

Severe acute respiratory syndrome (SARS), caused by a novel coronavirus (SARS-CoV) (1)(2)(3)(4)(5), has affected 8096 people and produced 774 deaths in 29 countries/regions (6). The vital step in preventing and controlling future epidemics is to block transmission of infection through an effective quarantine policy, which in turn hinges on early diagnosis and confirmation of the disease, particularly by laboratory tests (7). The need for rapid, ultrasensitive assays that can detect infection very early in the course of the disease is obvious.
The antibody response to SARS-CoV infection is detectable only after ∼10 days of illness (8); hence early laboratory diagnosis rests on early detection of the virus itself. Detection relies on reverse transcription followed by PCR (RT-PCR) (7). We designed a 1-step real-time quantitative RT-PCR assay for SARS-CoV with the use of 2 TaqMan probes, instead of 1 probe, hybridizing to the same PCR product to further improve the sensitivity. This simple modification using dual TaqMan probes for quantification has wide applications in areas in which ultrasensitivity is critically required.
Our 1-step assay was designed to amplify the ORF1b regions of the SARS-CoV by TaqMan EZ RT-PCR Kit in a 7500 Real Time PCR System (Applied Biosystems). We compared assays using 1 and 2 TaqMan probes (Fig. 1, A and B ). The 25-μL reaction mixture contained 1× TaqMan EZ Buffer, 3 mM manganese acetate, 0.3 mM each deoxynucleotide triphosphate (except 1.2 mM for dUTP), 0.25 U of AmpErase UNG, 2.5 U of rTth DNA polymerase, 0.8 μM each primer, 0.4 μM each probe, and 10 μL of extracted RNA (4 μL for the 1-probe assay). The 1-probe assay was based on a previous report (9), but with slight modifications. Reactions were started by incubation at 50 °C for 2 min, followed by reverse transcription at 60 °C for 30 min, denaturation at 95 °C for 5 min, and amplification comprising 50 cycles of 95 °C for 15 s and 58 °C for 1 min. Calibrators were prepared from a concentrated RNA stock extracted from a SARS-CoV culture with the QIAamp Viral RNA Mini Kit (Qiagen) and quantified by RealArt HPA-Coronavirus LC RT-PCR Kit (Artus) in the Public Health Laboratory Centre. A no-template control was included in each run.
Figure 1.
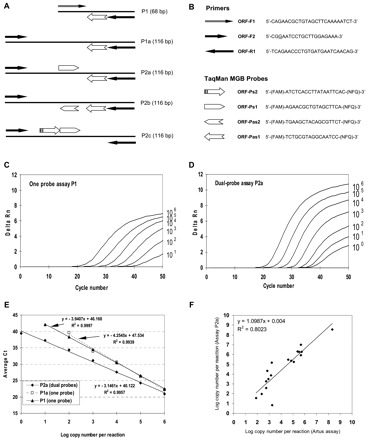
RT-PCR assay design and performance.
(A), 1-step RT-PCR assays with 1 (assay P1) or 2 TaqMan probes (assays P2a to P2c). The length of each amplicon is indicated in parentheses. The same forward primer was used for the 3 dual-probe assays and was upstream of that for the 1-probe assay. The arrowheads indicate the 3′ end of a primer or probe. (B), names and sequences of the primers and the TaqMan MGB probes shown together with the symbols used in A. Note that the TaqMan MGB probes (Applied Biosystems) are labeled with a 5′ reporter dye, 6-carboxyfluorescein (FAM), and a 3′ nonfluorescent quencher (NFQ) plus a minor grove binder (MGB) that stabilizes the probe–target duplex by binding the minor groove of double-stranded DNA (31). (C), amplification plot of FAM fluorescence intensity against the PCR cycle for the P1 one-probe assay. Delta Rn (y axis) indicates the magnitude of the signal intensity generated by a given set of PCR conditions and is obtained from the equation: delta Rn = (Rn+) − (Rn−). The Rn+ value is obtained as a ratio of FAM fluorescence intensity to the fluorescence intensity of the passive reference dye (ROX) included in the reaction mixture for a PCR with template. The Rn− value is similarly obtained as a ratio for a PCR without template (the no-template control). The RNA copy numbers per reaction are indicated on the right for each curve. (D), amplification plot of FAM fluorescence intensity against the PCR cycle for the P2a dual-probe assay. Assays P2b and P2c produced similar amplification plots (data not shown). (E), calibration curves for the P1 one-probe assay and the P2a dual-probe assay. Assays P2b and P2c produced calibration curves very similar to that for assay P2a (data not shown). (F), comparison of the RNA copy number per mL of input RNA sample determined by Artus assay (x axis) and the P2a dual-probe assay (y axis). The RNA samples were extracted from 18 confirmed SARS cases with 6 cases each providing stool, nasopharyngeal aspirate, and serum specimens.
As expected, the final fluorescence intensity was up to twice as high and the threshold cycle number (Ct) smaller in the dual-probe assay (P2a; Fig. 1A ) than in the 1-probe assay (P1; Fig. 1A ) for a given input RNA copy number per reaction (Fig. 1, C and D ). Comparison was made on the basis of copy number per reaction to account for the different input RNA volumes for the 2 assays. Moreover, the calibrator containing 1 RNA copy per reaction was detected 16 times out of 20 by the dual-probe assay but only 5 times out of 10 by the 1-probe assay (Fig. 1E ). The improved sensitivity was not attributable to the change in the forward primer. On the contrary, assay P1a (Fig. 1A ), which used the same forward primer as P2a but only the same 1 probe as in P1, hardly detected the calibrator containing 100 copies per reaction although it gave a calibration curve almost overlapping with that of P1 (Fig. 1E ). This simple modification of using dual probes, instead of 1 probe, increased the sensitivity of the assay.
We further investigated the effect on the assay when the 2 probes hybridized to complementary target strands or to the same strand. In the P2a dual-probe assay, the 2 probes hybridized to complementary strands. In assay P2b, the 2 antisense probes hybridized to the same sense target strand, whereas the 2 sense probes in assay P2c hybridized to the same antisense target strand (Fig. 1A ). Assays P2b and P2c gave the same results as assay P2a when the same series of calibrators was used. Thus, both cleavage efficiency and assay sensitivity were not affected whether the 2 probes were cleaved by the same polymerase molecule (as in assays P2b and P2c) or by 2 different polymerase molecules (as in assay P2a) for a given pair of complementary target strands in any single cycle. This in turn allows more flexible probe design even within a short stretch of sequences, provided that the 2 probes do not hybridize to each other.
We analyzed archived RNA samples from 18 SARS cases that were confirmed during the outbreak: 6 were extracted from stool (collected 3–21 days after onset of illness), 6 from nasopharyngeal aspirate (1–9 days after onset), and 6 from serum (1–10 days after onset). These RNA samples had previously been assayed by the Artus assay (concentrations, 7.7 × 101 to 2.1 × 108 per mL of RNA sample) in the Public Health Laboratory Centre, and all tested positive with our dual-probe assay P2a (concentrations, 6.9 × 101 to 3.7 × 108 per mL of RNA sample; Fig. 1F ). This suggests that our dual-probe assay is at least as sensitive as the Artus assay because both could detect the SARS-CoV in clinical specimens collected in the early days after disease onset. To further evaluate the specificity of the P2a dual-probe assay, we analyzed RNA samples extracted from confirmed cases of influenza A (n = 10 each for H3N2 and H1N1), norovirus (n = 5), and from 2 other human coronaviruses (OC43 and 229E). All of these samples tested negative.
With improved RNA extraction methods (10)(11), detection limits of 10 copies per RT-PCR reaction are common (3)(9)(11)(12)(13)(14), and a few studies have reported limits of detection of 5–8 copies per reaction (15)(16)(17). An additional nested PCR step after RT-PCR (18) allowed detection of 1 copy per reaction but with a much reduced dynamic detection range (19). Use of 2 TaqMan probes, instead of 1, increases the assay sensitivity without compromising the dynamic detection range. Even for an assay with a detection limit of 1 copy per reaction (20), we speculate that the dual-probe strategy will increase the signal strengths and hence the assay reproducibility at the low detection range.
The use of 2 TaqMan probes labeled with 2 different reporter dyes in a single PCR for qualitative purposes such as allelic discrimination is well established, in which each allele-specific probe hybridizes only to an allele-specific amplicon but not the nonallelic amplicon (21). As far as we are aware, this is the first report describing the use of 2 TaqMan probes labeled with the same reporter dye and hybridizing to the same amplicon for quantification purposes, which increases the sensitivity of the quantitative assay per se. Obviously, this strategy is applicable to both RNA and DNA as the input templates. This simple modification can also be applied to many areas in which ultrasensitivity or early detection of the target nucleic acids is of utmost importance, e.g., infections (SARS being just one example) and tumors. Increased assay sensitivity would counteract the dilution effect of pooling of donor plasmas for nucleic acid testing in the screening of transmissible infectious agents in blood donations (22)(23). Quantification of residual tumor cells, e.g., leukemic cells, at early remission requires a sensitive assay (24). Very limited amounts of target sequences are also encountered in paraffin-embedded tissue blocks (25), ancient specimens (26), forensic specimens taken from crime scenes (26)(27), circulating nucleic acids (28)(29), and engraftment of sex-mismatched organ transplantation (30).
Our preliminary data indicate that the use of 3 probes did not further enhance the assay sensitivity, but rather increased the variability between duplicate readings. On the other hand, it is worth investigating whether the dual-probe strategy for quantification purposes can be extended to molecular beacons and hybridization probes.
In conclusion, we report the use of dual TaqMan probes for quantification purposes and apply it to the detection of SARS-CoV with a detection limit of 1 copy RNA per reaction. This strategy is expected to be applicable to many areas requiring ultrasensitivity and/or early detection of target sequences.
Acknowledgments
This work was supported by a Hong Kong Research Grant Council Special Grant for SARS Research (PolyU 5520/03M). Purchase of the 7500 Real Time PCR System was supported by a Big Equipment Grant (G.53.27.D032) to S.P.Y., S.S.T.T., and P.H.M.L. by the Hong Kong Polytechnic University. We thank Lee Sau Yin for help in aligning the SARS-CoV genome sequences and Karen Anne Rocha for setting up the P1 assay in the initial stage.
References
- 1.Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W, et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003;361:1319-1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 2003;348:1953-1966. [DOI] [PubMed] [Google Scholar]
- 3.Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 2003;348:1967-1976. [DOI] [PubMed] [Google Scholar]
- 4.Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003;300:1394-1399. [DOI] [PubMed] [Google Scholar]
- 5.Marra MA, Jones SJ, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YS, et al. The genome sequence of the SARS-associated coronavirus. Science 2003;300:1399-1404. [DOI] [PubMed] [Google Scholar]
- 6.World Health Organization. Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003. http://www.who.int/csr/sars/country/table2004_04_21/en/index.html (accessed August 1, 2005)..
- 7.Poon LL, Chan KH, Peiris JS. Crouching tiger, hidden dragon: the laboratory diagnosis of severe acute respiratory syndrome. Clin Infect Dis 2003;38:297-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peiris JS, Chu CM, Cheng VC, Chan KS, Hung IF, Poon LL, et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 2003;361:1767-1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poon LL, Wong BW, Chan KH, Leung CS, Yuen KY, Guan Y, et al. A one-step quantitative RT-PCR for detection of SARS coronavirus with an internal control for PCR inhibitors. J Clin Virol 2004;30:214-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yam WC, Chan KH, Chow KH, Poon LL, Lam HY, Yuen KY, et al. Clinical evaluation of real-time PCR assays for rapid diagnosis of SARS coronavirus during outbreak and post-epidemic periods. J Clin Virol 2005;33:19-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poon LL, Chan KH, Wong OK, Yam WC, Yuen KY, Guan Y, et al. Early diagnosis of SARS coronavirus infection by real time RT-PCR. J Clin Virol 2003;28:233-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poon LLM, Wong OK, Luk W, Yuen KY, Peiris JSM, Guan Y. Rapid diagnosis of a coronavirus associated with severe acute respiratory syndrome. Clin Chem 2003;49:953-955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poon LL, Chan KH, Wong OK, Cheung TK, Ng I, Zheng B, et al. Detection of SARS coronavirus in patients with severe acute respiratory syndrome by conventional and real-time quantitative reverse transcription-PCR assays. Clin Chem 2004;50:67-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin HH, Wang SJ, Liu YC, Lee SS, Hwang CK, Chen YS, et al. Quantitation of severe acute respiratory syndrome coronavirus genome by real-time polymerase chain reaction assay using minor groove binder DNA probe technology. J Microbiol Immunol Infect 2004;37:258-265. [PubMed] [Google Scholar]
- 15.Houng HSH, Norwood D, Ludwig GV, Sun W, Lin M, Vaughn DW. Development and evaluation of an efficient 3′-noncoding region based SARS coronavirus (SARS-CoV) RT-PCR assay for detection of SARS-CoV infections. J Virol Methods 2004;120:33-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng EK, Hui DS, Chan KC, Hung EC, Chiu RW, Lee N, et al. Quantitative analysis and prognostic implication of SARS coronavirus RNA in the plasma and serum of patients with severe acute respiratory syndrome. Clin Chem 2003;49:1976-1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nitsche A, Schweiger B, Ellerbrok H, Niedrig M, Pauli G. SARS coronavirus detection. Emerg Infect Dis 2004;10:1300-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu Q, Xu Z, Wei T, Zeng H, Li J, Gang H, et al. Development of TaqMan RT-nested PCR system for clinical SARS-CoV detection. J Virol Methods 2004;119:17-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang SS, Chen TC, Yang JY, Hsiung CA, Su IJ, Liu YL, et al. Sensitive and quantitative detection of severe acute respiratory syndrome coronavirus infection by real-time nested-polymerase chain reaction. Clin Infect Dis 2004;38:293-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drosten C, Chiu LL, Panning M, Leong HN, Preiser W, Tam JS, et al. Evaluation of advanced reverse-transcription-PCR assays and an alternative PCR target region for detection of severe acute respiratory syndrome-associated coronavirus. J Clin Microbiol 2004;42:2043-2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Livak KJ. Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Genet Anal 1999;14:143-149. [DOI] [PubMed] [Google Scholar]
- 22.Tabor E, Epstein JS. NAT screening of blood and plasma donations: evolution of technology and regulatory policy. Transfusion 2002;42:1230-1237. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt M, Brixner V, Ruster B, Hourfar MK, Drosten C, Preiser W, et al. NAT screening of blood donors for severe acute respiratory syndrome coronavirus can potentially prevent transfusion associated transmissions. Transfusion 2004;44:470-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eckert C, Scrideli CA, Taube T, Songia S, Wellmann S, Manenti M, et al. Comparison between TaqMan and LightCycler technologies for quantification of minimal residual disease by using immunoglobulin and T-cell receptor genes consensus probes. Leukemia 2003;17:2517-2524. [DOI] [PubMed] [Google Scholar]
- 25.Specht K, Richter T, Muller U, Walch A, Werner M, Hofler H. Quantitative gene expression analysis in microdissected archival formalin-fixed and paraffin-embedded tumor tissue. Am J Pathol 2001;158:419-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alonso A, Martin P, Albarran C, Garcia P, Garcia O, de Simon LF, et al. Real-time PCR designs to estimate nuclear and mitochondrial DNA copy number in forensic and ancient DNA studies. Forensic Sci Int 2004;139:141-149. [DOI] [PubMed] [Google Scholar]
- 27.Alonso A, Martin P. A real-time PCR protocol to determine the number of amelogenin (X-Y) gene copies from forensic DNA samples. Methods Mol Biol 2005;297:31-44. [DOI] [PubMed] [Google Scholar]
- 28.Papadopoulou E, Davilas E, Sotiriou V, Koliopanos A, Aggelakis F, Dardoufas K, et al. Cell-free DNA and RNA in plasma as a new molecular marker for prostate cancer. Oncol Res 2004;14:439-445. [DOI] [PubMed] [Google Scholar]
- 29.Chan KC, Lo YM. Circulating DNA analysis: protocols and clinical applications using TaqMan assays. Methods Mol Med 2004;97:217-236. [DOI] [PubMed] [Google Scholar]
- 30.Wang LJ, Chen YM, George D, Smets F, Sokal EM, Bremer EG, et al. Engraftment assessment in human and mouse liver tissue after sex-mismatched liver cell transplantation by real-time quantitative PCR for Y chromosome sequences. Liver Transpl 2002;8:822-828. [DOI] [PubMed] [Google Scholar]
- 31.Kutyavin IV, Afonina IA, Mills A, Gorn VV, Lukhtanov EA, Belousov ES, et al. 3′-Minor groove binder-DNA probes increase sequence specificity at PCR extension temperatures. Nucleic Acids Res 2000;28:655-661. [DOI] [PMC free article] [PubMed] [Google Scholar]