

Graphical abstract
Abbreviations: AdV, adenovirus; ATP, adenosine triphosphate; APC, antigen presenting cell; CTLA-4, cytotoxic T lymphocyte antigen 4; CTL, cytotoxic T lymphocyte; CXCL/CCL, C-X-C motif ligands; DAMPs, damage-associated molecular pattern molecules; DC, dendritic cell; GITRL, glucocorticoid-induced TNFR-related protein; GM-CSF, granulocyte-macrophage colony-stimulating factor; HMGB1, high-mobility group protein B1; HPGD, hydroxyprostaglandin dehydrogenase; HSV, Herpes Simplex Virus; IAV, influenza A virus; IDO, indoleamine 2,3-dioxygenase; IFN, interferon; IL, interleukin; LAG-3, lymphocyte-activation gene 3 protein; MDSC, myeloid derived suppressor cell; MHC, major histocompatibility complex; MV, measles virus; MYXV, myxoma virus; NDV, Newcastle diseases virus; NK, natural killer; OV, oncolytic virus; PD-L1, programmed death ligand 1; PGE2, prostaglandin 2; TAA, tumor associated antigen; TGF, transforming growth factor; TIM-3, T cell immunoglobulin domain and mucin domain-3; TME, tumor microenvironment; TNF, tumor necrosis factor; VSV, vesicular stomatitis virus; VV, vaccinia virus
Keywords: Oncolytic viruses, Immune therapy, Transgenes
Highlights
-
•
Oncolytic viruses expressing immune modulators as transgene enhance the efficacy of anti-tumor therapies.
-
•
The enhanced efficacy is dependent on the combination of virus and immune modulator.
-
•
The localized expression of transgenic agonistic agents could reduce adverse effects in contrast to systemic administration.
-
•
Arming viruses with checkpoint inhibitors might not be the most effective combination strategy compared to agonistic agents.
Abstract
Oncolytic viruses (OVs), viruses that specifically result in killing tumor cells, represent a promising class of cancer therapy. Recently, the focus in the OV therapy field has shifted from their direct oncolytic effect to their immune stimulatory effect. OV therapy can function as a “kick start” for the antitumor immune response by releasing tumor associated antigens and release of inflammatory signals. Combining OVs with immune modulators could enhance the efficacy of both immune and OV therapies. Additionally, genetic engineering of OVs allows local expression of immune therapeutics, thereby reducing related toxicities. Different options to modify the tumor microenvironment in combination with OV therapy have been explored. The possibilities and obstacles of these combinations will be discussed in this review.
1. Introduction
1.1. The anti-tumor immune response and the immune profile of tumors
The innate and the adaptive immune system work together to detect transformed cells and remove them before they form a tumor [1]. The anti-tumor response starts with the release of tumor associated antigens (TAA) from dying cancer cells and accompanying signal molecules, which attract and activate cells of the innate immune system [[2], [3]]. Whereas NK and γd-T cells can recognize and kill tumor cells directly, antigen presenting cells (APCs), such as DCs and macrophages, take up TAAs to activate the adaptive immune system [[4], [5]]. The maturation of APCs by the accompanying danger signal molecules determines the skewing to a preferred T helper cell (Th) 1 response. These Th1 signals constitute of pro-inflammatory cytokines, such as interleukin (IL)-12, type I interferons (IFNs) and tumor necrosis factor (TNF), and damage-associated molecular pattern molecules (DAMPs), such as nuclear protein HMGB1, heat-shock proteins and ATP, [3]. The Th1 cytokines stimulate the generation of tumor specific cytotoxic CD8+ T cells (CTLs), which are crucial effector cells in the antitumor response [6]. Subsequently, effector T cells, including T helper cells and CTLs, are attracted to the tumor site via a gradient of T cell attracting chemokines, including chemokine (C-C motif) ligand 2 (CCL2), CCL5/RANTES, chemokine (C-X-C motif) ligand 9 (CXCL9), and CXCL10 [7]. At the site, CTLs recognize and kill tumor cells mediated by MHCI-T cell receptor interactions. If the immune system succeeds in destruction of the beginning tumor, the host remains free of cancer.
In some cases, tumor cells are reprogrammed to evade the immune system resulting in an equilibrium between dying tumor cells and tumor cells surviving the immune attack. As a consequence, a selection of immunosuppressive or less immunogenic tumor cell variants is introduced, which cannot be eliminated by the immune system [8]. These tumor cells establish a tumor microenvironment (TME), in which the function of anti-tumor immune cells is attenuated (Fig. 1 ) [9]. First of all, tumor cells and stromal cells (endothelial and epithelial cells and fibroblasts) produce factors such as transforming growth factor-β (TGF-β), prostaglandin E2 (PGE2) and IL-10, that disrupt APC maturation in the TME [[3], [7], [9]]. As a result, DCs isolated from the TME often display a partly matured, immune suppressive phenotype and secrete cytokines that induce non-favorable Th2 responses [7]. Secondly, tumors inhibit infiltration of effector T cells by repressing the production of T cell attracting chemokines CXCL9/10 and modification of CCL2 [[7], [10]]. Thirdly, effector T cells that can infiltrate the tumor are attenuated by expression of several immunosuppressive molecules and persistent exposure to tumor antigens. As a result, T helper cells and CTLs isolated from the TME often present an exhausted phenotype, characterized by high level expression of immune checkpoint receptors such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) [10]. Ligation of these receptors with their ligands expressed on tumor and stromal cells, but also immunosuppressed APCs, leads to inhibition of the tumor specific T cell response. Fourthly, regulatory immune cells such as CD4+ regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) are recruited to the tumor site. Similar to tumor cells, Tregs secrete IL-10, Indoleamine 2,3-dioxygenase (IDO) and TGF-β, leading to further attenuation of the T cell response [[7], [11]]. Furthermore, Tregs consume IL-2, which is indispensable for T cell activation [7]. MDSCs contribute to the suppression of effector T cells through production of arginase and nitric oxide, which deprives T cells from amino acids necessary for proliferation [12].
Fig. 1.

The immunosuppressive tumor micro environment. (A) Tumor cells (orange) and stromal cells (pink) secrete immune suppressive molecules, which inhibit the maturation of APCs. Maturated APCs migrate to the lymph node to activate the adaptive immune system. (B) As a result, activated T cells migrate to the tumor driven by a chemokine gradient. However, the secretion of chemokines is lowered in the tumor resulting in reduced T cell infiltration. (C) T cells that enter the TME to target the tumor cells are inhibited by immune suppressive receptors expressed by the tumor, stromal cell, but also immune suppressed APCs. (D) Tregs and MDSCs are recruited to the TME, which secrete more immune suppressive molecules and inhibit the T cell response even further.
Despite all these evasion mechanisms, CTLs and Th1 T helper cells are still considered to be the most crucial effector cells in anti-tumor immunity and their infiltration into the TME is associated with good prognosis in various types of cancer [13]. Proper activation of these cells is key for an effective antitumor response and the abundance of mechanisms used by tumors to suppress these cells offers many targets for cancer immunotherapy strategies.
At the moment, multiple strategies to target the TME are being explored. Recent successes have led to the FDA approval of checkpoint inhibitors anti-CTLA-4 (clinical responses in 10–15% of treated patients) and anti-PD1 (clinical responses in 30–40% of patients) for treatment of melanoma [[14], [15]]. Clinical trials have shown that dual, synergistic blockage improved antitumor responses against melanoma, indicating that it might take more than one approach to induce powerful and long lasting anti-tumor immunity [[16], [17]]. However, systemic administration of these checkpoint inhibitors, as well as other immunotherapies, often coincides with severe immune-related adverse effects similar to autoimmune diseases [[16], [17]]. A promising treatment option to potentially overcome this obstacle is oncolytic virotherapy.
1.2. Oncolytic viral therapy
Oncolytic virotherapy is an approach that uses oncolytic viruses (OVs), either with natural tropism for neoplastic cells or genetically modified to enhance selectivity for tumor cells [[18], [19]]. Tumor cells often lack an adequate antiviral response, making them more susceptible to OV infection than healthy cells. The viral infection leads to tumor regression through two distinct mechanisms: direct killing of tumor cells by replication dependent induced cell death and promotion of an antitumor response towards all tumor cells, including non-infected cells, by inducing immunogenic cell death. Types of immunogenic cell death, such as immunogenic apoptosis, necrosis and autophagic cell death, are characterized by the release of TAAs in combination with DAMPs and viral pathogen associated molecular patterns (PAMPs) [19]. Following the secretion of DAMPs and cytokines, more innate immune cells, such as macrophages, DCs, NK cells and neutrophils infiltrate the tumor environment. The immune stimulating cytokine secretion leads to maturation of APCs and hence presentation of TAAs and viral antigens to activate the adaptive immune system in the lymph nodes. Cytotoxic T cells will start infiltrating the tumor again and specifically eliminate cancer cells. Simultaneously, memory T cells are formed, which improves protection against new tumor challenges in mouse models [[20], [21]]. Therefore, it is evident that OV therapy can function as a ‘kick start’ for the antitumor immune response by providing TAAs in an immunogenic manner and inducing infiltration of immune cells.
Recently, the focus in the oncolytic virotherapy field has shifted from their oncolytic effect to their immune stimulatory effect. Recombinant OVs armed with immune modulators further enhance the activation of the immune system and overcome the immunosuppressive TME [18]. The first armed OV approved by the FDA is oncolytic Herpes-Simplex-Virus (HSV)-1 expressing GM-CSF showing improvement of melanoma treatment, but no cure yet (T-VEC) [22]. Therefore, many more immune modulator armed OV therapies followed and their obstacles and opportunities have come to light. This review will give an overview of the state-of-the-art therapies used in combination with immune modulators to treat cancer patients and give a hint on potential future directions.
2. Immune therapy
Immune therapies for treatment of cancer aim at overcoming the tumor immune suppressive environment and at increasing antitumor immunity. Most immune therapies target directly or indirectly the inhibitory or stimulatory receptors on immune cells and are often based on monoclonal antibodies. Other therapies intend to restore the intratumoral balance of cytokines and chemokines into a more favorable inflammatory TME to attract and activate immune cells. The most interesting noncellular therapies for combination with OV therapy for solid tumors are described below.
2.1. Cytokines
Cytokines are key players in stimulating and regulating antitumor immune responses. For this reason, one of the first immune therapeutic approaches in cancer treatment was the administration of recombinant cytokines. As described before, the most essential cytokines in the anti-tumor response are IL-12, GM-CSF, IL-2 and IL-2-related cytokines IL-15 and IL-21, and they all stimulate different parts of the immune system [23]: GM-CSF recruits APCs to the TME [17], IL-12 is normally expressed by APCs and stimulates polarization of T helper cells to a Th1 phenotype, IL-2 is a T cell growth factor and improves T cell expansion [9], and the IL-2-related cytokines promote survival of T cells, but also play an important role in NK cell activation [23]. Despite the their plethora of immune modulatory actions, cytokines have lost their popularity as a monotherapy, because of their low objective response rates and non-negligible side effects upon systemic administration [[24], [25]].
2.2. Inhibitory receptors
One of the most promising immune therapeutics are checkpoint inhibitors. Inhibitory receptors such as CTLA-4 and PD-1 act as checkpoints to avoid over activation and are expressed by T cells and inhibit the T cell activation in the lymph nodes and survival in the TME [26]. Several checkpoint inhibitors have been approved by the FDA of which the first were against CTLA-4 and PD-1. Treatment with these inhibitors results in reactivation of the suppressed immune cells [[14], [15]]. In addition, blocking of CTLA-4 can lead to depletion of Tregs [25]. Combinations of a-CTLA-4 antibody and a-PD-1 antibody have shown to increase responses to treatment of advanced melanoma. However, the frequency of severe immune-related adverse events was also enhanced in clinical studies (16.3 −27.3% in monotherapy group vs. 55% in combination therapy group) [16]. Another disadvantage of these therapies is the fact that tumors can become resistant towards checkpoint inhibitors [27]. As a result, a variety of other co-inhibitory receptors, such as lymphocyte activation gene 3 (LAG-3) and T cell immunoglobulin and mucin receptor protein 3 (TIM-3), have recently been identified. LAG-3 is normally activated by its ligand MHCII on APCs and indirectly reduces T cell proliferation [28]. Interaction of TIM-3 with its ligand galectin-9, expressed by Tregs, induces cell death in Th1 cells [28]. Blockage of these inhibitory receptors can unleash potent antitumor CTL responses and are now in clinical or preclinical development [[17], [28]].
2.3. Co-stimulatory receptors
Agonistic antibodies are also in development to activate co-stimulatory receptors expressed by T cells, such as 4-1BB and OX40 [17]. Stimulation of these two receptors by their ligands or monoclonal antibodies induces activation, proliferation and survival of T cells. Another interesting target is CD40, which is expressed on DCs. CD40 ligation with CD40L or therapeutic antibody stimulates DC maturation and presentation of antigens, which leads to efficient T cell priming [28]. More stimulatory co-receptors are explored today, such as B7.1 and GITR [[29], [30], [31]], which might benefit the effect of current therapies.
2.4. BiTEs
Another class of therapeutics constitute of dual specificity recombinant antibodies, also called bispecific T cell engagers (BiTEs), which have shown promise as anti-tumor therapeutics. These antibodies simultaneously bind to CTLs via the T cell receptor (TCR) and a tumor antigen expressed on the tumor cell resulting in bypassing MHC dependent antigen presentation [32]. An example of such therapy is Blinatumomab, which engages the CTL to CD19+ tumor cells. Currently, it is approved by the FDA for treating acute lymphoblastic leukemia, which is an hematologic malignancy [32].
3. Combination of immune therapy and oncolytic virotherapy
Therapeutic treatment of solid tumors could be enhanced by combination treatment of both immune and OV therapy. By introducing these discussed immune stimulators, checkpoint inhibitors and cytokines as immune modulators in viral vectors, adverse events can be reduced, resistance reverted and treatment responsiveness enhanced.
3.1. Combination of OVs with cytokines and chemokines
Several viruses have been engineered to express different cytokines or chemokines. Cytokines and chemokines are attractive transgenes, because they are encoded by small genes and are in general easy to build in a viral genome. Moreover, they often have pleiotropic effects, which means they can target different immune cells simultaneously [[23], [32]]. A complete overview of all cytokines and chemokines used in oncolytic virotherapy is given in Table 1 .
Table 1.
Combination therapy of armed oncolytic viruses and immune modulators.
| Transgene | Virus | Tumor | Additive immunologic effects | Toxicity |
|---|---|---|---|---|
|
Cytokines | ||||
| GM-CSF | HSV [[22], [48], [121]] | Adenocarcinoma [33], Metastised (phase I)a [[22], [34]] Breast cancer [121], Melanoma (phase I)a [[121], [122]] | Improved peripheral blood mononuclear cell response [124] | Grade 1 and 2 [[22], [34]], |
| AdV [[34], [110], [122]] | CD3+ T cell infiltration [33] | |||
| MV [33] | Long-term immunity against rechallenge with tumor cells [33] | |||
| VSV [123] | ||||
| NDV [124] | ||||
| IL-12 | AdV [108] | Neuroblastoma [45], Glioma [43], Prostate [48], Squamous Cell Carcinoma [[46], [47]], Melanoma [108] | Infiltration of macrophages, T helper, CTL and NK cells [[43], [45], [47], [48]] | No signs [[44], [46], [47]] |
| HSV [[30], [43], [44], [45], [47], [48]] | Improved survival [108] and protective against rechallenge [47] | |||
| VSV [46] | ||||
| IL-2 | NDV [[52], [53], [54], [55], [56]] | Melanoma [[54], [55]], Hepatoma [[52], [53]], Squamous Cell Carcinoma [51] | Infiltration of T helper and CTL [[51], [52], [53], [54], [55], [56]] | No signs [[53], [54]] |
| HSV [51] | Immunity against rechallenge with tumor cells [[51], [52], [53], [54]] | |||
| IL-15 | VSV [57] | Colon carcinoma [57], Melanoma [[58], [127]] | Increase in tumor specific CTLs in the blood [57] | IL-15 only detectable in tumor [58] |
| NDV [58] | Infiltration of T helper and CTL [58], [126] | |||
| IAV [125] | Immunity against rechallenge with tumor cells [58] | |||
| HSV [126] | Increases survival in mouse model [[58], [127]] | |||
| IAV [127] | ||||
| IFN-β | MV [61] | Non-small cell lung cancer [128], [129], | Improved survival mouse model [61] | Not reported |
| VV [128] | Mesothelioma [61], Pancreatic adenocarcinoma [59] | |||
| VSV [129] | ||||
| NDV [59] | ||||
| IFN-γ | NDV [55] | Melanoma [55], Mammary and colon carcinoma[62] | Increased cytokine expression and improved DC maturation [62] | Not Reported |
| VSV [62] | Increased T cell infiltration [55] | |||
| Others: IL-18[30], [108], IL-17[130], TNF[55], MIP1a[131], FLT3L[131] | NDV [55] | Improved T cell responses | Not reported | |
| VSV [130] | ||||
| AdV [108] | ||||
| HSV [30] | ||||
| Chemokines: CCL5[65], CCL2[67], CCL19[132], CXCL11[64], [133] | VV [[65], [66], [122], [123]] | Colon carcinoma | Improved DC maturation [65] | Not reported |
| HSV [67] | Improved infiltration T helper cells and CTLs [64], [65], [132], [133] | |||
| Induces a Th2 response, but reverts to a Th1 response in combination with DC vaccination [65] | ||||
|
Co-stimulatory ligands | ||||
| B7.1/CD80 | HSV [84] | Neuroblastoma[84], Melanoma (patients)a [86] | Immunity against rechallenge with tumor cells [84] | Low grade [86] |
| VV [86] | Response in 3/11 patients [86] | |||
| 4-1BBL/CD137L | VV [89] | Melanoma | No difference in DC maturation | Not reported |
| Infiltration of CTL | ||||
| CD40L | AdV [92] | Melanoma[93], | More Th1 cytokines [92] | No signs [92] |
| AdVdd [90] | Solid tumor (patients)a[92] | Infiltration of T helper, CTL, NK, DC, MDSC [93] | ||
| VV [93] | Improved survival, but not with armed VSV [90] | |||
| VSV [90] | ||||
| OX-40L | AdV [95] | Melanoma, lung carcinoma | Infiltration of T helper and CTL | Not reported |
| GITRL | AdV [31] | Melanoma | Infiltration of T helper and CTL | No signs |
| LIGHT | AdV [99] | Prostate | Recruitment of effector T cells Reduced Treg suppression |
Not reported |
| CD70 | VV [98] | Colon adenocarcinoma | Reduced tumor growth | Not reported |
|
Checkpoint inhibitors | ||||
| anti-CTLA4 | MV [75] | Melanoma | Infiltration of T helper and CTL [75], [108] | No signs [75], [108] |
| AdV [[74], [108]] | Decreased infiltration of Tregs [75] | |||
| anti-PD1 or PD-L1 | MV [75] | Melanoma | Infiltration of T helper and CTL [75], [83] | No signs [75] |
| VV [83] | Decreased infiltration of Tregs [75], [83] | |||
| MYXV [81] | Improved survival [81] | |||
|
Combinations | ||||
| GM-CSF+IL-12 | AdV [[104], [105]] | Melanoma | Secreted cytokine profile shifted from Th2 to Th2 response [105] | Not reported |
| Infiltration of T helper, CTL, NK and DC [104], [105] | ||||
| Immunity against rechallenge with tumor cells [105] | ||||
| IL-12 + IL-18 | AdV [106] | Melanoma | Infiltration of T helper, CTL, NK | Not reported |
| IL-12 + CCL2 | HSV [67] | Neuroblastoma | Reduced tumor growth | Not reported |
| B7.1 + IL-12 | AdV [41] | Melanoma | Infiltration of T helper, CTL and DC | Not reported |
| B7.1 + IL-18 | HSV [132] | Neuroblastoma, Prostate | Reduced tumor growth No significant difference in survival |
Not reported |
| B7.1 + GM-CSF | AdV [85] | Melanoma | Infiltration of T helper, CTL and DC Immunity against rechallenge with tumor cell | Not reported |
| 4-1BBL + IL-12 | AdV [42] | Melanoma | Infiltration DC, T helper and CTL | No signs |
|
Others | ||||
| HPGD | VV [100] | Several tumor models | Expression of Th1 cytokines, CXCL10/11, CCL5 | Not reported |
| Decreased infiltration MDSC | ||||
| More DCs secreting IL-12 in LN | ||||
| TRIF | VV [100] | Renal cell carcinoma | Increased immune stimulatory cytokine response | Not reported |
| Improved survival | ||||
| DAI | VV [103] | Melanoma | Improved CD8+ cell infiltration | Not reported |
| Reduced tumor growth | ||||
indicates patient studies, whereas other studies were performed in mouse models.
3.1.1. GM-CSF
The most extensively studied transgene is the cytokine GM-CSF. GM-CSF promotes DC recruitment and maturation. GM-CSF has been successfully used to arm HSV and this armed virus has been approved by the FDA under the name of T-VEC for treatment of metastatic melanoma patients [22]. Besides HSV, other viruses have also been armed with GM-CSF [[33], [34]]. Phase 1 clinical trials in patients with colorectal and hepatocellular carcinoma, neuroblastoma and Ewings sarcoma have proven the efficacy and safety of oncolytic vaccinia virus (VV) expressing GM-CSF (Pexa-Vec) [[35], [36], [37]]. In addition, two different adenovirus serotypes expressing GM-CSF induced long term survival of patients with, amongst others, ovarian, colon, pancreatic and breast cancer, with no severe side effects [[34], [38]]. Biopsies obtained from these patients with different metastatic tumors showed increased infiltration of T-cells and macrophages, which also correlated with patient survival and hence the additive effect of GM-CSF as vectorized immune modulator [38].
3.1.2. IL-12
IL-12 both activates and promotes survival of NK cells, but also Th1 effector cells [[39], [40]]. Several viruses have been armed with IL-12 and tested in different tumor models (Table 1) [[41], [42], [43], [44], [45], [46]]. HSV-IL-12 induces tumor infiltration of effector T cells, NK cells and APCs in neuroblastoma and glioma mouse models [[43], [45]]. When comparing HSV-IL-12 with HSV-GM-CSF, HSV-IL-12 was demonstrated most effective in tumor growth inhibition of injected tumors as well as metastases in a squamous cell carcinoma mouse model and a prostate cancer mouse model [[47], [48]]. In addition, mice were better protected against re-challenges with tumor cells in the HSV-IL-12 treated group, indicating the formation of a long term, anti-tumor response [47]. Similarly, adenovirus armed with IL-12 showed improved tumor reduction compared to adenovirus armed with GM-CSF in a thyroid cancer rat model [49]. In conclusion, multiple studies demonstrate that OVs armed with IL-12 yield better anti-tumor effects than vectorized GM-CSF and can even be further improved by combining different immune modulators.
3.1.3. IL-2 and IL-15
IL-2 and IL-15 both signal via cytokine receptors of the common γ chain family and are important for the stimulation, proliferation and survival of T cells and NK cells [[23], [50]]. Systemic treatment with IL-2 is associated with major adverse side effects in humans [24]. Therefore, local delivery of IL-2 by OVs has been tested by several research groups [[51], [52], [53], [54], [55], [56]]. In these murine studies, reduced tumor growth and increased T cell infiltration of the tumors was reported. No distress was observed in mice and IL-2 production was limited to the tumor site, which might indicate less side-effects when administrated to humans [[53], [54], [55]]. Also, mice were protected from re-challenge with tumor cells suggesting induction of long term tumor specific immunity and thus show promise as vectorized immune modulator [[52], [53], [54], [55]].
Despite the promising effects of IL-2, IL-15 showed to have several advantages over IL-2. In contrast to IL-2, Il-15 can stimulate only NK and effector T-cells, whereas IL-2 has also the undesirable effect of stimulating Tregs [50]. Even though systemic treatment with IL-15 induces less toxic effects local expression of IL-15 mediated by armed Vescilular Stomatitus virus (VSV) enhanced anti-tumor activity compared to VSV treatment combined with systemic IL-15 in murine models [57]. Vectorization by NDV demonstrated that IL-15 induced more CTL infiltration and increased activation of tumor-specific effector cells resulting in improved survival rates compared to IL-2 in a melanoma mouse model [58]. Altogether, these studies indicate that treatment with OVs expressing IL-15 is more efficient than systemic treatment with IL-15 or treatment with OVs expressing IL-2.
3.1.4. Type I and II interferons
IFN-α/β are important in antiviral responses, but also play a role in anti-cancer immunity by inducing DC maturation and CTL and NK cell activation [[23], [32]]. In addition, they result in upregulation of MHC I expression on tumor cells and can have direct effect on cell proliferation. Only a few studies have investigated OVs armed with type I interferons [[59], [60], [61]]. The direct anti-tumor effects of IFN-β expressed by the measles virus (MV) and NDV has been demonstrated in immune deficient mouse models leading to some improvement of the viral therapy. Buijs et al. showed that a virus expressing IFN-β interfered with oncolytic NDV replication, whereas Willmon et al. and Li et al. found that viral IFN-β expression did not interfere with VSV and MV replication [[59], [60], [61]]. However, the immune stimulating effect of virus mediated IFN-β expression still needs to be assessed in an immunocompetent model to determine the potency of IFN-β as transgene.
Type II interferon (IFN-γ) is an important Th1 effector cytokine, secreted by activated Th1 cells, CTLs and NK cells [23]. IFN-γ upregulates MHC I expression in tumor cells and promotes Th1 skewing via an autocrine loop [23]. A recent study describing VSV expressing IFN-γ suggested that this virus induces a stronger immune response by increasing MHC I antigen presentation on tumor cells, enhancing DC maturation and attracting T cells to the tumor site by inducing CCL2 expression in mice [62]. The authors did not observe any difference in viral replication, even though IFN-γ is also known for its antiviral activity. In another murine study, treatment with NDV- IFN-γ did not result in a significant beneficial effect compared to unarmed NDV [55]. In conclusion, arming OVs with IFN-γ has been shown to be effective, but efficacy may depend on the virus used.
3.1.5. Effector cell attracting chemokines
Insufficient infiltration of effector lymphocytes in the tumor often correlates with low efficacy of T cell stimulating immunotherapies [[7], [10]]. Therefore, OVs armed with chemokines, which attract effector cells, may improve antitumor efficacy in addition to the endogenous chemokine release upon viral infection. Important chemokines in the TME are CCL2, CCL5 (RANTES), CXCL9 and CXCL10, which attract Th1 cells and CTLs and CCL22, which attracts undesirable Tregs [63]. Combination of OV therapy with a chemokine modulating cocktail, which induces production of CCL5 and CXCL10 while reducing CCL22, was shown to promote trafficking of T helper cells and CTLs to the TME and resulted in improved survival in a murine colon cancer model [64]. Administration of viruses armed with CCL5 or CCL2 both resulted in increased numbers of infiltrating Th1 cells in colon cancer and neuroblastoma [[65], [66], [67]]. In conclusion, these studies have shown that the use of OVs armed with chemokines efficiently increased the infiltration of T cells into the tumor. However, none of the studies have assessed the infiltration of immune cells in distant tumors and tumor reduction has not been mentioned.
3.2. Combination of OVs with blocking of co-inhibitory receptors
The most popular immunotherapeutics are the checkpoint inhibitors, anti-CTLA-4 or anti-PD-1/PD-L1, yielding promising effects but coinciding with major adverse effects, as described above [68]. In addition, some tumors are resistant to these immunotherapies, dependent on the immunogenicity of the tumor and the suppression of the anti-tumor immune responses in the TME [69]. Combination of systemic immunotherapeutics with localized OV therapy may enhance their therapeutic efficacy and may overcome tumor resistance and reduce immune related adverse effects.
3.2.1. CTLA-4
CTLA-4 is a checkpoint inhibitor that inhibits early stages of T cell activation in the lymph nodes, but also stimulates undesirable Treg functions [[28], [68]]. Blockage of CTLA-4 signaling releases a brake on T cell activation and even depletes intratumoral Tregs [28]. Intratumoral administration of NDV combined with systemic treatment with anti-CTLA-4 (Ipilimumab) showed improved antitumor effect mediated by CTLs and NK cells in a murine melanoma model [70]. Studies using combinations of systemic CTLA-4 blockade with other OVs (VV [[71], [72]] and VSV [20]) showed prolonged survival in renal [71], lung [72] and mammary [20] tumor models, long term protection to re-challenge with tumor cells [[71], [72]], and even cured mice [20]. Moreover, T-VEC combined with systemic CTLA-4 blockade has been evaluated for therapy in melanoma patients, which yielded a tumor growth control that was significantly greater than observed after both monotherapies [73]. The incidence of severe adverse effects was similar to Ipilimumab monotherapy [68], [73]. Local expression of anti-CTLA-4 induced by administration of adenovirus armed with anti-CTLA-4 resulted in significantly higher concentrations of the antibody in the treated tumor, while plasma levels remained at concentrations indicated as safe in murine models [74]. However, Engeland et al. demonstrated that treatment with MV armed with anti-CTLA-4 was less efficient than intratumoral treatment with MV combined with systemic administration of anti-CTLA-4, presumably because the major site of action of CTLA-4 is in the lymph node and not in the periphery [75]. Though local expression may lead to transient expression of the transgene in tumor draining lymph nodes, this study indicated that systemic anti-CTLA-4 therapy may be required for optimal responses.
3.2.2. PD-1 and PD-L1
With a completely different mechanism of action, PD-1 is also a checkpoint inhibitor expressed by effector T cells [68]. While CTLA-4 inhibits T cells in early activation stages in the lymph nodes, PD-1 signaling limits the function of activated T cells at later stages of the immune response taking place in tumors and tissues [68]. Multiple OVs such as MV [76], reovirus [[77], [78]], and VSV [[79], [80]], have been combined with systemic PD-1 blockade in treatment of murine glioblastoma [76], melanoma [[77], [78]], and acute myeloid leukemia [79] tumor models, resulting in enhanced influx of CTLs in the tumors and prolonged survival in mice. Specifically CTLs and NK cells, but not T helper cells, were shown to mediate the beneficial effects [[77], [79], [81]]. Moreover, a phase 1b clinical trial with the FDA approved T-VEC combined with PD-1 resulted in good response rates in patients [82]. Arming Myxoma Virus (MYXV) with anti-PD-1 did result in improved survival in mice as well [81]. However, local expression induced by administration of MV or VV armed with anti-PD-1 or anti-PD-L1 demonstrated to be as efficient as therapy with unarmed VV combined with systemic anti-PD-1/anti-PD-L1 treatment in murine tumor models [[75], [83]]. In conclusion, these studies suggest that OVs combined with checkpoint inhibitors improve treatment, but the benefits of vectorization differ per oncolytic virus.
3.3. Combination of OVs with activation of co-stimulatory receptors
3.3.1. B7-1
B7-1 (CD80) is expressed by APCs and a potent co-stimulatory molecule for T cells. B7-1 provides co-stimulation via interaction with CD28 on T cells, but inhibits T cells through interaction with CTLA-4 [28]. Nevertheless, several OVs armed with B7-1 have been tested in (pre)- clinical trials for the treatment of melanoma [[41], [84], [85], [86]]. Replication defective HSV expressing soluble B7-1 induced a prolonged tumor specific immune response in mice bearing neuroblastoma tumors [84]. VV expressing B7-1 was eventually evaluated in melanoma patients, where the treatment was well tolerated, but only few patients responded [86]. Overall, OV expression of B7-1 seems to have additional benefits in combination with other transgenes, but not as a single transgene.
3.3.2. 4-1BB
4-1BB is a surface protein primarily present on activated T cells and NK cells. 4-1BB signaling promotes Th1 skewing over Th2, protects T cells from activation induced cell death and enhances cytotoxic activity of T cells and NK cells [28]. 4-1BB signaling has been shown to be more potent in T cell activation compared to the CD28 co-stimulation by B7-1 [87]. A combination of oncolytic VV with systemic anti-4-1BB administration in a breast cancer model resulted in increased survival and tumor infiltration by CTLs compared to both monotherapies in patients (40% survival vs. 0% survival) [88]. Local expression of 4-1BB ligand (4-1BBL) by oncolytic VV in mice has demonstrated to enhance tumor regression and this effect was enhanced even more in combination with lymph node depletion in order to slow down viral clearance [89]. Local injection of the armed virus resulted in an improved CTL/Treg ratio in the TME. In another study, vectorization of both IL-12 and 4-1BBL by adenovirus increased T helper, CTL and DC infiltration, which resulted in improved survival of the mice [42]. In other murine models, DC vaccination combined with the armed adenovirus combined treatment yielded even better results and DCs showed enhanced migration to the tumor draining lymph node, where they activated T cells [42]. These studies have shown that vectorization of this immune modulator in OVs enhances its therapeutic efficacy.
3.3.3. CD40
The maturation and activation status of DCs is often a limiting factor in the induction of antitumor immune responses. Ligation of the CD40 receptor provides a strong activating signal to DCs, resulting in upregulation of MHC II and co-stimulatory molecules and production of IL-12, which is important for skewing of T cells towards a Th1 phenotype [28]. As DC activation takes place in the TME, treatment with OVs armed with a stimulatory CD40 antibody or CD40 ligand (CD40L) may lead to enhanced therapeutic efficacy.
Adenoviruses armed with CD40L have been tested in melanoma mouse models and patients with different types of cancer [[90], [91], [92]]. Treatment with a CD40L expressing VV resulted in tumor growth inhibition and increased infiltration of effector T cells, NK cells, DCs in a melanoma mouse model. However, also numbers of Myeloid derived suppressor cells increased of which its effects are unknown [93]. When evaluated in a clinical trial, patients showed disease control for 3–6 months and a systemic tumor specific immune response was induced [92].
3.3.4. OX40
Similar to checkpoint inhibitors, more and more co-stimulatory targets, such as OX40 and GITR, are being discovered as anti-tumor targets. The OX40 receptor is expressed by activated T cells and induces the production of Th1 and Th2 cytokines upon interaction with OX40 ligand (OX40L) [[28], [94]]. OX40 ligation also directly blunts the suppressive effects of Tregs [[28], [94]]. Arming of OV with OX40L has been described once: adenovirus expressing OX40L led to suppression of melanoma, lung and colon tumor growth in mice. This effect was mediated by Th1 cells and CTLs and an increase in Th1 rather than Th2 cytokine expression [95]. A study combining systemic OX40L therapy with systemic 4-1BBL therapy and IL-12 expressing adenovirus, revealed increased expression of Th1 cytokines. thereby the antitumor CTL response were enhanced leading to tumor rejection in a colorectal cancer model involving liver metastases compared to OX40L monotherapy and combined 4-1BBL and IL-12 armed OV treatment [96]. This makes OX40 an interesting transgene, but more research needs to be performed to elucidate its beneficial effects in other tumor models and OVs.
3.3.5. GITR
GITR is a stimulatory receptor expressed on activated T cells, but it is also constitutively expressed on Tregs [[28], [94]]. Activation of the receptor promotes proliferation and cytokine production in T effector cells, whereas it inhibits Tregs [[28], [94]]. Expression of GITR ligand itself by an adenoviral vector resulted in increased infiltration of T helper cells and CTLs and suppression of tumor growth, leading to prolonged survival [31]. Systemic administration of a stimulating GITR antibody together with intratumoral injection of an adenoviral vector armed with IFN-α resulted in enhanced tumor growth inhibition in injected and distant tumors compared to single treatment in colon and pancreatic murine cancer models [97]. This makes GITR ligand attractive as a transgene, but similar to OX40L, more research needs to be performed. More research needs to be done towards these targets, just like for the newly discovered LIGHT and CD27 anti-tumor targets [[98], [99]], (Table 1).
3.4. Combination of OVs with other immune stimulatory approaches
3.4.1. Prostaglandin E2 blockade
Prostaglandin E2 (PGE2) is a principal mediator of inflammation expressed by tumor cells and immune cells and stimulates accumulation of MDSCs in the TME. Moreover, PGE2 induces IDO and IL-10 expression in DCs, whereas it reduces IL-12 expression, which is necessary to induce a Th1 response [[7], [10]]. A recent study, revealed PGE2 as an important mediator of resistance to OV therapy and other immunotherapies [100]. Treatment of mice with VV expressing a PGE2 inactivating enzyme named hydroxyprostaglandin dehydrogenase (HPGD), resulted in a decreased number of MDSCs in the tumor and yielded a better response than other PGE2 blocking agents [100]. The virus was also shown to sensitize an otherwise resistant renal tumor to anti-PD-1 treatment [100]. Thus, blockade of PGE2 showed promising results but this approach needs further validation in other tumor models and with other OVs.
3.4.2. Pathogen receptors
Toll like receptors (TLRs) are part of the innate immune system and recognize pathogenic molecules such as bacterial lipids and proteins or viral DNA and RNA [101]. The type of TLR that is activated upon infection is an important determinant for the skewing of the subsequent adaptive immune response [101]. Manipulation of TLR signaling can therefore switch the induced immune response from Th2 to Th1.
TRIF mediates TLR3 signaling, whereas other TLRs signal via MyD88 [101]. Moreover, this pathway induces the production of Th1 cytokines instead of Th2 cytokines. To manipulate TLR3 signaling, a VV was engineered to express TRIF to increase TLR3 signaling [102]. In addition, the viral particle was deglycosylated, resulting in reduced Th2 responses. Treatment of mice with deglycosylated VV-TRIF resulted in increased production of Th1 but not Th2 cytokines in the TME. Also, the virus induced infiltration of T helper cells and CTLs. These CTLs were both virus and tumor cell specific. VV-TRIF showed improved anti-tumor efficacy in colon and renal tumor models compared to VV-GM-CSF (Pexa-Vec). Similar results were obtained using intracellular pattern recognition receptor DNA-dependent activator of IFN-regulatory factors (DAI) as an vectorized immune modulator [103]. These studies show that the antitumor immune response can be switched from Th2 to Th1 by manipulating intracellular pathogen receptor signaling pathways in tumor cells.
3.4.3. BiTEs
BiTEs (bispecific T cell engagers) are a class of bispecific monoclonal antibodies that have shown promising anti-tumor effects [104]. However, they have a very short half-life and therefore require continuous infusion. Vaccinia virus (VV) armed with a BiTE with specificity for the TCR and a TAA was tested in a xenograft lung cancer mouse model. The study showed increased tumor cell killing through T cell activation and skewing towards Th1 responses [105]. A disadvantage of BiTEs is that they target only one TAA and therefore stimulate the immune response directed to only that TAA, while other immune-modulatory transgenes induce immunity to multiple TAAs that are released by dying tumor cells.
3.5. Combining different immune modulators
To further improve the efficacy of OV treatment, different combinations of therapies are being exploited. OVs armed with several cytokine combinations have been tested with promising results. Adenovirus expressing both GM-CSF and IL-12 induced infiltration of effector T cells, NK cells and activated APCs combined with DC vaccination in a melanoma mouse model, as well as long-term protection to re-challenge with tumor cells [106]. Moreover, viral expression of GM-CSF and IL-12 in the TME shifted the intratumoral cytokine profile from a Th2 to a preferred Th1 response and improved DC migration to the tumor site [[106], [107]]. Similarly, viral co-expression of IL-12 and IL-18 showed enhanced therapeutic efficacy and increased infiltration of DCs and effector T cells compared to mono-expression of IL-12 in murine models [[41], [42], [108]]. The combination of IL-12 and IL-18 showed to have synergistic effects on IFN-γ production, an important Th1 effector cytokine, by T cells and NK cells and polarization towards a Th1 response [[28], [109]]. Combining IL-12 expression with CCL2, an important T-cell chemokine, resulted in enhanced neuroblastoma growth inhibition compared to administration of armed HSV alone demonstrating the potential of combining different cytokines and chemokines in mice [67].
In addition, cytokines have also been combined with immune stimulatory receptor ligands. Treatment with adenovirus armed with GM-CSF and B7-1 in a melanoma mouse model resulted in prolonged survival and resistance to tumor re-challenge compared to treatment with unarmed adenovirus [85]. Similarly, treatment with oncolytic adenovirus armed with IL-12 as well as B7-1 resulted more often in complete regression in a melanoma mouse model compared to adenovirus armed with IL-12 alone [41]. Combination of three armed OVs with IL-12, IL-18, and B7-1 yielded significant better results in inhibition of local and distant tumor growth in a neuroblastoma mouse model, compared to single vector treatment demonstrating the possible synergistic effects between cytokines and co-stimulatory receptors [30].
The combination of checkpoint inhibitors and cytokines work equally well. Co-administration of oncolytic adenovirus expressing GM-CSF and oncolytic adenovirus expressing anti-CTLA-4 has been shown to yield additive antitumor activity in a lung cancer mouse model [110]. Furthermore, T-VEC in combination with systemic treatment with anti-CTLA-4 resulted in significantly improved tumor growth control rates in patients with advanced melanoma compared to both monotherapies alone [73]. Currently T-VEC in combination with anti-PD-1 is being evaluated in clinical trials [111], but the results are still unknown. In addition, Sorensen et al. engineered a replication deficient adenovirus expressing a TAA. By combining administration of this virus with systemic administration of CD40 stimulatory antibodies and CTLA-4 blockage, tumor growth of melanoma was reduced and long term survival was observed in 30–40% of mice in contrast to the monotherapies [112]. Thus, stimulation of both DCs and T cells is necessary for long term anti-tumor immune response. Therefore, these (pre-)clinical trials with combination-therapies demonstrate the potential to improve current therapies.
Taken together, these studies provide a strong rationale for further evaluation of OVs as a local delivery vector for immunotherapies targeting co-stimulatory and co-inhibitory receptors on anti-tumor effector cells or other parts of the TME.
4. Discussion and future directions
OVs represent a class of promising agents to treat cancer. Besides their direct oncolytic effects, they can function as a ‘kick-start’ for anti-tumor immunity. Combining OV therapy with existing immune therapies enhances the potential of both therapies by synergizing their effects. Many different combinations have been tested and are summarized in this review (Table 1). These studies have broadened our understanding of the strengths of OV immune therapies, but also of its limitations. As a result, these learning lessons enable us to discuss the potential future directions and further considerations in deciding on effective immune modulators and viral combinations and reducing risks.
4.1. Effective immune modulators
One of the greatest advantages of OVs armed with immune modulators is that this therapy often induces safer systemic and/or more effective localized concentrations of the modulator than systemic monotherapy [[58], [86], [92]]. Nevertheless, vectorization of checkpoint inhibitors can also limit the potency of the immune therapies by incorrect localization and timing and no resilience in case of tumor resistance. With regard to localization, a checkpoint receptor, such as CTLA-4, is mainly functional in the lymph node and requires systemic delivery of the blocking antibodies [72]. For instance, the anti-CTLA-4 armed MV proved to be less effective than monotherapy with CTLA-4 inhibitors [75]. Similar results were found for PD-L1 inhibitors delivered by MV and VV [[75], [83]], illustrating the second disadvantage of OVs armed with checkpoint inhibitors: the timing of administration of combined therapies. Rojas et al. and Gao et al. demonstrated that systemic administration of immune therapeutics given shortly (1–3 days) after OV therapy resulted in additive efficacy of the combination therapy in contrast to immune therapeutics given prior to OV administration [[71], [113]]. However, timing strategies may differ per virus as was seen with the armed MYXV where vectorization of anti-PD-1 did result in additive effects [81]. In addition to localization and timing, multiple studies have shown the development of resistance towards these checkpoint inhibitors over time in which new inhibitory molecules were upregulated on the cancer cells rendering the therapy ineffective [27]. Systemic administration of checkpoint inhibitors would allow for an easier transition between checkpoint therapies if necessary. Therefore, we think that arming viruses with checkpoint inhibitors might not be the most effective combination strategy.
An alternative approach to consider would be the targeting of APCs, NK cells and CTLs by arming viruses with immune stimulating agonists and cytokines. These immune cells express a known and stable subset of cytokine and co-stimulatory receptors and hence circumvent the treatment resistance as is seen with checkpoint inhibitors. Several studies have shown the additive effects of OVs armed with these agonistic agents compared to OV therapy alone (Table 1). In addition, the localized expression of agonistic agents reduces adverse effects in contrast to systemic administration and still allow combination therapy with checkpoint inhibitors [[58], [114], [115]]. Based on the reviewed studies, we reason that both cytokines and agonists targeting co-stimulatory receptors are very promising as immune modulators in the OV treatment against solid tumors.
4.2. Viral combinations
Another point of consideration is the combination of the virus and the immune modulator. The virus determines both the ‘kick-start’ effect, which initiates the inflammation, and the concentration and duration of expression of the vectorized immune modulators. However, viruses differ in the effectiveness in killing the target cell or in expressing the immune modulator. For example, in a study comparing VSV with a replication defective adeno virus, both expressing CD40L, it was shown that the replication defective virus was superior in increasing survival rates over the replicative virus [116]. This is probably because of the low immunogenicity of the replication defective virus resulting in reduced viral clearance by the immune system and hence more immune modulator secretion. However, whether the benefits of a weaker immune response hold true for all therapies remains unknown and probably depends on the virus.
In addition to oncolytic activity, the efficacy of the OV immune therapy is influenced by the efficiency of a virus to express the immune modulators. For instance, for NDV it has been shown that the insertion site in the virus determines the expression levels of the transgene [56]. The effect of the insertion site may also hold true for other OVs, but is often not discussed. In addition, the size of the immune modulators is also of importance. Studies using a complete monoclonal antibody, only the light chain or a single-chain variable fragment showed that smaller proteins were more effectively produced than the larger proteins by cells infected with VV. However, the single chain fragment without an IgG domain was degraded faster in the TME, resulting in similar antitumor activity as the monoclonal antibody [83]. As not all viruses tolerate large insertions, this information should be considered when deciding which modulator to incorporate. However, predicting the efficiency of the modulator expression remains difficult, because differences in expression levels can occur between tumors independent of the virus [110].
4.3. Reducing risks
While moving forward with OV immune therapies to the clinic, safety ought to be considered once more. So far, no adverse effects have been reported on the therapies combining OV with immune therapies in murine models or clinical trials. Nevertheless, adverse effects of the viral infection, such as excessive viral replication or expression of the immune modulators leading to an overreaction of the immune system cannot be excluded yet. A possibility to reduce the risks on adverse effects would be the incorporation of a fail-safe mechanism into the OV therapy to abort viral replication if necessary. Only for HSV, drugs are available to inhibit viral replication [117]. Alternatively, virus with attenuated virulence or viruses with a different host range may be used. In addition, incorporation of suicide genes, such as thymidine kinase [117], rat cytochrome P450 [118] or cytosine deaminase [119], would allow inhibition of viral replication by using drugs as well. Moreover, it could add to the tumor lytic efficacy of the therapy [120]. During OV therapy, the immune modulator is not incorporated into the host genome and therefore expression levels are dependent on viral replication which is usually transient. Thus, controlling the viral replication would reduce the risks on severe adverse effects of the immune modulators.
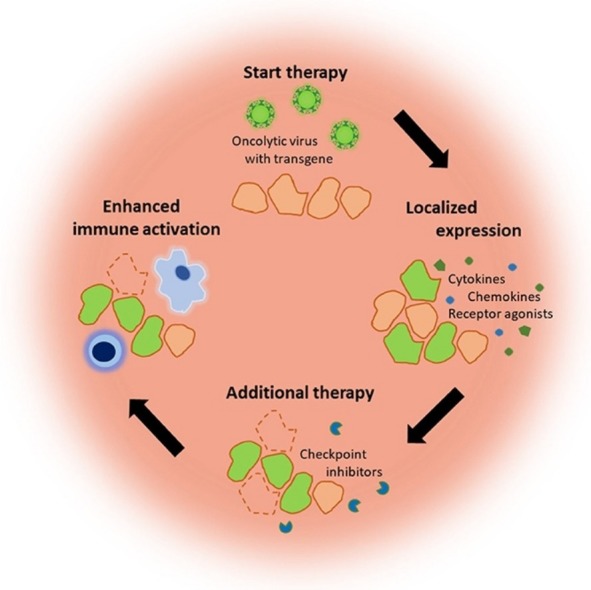
In conclusion, the overall effect of the oncolytic immune therapy depends on the interplay between virus, immune modulator and tumor. Not only tumors develop over time, but the immune system as well. This means that every stage of immune activation should be considered while deciding on the incorporation of an immune modulator (Fig. 2 ). Weak immunogenic tumors in an immunosuppressive TME will likely benefit from potent oncolytic viruses, cytokines and innate stimulating agonists to activate the initial innate immune response. If eventually the tumor is inflamed, effector T cells and NK cells responses can be improved by immune activating agonists, such as 4-1BBL, and checkpoint inhibitors. The vectorization of both cytokines and immune activating agonists could reduce possible adverse effects compared to systemic administration, whereas systemic delivery of checkpoint inhibitors improves the timing and localization of the treatment. Future directions will have to explore multiple combinations of dually armed OVs allowing to overcome tumor heterogeneity or at least to use both OVs and immune modulators to their full potential.
Fig. 2.

Combining oncolytic viral therapy with immune modulators. (A) Weak immunogenic tumor cells in an immunosuppressive TME are infected by oncolytic viruses (green) armed with immune modulators. (B) Tumor cells start secreting viral induced cytokines and chemokines, but also the immune modulators, which improves the immune activation. As a result, immune cells start to infiltrate the TME. (C) Occasionally, the virus will induce an immunogenic cell death. The TAAs and immune stimulating environment will result in the maturation of APCs. (D) The APCs will activate the adaptive immune system upon which T cells will start infiltrating the tumor attracted by the secreted chemokines. Possible activity of immune suppressive ligands, such as PD-L1, will be prevented by the circulating immune checkpoint inhibitors resulting in clearance of the tumor.
Conflicts of interest
The authors declare no conflict of interest.
The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Funding
This work was supported by Stichting Technische Wetenschappen, [#15414] and the foundation “overleven met alvleesklierkanker” (www.supportcasper.nl).
Biographies

Ms. J. Fréderique de Graaf obtained her Master’s degree in Infection and Immunity at the university of Utrecht, The Netherlands, during which she obtained research training at the Biochemistry Department of the University of Ottawa, Canada and the Laboratory of Translational Immunology of Utrecht Medical Institute, The Netherlands. Her PhD studies at the department of Viroscience of Erasmus MC, Rotterdam The Netherlands involves avian paramyxoviruses and their application in oncolytic viro-immunotherapy for treatment of pancreatic cancer patients.

Ms. Lisanne de Vor received her Master’s degree in Infection and Immunity in Utrecht, The Netherlands, and followed research training at the Viroscience Department of Erasmus MC Rotterdam, The Netherlands and the Karolinska Institute, Sweden.She currently works at the department of Microbiology at Utrecht Medical Institute, The Netherlands.

Prof. Ron A.M. Fouchier received a PhD in Medicine from the University of Amsterdam in 1995 for studies on HIV and continued to study HIV as a post-doctoral fellow at the Howard Hughes Medical Institute, University of Pennsylvania School of Medicine in Philadelphia, from 1995 to 1998. He is currently Professor at the Viroscience Department of Erasmus MC Rotterdam, The Netherlands, where his work focuses on influenza virus zoonoses and pathogenicity and oncolytic viruses. Achievements of his team include the identification and characterization of several “new” viruses; the human metapneumovirus (hMPV), human coronavirus NL63, the SARS coronavirus (SARS-CoV), the MERS coronavirus (MERS-CoV), and a new influenza A virus subtype (H16).

Dr. Bernadette G. van den Hoogen started research training in the group of Prof. S. Ross, at the Pennsylvania School of Medicine in Philadelphia from 1995 to 1998. Subsequently, Bernadette obtained her PhD at the department of Viroscience, Erasmus MC, Rotterdam, The Netherlands on “the Discovery and Characterization of the Human Metapneumovirus (HMPV)". Since then, Bernadette has been investigating the interaction between HMPV and the human immune system, but the use of paramyxoviruses as oncolytic viruses. Her oncolytic research focuses on avian paramyxoviruses and their application in oncolytic viro-immunotherapy for treatment of pancreatic cancer patients.
Contributor Information
J.F. de Graaf, Email: J.F.deGraaf@erasmusmc.nl.
L. de Vor, Email: L.deVor-2@umcutrecht.nl.
R.A.M. Fouchier, Email: R.A.M.Fouchier@erasmusmc.nl.
B.G. van den Hoogen, Email: b.vandenhoogen@erasmusmc.nl.
References
- 1.Dunn G.P., Bruce A.T., Ikeda H., Old L.J., Schreiber R.D. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 2002;3(no. 11):991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 2.Chen D.S., Mellman I. Oncology meets immunology: the cancer-Immunity cycle. Immunity. 2013;39(1):1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 3.Kapsenberg M.L. Dendritic-cell control of pathogen-driven T-cell polarization. Nat. Rev. Immunol. 2003;3(no. 12):984–993. doi: 10.1038/nri1246. [DOI] [PubMed] [Google Scholar]
- 4.Waldhauer I., Steinle A. NK cells and cancer immunosurveillance. Oncogene. 2008;27(no. 45):5932–5943. doi: 10.1038/onc.2008.267. [DOI] [PubMed] [Google Scholar]
- 5.Zou C., Zhao P., Xiao Z., Han X., Fu F., Fu L. T cells in cancer immunotherapy. Oncotarget. 2017;8(no. 5):8900–8909. doi: 10.18632/oncotarget.13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raphael I., Nalawade S., Eagar T.N., Forsthuber T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. 2015;74(1):5–17. doi: 10.1016/j.cyto.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motz G., Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013;39(no. 1):61–73. doi: 10.1016/j.immuni.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schreiber R.D., Old L.J., Smyth M.J. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science (80) 2011;331(no. 6024):1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 9.van der Burg S.H., Arens R., Ossendorp F., van Hall T., Melief C.J.M. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat. Rev. Cancer. 2016;16(4):219–233. doi: 10.1038/nrc.2016.16. [DOI] [PubMed] [Google Scholar]
- 10.Pitt J.M., Marabelle A., Eggermont A., Soria J. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. Adv. 2016;(April):1–43. doi: 10.1093/annonc/mdw168. (Access, no) [DOI] [PubMed] [Google Scholar]
- 11.Yu J. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with Breast cancer. J. Immunol. 2013;190(March no. (7)):3783. doi: 10.4049/jimmunol.1201449. (LP-3797, 2013) [DOI] [PubMed] [Google Scholar]
- 12.Bronte V., Serafini P., Mazzoni A., Segal D.M., Zanovello P. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol. 2003;24(no. 6):301–305. doi: 10.1016/s1471-4906(03)00132-7. [DOI] [PubMed] [Google Scholar]
- 13.Fridman W.H., Pagès F., Sautès-Fridman C., Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer. 2012;12(4):298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 14.Robert C. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015;372(no. 4):320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 15.Robert C. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011;364(no. 26):2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 16.Larkin et al J. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015;373(no. 1):23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farkona S., Diamandis E.P., Blasutig I.M. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14(no. 1) doi: 10.1186/s12916-016-0623-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaufman H.L., Kohlhapp F.J., Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015;14(no. 9):642–662. doi: 10.1038/nrd4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartlett D.L. Oncolytic viruses as therapeutic cancer vaccines. Mol. Cancer. 2013;12(September no. (1)):103. doi: 10.1186/1476-4598-12-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao Y., Whitaker-Dowling P., a Griffin J., a Barmada M., Bergman I. Recombinant vesicular stomatitis virus targeted to Her2/neu combined with anti-CTLA4 antibody eliminates implanted mammary tumors. Cancer Gene Ther. 2009;16(no. 1):44–52. doi: 10.1038/cgt.2008.55. [DOI] [PubMed] [Google Scholar]
- 21.Cuadrado-Castano S., Sanchez-Aparicio M.T., García-Sastre A., Villar E. The therapeutic effect of death: newcastle disease virus and its antitumor potential. Virus Res. 2015;209:56–66. doi: 10.1016/j.virusres.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andtbacka R.H.I. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015;33(no. 25):2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 23.Smyth M.J., Cretney E., Kershaw M.H., Hayakawa Y. Cytokines in cancer immunity and immunotherapy. Immunol. Rev. 2004;202(December):275–293. doi: 10.1111/j.0105-2896.2004.00199.x. [DOI] [PubMed] [Google Scholar]
- 24.Vacchelli E. Trial Watch—Immunostimulation with cytokines in cancer therapy. Oncoimmunology. 2016;5(no. 2):e1115942. doi: 10.1080/2162402X.2015.1115942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papaioannou N.E., Beniata O.V., Vitsos P., Tsitsilonis O., Samara P. Harnessing the immune system to improve cancer therapy. Ann. Transl. Med. 2016;4(no. 14):261. doi: 10.21037/atm.2016.04.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pitt J.M., Marabelle A., Eggermont A., Soria J.C., Kroemer G., Zitvogel L. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 2016;27(no. 8):1482–1492. doi: 10.1093/annonc/mdw168. [DOI] [PubMed] [Google Scholar]
- 27.Koyama S. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016;7:1–9. doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khalil D.N., Smith E.L., Brentjens R.J., Wolchok J.D. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016;13(no. 5):273–290. doi: 10.1038/nrclinonc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaufman H.L. Local delivery of vaccinia virus expressing multiple costimulatory molecules for the treatment of established tumors. Hum. Gene Ther. 2006;244(February):239–244. doi: 10.1089/hum.2006.17.239. [DOI] [PubMed] [Google Scholar]
- 30.Ino Y., Saeki Y., Fukuhara H., Todo T. Triple combination of oncolytic herpes simplex virus-1 vectors armed with interleukin-12, interleukin-18, or soluble B7-1 results in enhanced antitumor efficacy. Clin. Cancer Res. 2006;12(no. 2):643–652. doi: 10.1158/1078-0432.CCR-05-1494. [DOI] [PubMed] [Google Scholar]
- 31.Calmels B., Paul S., Futin N., Ledoux C., Stoeckel F., Acres B. Bypassing tumor-associated immune suppression with recombinant adenovirus constructs expressing membrane bound or secreted GITR-L. Cancer Gene Ther. 2005;12(no. 2):198–205. doi: 10.1038/sj.cgt.7700781. [DOI] [PubMed] [Google Scholar]
- 32.Lee S., Margolin K. Cytokines in cancer immunotherapy. Cancers (Basel). 2011;3(no. 4):3856–3893. doi: 10.3390/cancers3043856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grossardt C. Granulocyte-macrophage colony-stimulating factor-armed oncolytic measles virus is an effective therapeutic cancer vaccine. Hum. Gene Ther. 2013;24(no. 7):644–654. doi: 10.1089/hum.2012.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cerullo V. Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoral immunity in cancer patients. Cancer Res. 2010;70(no. 11):4297–4309. doi: 10.1158/0008-5472.CAN-09-3567. [DOI] [PubMed] [Google Scholar]
- 35.Park S.H. Phase 1b trial of biweekly intravenous pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus in colorectal cancer. Mol. Ther. 2015;23(no. 9):1532–1540. doi: 10.1038/mt.2015.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cripe T.P. Phase 1 study of intratumoral pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol. Ther. 2014;23(no. 3):602–608. doi: 10.1038/mt.2014.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heo J. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013;19(no. 3):329–336. doi: 10.1038/nm.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hemminki O. Immunological data from cancer patients treated with Ad5/3-E2F-Δ24-GMCSF suggests utility for tumor immunotherapy. Oncotarget. 2015;6(no. 6):4467. doi: 10.18632/oncotarget.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Colombo M.P., Trinchieri G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002;13(no. 2):155–168. doi: 10.1016/s1359-6101(01)00032-6. [DOI] [PubMed] [Google Scholar]
- 40.Del Vecchio M. Interleukin-12: Biological properties and clinical application. Clin. Cancer Res. 2007;13(no. 16):4677–4685. doi: 10.1158/1078-0432.CCR-07-0776. [DOI] [PubMed] [Google Scholar]
- 41.Lee Y.S. Enhanced antitumor effect of oncolytic adenovirus expressing interleukin-12 and B7-1 in an immunocompetent murine model. Clin. Cancer Res. 2006;12(no. 19):5859–5868. doi: 10.1158/1078-0432.CCR-06-0935. [DOI] [PubMed] [Google Scholar]
- 42.Huang J.-H. Therapeutic and tumor-specific immunity induced by combination of dendritic cells and oncolytic adenovirus expressing IL-12 and 4-1BBL. Mol. Ther. 2010;18(no. 2):264–274. doi: 10.1038/mt.2009.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hellums E.K. Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro. Oncol. 2005;7:213–224. doi: 10.1215/S1152851705000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Markert J.M. Preclinical evaluation of a genetically engineered herpes simplex virus expressing interleukin-12. J. Virol. 2012;86(no. 9):5304–5313. doi: 10.1128/JVI.06998-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parker J.N., Gillespie G.Y., Love C.E., Randall S., Whitley R.J., Markert J.M. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. U. S. A. 2000;97(no. 5):2208–2213. doi: 10.1073/pnas.040557897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shin E.J. Interleukin-12 expression enhances vesicular stomatitis virus oncolytic therapy in murine squamous cell carcinoma. Laryngoscope. 2007;117(no. 2):210–214. doi: 10.1097/01.mlg.0000246194.66295.d8. [DOI] [PubMed] [Google Scholar]
- 47.Wong R.J. Cytokine gene transfer enhances herpes oncolytic therapy in murine squamous cell carcinoma. Hum. Gene Ther. 2001;12(no. 3):253–265. doi: 10.1089/10430340150218396. [DOI] [PubMed] [Google Scholar]
- 48.Varghese S., Rabkin S.D., Liu R., Nielsen P.G., Ipe T., Martuza R.L. Enhanced therapeutic efficacy of IL-12, but not GM-CSF, expressing oncolytic herpes simplex virus for transgenic mouse derived prostate cancers. Cancer Gene Ther. 2006;13(no. 3):253–265. doi: 10.1038/sj.cgt.7700900. [DOI] [PubMed] [Google Scholar]
- 49.Tanaka K. Thyroid cancer immuno-therapy with retroviral and adenoviral vectors expressing granulocyte macrophage colony stimulating factor and interleukin-12 in a rat model. Clin. Endocrinol. 2003;59(no. 6):734–742. doi: 10.1046/j.1365-2265.2003.01915.x. [DOI] [PubMed] [Google Scholar]
- 50.Jakobisiak M., Golab J., Lasek W. Interleukin 15 as a promising candidate for tumor immunotherapy. Cytokine Growth Factor Rev. 2011;22(no. 2):99–108. doi: 10.1016/j.cytogfr.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 51.Carew J. A novel approach to cancer therapy using an oncolytic herpes virus to package amplicons containing cytokine genes. Mol. Ther. 2001;4(no. 3):250–256. doi: 10.1006/mthe.2001.0448. [DOI] [PubMed] [Google Scholar]
- 52.Bai F. Genetically engineered Newcastle disease virus expressing interleukin 2 is a potential drug candidate for cancer immunotherapy. Immunol. Lett. 2014;159(no. 1–2):36–46. doi: 10.1016/j.imlet.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 53.Wu Y. Recombinant Newcastle disease virus (NDV/Anh-IL-2) expressing human IL-2 as a potential candidate for suppresses growth of hepatoma therapy. J. Pharmacol. Sci. 2016;132(no. 1):24–30. doi: 10.1016/j.jphs.2016.03.012. [DOI] [PubMed] [Google Scholar]
- 54.Zamarin D., Vigil a, Kelly K., García-Sastre a, Fong Y. Genetically engineered Newcastle disease virus for malignant melanoma therapy. Gene Ther. 2009;16(no. 6):796–804. doi: 10.1038/gt.2009.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vigil A. Use of reverse genetics to enhance the oncolytic properties of newcastle disease virus. Cancer Res. 2007;67(no. 17):8285–8292. doi: 10.1158/0008-5472.CAN-07-1025. [DOI] [PubMed] [Google Scholar]
- 56.Pan Z. Identification of optimal insertion site in Recombinant Newcastle Disease Virus (rNDV) vector expressing foreign gene to enhance its anti-tumor effect. PLoS One. 2016;11(10):1–13. doi: 10.1371/journal.pone.0164723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stephenson K.B., Barra N.G., Davies E., Ashkar A.A., Lichty B.D. Expressing human interleukin-15 from oncolytic vesicular stomatitis virus improves survival in a murine metastatic colon adenocarcinoma model through the enhancement of anti-tumor immunity. Cancer Gene Ther. 2012;19(no. 4):238–246. doi: 10.1038/cgt.2011.81. [DOI] [PubMed] [Google Scholar]
- 58.Niu Z. Recombinant Newcastle disease virus expressing IL15 demonstrates promising antitumor efficiency in melanoma model. Technol. Cancer Res. Treat. 2015;14(no. 5):607–615. doi: 10.7785/tcrt.2012.500414. [DOI] [PubMed] [Google Scholar]
- 59.Buijs P., van Nieuwkoop S., Vaes V., Fouchier R., van Eijck C., Van Den Hoogen B. Recombinant immunomodulating lentogenic or mesogenic oncolytic newcastle disease virus for treatment of pancreatic adenocarcinoma. Viruses. 2015;7(6):2980–2998. doi: 10.3390/v7062756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Willmon C.L. Expression of IFN-?? enhances both efficacy and safety of oncolytic vesicular stomatitis virus for therapy of mesothelioma. Cancer Res. 2009;69(no. 19):7713–7720. doi: 10.1158/0008-5472.CAN-09-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H., Peng K.-W., Dingli D., Kratzke R.A., Russell S.J. Oncolytic measles viruses encoding interferon beta and the thyroidal sodium iodide symporter gene for mesothelioma virotherapy. Cancer Gene Ther. 2010;17(no. 8):550–558. doi: 10.1038/cgt.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bourgeois-Daigneault M.-C. Oncolytic vesicular stomatitis virus expressing interferon-γ has enhanced therapeutic activity. Mol. Ther. 2015;3(December):16001. doi: 10.1038/mto.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Franciszkiewicz K., Boissonnas A., Boutet M., Combadière C., Mami-Chouaib F. Role of chemokines and chemokine receptors in shaping the effector phase of the antitumor immune response. Cancer Res. 2012;72(no. 24):6325–6332. doi: 10.1158/0008-5472.CAN-12-2027. [DOI] [PubMed] [Google Scholar]
- 64.Francis L. Modulation of chemokines in the tumor microenvironment enhances oncolytic virotherapy for colorectal cancer. Oncotarget. 2016;7(16):22174–22185. doi: 10.18632/oncotarget.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li J. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol. Ther. 2011;19(no. 4):650–657. doi: 10.1038/mt.2010.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nishio N. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014;74(no. 18):5195–5205. doi: 10.1158/0008-5472.CAN-14-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parker J.N., Meleth S., Hughes K.B., Gillespie G.Y., Whitley R.J., Markert J.M. Enhanced inhibition of syngeneic murine tumors by combinatorial therapy with genetically engineered HSV-1 expressing CCL2 and IL-12. Cancer Gene Ther. 2005;12(no. 4):359–368. doi: 10.1038/sj.cgt.7700784. [DOI] [PubMed] [Google Scholar]
- 68.Postow M.A., Callahan M.K., Wolchok J.D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015;33(no. 17) doi: 10.1200/JCO.2014.59.4358. (p. JCO.2014.59.4358-) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kelderman S., Schumacher T.N.M., Haanen J.B.A.G. Acquired and intrinsic resistance in cancer immunotherapy. Mol. Oncol. 2014;8(no. 6):1132–1139. doi: 10.1016/j.molonc.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zamarin D. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014;6(no. 226) doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rojas J.J., Sampath P., Hou W., Thorne S.H. Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin. Cancer Res. 2015;21(no. 24):5543–5551. doi: 10.1158/1078-0432.CCR-14-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Foy S.P. Poxvirus-based active immunotherapy synergizes with CTLA-4 blockade to increase survival in a murine tumor model by improving the magnitude and quality of cytotoxic T cells. Cancer Immunol. Immunother. 2016;65(no. 5):537–549. doi: 10.1007/s00262-016-1816-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Puzanov I. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J. Clin. Oncol. 2016;34(no. 22):2619–2626. doi: 10.1200/JCO.2016.67.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dias J.D. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012;19(10):988–998. doi: 10.1038/gt.2011.176. [DOI] [PubMed] [Google Scholar]
- 75.Engeland C.E. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. 2014;22(11):1949–1959. doi: 10.1038/mt.2014.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hardcastle J. Immunovirotherapy with measles virus strains in combination with anti–PD-1 antibody blockade enhances antitumor activity in glioblastoma treatment. Neuro. Oncol. 2016;(July) doi: 10.1093/neuonc/now179. (p. now 79) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rajani K. Combination therapy with reovirus and anti-PD-1 blockade controls tumor growth through innate and adaptive immune responses. Mol. Ther. 2015;24(no. 1):1–9. doi: 10.1038/mt.2015.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ilett E. Prime-boost using separate oncolytic viruses in combination with checkpoint blockade improves anti-tumor therapy. Gene Ther. 2017;24(January no. (1)):21–30. doi: 10.1038/gt.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shen W., Patnaik M.M., Ruiz A., Russell S.J., Peng K.W. Immunovirotherapy with vesicular stomatitis virus and PD-L1 blockade enhances therapeutic outcome in murine acute myeloid leukemia. Blood. 2016;127(11):1449–1458. doi: 10.1182/blood-2015-06-652503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cockle J.V. Combination viroimmunotherapy with checkpoint inhibition to treat glioma, based on location-specific tumor profiling. Neuro. Oncol. 2016;18(no. 4):518–527. doi: 10.1093/neuonc/nov173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bartee M.Y., Dunlap K.M., Bartee E. Tumor-Localized secretion of soluble PD1 enhances oncolytic virotherapy. Cancer Res. 2017;77(11):2952–2964. doi: 10.1158/0008-5472.CAN-16-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ribas A. Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2017;170(no. 6):1109–1119. doi: 10.1016/j.cell.2017.08.027. (e10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kleinpeter P. Vectorization in an oncolytic vaccinia virus of an antibody, a Fab and a scFv against programmed cell death −1 (PD-1) allows their intratumoral delivery and an improved tumor-growth inhibition. Oncoimmunology. 2016;1(no. 10):e1220467. doi: 10.1080/2162402X.2016.1220467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Todo T., Martuza R.L., Dallman M.J. In situ expression of soluble B7-1 in the context of oncolytic herpes simplex virus induces potent antitumor immunity In situ expression of soluble B7-1 in the context of oncolytic herpes simplex virus induces potent antitumor immunity 1. Cancer Res. 2001;61:153–161. [PubMed] [Google Scholar]
- 85.Choi K.-J. Concurrent delivery of GM-CSF and B7-1 using an oncolytic adenovirus elicits potent antitumor effect. Gene Ther. 2006;13(no. 13):1010–1020. doi: 10.1038/sj.gt.3302759. [DOI] [PubMed] [Google Scholar]
- 86.Kaufman H.L. Targeting the local tumor microenvironment with vaccinia virus expressing B7. 1 for the treatment of melanoma. J. Clin. Invest. 2005;115(no. 7) doi: 10.1172/JCI24624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yan X., Johnson B.D., Orentas R.J. Murine CD8 lymphocyte expansion in vitro by artificial antigen-presenting cells expressing CD137L (4-1BBL) is superior to CD28, and CD137L expressed on neuroblastoma expands CD8 tumour-reactive effector cells in vivo. Immunology. 2004;112(1):105–116. doi: 10.1111/j.1365-2567.2004.01853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.John L.B. Oncolytic virus and anti-4-1BB combination therapy elicits strong antitumor immunity against established cancer. Cancer Res. 2012;72(no. 7):1651–1660. doi: 10.1158/0008-5472.CAN-11-2788. [DOI] [PubMed] [Google Scholar]
- 89.Hong S.K., Seunghee K.S., Dae W.K., Kaufman H.L. Host lymphodepletion enhances the therapeutic activity of an oncolytic vaccinia virus expressing 4-1BB ligand. Cancer Res. 2009;69(no. 21):8516–8525. doi: 10.1158/0008-5472.CAN-09-2522. [DOI] [PubMed] [Google Scholar]
- 90.Galivo F. Interference of CD40L-mediated tumor immunotherapy by oncolytic vesicular stomatitis virus. Hum. Gene Ther. 2010;21(no. 4):439–450. doi: 10.1089/hum.2009.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang Y.F. Antitumor effects of oncolytic adenovirus armed with PSA-IZ-CD40L fusion gene against prostate cancer. Gene Ther. 2014;21(no. 8):723–731. doi: 10.1038/gt.2014.46. [DOI] [PubMed] [Google Scholar]
- 92.Pesonen S. Oncolytic immunotherapy of advanced solid tumors with a CD40L-expressing replicating adenovirus: assessment of safety and immunologic responses in patients. Cancer Res. 2012;72(no. 7):1621–1631. doi: 10.1158/0008-5472.CAN-11-3001. [DOI] [PubMed] [Google Scholar]
- 93.Parviainen S. CD40 ligand and tdTomato-armed vaccinia virus for induction of antitumor immune response and tumor imaging. Gene Ther. 2014;21(no. 2):195–204. doi: 10.1038/gt.2013.73. [DOI] [PubMed] [Google Scholar]
- 94.Vilgelm A.E., Johnson D.B., Richmond A. Combinatorial approach to cancer immunotherapy: strength in numbers. J. Leukoc. Biol. 2016;100(no. 2):275–290. doi: 10.1189/jlb.5RI0116-013RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Andarini S. Adenovirus vector-mediated in vivo gene transfer of OX40 ligand to tumor cells enhances antitumor immunity of tumor-bearing hosts. Cancer Res. 2004;64(no. 9):3281–3287. doi: 10.1158/0008-5472.can-03-3911. [DOI] [PubMed] [Google Scholar]
- 96.Pan P.-Y., Zang Y., Weber K., Meseck M.L., Chen S.-H. OX40 ligation enhances primary and memory cytotoxic T lymphocyte responses in an immunotherapy for hepatic colon metastases. Mol. Ther. 2002;6(no. 4):528–536. doi: 10.1006/mthe.2002.0699. [DOI] [PubMed] [Google Scholar]
- 97.Aida K. Suppression of Tregs by anti-glucocorticoid induced TNF receptor antibody enhances the antitumor immunity of interferon-a gene therapy for pancreatic cancer. Cancer Sci. 2014;105(no. 2):159–167. doi: 10.1111/cas.12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lorenz M.G.O., Kantor J.A., Schlom J., Hodge J.W. Anti-tumor immunity elicited by a recombinant vaccinia. Hum. Gene Ther. 1999;1103:1095–1103. doi: 10.1089/10430349950018094. [DOI] [PubMed] [Google Scholar]
- 99.Yan L. Forced LIGHT expression in prostate tumors overcomes Treg mediated immunosuppression and synergizes with a prostate tumor therapeutic vaccine by recruiting effector T lymphocytes. Prostate. 2015;75(February (3)):280–291. doi: 10.1002/pros.22914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hou W., Sampath P., Rojas J.J., Thorne S.H. Oncolytic virus-mediated targeting of PGE2 in the tumor alters the immune status and sensitizes established and resistant tumors to immunotherapy. Cancer Cell. 2016;30(1):108–119. doi: 10.1016/j.ccell.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dajon M., Iribarren K., Cremer I. Toll-like receptor stimulation in cancer: a pro- and anti-tumor double-edged sword. Immunobiology. 2017;222(1):89–100. doi: 10.1016/j.imbio.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 102.Rojas J.J. Manipulating TLR signaling increases the anti-tumor t cell response induced by viral cancer therapies. Cell Rep. 2016;15(no. 2):264–273. doi: 10.1016/j.celrep.2016.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hirvinen M. Expression of DAI by an oncolytic vaccinia virus boosts the immunogenicity of the virus and enhances antitumor immunity. Mol. Ther. Oncolytics. 2016;3(March):16002. doi: 10.1038/mto.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang S.N., Choi I.K., Huang J.H., Yoo J.Y., Choi K.J., Yun C.O. Optimizing DC vaccination by combination with oncolytic adenovirus coexpressing IL-12 and GM-CSF. Mol. Ther. 2011;19(8):1558–1568. doi: 10.1038/mt.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Choi K.-J., Zhang S.-N., Choi I.-K., Kim J.-S., Yun C.-O. Strengthening of antitumor immune memory and prevention of thymic atrophy mediated by adenovirus expressing IL-12 and GM-CSF. Gene Ther. 2012;19(no. 7):711–723. doi: 10.1038/gt.2011.125. [DOI] [PubMed] [Google Scholar]
- 106.Choi I.-K. Oncolytic adenovirus co-expressing IL-12 and IL-18 improves tumor-specific immunity via differentiation of T cells expressing IL-12Rβ(2) or IL-18Rα. Gene Ther. 2011;18(no. 9):898–909. doi: 10.1038/gt.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li Q. Synergistic effects of IL-12 and IL-18 in skewing tumor-reactive T-cell responses towards a type 1 pattern. Cancer Res. 2005;65(no. 3):1063–1070. [PubMed] [Google Scholar]
- 108.Du T. Tumor-specific oncolytic adenoviruses expressing granulocyte macrophage colony-stimulating factor or anti-CTLA4 antibody for the treatment of cancers. Cancer Gene Ther. 2014;21(no. 8):340–348. doi: 10.1038/cgt.2014.34. [DOI] [PubMed] [Google Scholar]
- 109.Long G.V. A Phase I/III, multicenter, open-label trial of talimogene laherparepvec (T-VEC) in combination with pembrolizumab for the treatment of unresected, stage IIIb-IV melanoma (MASTERKEY-265) J. Immunother. Cancer. 2015;3(January):P181. [Google Scholar]
- 110.Sorensen M.R., Holst P.J., Steffensen M.A., Christensen J.P., Thomsen A.R. Adenoviral vaccination combined with CD40 stimulation and CTLA-4 blockage can lead to complete tumor regression in a murine melanoma model. Vaccine. 2010;28(41):6757–6764. doi: 10.1016/j.vaccine.2010.07.066. [DOI] [PubMed] [Google Scholar]
- 111.Tysome J.R. A novel therapeutic regimen to eradicate established solid tumors with an effective induction of tumor-specific immunity. Clin. Cancer Res. 2012;18 doi: 10.1158/1078-0432.CCR-12-0979. [DOI] [PubMed] [Google Scholar]
- 112.Wei H. Combinatorial PD-1 blockade and CD137 activation has therapeutic efficacy in murine cancer models and synergizes with cisplatin. PLoS One. 2013;8(December no. 12):e84927. doi: 10.1371/journal.pone.0084927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dai M. Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation. J. Immunother. 2013;36(no. 4):248–257. doi: 10.1097/CJI.0b013e3182943549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Galivo F. Interference of CD40L-mediated tumor immunotherapy by oncolytic vesicular stomatitis virus. Hum. Gene Ther. 2010;21 doi: 10.1089/hum.2009.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mathis J.M. Cancer-specific targeting of an adenovirus-delivered herpes simplex virus thymidine kinase suicide gene using translational control. J. Gene Med. 2006;8(September no. (9)):1105–1120. doi: 10.1002/jgm.935. [DOI] [PubMed] [Google Scholar]
- 116.Chase M., Chung R.Y., Chiocca E.A. An oncolytic viral mutant that delivers the CYP2B1 transgene and augments cyclophosphamide chemotherapy. Nat Biotech. 1998;16(May):444–448. doi: 10.1038/nbt0598-444. [DOI] [PubMed] [Google Scholar]
- 117.Nakamura H., Mullen J.T., Chandrasekhar S., Pawlik T.M., Yoon S.S., Tanabe K.K. Multimodality therapy with a replication-conditional herpes simplex virus 1 mutant that expresses yeast cytosine deaminase for intratumoral conversion of 5-Fluorocytosine to 5-Fluorouracil. Cancer Res. 2001;61(July no. (14)):5447. (LP-5452) [PubMed] [Google Scholar]
- 118.Zhang J. Potent anti-tumor activity of telomerase-dependent and HSV-TK armed oncolytic adenovirus for non-small cell lung cancer in vitro and in vivo. J. Exp. Clin. Cancer Res. 2010:1–7. doi: 10.1186/1756-9966-29-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hu J.C.C. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. 2006;12(no. 22):6737–6747. doi: 10.1158/1078-0432.CCR-06-0759. [DOI] [PubMed] [Google Scholar]
- 120.Bramante S. Treatment of melanoma with a serotype 5/3 chimeric oncolytic adenovirus coding for GM-CSF: Results in vitro, in rodents and in humans. Int. J. Cancer. 2015;137(7):1775–1783. doi: 10.1002/ijc.29536. [DOI] [PubMed] [Google Scholar]
- 121.Lemay C.G. Harnessing oncolytic virus-mediated antitumor immunity in an infected cell vaccine. Mol. Ther. 2012;20(no. 9):1791–1799. doi: 10.1038/mt.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Janke M. Recombinant Newcastle disease virus (NDV) with inserted gene coding for GM-CSF as a new vector for cancer immunogene therapy. Gene Ther. 2007;14(no. 23):1639–1649. doi: 10.1038/sj.gt.3303026. [DOI] [PubMed] [Google Scholar]
- 123.van Rikxoort M. Oncolytic effects of a novel influenza a virus expressing interleukin-15 from the NS reading frame. PLoS One. 2012;7(no. 5) doi: 10.1371/journal.pone.0036506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gaston D.C. Production of bioactive soluble interleukin-15 in complex with interleukin-15 receptor alpha from a conditionally-replicating oncolytic HSV-1. PLoS One. 2013;8(11):1–13. doi: 10.1371/journal.pone.0081768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hock K., Laengle J., Kuznetsova I., Egorov A. Oncology Oncolytic influenza A virus expressing interleukin-15 decreases tumor growth in vivo. Surgery. 2017;161(3):735–746. doi: 10.1016/j.surg.2016.08.045. [DOI] [PubMed] [Google Scholar]
- 126.Wang L.-C.S. Treating tumors with a vaccinia virus expressing IFNβ illustrates the complex relationships between oncolytic ability and immunogenicity. Mol. Ther. 2012;20(no. 4):736–748. doi: 10.1038/mt.2011.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Patel M.R. Vesicular stomatitis virus expressing interferon-β is oncolytic and promotes antitumor immune responses in a syngeneic murine model of non-small cell lung cancer. Oncotarget. 2015;6(no. 32):33165–33177. doi: 10.18632/oncotarget.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pulido J. Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma. Nat. Biotechnol. 2012;30(March no. (4)):337–343. doi: 10.1038/nbt.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ramakrishna E. Antitumoral immune response by recruitment and expansion of dendritic cells in tumors infected with telomerase-dependent oncolytic viruses. Cancer Res. 2009;69(no. 4):1448–1458. doi: 10.1158/0008-5472.CAN-08-1160. [DOI] [PubMed] [Google Scholar]
- 130.Li J., O’Malley M., Sampath P., Kalinski P., Bartlett D.L., Thorne S.H. Expression of CCL19 from oncolytic vaccinia enhances immunotherapeutic potential while maintaining oncolytic activity. Neoplasia. 2012;14(12):1115–1121. doi: 10.1593/neo.121272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jonas B.A. Combination of an oncolytic virus with PD-L1 blockade keeps cancer in check. Sci. Transl. Med. 2017;9(no. 386) doi: 10.1126/scitranslmed.aan2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fukuhara H., Ino Y., Kuroda T., Martuza R.L., Todo T. Triple gene-deleted oncolytic herpes simplex virus vector double-armed with interleukin 18 and soluble B7-1 constructed by bacterial artificial chromosome-mediated system. Cancer Res. 2005;65(no. 23):10663–10668. doi: 10.1158/0008-5472.CAN-05-2534. [DOI] [PubMed] [Google Scholar]