions for software contained in medical devices. Available at: http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm089593.pdf. Accessed May 2005. [Google Scholar]
- 11.FDA guidance for industry, FDA reviewers and compliance on off-the-shelf software use in medical devices. Available at: http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm073779.pdf. Accessed September 1999. [Google Scholar]
- 12.FDA draft guidance for industry and FDA staff: in vitro diagnostic (IVD) device studies—frequently asked questions. Available at: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/IVDRegulatoryAssistance/ucm123682.htm#8. Accessed October 2007. [Google Scholar]