

Abstract
Background: A novel coronavirus (CoV) was recently identified as the agent for severe acute respiratory syndrome (SARS). We compared the abilities of conventional and real-time reverse transcription-PCR (RT-PCR) assays to detect SARS CoV in clinical specimens.
Methods: RNA samples isolated from nasopharyngeal aspirate (NPA; n = 170) and stool (n = 44) were reverse-transcribed and tested by our in-house conventional RT-PCR assay. We selected 98 NPA and 37 stool samples collected at different times after the onset of disease and tested them in a real-time quantitative RT-PCR specific for the open reading frame (ORF) 1b region of SARS CoV. Detection rates for the conventional and real-time quantitative RT-PCR assays were compared. To investigate the nature of viral RNA molecules in these clinical samples, we determined copy numbers of ORF 1b and nucleocapsid (N) gene sequences of SARS CoV.
Results: The quantitative real-time RT-PCR assay was more sensitive than the conventional RT-PCR assay for detecting SARS CoV in samples collected early in the course of the disease. Real-time assays targeted at the ORF 1b region and the N gene revealed that copy numbers of ORF 1b and N gene sequences in clinical samples were similar.
Conclusions: NPA and stool samples can be used for early diagnosis of SARS. The real-time quantitative RT-PCR assay for SARS CoV is potentially useful for early detection of SARS CoV. Our results suggest that genomic RNA is the predominant viral RNA species in clinical samples.
Severe acute respiratory syndrome (SARS) 1 is an emerging disease that has affected many countries (1). This disease was first recognized in Guangdong Province, China, in November 2002. Subsequent to its introduction to Hong Kong in mid-February 2003, the virus spread across five continents. By May 17, 2003, a cumulative total of 8464 cases and 799 deaths had been reported from 31 countries (2). A novel coronavirus (SARS CoV), which is genetically related distantly to other groups of coronaviruses, was isolated from SARS patients by us (3) and others(4)(5). Seropositivity to SARS CoV has been observed in the majority of SARS patients (3)(6). By contrast, none of the patients with other respiratory diseases and healthy blood donors have had detectable antibodies (3)(6). Furthermore, experimental infection of cynomolgus macaques (Macaca fascicularis) confirmed the hypothesis that this newly discovered SARS CoV is the etiologic agent of this disease (7). Thus, the development and validation of a rapid noninvasive laboratory diagnostic test for this virus is an urgent clinical need for better management and control of this disease.
Currently, there are two diagnostic options for the laboratory diagnosis of SARS. The first is based on detecting antibodies against SARS CoV after infection (3). More than 93% of SARS patients were reported to have seroconverted by day 28 after disease onset (6). However, this is a retrospective diagnosis and is less useful for clinical management. The second option for SARS diagnosis is the detection of SARS CoV RNA in clinical specimens (3)(4)(5)(6)(8). SARS CoV RNA was reported to be detected in SARS patients before seroconversion (8). Thus, reverse transcription-PCR (RT-PCR) assays may be a better tool for the early diagnosis of SARS. Rapid identification of SARS patients allows prompt and appropriate clinical management, isolation, and quarantine, thereby minimizing the risk of cross-infection in hospitals or in the local community (9).
Recently, we and others have reported the use of conventional and real-time RT-PCR assays for SARS CoV detection (3)(4)(5)(6)(8). These assays, however, were evaluated on only a limited number of clinical samples. Previously, we demonstrated that the viral load of SARS CoV in SARS patients peaks at the second week of the disease (6). Here we focus on samples collected within the first 10 days of disease. Using a conventional RT-PCR assay, we determined the detection rates of the SARS CoV within the first 10 days after disease onset. In addition, real-time quantitative RT-PCR assays were developed to increase the detection rate of SARS CoV in these clinical specimens. We also determined the nature of the SARS CoV RNA species in these clinical samples.
Materials and Methods
patients and sample collection
Specimens from clinically confirmed SARS patients fulfilling the WHO case definition (10) admitted to three regional hospitals in Hong Kong between February 26, 2003, and April 7, 2003, were studied. Nasopharyngeal aspirate (NPA) and stool specimens were collected as described previously (8). Using our previously published serologic test (6), we confirmed that all of these patients were seropositive for SARS CoV. NPA and stool samples from patients with unrelated diseases were recruited as controls.
rna extraction and reverse transcription
RNA from 140 μL of the clinical sample was extracted by use of the QIAamp virus RNA mini reagent set (Qiagen) as instructed by the manufacturer. Extracted RNA was eluted in 50 μL of RNase-free water and stored at −20 °C. Complementary DNA was generated as described previously (8). Briefly, 10 μL of eluted RNA samples was reverse-transcribed by 200 U of Superscript II reverse transcriptase (Invitrogen) in a 20-μL reaction containing 0.15 μg of random hexamers, 10 mM dithiothreitol, and 0.5 mM deoxynucleotide triphosphates. Reactions were incubated at 42 °C for 50 min, followed by a heat inactivation step (72 °C for 15 min). Reverse-transcribed products were stored at −20 °C.
conventional pcr for sars CoV
Conventional PCR was performed as described previously (3).
real-time quantitative pcr assays for sars CoV
We used published sequences (GenBank accession no. AY278491) to develop quantitative RT-PCR assays specific for the open reading frame (ORF) 1b region and for the N gene of SARS CoV to quantify the amount of these viral cDNA sequences in samples. The starting material was cDNA from the original samples, which was amplified by TaqMan PCR Core Reagent in a 7000 Sequence Detection System (Applied Biosystems). Briefly, 2 μL of cDNA was amplified in a 25-μL reaction containing 0.625 U of AmpliTaq Gold polymerase (Applied Biosystems), 2.5 μL of 10× TaqMan buffer A, 0.2 mM deoxynucleotide triphosphates, 5.5 mM MgCl2, 2.5 U of AmpErase UNG, and 1× primers–probe mixture (Assays by Design; Applied Biosystems). For ORF 1b gene detection, the primer sequences were 5′-CAGAACGCTGTAGCTTCAAAAATCT-3′ (corresponding to nucleotides 17718–17742 of the SARS CoV genome) and 5′-TCAGAACCCTGTGATGAATCAACAG-3′ (complementary to nucleotides 17761–17785), and the probe was 5′-(FAM)TCTGCGTAGGCAATCC(NFQ)-3′, where FAM is 6-carboxyfluorescein and NFQ is nonfluorescent quencher (complementary to nucleotides 17745–17760). For N gene detection, the primer sequences were 5′-ACCAGAATGGAGGACGCAATG-3′ (corresponding to nucleotides 28202–28222) and 5′-GCTGTGAACCAAGACGCAGTATTAT-3′ (complementary to nucleotides 28262–28286), and the probe was 5′-(FAM)ACCCCAAGGTTTACCC(NFQ)-3′ (corresponding to nucleotides 28245–28260). Reactions were first incubated at 50 °C for 2 min, followed by 95 °C for 10 min. Reaction were then thermal-cycled for 45 cycles (95 °C for 15 s, 60 °C for 1 min). Serially diluted plasmids containing the target sequences were used as calibrators in each run.
infection
Confluent Vero cells were infected with SARS CoV at 0.1 multiplicity of infection. Infected cells were incubated for 60 h, and total RNA from these cells was extracted by use of TRIzol reagent (Invitrogen) as instructed by the manufacturer. To harvest pure genomic RNA, viruses were purified through a sucrose cushion by standard laboratory procedures.
statistical analysis
The Student t-test was used for the comparison of the threshold cycle (ΔCt) values between different types of samples. McNemar’s test was used to compare the detection rates of SARS CoV from different RT-PCR assays.
Results
detection of sars CoV in clinical samples by a conventional rt-pcr assay
Clinical specimens collected within the first 10 days of disease onset were studied. RNA was extracted from NPA (n = 170) and stool (n = 44) samples from serologically confirmed SARS patients. A primer pair specific for the ORF 1b region of SARS CoV (3) was used for SARS CoV detection. Positive signals were observed in 44% of NPA and 57% of stool samples.
To determine the rate of detection of SARS CoV at different stages of the disease, data from NPA and stool samples were further analyzed by subdividing these samples into different days of disease onset. As indicated in Table 1A , 10 NPA samples (26%) from early onset (days 1–3) were positive. The detection rate increased as the disease progressed. At days 7–10, 32 of 53 NPA samples were positive in the assay (Table 1A ). Similarly, the detection rates for SARS CoV in stool samples increased as the disease progressed. At days 1–3, two of eight stool samples were positive in the assay (Table 1A ). By contrast, 13 of 19 stool samples collected at between days 7 and 10 after disease onset were positive (Table 1A ).
Table 1.
Detection of SARS CoV by conventional and real-time quantitative RT-PCR assays.
| A. Conventional RT-PCR assay. | ||||
|---|---|---|---|---|
| Days after onset | No. of samples | No. of positive samples in conventional RT-PCR | ||
| NPA | ||||
| 1–3 | 39 | 10 | ||
| 4–6 | 78 | 32 | ||
| 7–10 | 53 | 32 | ||
| Stool | ||||
| 1–3 | 8 | 2 | ||
| 4–6 | 17 | 10 | ||
| 7–10 | 19 | 13 | ||
| B. Conventional and real-time quantitative RT-PCR assays. | ||||||
|---|---|---|---|---|---|---|
| Days after onset | No. of samples | No. of positive samples | ||||
| Conventional RT-PCR | Quantitative RT-PCR | |||||
| NPA1 | ||||||
| 1–3 | 32 | 10 | 16 | |||
| 4–6 | 35 | 9 | 11 | |||
| 7–10 | 31 | 14 | 16 | |||
| Stool | ||||||
| 1–3 | 6 | 2 | 4 | |||
| 4–6 | 15 | 10 | 12 | |||
| 7–10 | 16 | 10 | 10 | |||
The overall detection rate of the quantitative RT-PCR assay is statistically different from that of the conventional RT-PCR assay (McNemar test, P <0.01; 95% confidence interval, 4.2–16.2%).
detection of orf 1b gene by real-time quantitative assay
The relatively low detection rate early in the disease prompted us to establish a more sensitive real-time quantitative RT-PCR for SARS CoV detection. We first targeted at the ORF 1b region of the SARS CoV genomic RNA (11)(12). As shown in Fig. 1A , the assay had a dynamic range from 106 to at least 10 copies per reaction, and the water control was negative in the assay. To evaluate the specificity of the assay, NPA samples from 19 healthy individuals and from patients with adenovirus (n = 10), respiratory syncytial virus (n = 9), human metapneumovirus (n = 9), and influenza A virus (n = 10) infection were tested in the assay; no positive signals were observed among these control samples.
Figure 1.
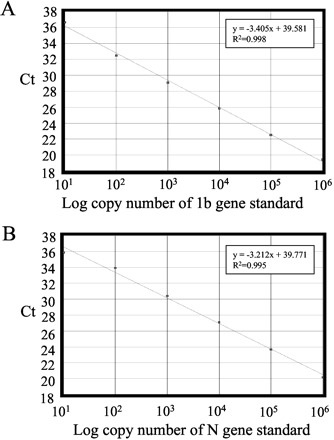
Real-time quantitative RT-PCR assays for SARS CoV.
(A), calibration curve for quantitative analysis of ORF 1b gene of SARS CoV. (B), calibration curve for quantitative analysis of N gene of SARS CoV. The Ct is the number of PCR cycles required for the fluorescence intensity of the reaction to reach a predefined threshold. The Ct is inversely proportional to the logarithm of the starting concentration of plasmid DNA. Equations for the regression lines and correlation coefficients are indicated.
Because of the limited availability of clinical samples, we could not afford to test all available NPA (n = 170) and stool samples (n = 44). In total, 98 NPA and 36 stool samples, which were previously tested by the above conventional RT-PCR assay, were randomly selected for the quantitative RT-PCR analysis. At days 1–3, the quantitative RT-PCR assay was able to detect SARS CoV in one-half of NPA samples and two-thirds of stool samples (Table 1B ). By contrast, only approximately one-third of these samples were positive in the conventional assay (Table 1B , days 1–3). At days 7–10, the detection rates of the quantitative assay became comparable to those for the conventional RT-PCR assay. These results indicated that the real-time quantitative assay is a better diagnostic method for early SARS diagnosis.
Using this quantitative assay, we also determined the amount of ORF 1b cDNA in these specimens. Serially diluted plasmid DNA containing the target regions was used to generate a calibration curve to estimate the copy number of target gene in cDNA samples. As shown in Fig. 2 , the progression of SARS within the first 10 days was reflected in the increased viral loads in the NPA and stool samples.
Figure 2.
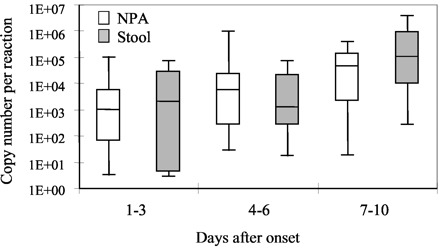
Change of SARS CoV viral load in samples collected at different days after onset.
The upper and lower limits of the boxes and the lines across the boxes indicate the 75th and 25th percentiles and the median, respectively. The upper and lower horizontal bars indicate the 90th and 10th percentiles, respectively. □, NPA samples; ▦, stool samples.
detection of n gene by real-time quantitative assay
In infected cells, coronaviruses are known to generate subgenomic mRNA for the synthesis of structural viral protein by discontinuous transcription (13). Because of the genomic organization of SARS CoV, all polycistronic viral mRNA molecules contain the SARS N gene (11)(12). Thus, by targeting the N gene sequence, we might generate a more sensitive assay for SARS diagnosis. To address this question, we developed a quantitative RT-PCR assay specific for the N gene of SARS CoV (Fig. 1B ). The assay had a dynamic range from 106 to at least 10 copies per reaction, and no signal was observed in water controls. No positive signal was observed in samples containing cDNA from a healthy individual (n = 19) and from patients with adenovirus (n = 10), respiratory syncytial virus (n = 9), human metapneumovirus (n = 9), or influenza A virus (n = 10) infections.
Sixty-eight NPA samples that were tested by the real-time assay for the ORF 1b region were randomly selected for testing by the N gene real-time PCR assay. Interestingly, the detection rates of these two assays were very similar. When the detection rates of these assays were compared (Table 2 ), we observed no statistically significant difference (McNemar’s test, P >0.05).
Table 2.
Detection rates of SARS-CoV in real-time quantitative RT-PCR assays.
| Assay for the ORF 1b sequence of SARS CoV | Total | |||
|---|---|---|---|---|
| Positive | Negative | |||
| Assay for the N gene of SARS CoV | 18 | 3 | 21 | |
| Positive | ||||
| Negative | 1 | 46 | 47 | |
| Total | 19 | 49 | 68 | |
We confirmed the above findings by comparing the Ct value of the ORF 1b assay (Ct1b) and the Ct value of the N gene assays (CtN). Samples that were positive in both assays were used for the analysis. We first determined the difference between the Ct1b and CtN from the same sample (i.e., ΔCt = Ct1b − CtN). If the N gene assay was more sensitive than the ORF 1b assay, one would expect that the ΔCt of a sample would be much greater than the ΔCtcalibrator. We first tested this hypothesis using RNA isolated from infected cells. Indeed, even when virus-infected cells were harvested at the late phase of viral replication (60 h postinfection), the ΔCt from infected cells was statistically greater than the ΔCtcalibrator (Table 3 ; P <0.02). By contrast, the ΔCt values for NPA samples were not statistically different from the ΔCtcalibrator (P = 0.89). In addition, we also determined the ΔCt value for stool samples. Again, the ΔCt of stool samples was not statistically different from the ΔCtcalibrator (Table 3 ; P = 0.64). Thus, the ORF 1b and the N gene quantitative assays are equally sensitive for SARS CoV detection in clinical specimens.
Table 3.
Mean (SD) differences in Ct values between quantitative assays for ORF1b and N gene.
| Calibrator (n = 14) | Infected cells (n = 9) | NPA (n = 18) | Stool (n = 11) | |
|---|---|---|---|---|
| ΔCt1 | 0.29 (1.03) | 1.32 (0.67)2 | 0.34 (0.79)c | 0.14 (0.79)d |
ΔCt, Ct value of a sample in the ORF 1b assay − Ct value of the same sample in the N gene assay.
ΔCt statistically different from that of the calibrator (Student t-test, P <0.02; 95% confidence interval, 0.39–1.67).
c,d ΔCt not statistically different from that of the calibrator (Student t-test): cP = 0.89; dP = 0.64.
nature of viral rna in clinical samples
With the data from the ΔCt analysis (Table 3 ), the overall results indicated that the amounts of 1b and N gene sequences in these samples were similar. These results highly suggested that genomic RNA is the predominant viral RNA species in these clinical samples. We tested this hypothesis with serially diluted cDNA samples derived from purified genomic RNA. Samples were subjected to both the ORF 1b and N gene quantitative assays, and Ct values obtained from these assays were used to deduce a linear regression line. As shown in Fig. 3 , Ct values from NPA and stool samples correlated with the regression line deduced from the genomic RNA. In addition, the ΔCt of genomic RNA (ΔCtgenomicRNA = 0.29 ± 0.40) was not statistically different from the ΔCt of NPA (P = 0.86) or the ΔCt of stool (P = 0.26). These results clearly suggest that genomic RNA is the predominant viral RNA species in these clinical samples.
Figure 3.
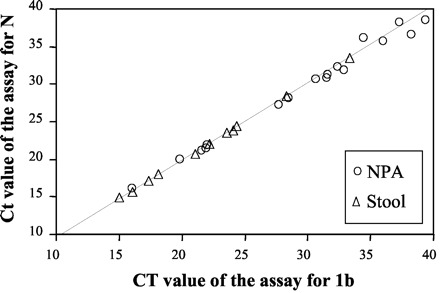
Correlation between Ct values of the RT-PCR assay for the ORF 1b region and Ct values of the RT-PCR assay for the N gene.
Serially diluted cDNA samples of purified viral genomic RNA were subjected to the ORF 1b and N gene assays, and the linear regression line was deduced from these Ct values is shown as a dotted line in the plot. ○, Ct values for a NPA sample in the assays; ▵, Ct values for a stool sample in the assays.
Discussion
Detection of SARS CoV early in the course of the disease is important for several reasons. The first is that the rapid identification of SARS CoV allows prompt patient management and quarantine, thereby minimizing the risk of large outbreaks in hospitals or in local communities (9). In addition, early detection of this virus might allow early interventions to control the replication of this pathogen. In view of the potential use of proteinase inhibitor to inhibit viral replication (14), applying quantitative RT-PCR technologies for SARS diagnosis might provide valuable information for prompt clinical treatment.
Our results demonstrated the potential use of NPA and stool samples for early SARS diagnosis. At days 1–3 after disease onset, approximately one-fourth of NPA and stool samples from SARS patients were positive in the conventional RT-PCR assay. The detection rates for these samples increased as the disease progressed. Because SARS is a respiratory disease, the low detection rate of SARS CoV in stool samples early after disease onset was not surprising. It is, however, striking that the detection rate of SARS CoV in NPA samples isolated during the first 3 days after disease onset was surprisingly low (26%). A possible explanation may be that the replicating site of SARS CoV in the early stage of the disease is restricted to the lower respiratory tract (15)(16). Currently, for those SARS patients who present early in the course of their disease, we routinely use multiple stool and NPA samples collected at different time points to increase the chance of detecting SARS CoV.
The low detection rate of the SARS CoV at the early stage of the disease prompted us to develop a more sensitive detection assay. In this study, we focused primarily on improving the detection of SARS CoV cDNA. This could be achieved by use of radioactively labeled probes to detect PCR amplicons. However, these approaches often require further downstream processing and are less practical in a routine clinical setting. Therefore, we established a real-time quantitative RT-PCR assay to target the ORF 1b region of SARS CoV. Our results demonstrated that early in the course of the disease, the real-time assay for the ORF 1b gene was more sensitive than the conventional RT-PCR assay (Table 1 ). However, the detection rates of the conventional and real-time assays became very similar as the disease progressed. This observation could be explained by the fact that there was more viral RNA in specimens collected at later time points (Fig. 2 ). Thus, for most of the samples collected at the second week after disease onset, the amount of viral RNA might well be above the detection limits of both assays.
Previously, we developed a SYBR Green detection assay for SARS diagnosis (8). Unlike the SYBR Green assay, the current assay could not reveal possible sequence variations within target sequences. However, the use of addition 5′-nuclease probes in the assay can minimize false-positive results. More importantly, the working platform used in this study could increase the throughput to 96 reactions per run.
Several conventional and real-time RT-PCR assays have been developed for SARS CoV (3)(4)(5)(6)(8). These assays, however, mainly targeted the ORF 1b region of SARS CoV. Several lines of evidence (11)(12) suggested that the detection rate of SARS CoV might be increased by targeting subgenomic mRNA of SARS CoV. However, our results indicated that, at least in NPA and stool samples, there is no particular advantage of targeting the N gene for clinical diagnosis. On the contrary, if one considers the fact that polymerase genes of RNA viruses are genetically more stable than the other viral genes (17)(18), the ORF 1b region of SARS CoV might be a better candidate for SARS diagnosis.
The isolation of viable SARS CoV from clinical samples demonstrated the presence of viral particles in clinical samples (19), indicating that genomic RNA exists in specimens. Our results further showed that genomic RNA might be the predominant viral RNA species in clinical samples. It is possible that most subgenomic mRNA that might present in clinical samples was unprotected and was rapidly degraded by endogenous RNases. By contrast, encapsulated genomic viral RNA was protected by viral particles from degradation.
In summary, we determined the detection rate of SARS CoV in NPA and stool samples from SARS patients. We also developed two real-time quantitative assays for the ORF 1b and N gene sequences of this pathogen. Our results showed that real-time assays for SARS CoV were particularly useful for early SARS diagnosis. Results from this study also indicated that SARS CoV RNA molecules in NPA and stool samples might be of genomic RNA origin. These results take us closer to the goal of prompt identification of SARS CoV in clinical specimens early in the disease, thereby leading to better control and clinical management of this disease.
Acknowledgments
We acknowledge research funding from Public Health Research Grant A195357 from the National Institute of Allergy and Infectious Diseases (US), The Wellcome Trust (Grant GR067072/D/02/Z), The Research Grant Council of Hong Kong (HKU 7543/03M), The University of Hong Kong, and the Hospital Authority of Hong Kong SAR.
Note Added in Proof:We recently demonstrated that with an optimized RNA extraction protocol, 80% of NPA samples collected at days 1–3 after disease onset were positive in the real-time quantitative RT-PCR assay (J Clin Virol2003;28:233–8).
Footnotes
Previously published online at DOI: 10.1373/clinchem.2003.023663
Nonstandard abbreviations: SARS, severe acute respiratory syndrome; CoV, coronavirus; RT-PCR, reverse transcription-PCR; NPA, nasopharyngeal aspirate; ORF, open reading frame; and Ct, threshold cycle.
References
- 1. World Health Organization. Acute respiratory syndrome. Wkly Epidemiol Rec 2003;78:73-74.12674023 [Google Scholar]
- 2.World Health Organization. Communicable disease surveillance & response (CSR). Cumulative number of reported probable cases. http://www.who.int/csr/sars/country/2003_06_17/en/ (Accessed June 18, 2003).. [Google Scholar]
- 3.Peiris JSM, Lai ST, Poon LLM, Guan Y, Yam LYC, Lim W, et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003;361:1319-1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 2003;348:1953-1966. [DOI] [PubMed] [Google Scholar]
- 5.Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 2003;348:1967-1976. [DOI] [PubMed] [Google Scholar]
- 6.Peiris JSM, Chu CM, Cheng VCC, Chan KS, Hung IF, Poon LLM, et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 2003;361:1767-1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fouchier RA, Kuiken T, Schutten M, van Amerongen G, van Doornum GJ, van den Hoogen BG, et al. Aetiology: Koch’s postulates fulfilled for SARS virus. Nature 2003;423:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poon LLM, Wong OK, Luk W, Yuen KY, Peiris JS, Guan Y. Rapid diagnosis of a coronavirus associated with severe acute respiratory syndrome (SARS). Clin Chem 2003;49:953-955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riley S, Fraser C, Donnelly CA, Ghani AC, Abu-Raddad LJ, Hedley AJ, et al. Transmission dynamics of the etiological agent of SARS in Hong Kong: impact of public health interventions. Science 2003;300:1961-1966. [DOI] [PubMed] [Google Scholar]
- 10.World Health Organization. Communicable disease surveillance & response (CSR). Case definitions for surveillance of severe acute respiratory syndrome (SARS).http://www.who.int/csr/sars/casedefinition/en/ (Accessed May 5, 2003).. [Google Scholar]
- 11.Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003;300:1394-1399. [DOI] [PubMed] [Google Scholar]
- 12.Marra MA, Jones SJ, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YS, et al. The genome sequence of the SARS-associated coronavirus. Science 2003;300:1399-1404. [DOI] [PubMed] [Google Scholar]
- 13.Enjuanes L, Sola I, Almazan F, Ortego J, Izeta A, Gonzalez JM, et al. Coronavirus derived expression systems. J Biotechnol 2001;88:183-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 2003;300:1763-1767. [DOI] [PubMed] [Google Scholar]
- 15.Tsang KW, Ho PL, Ooi GC, Yee WK, Wang T, Chan-Yeung M, et al. A cluster of cases of severe acute respiratory syndrome in Hong Kong. N Engl J Med 2003;348:1977-1985. [DOI] [PubMed] [Google Scholar]
- 16.Lee N, Hui D, Wu A, Chan P, Cameron P, Joynt GM, et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N Engl J Med 2003;348:1986-1994. [DOI] [PubMed] [Google Scholar]
- 17.Quinones-Mateu ME, Holguin A, Dopazo J, Najera I, Domingo E. Point mutant frequencies in the pol gene of human immunodeficiency virus type 1 are two- to threefold lower than those of env. AIDS Res Hum Retroviruses 1996;12:1117-1128. [DOI] [PubMed] [Google Scholar]
- 18.Toja M, Escarmis C, Domingo E. Genomic nucleotide sequence of a foot-and-mouth disease virus clone and its persistent derivatives. Implications for the evolution of viral quasispecies during a persistent infection. Virus Res 1999;64:161-171. [DOI] [PubMed] [Google Scholar]
- 19.Ruan YJ, Wei CL, Ee AL, Vega VB, Thoreau H, Su ST, et al. Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection. Lancet 2003;361:1779-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]