

Abstract
When unexpected diseases such as the severe acute respiratory syndrome (SARS) and avian influenza become a serious threat to public health, an immediate response is imperative. This should take into consideration existing licensed antiviral drugs against other viral diseases already known to be safe for use in humans. In this report, evidence is presented that HIV-1 protease inhibitors (PIs) currently used in anti-HIV-1 therapies might exert some effects on SARS and perhaps, on avian influenza. Evidence for the potential benefits of PIs against the SARS coronavirus (SARS-CoV) is provided by empirical clinical studies, in vivo viral inhibition assays and computational simulations of the docking of these compounds to the active site of the main SARS-CoV protease. As suggested by in silico docking of these molecules to a theoretical model of a subunit of type A influenza virus RNA-dependent RNA polymerase, there also exists a remote possibility that these PIs may have an effect on avian influenza viruses. Although this evidence is still far from being definitive, the results so far obtained suggest that PIs should be seriously taken into consideration for further testing as potential therapeutic agents for SARS and avian influenza.
Keywords: SARS, Type-A influenza, Coronavirus, Protease, Antiretroviral, RNA-dependent RNA polymerase
1. Introduction
The epidemic of the newly described severe acute respiratory syndrome (SARS) in 2003 confronted the world with a life-threatening disease for which no definitive treatment protocol existed. When such unexpected diseases become a serious threat to public health, immediate action has to be taken, and there is no time for specific drug design and safety testing. Therefore, antiviral drugs already licensed for other viral diseases and known to be safe to humans have to be taken into consideration as part of the immediate response. In this regard, the example of SARS is revealing: apart from palliative treatments such as corticosteroids, the most promising anti-SARS coronavirus (SARS-CoV) molecules to date are “recycled” drugs known to be effective against other viral diseases. These include interferons, ribavirin, chloroquine and HIV-1 protease inhibitors (PIs). The first are broad spectrum antiviral drugs inducing the degradation of viral RNAs. Ribavirin, often used in combination with interferons (Stroher et al., 2004), exerts its antiviral activity directly through lethal mutagenesis of the viral genetic material (Crotty et al., 2001) and has been reported in some, but not all studies to inhibit SARS-CoV replication (Chen et al., 2004, Chu et al., 2004, Stroher et al., 2004). Chloroquine was a first choice antimalarial before the malaria parasite Plasmodium falciparum developed widespread resistance. Later on, this drug found an application in rheumatic diseases and was shown to exert some anti-HIV-1 effects, likely to be due to inhibition of glycosylation of the viral envelope glycoproteins (a review on the anti-HIV-1 effects of chloroquine was published on the February 2001 issue of this journal) (Savarino et al., 2001, Savarino et al., 2004a, Savarino et al., 2004b). Chloroquine's capacity to interfere with viral particle maturation opens potential applications in other diseases caused by enveloped viruses. Following our suggestion that chloroquine might be effective against SARS-CoV (Savarino et al., 2003), Keyaerts et al. (2004) showed that the drug is an effective inhibitor of SARS-CoV replication in vitro. PIs are effective treatments for HIV-1/AIDS, and their effects on other viral diseases will be the central topic of this paper. I will first review what is known on the effects of these drugs on SARS. Then, I will discuss the grounds for considering these drugs in the context of avian influenza.
2. Potential anti-SARS effects of HIV-1 protease inhibitors: clinical and laboratory findings
The anti-HIV-1 activity of PIs is based on inhibition of the HIV-1 aspartic protease, responsible for the cleavage of the Gag/Pol polypeptide containing the main viral enzymes and the structural viral core proteins. Inhibition of the HIV-1 protease leads to the production of immature viral particles and inhibition of viral replication. Although PIs were designed to be effective inhibitors of HIV-1 replication, they showed inhibitory effects against a wide spectrum of pathogens. Indeed, inhibitory effects of PIs were shown on Candida albicans (Cassone et al., 1999) and, more recently, on P. falciparum (Savarino et al., 2004a). These activities seem to be due to inhibition of some non-retroviral aspartic proteases, such as the C. albicans secretory aspartic proteases (Saps) and P. falciparum plasmepsins, as the three-dimensional (3D) architecture of these microbial proteases resembles that of the HIV-1 protease (Tacconelli et al., 2004, Savarino et al., in press).
That the anti-HIV-1 drug Kaletra (a combination of the PIs ritonavir and lopinavir) was found to be of some benefit for individuals with SARS was rather surprising (Cheng et al., 2004), as the SARS-CoV genome encodes for no aspartic proteases. Despite the lack of double-blind placebo-controlled clinical trials, the clinical experience of physicians involved in the care of people with SARS during the great epidemic of 2003 strongly suggests that lopinavir/ritonavir should be taken into consideration in the clinical management of this disease (Cheng et al., 2004). A retrospective matched cohort study of Chan et al. (2003) found that the addition of lopinavir/ritonavir to standard initial treatment protocols was associated with a reduction in the overall death rate (2.3%) and intubation rate (0%), when compared with a matched cohort who received standard treatment (15.6% and 11.0%, respectively, P < 0.05), and with a lower necessity of methylprednisolone at high doses. In another study, Chu et al. (2004) evaluated the effects of Kaletra in association with ribavirin. The adverse clinical outcome (acute respiratory distress syndrome or death) was significantly lower in the treatment group than in the historical controls treated with ribavirin alone (2.4% versus 28.8%, P < 0.001) at day 21 after the onset of symptoms. Both studies conclude that a randomised trial would be needed to validate these results, but the possibility of using lopinavir/ritonavir as a SARS treatment is an option if SARS were to return. Other considerations on the potential anti-SARS benefits of anti-HIV-1 substances were made in an interesting paper by Chen and Cao (2004). These authors observed that none of 19 people with HIV-1/AIDS, hospitalised together with 95 individuals with SARS on the same hospital floor, contracted SARS (Chen et al., 2003, Chen and Cao, 2004). However, only 11 of these subjects were under antiretroviral regimens, and only 2 were receiving a protease inhibitor (indinavir), the remaining being under different combinations of reverse transcriptase inhibitors (Chen et al., 2003). Attributing this observation to casualty is difficult: according to the report of Chen and Cao, the individuals with HIV-1/AIDS indeed had close contacts with those with SARS, and 6 of 28 medical workers who served on the same floor contracted SARS. It is possible that these effects are due to viral interference and/or to non-specific antiviral factors induced by HIV-1; however, the report of Chen and Cao also raised the hypothesis that anti-SARS effects might be an unexpected property of antiretroviral drugs in general and not only a property of PIs.
The clinical effects of antiretroviral drugs on SARS were initially attributed to non-specific anti-inflammatory effects, but later on, in vitro studies showed that members of the PI class could indeed exert direct antiviral effects against SARS-CoV. Yamamoto et al. (2004) showed that the protease inhibitor nelfinavir (10 μM), but not ritonavir, significantly and efficiently inhibited viral antigen expression (measured by immunofluorescence), the production of virions (measured by real-time RT-PCR) and the cytopathic effect (measured by the methyl tetrazolium assay) in Vero E6 cells infected with SARS-CoV at a multiplicity of infection of 0.01. Moreover, time-of-addition experiments showed that nelfinavir inhibited SARS-CoV replication at a post-entry step (Yamamoto et al., 2004). Chu et al. (2004) showed that lopinavir (6.4 μM) inhibited the cytopathic effect of SARS-CoV (measured by plaque-reduction assay) in foetal rhesus kidney-4 (fRHK-4) cells. Of note, the effects of lopinavir were synergistic to those of ribavirin (Chu et al., 2004). Therefore, Chu and coworkers attribute to this antiviral synergism the clinical benefits observed in individuals with SARS and treated with ribavirin plus Kaletra. Chen et al. (2004) screened the effects of different antiviral compounds against 10 clinical isolates of SARS-CoV. These compounds included the nucleosidic reverse transcriptase inhibitors (NRTIs) zidovudine and stavudine, the non-nucleosidic reverse transcriptase inhibitor (NNRTI) nevirapine, and the PIs ritonavir and lopinavir. Again, only lopinavir resulted to exert detectable effects against SARS-CoV. The EC50 of lopinavir ranged from 1.6 to 6.4 μM at 48 h post-infection and from 6.4 to 12.8 μM at 72 h post-infection in the fRHK-4 cell line (Chen et al., 2004). However, they warn that the antiviral effects are variable and cell line dependent. For example, the EC50 of lopinavir against the prototype SARS-CoV strain 39849 ranged from 3.2 to 6.4 μM in fRHK-4 cells and from 6.4 to 12.8 μM in Vero cells (Chen et al., 2004). Therefore, it is likely that the antiviral effects of some anti-SARS compounds reported in the literature have been exaggerated or underestimated depending on the assay and cell line used by the different groups. In this regard, the example of ribavirin is interesting: some report that it is an inhibitor SARS-CoV replication, others report that it exerts its antiviral effects only at concentrations too high to be reached in vivo (Chen et al., 2004, Chu et al., 2004, Stroher et al., 2004).
3. The SARS coronavirus main protease: grounds for the potential anti-SARS effects of HIV-1 protease inhibitors
As a member of the Coronaviridae family, SARS-CoV encodes for three different proteases, one of which, the 3C-like protease (3CLpro), or main coronavirus protease (Fan et al., 2004, Sun et al., 2003, Yang et al., 2003), is likely to be a target for PIs (Jenwitheesuk and Samudrala, 2003a, Jenwitheesuk and Samudrala, 2003b). Preliminary data of Yamamoto et al. (2004) indicate that 3CLpro can be inhibited, though only partially, by lopinavir, which is a derivative of ritonavir. This is apparently surprising, because 3CLpro is a cysteine protease rather than an aspartic protease. In the active site of SARS-CoV 3CLpro, Cys145 and a His41 form a “catalytic dyad” (Anand et al., 2003). Cysteine proteases usually display a catalytic triad consisting of serine, histidine and aspartate; however, the 3D structure of SARS-CoV 3CLpro shows no aspartate residue in the vicinity of Cys145 and His41. SARS-CoV 3CLpro is a homodimer with each monomer comprising three domains (Anand et al., 2003). The first two domains show a chymotrypsin-like folding (root mean square deviation from bovine α-chymotrypsin: 2.6 Å; P < 0.05; Fig. 1 ), which is responsible for the catalytic reaction, and the third domain is α-helical and plays a critical role in enzyme dimerisation (Shi et al., 2004). The chymotrypsin-like folding of 3CLpro is intriguing because the overall sequence identity between the two proteins is quite low (∼11%). As chymotrypsin and other proteins with similar 3D folding are serine proteases, it raised the hypothesis that 3CLpro was originally a chymotrypsin-like serine protease that later evolved in a cysteine protease (Shan et al., 2004). That a chymotrypsin-fold protease may be a target for PIs is supported by in vitro experiments of independent groups, showing that PIs also inhibit chymotrypsin-like components of the mammalian proteasome (Piccinini et al., 2002, Schmidtke et al., 1999).
Fig. 1.
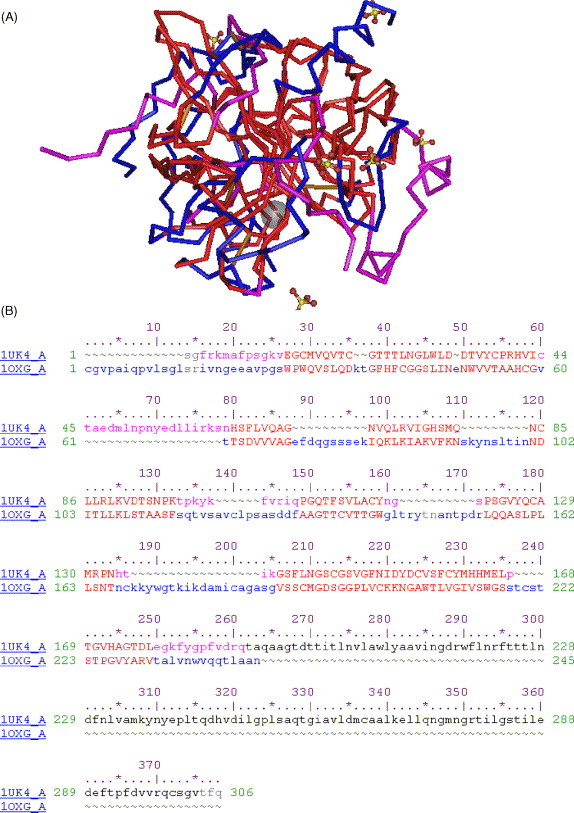
Structural similarity between the SARS coronavirus (SARS-CoV) main protease (3CLpro) and bovine α-chymotrypsin. (A) Three-dimensional structural superimposition between the HIV-1 protease (accession number in the Protein Data Bank: 1UK4) and α-chymotrypsin (accession number: 1OXG). The residues significantly aligned and corresponding to the catalytic site of both molecules are shown in red. The other regions of the catalytic domains of 3CLpro are shown in violet. Unaligned regions of α-chymotrypsin are shown in blue. (B) Sequence alignment corresponding to the structural alignment in (A). The colours of the residues in (B) strictly correspond to those of (A). This alignment was obtained using the VAST algorithm and visualised using the Cn3d 4.1 program (www.ncbi.nlm.nih.gov).
The limited experimental evidence available for an inhibitory effect of PIs on 3CLpro is supported by computational simulations revealing that the catalytic site of 3CLpro allows the docking of several PIs. The in silico simulation that I have adopted, i.e., the genetic algorithm GOLD, uses ligand/protein docking techniques which have been used to successfully predict the susceptibility of P. falciparum to PIs (Savarino et al., 2004a, 2005, manuscript in preparation). As shown in Fig. 2 , clinically used anti-HIV-1 PIs only partially fill the binding cavity of 3CLpro, but the GOLD fitness scores obtained (ritonavir = 62.77; saquinavir = 48.07; indinavir = 48.93) indicate stable ligand/protein interactions. Only ritonavir, however, was found to display significant hydrogen bonding with 3CLpro (data not shown). These observations are in line with previous calculations of independent groups using 3CLpros of SARS-CoV and transmissible gastroenteritis virus (TGEV) (Jenwitheesuk and Samudrala, 2003a, Zhang and Yap, 2004). Among the various compounds that they tested, Zhang and Yap (2004) indicated ritonavir as the compound with the highest binding affinity (K i = 5.6 × 10−25 M). These authors attribute to the other Kaletra component, lopinavir, a K i of 8.7 × 10−20 M (Zhang and Yap, 2004). Instead, Jenwitheesuk and Samudrala (2003b) attribute to ritonavir a K i of ∼10−7 M. My calculations attribute to ritonavir a K i of ∼10−5 M. The reason for these discrepancies are due to the different methods adopted. The above-mentioned authors use different molecular dynamics (MD) techniques to calculate the binding affinities. Instead, I estimated the theoretical binding affinity of PIs to 3CLpro based on a correlation between docking fitness and experimental data embedded in the GOLD program. This dataset compares the GOLD fitness scores obtained for a panel of potential α-chymotrypsin inhibitors to their experimentally determined K i's. It may be argued that the SARS-CoV 3CLpro is a protein with chymotrypsin folding but not α-chymotrypsin itself. In any event, the K i that I estimated is more realistic than those previously reported, given that PIs, at 10 μM, have been determined to inhibit the 3C-like protease only partially (Yamamoto et al., 2004).
Fig. 2.
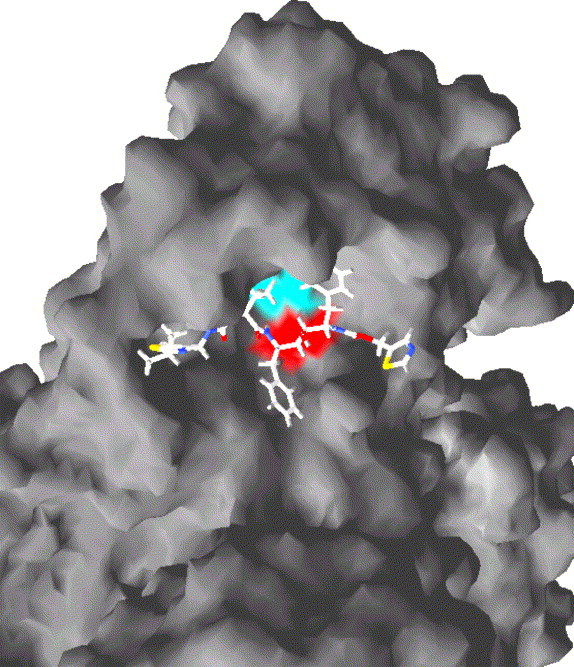
Docking of the HIV-1 protease inhibitor (PI) saquinavir to the active site of the severe acute respiratory syndrome coronavirus (SARS-CoV) main protease. The molecular surface corresponding to the catalytic His41 and Cys145 residues is shown in blue and red, respectively. Molecular docking was computed using the genetic algorithm GOLD (Cambridge Crystallographic Data Centre, Cambridge, UK) and visualised using the program SWISSpdbviewer (available at www.expasy.org). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Although experiments providing a causal link between 3CLpro inhibition and impairment of SARS-CoV replication are still lacking, 3CLpro is indicated by most groups as the main target for PIs in SARS-CoV, as virological, biochemical and bioinformatic data seem to agree in this regard. The fact that, experimentally, lopinavir and ritonavir only partially inhibit 3CLpro does not exclude a possible causal link between 3CLpro and SARS-CoV inhibition. Indeed, the extent of 3CLpro inhibition necessary for exerting antiviral effects is still not known. However, it cannot be excluded that inhibition of any other of the post-entry steps of the viral life cycle may contribute to the anti-SARS-CoV effects of PIs.
The possibility that PIs may dock to the active site of 3CLpro is intriguing as it extends the potential effects of these drugs beyond those organisms possessing aspartic proteases resembling the HIV-1 protease.
4. Expanding the applications of HIV-1 protease inhibitors: theoretical effects on influenza A viruses
Another target for PIs might be a putative protease component of the RNA-directed RNA polymerase (RdRp) of the type A influenza virus. Orthomyxoviridae had long been believed not to encode for any proteases, until the observations of Hara et al., 2001a, Hara et al., 2001b suggested that a chymotrypsin-like protease might be hidden in the carboxy-terminal region of the PA subunit of RdRp (Hara et al., 2001a, Hara et al., 2001b). Their conclusion is based on the observed capacity of PA to cleave chymotrypsin-specific substrates and on its amidolytic activity, which is a peculiar feature of chymotrypsins (Hara et al., 2001a). Hara et al. (2001a) showed by mutational analysis that PA could be a serine protease with a Ser624 in the active site. Abolition of this putative chymotrypsin-like protease activity of PA by substitution of the catalytic Ser624 residue with Ala significantly decreased the yield of viral progeny (Toyoda et al., 2003). Hara et al. (2001a) identified the other active residues of the catalytic triad as His510 and Asp547 because of their level of conservation in the PAs of type A influenza viruses. However, a survey of public sequence databases has shown that Asp547 is not conserved in all PAs of influenza A viruses. Theoretically, this observation does not contradict the theory of Hara et al. because chymotrypsin-like proteases can be functionally active proteases without the aspartate residue. One example for this is the above-described catalytic dyad of 3CLpro. Alternatively, other Lewis bases in the vicinity of His510 might replace the Asp residue as a third component of the triad.
In line with the observations of Hara et al., the carboxy-terminal region of PA, believed to be responsible for its chymotrypsin-like activity, in my analysis showed a significant though moderate similarity with α-chymotrypsin (∼21% identity), according to CLUSTALW calculations (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_clustalw.html), and His510 and Ser624 might correspond to His57 and Ser195 of chymotrypsin (Hara et al., 2001a). According to my calculations, the second most similar protein to the carboxy-terminal region of PA was, among the proteins analysed, SARS-CoV 3CLpro. As shown in Fig. 3 , PA shares 43% sequence similarity and 19.1% identity with 3CLpro. This level of similarity is one of the highest of those found with the other chymotrypsin-like proteases analysed (data not shown).
Fig. 3.

Sequence alignment between the COOH-terminal region of the PA subunit of the RNA-dependent RNA polymerase of a H5N1 avian influenza virus (NCBI accession number: AAV48550), and the first two domains of the severe acute respiratory syndrome coronavirus (SARS-CoV) main protease (3CLpro; NCBI accession number: P59641). The alignment was generated using the CLUSTALW software (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_clustalw.html). The boxes evidence the perfect alignment between the catalytic His41 and Cys145 of SARS-CoV 3CLpro and the putatively catalytic His510 and Cys145 of influenza A PA.
The sequence similarity between PA and 3CLpro raises the hypothesis that PI molecules might bind PA similarly to 3CLpro. Although substitution of the putative catalytic Ser624 residue of the protease activity of PA was differently reported to affect viral replication (Fodor et al., 2002, Toyoda et al., 2003), the log reduction of viral replication in mice intranasally infected with a virus expressing a Ser624 > Ala PA mutant lacking protease activity (9.3 × 103 PFU in mutant virus-infected mice versus 4.0 × 104 PFU in controls) observed by Toyoda et al. (2003) suggests that this protein merits attention as a target for pharmacological interventions. Similar mutants constructed by Fodor et al. in a previous study (2002) had displayed no reduced replicative capacity in cell cultures. These different results are likely to be attributed to the fact that Toyoda et al. conducted their tests in a mouse model whereas Fodor and coworkers monitored viral replication in a cell line model. A cell line may deviate from in vivo conditions in any of a number of molecular characteristics The different results between in vitro and in vivo models are reminiscent of the results on SARS-CoV 3CLpro inhibition described in the previous sections of this paper and reflect the need of further research. Moreover, Fodor et al. report in the same paper that the His510 > Ala substitution in PA produces replication-defective viruses. Of note, His510 is a putative catalytic residue of the possible chymotrypsin activity. These authors attribute their observation to an impaired capacity of the mutated RdRp to transcribe the viral RNA (vRNA) into mRNA because of a defect in the endonucleolytic activity required to exert this function. Indeed, all three RdRp subunits (PB1, PB2 and PA) are required for efficient synthesis of RNA transcripts. PB1 interacts both with PB2 and PA, and a low resolution structural model of the influenza ribonucleoprotein particle generated by electron microscopy suggests a rather compact structure for the polymerase complex (Martín-Benito et al., 2001). The observations of Toyoda and Fodor are not necessarily mutually exclusive. Indeed, His510 does not seem to be directly involved in the endonucleolytic reaction (Fodor et al., 2002).
In any event, if the reduction of viral replication observed in vivo by Toyoda and coworkers should become achievable in humans treated with PA inhibitors, it might contribute to attenuate the life-threatening manifestations of avian influenza. Alternatively, PA inhibitors might be used in combination strategies in order to impair the viral life cycle at multiple steps.
The 3D structure of RdRp has not been crystallographically solved yet, and such studies will be necessary to develop specific inhibitors. As the availability of an X-ray structure of PA is likely to take a long time, I have modelled the chymotrypsin-like component of the PA of a H5N1 chicken influenza virus isolate (NCBI accession number: AAV48550) based on the X-ray solved structure of bovine α-chymotrypsin (Protein Data Bank accession number: 1OXG). Although it is usually difficult to predict protein folding based on the 3D structure of homologues with less than 50% of sequence identity, the approach of homology modelling is validated in this case by experimental observations showing that chymotrypsin-like proteases show an extremely conserved 3D folding, even when homology in the primary structure is quite low. The 3D architecture of SARS-CoV 3CLpro described in Fig. 1 constitutes such an example. Despite its chymotrypsin structure, 3CLpro shares only 10.5% sequence identity with α-chymotrypsin (Fig. 1), which is lower than that shared between α-chymotrypsin and PA.
This extremely conserved 3D folding is probably forced by evolution in order to maintain the activities of chymotrypsin-like enzymes. It is moreover worth to be noted that PA sequences may vary a lot even among influenza A viruses. However, according to my estimations, their level of sequence similarity with α-chymotrypsin resulted to be similar, independently of the viral variants analysed (data not shown). Since above-mentioned research of Fodor et al. and Tayada and coworkers both suggest that the carboxy-terminal region of PA plays a role in viral replication, it is expected to maintain some conserved 3D architecture. Therefore, it is possible to hypothesise that there are positions that allow mutations and positions which do not, or at least, are compatible with some kinds of mutations only. As the types of mutations allowed often depend on mutations occurring in other positions, this is a very complex matter, not resolvable by simple sequence alignments. A specific algorithm will be necessary to clarify this point, and this could be an interesting aim for future research.
Computational simulations of the docking of three well-known PIs (ritonavir, saquinavir and indinavir) to PA indicate ritonavir as the compound with the highest GOLD fitness score (i.e., 64.67), slightly higher that obtained with SARS-CoV 3CLpro, and estimated to correspond to a K i of ∼10−5 M, well within the steady-state concentrations reachable in vivo at a dosage used in the treatment for HIV-1/AIDS. Similar results have been obtained by evaluating the docking of ritonavir to a PA structure calculated by the software ROSETTA, which predicts the 3D structure ab initio based on protein sequence (http://www.bioinfo.rpi.edu/∼bystrc/hmmstr/server.php). Fig. 4 shows the theoretical docking of ritonavir inside the binding cavity of PA. According to this model, significant hydrogen bonding might occur with both the putatively catalytic His510 and Ser624. Therefore, if the above reviewed observations of both Toyoda et al. and Fodor et al. are correct, it is possible to speculate that PIs might inhibit either the proteolytic or the endonucleolytic function of PA.
Fig. 4.
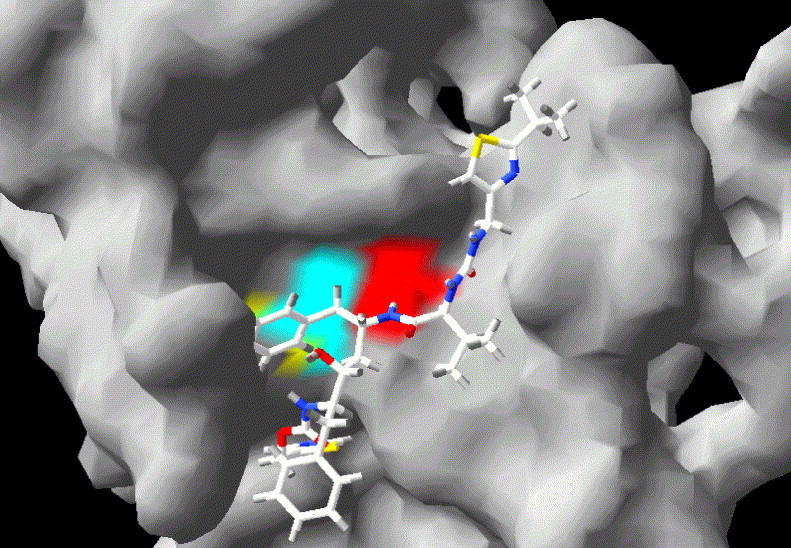
Theoretical docking of an HIV-1 protease inhibitor (ritonavir) to the active site of the chymotrypsin-like protease domains of the PA subunit of the RNA-dependent RNA polymerase of an H5N1 avian influenza virus. The three-dimensional structure of the COOH-terminal portion of PA was obtained by homology molecular modelling using the software tool SWISS MODEL (available at www.expasy.org). The model was then adjusted manually so as to match the distances between the catalytic amino acids determined by X-ray in bovine α-chymotrypsin (Protein Data Bank accession number: 1OXG). The molecular surface corresponding to His510, Asp547 and Ser624 is shown in blue, yellow and violet, respectively. Molecular docking and its visualisation were obtained as described in the caption for Fig. 2. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
5. Concluding remarks
The results so far obtained suggest that PIs should be taken into consideration for further study in the context of SARS and, maybe, influenza, including avian influenza viruses. The evidence for an inhibitory effect of PIs against the etiologic agents of these diseases is still very preliminary in the case of SARS and only theoretical in the case of type A influenza viruses. Anyway, a demonstration of effects of PIs on chymotrypsin-like viral proteases might open new avenues to antiviral research. As for the potential effects of PIs on SARS, more extensive studies on the inhibitory effects of PIs on 3CLpro, as well as studies on the causal link between inhibition of 3CLpro and antiviral effects, should be necessary. Furthermore, from the literature analysed, the need of standardisation of the in vitro testing of anti-SARS compounds is apparent, in order to better compare the results obtained by independent groups. Moreover, more studies evaluating the effects of PIs in combination with other anti-SARS drugs are needed. We recently demonstrated a synergistic anti-HIV-1 effect of PIs in combination with chloroquine (Savarino et al., 2004b). This synergism is associated to combined inhibitory effects of chloroquine and PIs on the drug-extruding pumps P-glycoprotein and multidrug resistance associated protein 1 (MRP1), likely to result in increased drug concentrations (Savarino et al., 2004b). As chloroquine is emerging as a new promising anti-SARS compound in vitro (Keyaerts et al., 2004), PI/chloroquine combinations should be tested in SARS-CoV-infected cell cultures, in order to increase the potential pharmacological combinations available. Alternatively, PIs might be regarded as a lead for the design of new anticoronavirus compounds.
As for other possible antiviral applications of PIs, the possible chymotrypsin-like nature of the carboxy-terminal region of the PA subunit of type A influenza virus RdRp, a property shared with enzymes known to be susceptible to PIs, as well as the theoretical possibility that PIs might successfully dock to its putative active site, should constitute a basis for immediate testing of these antiretroviral drugs against the influenza viruses in vitro. Although in other times this research should be considered to be at an embryonic stage, the recent human cases of avian influenza in Vietnam and Cambodia should prompt the scientific community to consider this as well as any other possible pharmacological strategy to combat this disease. The World Health Organization (WHO) has recently issued an alert on the emergence of a possible pandemic of the H5N1 subtype influenza A virus (www.who.int). This virus is currently causing large epidemics in poultry and has acquired the ability to transmit to humans. It may in future acquire the ability to spread by direct inter-human transmission. As the mortality of infected people is very high (76% since January 2004), many millions of deaths may be expected in case of a global outbreak. For these reasons, if PIs should prove to be effective against H5N1 viruses in vitro, it may be immediately available for in vivo testing in affected individuals as an alternative to the one available group of antivirals presently available, viz. the neuraminidase inhibitors.
Meanwhile, the importance of studying PA should not be underestimated. Further research is needed to confirm the observations of Hara and coworkers on the chymotrypsin-like activity of PA, determine its 3D architecture by crystallographic analysis, and define its role in the Orthomyxoviridae life cycle. These researches might disclose new targets for future pharmacological interventions against influenza.
References
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Cassone A., De Bernardis F., Torosantucci A., Tacconelli E., Tumbarello M., Cauda R. In vitro and in vivo anticandidal activity of human immunodeficiency virus protease inhibitors. J Infect Dis. 1999;180:448–453. doi: 10.1086/314871. [DOI] [PubMed] [Google Scholar]
- Chan K.S., Lai S.T., Chu C.M., Tsui E., Tam C.Y., Wong M.M. Treatment of severe acute respiratory syndrome with lopinavir/ritonavir: a multicentre retrospective matched cohort study. Hong Kong Med J. 2003;9:399–406. [PubMed] [Google Scholar]
- Chen X.P., Cao Y. Consideration of highly active antiretroviral therapy in the prevention and treatment of severe acute respiratory syndrome. Clin Infect Dis. 2004;38:1030–1032. doi: 10.1086/386340. [DOI] [PubMed] [Google Scholar]
- Chen X.P., Li G.H., Tang X.P., Xiong Y., Chen X.J., Cao Y. Lack of severe acute respiratory syndrome in 19 AIDS patients hospitalised together. J Acquir Immun Defic Syndr. 2003;34:242–243. doi: 10.1097/00126334-200310010-00016. [DOI] [PubMed] [Google Scholar]
- Chen F., Chan K.H., Jiang Y., Kao R.Y., Lu H.T., Fan K.W. In vitro susceptibility of 10 clinical isolates of SARS coronavirus to selected antiviral compounds. J Clin Virol. 2004;31:69–75. doi: 10.1016/j.jcv.2004.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng V.C., Tang B.S., Wu A.K., Chu C.M., Yuen K.Y. Medical treatment of viral pneumonia including SARS in immunocompetent adult. J Infect. 2004;49:262–273. doi: 10.1016/j.jinf.2004.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C.M., Cheng V.C., Hung I.F., Wong M.M., Chan K.H., Chan K.S., HKU/UCH SARS Group Role of lopinavir/ritonavir in the treatment of SARS: initial virological and clinical findings. Thorax. 2004;59:252–256. doi: 10.1136/thorax.2003.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S., Cameron C.E., Andino R. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc Natl Acad Sci USA. 2001;98:6895–6900. doi: 10.1073/pnas.111085598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan K., Wei P., Feng Q., Chen S., Huang C., Ma L. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J Biol Chem. 2004;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodor E., Crow M., Mingay L.J., Deng T., Sharps J., Fecther P. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J Virol. 2002;76:8989–9001. doi: 10.1128/JVI.76.18.8989-9001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K., Shiota M., Kido H., Ohtsu Y., Kashiwagi T., Iwahashi J. Influenza virus RNA polymerase PA subunit is a novel serine protease with Ser624 at the active site. Genes Cells. 2001;6:87–97. doi: 10.1046/j.1365-2443.2001.00399.x. [DOI] [PubMed] [Google Scholar]
- Hara K., Shiota M., Kido H., Othsu Y., Toyoda T. Protease activity of influenza virus RNA polymerase PA subunit. Int Congr Ser. 2001;1219:479–485. [Google Scholar]
- Jenwitheesuk E., Samudrala R. Identifying inhibitors of the SARS coronavirus proteinase. Bioorg Med Chem Lett. 2003;13:3989–3992. doi: 10.1016/j.bmcl.2003.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenwitheesuk E., Samudrala R. Improved prediction of HIV-1 protease-inhibitors binding energies by molecular dynamics simulations. BMC Struct Biol. 2003;3:2–10. doi: 10.1186/1472-6807-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyaerts E., Vijgen L., Maes P., Neyts J., Van Ranst M. In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine. Biochem Biophys Res Commun. 2004;323:264–268. doi: 10.1016/j.bbrc.2004.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Benito J., Area E., Ortega J., Liorca O., Valpuesta J.M., Carrascosa J.L. Three-dimensional reconstruction of a recombinant influenza virus ribonucleoprotein particle. EMBO Rep. 2001;2:313–317. doi: 10.1093/embo-reports/kve063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccinini M., Rinaudo M.T., Chiapello N., Ricotti E., Baldovino S., Mosert M. The human 26S proteasome is a target of antiretroviral agents. AIDS. 2002;16:693–700. doi: 10.1097/00002030-200203290-00004. [DOI] [PubMed] [Google Scholar]
- Savarino A., Gennero L., Sperber K., Boelaert J.R. The anti-HIV-1 activity of chloroquine. J Clin Virol. 2001;20:131–135. doi: 10.1016/s1386-6532(00)00139-6. [DOI] [PubMed] [Google Scholar]
- Savarino A., Boelaert J.R., Cassone A., Majori G., Cauda R. Effects of chloroquine in viral infections: an old drug against today's diseases? Lancet Infect Dis. 2003;3:722–727. doi: 10.1016/S1473-3099(03)00806-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarino A., Lucia M.B., Rastrelli E., Rutella S., Golotta C., Morra E. Anti-HIV effects of chloroquine: inhibition of viral particle glycosylation and synergism with protease inhibitors. J Acquir Defic Syndr. 2004;35:223–232. doi: 10.1097/00126334-200403010-00002. [DOI] [PubMed] [Google Scholar]
- Savarino A, Sannella A, Spaccapelo R, Lucia MB, Severini C, Boelaert JR, et al. HIV-1 protease inhibitors exert antimalarial effects both in vitro and in vivo and partially revert Plasmodium falciparum resistance to chloroquine in vitro. Med Gen Med 2004b;6:ThPeA6940 [eJIAS. 2004 Jul 11;1(1):ThPeA6940].
- Savarino A, Cauda R, Cassone A. Aspartic proteases of Plasmodium falciparum as target of HIV protease inhibitors. J Infect Dis, in press. [DOI] [PubMed]
- Schmidtke G., Holzhutter H.G., Bogyo M., Kairies N., Groll M., de Giuli R. How an inhibitor of the HIV-1 protease modulates proteasome activity. J Biol Chem. 1999;274:35734–35740. doi: 10.1074/jbc.274.50.35734. [DOI] [PubMed] [Google Scholar]
- Shan Y.F., Li S.F., Xu G.J. A novel auto-cleavage assay for studying mutational effects of the active site of severe acute respiratory syndrome coronavirus 3C-like protease. Biochem Biophys Res Commun. 2004;324:579–583. doi: 10.1016/j.bbrc.2004.09.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Wei Z., Song J. Dissection study on the severe acute respiratory syndrome 3C-like protease reveals the critical role of the extra domain in dimerization of the enzyme: defining the extra domain as a new target for design of highly specific protease inhibitors. J Biol Chem. 2004;279:24765–24773. doi: 10.1074/jbc.M311744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroher U., DiCaro A., Li Y., Strong J.E., Aoki F., Plummer F. Severe acute respiratory syndrome-related coronavirus is inhibited by interferon-alpha. J Infect Dis. 2004;189:1164–1167. doi: 10.1086/382597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H., Lou H., Yu C., Sun T., Chen T., Peng S. Molecular cloning, expression, purification, and mass spectrometric characterization of 3C-like protease of SARS coronavirus. Protein Exp Purif. 2003;32:302–308. doi: 10.1016/j.pep.2003.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacconelli E., Savarino A., De Bernardis F., Cauda R., Cassone A. Candidiasis and HIV-protease inhibitors: the expected and unexpected. Curr Med Chem Immunol Endocr Metab Agents. 2004;4:49–59. [Google Scholar]
- Toyoda T., Hara K., Imamura Y. Ser624 of the PA subunit of influenza A virus is not essential for viral growth in cells and mice, but required for the maximal viral growth. Arch Virol. 2003;148:1687–1696. doi: 10.1007/s00705-003-0140-7. [DOI] [PubMed] [Google Scholar]
- Yamamoto N., Yang R., Yoshinaka Y., Amari S., Nakano T., Cinatl J. HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem Biophys Res Commun. 2004;318:719–725. doi: 10.1016/j.bbrc.2004.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. PNAS. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.W., Yap Y.L. Old drugs as lead compounds for a new disease? Binding analysis of SARS coronavirus main proteinase with HIV, psychotic and parasite drugs. Bioorg Med Chem. 2004;12:2517–2521. doi: 10.1016/j.bmc.2004.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]