

Abstract
The sudden emergence of severe acute respiratory syndrome (SARS) has boosted research on innate immune responses to coronaviruses. It is now well established that the causative agent, a newly identified coronavirus termed SARS-CoV, employs multiple passive and active mechanisms to avoid induction of the antiviral type I interferons in tissue cells. By contrast, chemokines such as IP-10 or IL-8 are strongly upregulated. The imbalance in the IFN response is thought to contribute to the establishment of viremia early in infection, whereas the production of chemokines by infected organs may be responsible for (i) massive immune cell infiltrations found in the lungs of SARS victims, and (ii) the dysregulation of adaptive immunity. Here, we will review the most recent findings on the interaction of SARS-CoV and related Coronaviridae members with the type I interferon and cytokine responses and discuss implications for pathogenesis and therapy.
Keywords: SARS, Coronaviruses, Interferon, Chemokines, Viral countermeasures
1. The Coronaviridae family
Coronaviruses are enveloped, positive-stranded RNA viruses that can infect a variety of vertebrates and are mainly associated with respiratory and enteric diseases. They have long been recognized as important pathogens of livestock and companion animals, and coronaviruses are a common cause of respiratory tract infections in man [1], [2]. In 2003, a coronavirus has been identified as the causative agent of a new human disease, the severe acute respiratory syndrome (SARS) [3], [4], [5]. The SARS coronavirus (SARS-CoV) spread within few months to more than 30 countries and caused the first epidemic of the new millennium. This event not only highlighted the potential of coronaviruses to seriously affect human health, but also gave a strong impetus on coronavirus research. Since then, bats were identified as a possible reservoir species of SARS-CoV [6], and a wealth of knowledge about coronavirus replication and pathogenesis has been gained [7], [8], [9].
The family Coronaviridae comprise two genera, Coronaviruses and Toroviruses, and is grouped together with two other families, the Arteriviridae and the Roniviridae, into the order Nidovirales [1], [10]. Although Nidoviruses differ in their genome sizes, structural proteins and morphology, they share a common genome organization and common mechanisms of RNA replication [1], [8], [9], [10], [11]. The name Nidovirus (from the Latin “nido” - nest) refers to the ability to transcribe a so-called “nested set” of subgenomic mRNAs [12]. Coronaviruses have genomes of approximately 30,000 nt, a length that is unprecedented amongst RNA viruses. The extreme genome size requires complex and only incompletely understood mechanisms of RNA replication, transcription, modification, and recombination, which are conducted by a multi-enzyme complex encoded by the replicase gene and the nucleocapsid gene [13], [14], [15]. It is speculated that coronaviruses have evolved this complex replication machinery to stably maintain their large genomic RNA and to encode additional functions that impact on virus–host interactions [10]. Furthermore, the unique transcription strategy involves a discontinuous step during negative strand RNA synthesis that mechanistically reflects similarity-assisted RNA recombination. Thus, coronaviruses are well equipped for high frequency RNA recombination which facilitates rapid adaptation to new hosts.
Coronavirus particles are enveloped and display a typical solar or crown-like (“corona”) appearance in electron microscopy. Historically, the family has been divided into three groups based on serological cross-reactivity. Later, this grouping has been confirmed by phylogenetic analyses based on genome sequencing data [8], [10], [16]. Soon after SARS-CoV has been recognized as a coronavirus, phylogenetic analyses revealed a relationship to group 2 coronaviruses, for which the mouse hepatitis virus (MHV) is the prototype. However, SARS-CoV also has unique features, suggesting that SARS-CoV represents an early split-off from the coronavirus group 2 lineage [8], [16].
2. Diseases caused by coronaviruses
Coronaviruses can cause a variety of diseases in animals and humans [2]. Of economical importance are coronaviruses such as porcine transmissible gastroenteritis virus, bovine coronavirus, feline infectious peritonitis virus, and avian infectious bronchitis virus. MHV, a natural mouse pathogen, has been extensively studied in the context of host immune responses and pathogenesis [2], [17], [18]. There are many different and well-characterized strains of MHV which, depending on organ tropism, virulence and host strain, can cause a wide array of diseases ranging from hepatitis, respiratory symptoms and gastroenteritis to CNS infection, demyelination, and acute meningitis [2], [17], [19], [20], [21], [22], [23]. Therefore, by using appropriate combinations of virus and mouse strains, MHV infections provide suitable models for a number of diseases that are of medical importance, such as encephalitis, immune-mediated demyelination (e.g. multiple sclerosis), hepatitis and acute respiratory infections (e.g. SARS).
Besides SARS-CoV, there are several human coronaviruses (HCoVs) which cause mainly mild respiratory tract infections (common cold; HCoV-229E, HCoV-OC43, HCoV-NL63 and HCoV-HKU) and sometimes enteric infections [2]. HCoV infections are prevalent in children, but more severe symptoms have been observed in immunocompromised individuals and occasionally in the elderly [24], [25]. Notably, HCoV-NL63 appears to cause more severe respiratory symptoms and has been associated with croup in children [26].
3. Innate immunity—the interferon system
The most efficient and rapid host response against viruses consists of the production of type I IFNs (IFN-α/β), an essential part of the antiviral innate immune system. As far as it is known, all nucleated cells of the mammalian body are able to synthesize and secrete type I IFNs. The mode of induction and the type of IFN being secreted, however, can differ among cell types. Secreted IFNs stimulate neighbouring cells to express potent antiviral proteins [27], [28], [29]. Besides their role as direct antiviral messengers, IFNs posses a wide range of other biological activities including inhibition of cell proliferation, regulation of apoptosis, and, importantly, immunomodulation [30], [31]. Thus, the IFN production triggered by the first contact with the viral intruder slows down or even stops virus multiplication, buys the organism time, and helps to establish an adaptive immune response.
Type I IFNs are classified according to their amino acid sequence and comprise a large number (at least 13) of IFN-α subtypes and a single IFN-β [32], as well as some additional family members [33], [34], [35]. Expression patterns, i.e. which IFNs will be synthesized at which time point, mostly depend on the particular cell type.
3.1. Interferon induction
Epithelial cells, fibroblasts and neurons mainly secrete IFN-β as an initial response to infection but switch to IFN-α during the subsequent amplification phase of the IFN response [36], [37]. By contrast, dendritic cells, which play an important role in immunosurveillance and provide an interface between innate and adaptive immunity, directly produce high levels of IFN-α subtypes [38], [39].
IFN induction in fibroblasts occurs mainly by an intracellular pathway (Fig. 1A). Hallmark molecules of RNA viruses such as double-stranded (ds) RNA and 5′-triphosphorylated single-stranded (ss) RNA trigger a signaling chain which activates IFN-β gene expression [40], [41], [42], [43]. Two RNA helicases, RIG-I and MDA-5, are the main intracellular receptors of viral RNA [44], [45], [46], [47]. RIG-I and MDA-5 recognize different and non-overlapping sets of viruses, suggesting a degree of specificity in RNA recognition [45]. Indeed, it was recently found that RIG-I has the unique ability to bind the triphosphate groups on the 5′-end of uncapped viral ssRNA [40], [41], [42]. MDA-5, by contrast, is apparently more dependent on dsRNA structures since it is required for the IFN response against picornaviruses which have genomic RNAs with a protein-protected 5′-end [45], [48] and produce high levels of dsRNA [43]. The binding of a viral RNA to RIG-I and MDA-5 induces a signaling chain which eventually results in the phosphorylation of the transcription factor IRF-3 [49], [50]. IRF-3 is a member of the IFN regulatory factor (IRF) family [51], [52] and plays a central role in the activation of the IFN-β promoter [53]. Phosphorylated IRF-3 homo-dimerizes and moves into the nucleus where it recruits the transcriptional coactivators p300 and CREB-binding protein (CBP) to initiate IFN-β mRNA synthesis. This first-wave IFN triggers expression of a related factor, IRF-7, which in fibroblasts is only present in low amounts [54]. IRF-7 can be activated the same way as IRF-3 [55], [56], leading to a positive-feedback loop that initiates the synthesis of several IFN-α subtypes as the second-wave IFNs [37], [54]. In addition, the transcription factors NF-κB (activated by RIG-I, MDA-5 and the dsRNA-dependent kinase PKR) and AP-1 (activated by stress-induced Jun kinase) are triggered by viral replication [57], [58] to enhance IFN-β gene expression.
Fig. 1.
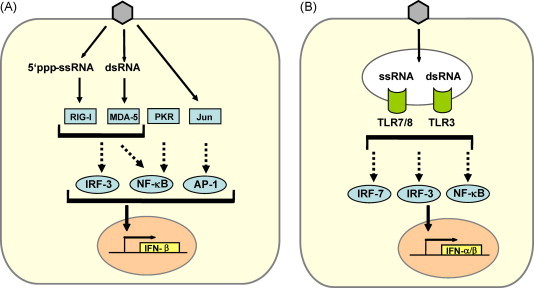
Parallel pathways of type I IFN induction by RNA viruses. (A) Intracellular pathway. Characteristic by-products of virus replication such as dsRNA or 5′triphosphorylated ssRNA lead to activation of the transcription factor IRF-3. Cooperative action with NF-κB and AP-1 is required for full activation of the IFN-β promoter. IRF-3 is phosphorylated by the kinases TBK-1 and IKKɛ (not shown) which in turn are activated by the RNA-sensing molecules RIG-I (recognizing 5′ triphosphorylated ssRNA) and MDA-5 (recognizing dsRNA). PKR, which also recognizes dsRNA, is important for activating NF-κB. AP-1 is activated by the stress-responsive kinase Jun. (B) Endosomal pathway. TLR7/8 and TLR3 recognize viral ssRNA and dsRNA, respectively, and activate IFNα/β transcription via the transcription factors IRF-7, IRF-3, and NF-κB. Only those parts of the pathways are depicted which are relevant for the discussion of coronaviral interactions (see main text). For comprehensive representations see recent reviews [53], [61], [181].
Myeloid dendritic cells (mDCs) [39] and, most prominently, plasmacytoid dendritic cells (pDCs) [38] are the main IFN producers of the lymphatic system. mDCs can sense dsRNA by the classic intracellular pathway [39] and, in addition, by the endosomal toll-like receptor (TLR) 3 [59]. pDCs predominantly monitor RNA virus infections by the endosomal TLR7 and TLR8 which recognize ssRNA [60]. Activated TLRs signal through different intracellular adaptor molecules to induce IRF- and NF-κB-dependent IFN transcription [61] (Fig. 1B). Interestingly, in contrast to other cell types, pDCs contain considerable amounts of constitutively expressed IRF-7 [62], [63]. IRF-7 is further upregulated in response to IFN and generates a positive-feedback loop for high IFN-α and IFN-β production [64], [65]. In addition, TLR7 and TLR9 are retained in the endosomes of pDCs to allow prolonged IFN induction signaling [66].
3.2. Interferon signaling
All IFN-α/β subtypes bind to and activate a common type I IFN receptor which is present on virtually all host cells [28], [67]. Binding of IFN-α/β leads to conformational changes in the intracellular parts of the receptor which activate the so-called JAK-STAT signaling pathway. The signal transducer and activator of transcription (STAT) proteins are latent cytoplasmic transcription factors which become phosphorylated by the Janus kinase (JAK) family members JAK-1 and TYK-2 [68], [69]. Phosphorylated STAT-1 and STAT-2 recruit a third factor, IRF-9 (also called p48), to form a complex known as IFN stimulated gene factor 3 (ISGF-3). The ISGF-3 heterotrimer translocates to the nucleus and binds to IFN-stimulated response elements (ISRE) in the promoter regions of IFN-stimulated genes (ISGs), thereby inducing their transcription.
3.3. Interferon effector proteins
IFN-α/β activate the expression of more than 300 IFN-stimulated genes (ISGs) which have antiviral, antiproliferative, and immunomodulatory functions [70]. IFN-induced proteins include enzymes, transcription factors, cell surface glycoproteins, cytokines, chemokines and a large number of factors with unknown function. Up to now, only a few proteins with antiviral activity have been characterized in detail. These are the Mx GTPases, the protein kinase R (PKR), the 2′-5′ oligoadenylate synthetases (2-5 OAS)/RNaseL system, the RNA-specific adenosine deaminase 1 (ADAR 1), and the products of the ISG56 (p56) and ISG20 genes. Mx proteins belong to the superfamily of dynamin-like large GTPases and have been discovered as mediators of genetic resistance against orthomyxoviruses in mice. The human MxA protein blocks replication of the infecting virus soon after cell entry by targeting and missorting viral ribonucleoprotein particles [71], [72], [73], [74]. PKR, 2-5 OAS and ADAR are constitutively expressed in a latent, inactive form. Basal mRNA levels are upregulated by IFN-α/β and these enzymes need to be activated by viral dsRNA. PKR is a serine-threonine kinase that phosphorylates the alpha subunit of the eukaryotic translation initiation factor eIF2 [75], [76], thus blocking translation of cellular and viral mRNAs. The 2-5 OAS catalyses the synthesis of short 2′-5′ oligoadenylates [77] that activate the latent endoribonuclease RNaseL which in turn degrades both viral and cellular RNAs [78]. ADAR 1 catalyzes the deamination of adenosine on target dsRNAs to yield inosine. As a result the secondary structure is destabilized due to a change from an AU base pair to the less stable IU base pair and mutations accumulate within the viral genome [28]. P56 binds the eukaryotic initiation factor 3e (eIF3e) subunit of the eukaryotic translation initiation factor eIF3. It functions as an inhibitor of translation initiation at the level of eIF3 ternary complex formation and is likely to suppress viral RNA translation [79], [80]. ISG20 is an IFN-induced 3′-5′ exonuclease that specifically degrades ssRNA in vitro. In cell culture, expression of ISG20 leads to a reduction of vesicular stomatitis virus (VSV), influenza virus and retrovirus replication [81], [82], [83].
4. Coronaviruses: protective role of the interferon system
MHV and several animal coronaviruses were shown to be sensitive to the antiviral action of type I IFNs [20], [84], [85], [86]. Growth of SARS-CoV can also be inhibited by exogenously added IFN-α/β [87], [88], [89], [90], [91], and mice lacking STAT1 or the type I IFN receptor are more prone to SARS-CoV- or MHV-induced organ damages [92], [93]. Direct IFN treatment of experimentally infected mice or macaques has a protective effect against SARS-CoV or MHV-1, respectively [20], [94], [95]. IFNs may also alleviate symptoms in SARS patients, but case numbers are too low for definite conclusions [96], [97]. In the animal models for SARS, IFNs were most efficient if given before infection, but still have a certain antiviral effect if given after exposure to virus [94], [95]. Thus, IFNs, which are an approved medication against several viral and malignant diseases [28], [98], may offer the possibility both of prevention and treatment of SARS with a licensed drug.
Although IFN treatment has clear beneficial effects, the identification of the responsible effector protein(s) is still out. The moderate inhibiting effect of IFN-γ is most likely caused by inducible NO synthetase [99], but which ISG confers the much stronger effect of IFN-α/β is unknown. It is however established that MxA plays no role as an anti-SARS-CoV factor [89].
5. How do coronaviruses cope with the IFN system?
SARS-CoV, MHV, and a number of other coronaviruses are highly pathogenic despite their sensitivity to IFN-α/β. Moreover, SARS-CoV [43] as well as MHV [100] were shown to generate substantial amounts of the IFN inducer dsRNA during infection. This implies that coronaviruses somehow avoid or inhibit the production of IFN in a manner similar to other viruses [27], [29], [101]. Indeed, in fibroblasts productively infected with SARS-CoV or MHV no detectable induction of IFN-β occurs [102], [103], [104]. Human macrophages, which become only non-productively infected with SARS-CoV, are also unable to launch an IFN response [105], [106]. In fibroblasts, a lack of transcriptional induction was also observed for IFN-α, IFN-λ, RANTES and IL-6 [107], suggesting that SARS-CoV-infected tissue cells are severely impaired in the production of a wide range of cytokines. In line with this, we have recently shown that in cells infected with SARS-CoV, no phosphorylation, dimerisation or CBP-binding of IRF-3 occur [102].
One possible mechanism that may at least in part account for the absence of IRF-3 activation and type I IFN expression is impaired sensing of coronaviruses by host cell-encoded pathogen recognition receptors (PRRs), such as TLRs and the intracellular RNA sensors. Cytoplasmic viral RNAs could be recognized by RIG-I, MDA-5, or PKR (Fig. 2 ). As RIG-I is triggered by 5′-triphosphates on ssRNA, it might not recognize the 5′-capped genomic and subgenomic mRNAs. However, like all positive stranded RNA viruses, coronaviruses replicate their genome via negative-stranded RNA intermediates containing (most likely) 5′-triphosphate ends. Moreover, the dsRNAs which are detectably formed during virus replication [43], [100] should be recognized intracellularly by MDA-5 and PKR and extracellularly by TLR-3. Coronaviruses may escape cellular RNA sensing by creating a microenvironment that is not accessible to cytoplasmic PRRs. Indeed, it has been shown that infection induces formation of double membrane vesicles (DMV) at perinuclear sites within the cytoplasm where RNA synthesis takes place [108], [109], [110], [111] (Fig. 2). It is tempting to speculate that dsRNA replication intermediates containing 5′-triphosphorylated negative strands are located within DMVs and therefore protected from PRR sensing. In line with this, unimpeded IFN-β mRNA production is observed in co-infections with Sendai Virus (SeV) and MHV or SARS-CoV [100], [112]. Similar findings were reported for MHV-infected cells treated with the IFN inducer poly I:C [104]. Apparently, the sensing of SeV RNA or poly I:C results in the transcription of IFN-β mRNA, and coronaviruses are unable to interrupt this process. Noteworthy, despite significant IFN-β mRNA transcription, only markedly reduced levels of IFN-β protein are secreted from in SeV/MHV-infected cells [112]. This indicates that MHV (and possibly other coronaviruses) counteract IFN-β mRNA nuclear export and/or translation or affect downstream events such as IFN-β protein stability and secretion.
Fig. 2.
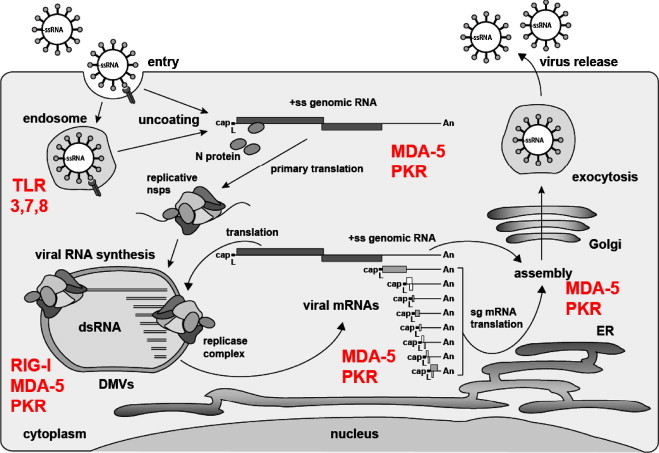
Coronavirus life cycle and RNA-specific pathogen recognition receptors. The coronavirus life cycle is illustrated together with PRRs with the potential to sense viral RNA. Coronaviruses enter their host cells either on the plasma membrane or via endosomes where they could be recognized by TLR 3, 7, or 8. Note that MHV is shown to be recognized by TLR7 in pDCs [92]. Upon uncoating the capped viral ssRNA is released into the host cell cytoplasm and could be sensed by MDA-5 or PKR due to secondary structures containing dsRNA domains. Viral RNA synthesis takes place in or at double membrane vesicles (DMVs) and involves the appearance of dsRNA [43], again potentially recognized by MDA-5 and PKR. The negative-sense RNAs arising as an intermediate of DMV-associated genome replication and transcription are possibly 5′-triphosphorylated and thus could be recognized by RIG-I. Finally, a nested set of viral mRNAs are released into the cytoplasm (putative sensors: MDA-5 or PKR) where they are translated. The full-length genomic RNA can also be translated and is eventually packaged into progeny virus particles which are released from the host cell via the exocytosis pathway.
In addition to those mechanisms, expression studies using cDNA plasmids of SARS-CoV have shown that the proteins encoded by ORF3b, ORF6 as well as the nucleocapsid (N) protein are capable of inhibiting activation of IRF-3, and the ORF3b and ORF6 gene products additionally inhibit IFN signaling [113]. The mechanism of IFN signaling suppression by the SARS-CoV ORF6 gene product has been characterized in detail and it was shown that it tethers karyopherin alpha 2 and karyopherin beta 1 to the ER/Golgi membrane to disrupt nuclear import of STAT1 [114]. Additionally, the ORF7a protein has been shown to inhibit cellular protein synthesis [115], and the nsp 1 gene product has been suggested to promote host cell mRNA degradation [116]. The role of nsp1 in counteracting host innate immune responses was further studied in the MHV system [117]. A mutant virus containing a deletion in nsp1 was shown to replicate like wild-type virus in vitro but was strongly attenuated in mice, demonstrating that nsp1 is a major pathogenicity factor. In type I IFN receptor-deficient mice, however, replication of the nsp1 mutant virus was restored almost to the level of wild-type virus. Detailed phenotypic analysis revealed that nsp1 mutant replication was particularly reduced in IFN-α-treated macrophages, indicating that nsp1 mainly affects IFN signaling or downstream events of the type I IFN response.
Thus, the picture emerges that coronaviruses counter the antiviral IFN response not by relying on one single IFN antagonistic factor, as many other RNA viruses do [27], [29], [101], [118], but by using a multitude of passive and active mechanisms. Passive mechanisms include the induction of DMVs that may help to hide and protect RNA replication intermediates from getting sensed by intracellular PRRs. Active mechanisms include functions provided by the ORF3b, ORF6, N, nsp1 and ORF7a gene products. Their combined effects may provide an explanation for the absence of the IRF-3-dependent IFN-β and RANTES expression and the STAT-dependent transcription of the IFN-α genes in coronavirus-infected cells. Furthermore, on the effector side, the N protein of MHV was shown to contribute to viral IFN resistance by interfering with the 2-5 OAS pathway [119].
Interestingly, despite this multi-pronged IFN escape strategy, the fact that superinfection with SeV unleashes a strong transcriptional IFN response in SARS-CoV-infected cells suggests an unexpectedly high degree of coronavirus-specific inhibition of IRF-3 by ORF3b, ORF6 and N, which may be explained by the strong compartimentalization of the coronavirus factories in infected cells. Also, it has to be kept in mind that viral IFN antagonism is usually not perfect and serves to delay rather than completely suppress IFN induction [27].
A notable exception from the general picture of an impeded IFN response in coronavirus-infected cells is provided by pDCs. Whereas mDCs infected with SARS-CoV or MHV have no detectable IFN synthesis (similar to fibroblasts), pDCs secrete substantial amounts of IFN in response to these coronaviruses [92]. For MHV it was further shown that this response was dependent on the presence of TLR-7 and MyD88, which induce IFN-α expression via constitutively expressed IRF-7. Apparently, in contrast to the ubiquitous IRF-3 signaling chain, the pDC-restricted TLR-7/IRF-7 pathway is not affected by the virus. TLR-7 is located in endosomes of pDCs and may sense the viral genome during virus entry, or sense viral RNA that has been shuttled to endosomes by autophagosomes [120]. The TLR-7-dependent activation of pDCs most probably provides an important protective mechanism from coronavirus infection. Indeed, depletion of pDCs in mice significantly reduced serum IFN-α levels, and led to increased virus replication, virus spread to multiple organs, and severe clinical signs of disease [92]. Thus, a weak type I IFN response early during coronavirus infection may explain why the prognosis for SARS worsens with increasing age, and it is feasible to suspect that the functionality and responsiveness of these professional IFN producers plays an important role in the protection from severe coronavirus-induced disease.
Interestingly, in an MHV-1-based in vivo model for SARS, it was shown that a mouse strain which develops severe SARS-like symptoms has little IFN production after infection, whereas another mouse strain which was protected from disease produces high amounts of type I IFNs [20]. Moreover, it is well known that the virulence and IFN resistance of MHV strains correlate [121]. In line with this, a recent study using SARS patient materials suggests the presence of high levels of type I IFNs which might be responsible for the recovery of the majority of patients [122]. Thus, the degree of IFN escape by coronaviruses may be host-specific as well as strain-specific and determine viral pathogenesis in a manner similar to what was observed, for e.g. Ebola virus [123], [124].
6. Cytokines and chemokines induced by SARS-CoV and MHV
IRF-3 is not only crucial for IFN induction, but also participates in transactivation of the genes for RANTES [125] and IP-10 [126]. It could therefore be expected that coronaviruses also suppress production of these chemokines (which are also termed CCL5 and CXCL10, respectively). Recent data indicate that RANTES transcripts are indeed absent in tissue cells productively infected with SARS-CoV [107], [127] or MHV [103]. IP-10 transcription, however, is upregulated in some SARS-CoV-infected fibroblast cell lines [107], [127] and in macrophages [105]. Similarly, MHV induces a strong IP-10 response in mouse brain [128], mDCs and in pDCs [92], whereas type I IFNs are only produced in pDCs [92]. This suggests that transcription of IP-10 is (i) less dependent on IRF-3 than RANTES and IFN-β are, and (ii) therefore largely unaffected by coronaviral inhibition mechanisms. Transactivation of the IP-10 gene is not only triggered by IRF-3, but also by the important transcription factor NF-κB [126]. There are some reports that SARS-CoV [127], [129] and MHV [103], [130] trigger an NF-κB response, although other groups could not verify these finding in their systems [104], [131]. Moreover, the SARS-CoV N protein is a strong antagonist of NF-κB [113] and the NF-κB-dependent proinflammatory TNF-α is suspiciously absent in the SARS cytokine profile (see below). Activation of NF-κB may therefore not be the final explanation for the upregulation of the IP-10 gene in SARS-CoV-infected cells. Indeed, recent reports indicate that IRF-5 can be activated by virus infection in a manner similar to IRF-3 [132], and that IRF-5 participates in induction of proinflammatory cytokines rather than type I IFNs [52], [133]. It can be speculated that IRF-5 is involved in the prominent upregulation of the IP-10 gene by SARS-CoV.
Some SARS-CoV-infected cell lines also produce significant amounts of the chemokine IL-8 (CXCL8) [107], [127], an activity which was traced back to the viral spike and nucleocapsid proteins [134], [135]. Similarly, the mouse counterpart of human IL-8, CXCL2, is upregulated in fibroblasts after MHV infection [103]. Expression of IL-8 is dependent on the transcription factor AP-1, and molecular analyses revealed that SARS-CoV and MHV strongly activate AP-1 [103], [129], [135]. Interestingly, human IL-8 was shown to inhibit the antiviral action of IFN [136]. Therefore, besides the direct inhibition of IFN induction by viral proteins (see above), secreted human IL-8 (and possibly mouse CXCL2 as well) might contribute to diminish the IFN response in coronavirus infections.
Taken together, the in vitro cytokine profiles of SARS-CoV and MHV infection appear to be mainly based on the transcriptional activation of IP-10 and IL-8, possibly mediated by NF-κB or IRF-5, and AP-1. IRF-3-depending genes for antiviral cytokines such as type I IFNs, by contrast, remain mostly silenced during the initial phase of infection.
IP-10 is a chemoattractant causing T cell infiltration into coronavirus-infected organs [137]. Initially identified as an IFN-γ-responsive gene, it was later shown to be induced by IFN-α/β [138] and virus infections including those with SARS-CoV [129], [139]. Interestingly, IP-10 is an excellent prognostic marker for SARS disease progression [140], [141], [142]. This implicates that the findings in cell culture reflect the in vivo situation to a considerable extent, and that IP-10-mediated lymphocyte infiltrations may play a major part in SARS pathology in a manner similar to other viral diseases [143].
7. In vivo cytokine profile and SARS pathology
Patient studies can rarely be standardized and controlled with the same accuracy as in vitro studies. Moreover, the kinetics and interrelation of cytokine production and SARS-CoV spread, a critical point for data interpretation, cannot be properly investigated with human subjects. Nonetheless data obtained from ex vivo peripheral blood mononuclear cells or from SARS patients’ sera are largely in agreement with the above-discussed findings in cell culture.
Most studies involving patient materials found no significant upregulation for α/β-IFNs or for IFN-induced genes [139], [140], [144], [145], [146], [147], [148]. Interestingly, however, a recent study investigating immune responses of 40 clinically well-defined SARS cases revealed high levels of plasma IFN-α (but not IFN-β) and an untypical ISG expression profile in pre-crisis patients, but not in the crisis patients [122]. Possibly, these significant amounts of IFN-α early in infection are produced by infected pDCs, as those cells are capable of a full response to SARS-CoV [92].
High levels of the chemokines IL-8 and IP-10 along with the proinflammatory cytokine IL-6 [139], [140], [146], [148], [149], [150], [151], [152], [153] were often detected in patients. Given the cell culture results for SARS-CoV, at least for IL-8 and IP-10 a direct production by virus-infected cells is conceivable. IL-6, however, is not or only weakly induced in productively infected tissue cells [107], [127], [154], [155] but moderately upregulated in abortively infected macrophages and DCs [156]. Interestingly, IL-6 induction by SARS-CoV could be strongly boosted by priming of macrophages with bacterial LPS [157]. Thus, it can be speculated that IL-8 and IP-10 in SARS patients are directly produced by virus-infected cells, whereas upregulation of the proinflammatory cytokine IL-6 is more likely a secondary response due to an activation of the immune system.
High viral load, systemic and multiorgan infection, massive lung infiltrations by monocytes and macrophages, and rapid depletion of T cells are the hallmarks of full-blown SARS [2], [7], [158], [159], [160], [161], [162], [163], [164], [165], [166]. It is debated whether the disease is caused by the virus or is the result of a dysregulated immune response. After primary infection, SARS-CoV grows at a fast rate and spreads to different organs, including the lungs [161], [164], [167]. Autopsies from deceased patients revealed severe damage of the lungs and lymphatic tissues, accompanied by infiltrations of monocytic cells [168], [169], [170]. This may indicate that immunopathogenesis is involved in the severe outcome of the disease, providing the rationale for SARS therapy with immunosuppressant corticosteroids [171]. On the other hand, cell damages could have been directly caused by the virus, as SARS-CoV is cytolytic [172], and high titers of virus have been found in several organs of deceased patients [160], [161], [173]. In addition, signs of necrosis were found besides virus particles in affected tissues [170], and high viral loads are predictive of adverse clinical outcome [174].
The cytokine profile outlined above most probably plays a significant role in SARS pathology. In the initial phase of infection, dampening and misregulating the antiviral IFN response may allow the virus to grow rapidly and spread to different organs, including the lungs [161], [175]. The early IFN-α detected in patients before the onset of disease [122] may be derived from pDCs which are capable of responding to SARS-CoV [92], whereas IFN-β production by tissue cells is suppressed [102]. Thus, the virus buys time during the initial, critical phase of infection in order to establish itself in the host. At the same time, the virus-induced chemokines IP-10 and IL-8 attract immune cells. These invading cells can themselves be infected [161] and might produce even more chemokines and cytokines such as the proinflammatory cytokine IL-6 [142], [156] and possibly also IFN-γ (which can induce even more IP-10) and the anti-inflammatory cytokine TGF-β [139], [140], [146], [176]. This mixture of high-level virus replication followed by the invasion of activated immune cells and production of both pro- and anti-inflammatory cytokines may result in a cytokine storm which leads to organ destruction and exhaustion of the immune system, eventually culminating in the severe and often fatal respiratory distress, the hallmark of full-blown SARS.
8. Concluding remarks
Much has been learned about coronaviruses and their interactions with the IFN and cytokine responses, but a lot of questions still remain to be answered:
-
•
How do coronaviruses escape from getting sensed by cytoplasmic PRRs?
-
•
To which extend do the recently discovered IFN antagonists contribute to coronavirus-induced disease and pathology?
-
•
Plasmid-expressed ORF3b, ORF6 and N of SARS-CoV are all able to inhibit IRF-3 [113], but coronavirus-infected cells fail to block IRF-3 activation by heterologous inducers [100], [104], [112]. How is this specificity of the coronaviral IRF-3 antagonists achieved?
-
•
Are there other, evolutionary conserved IFN antagonists encoded by all coronaviruses, and do they target the same signaling pathway(s)?
-
•
Are other IRFs, such as IRF-7 or IRF-5, inhibited to the same extent as IRF-3?
-
•
How important is the early type I IFN response in vivo? Data in the MHV system indicate that early, mainly pDC-mediated, type I IFN responses are essential to control MHV infections [92]. In agreement with this it has recently been shown that SARS-CoV infection triggers an early type I IFN response in cynomolgus macaques [177]. Interestingly, STAT 1 nuclear import was impaired in SARS-CoV infected cells, but not in surrounding non-infected cells.
-
•
What are the main type I IFN responder cells and how important are they to control coronavirus infection in vivo?
-
•
Which ISG(s) is/are responsible for inhibiting coronavirus replication? Given the high amount of dsRNA in infected cells [43], [100], PKR, RNaseL and ADAR qualify as the prime candidates.
-
•
How do the observed cell type-specific IFN and cytokine expression patterns impact on coronavirus disease and pathology?
To address these questions, robust and reliable animal models of coronavirus infections are needed. MHV as a natural mouse pathogen and the recently developed murine systems for SARS-CoV infection [178], [179], [180] will certainly be of advantage in that context, since they allow for the use of genetically modified virus and host strains. Although the translation of our knowledge of coronavirus–host interactions from the cellular level to the level of the host organism will be a challenging task, it will certainly improve our knowledge of SARS pathogenesis and open up new ways for the prevention and treatment of coronaviral diseases.
Conflict of interest
None.
Acknowledgements
Our own work described in the text was supported by grants from the Deutsche Forschungsgemeinschaft, the Sino-German Center for Research promotion, the german Bundesministerium für Bildung und Forschung, the Swiss National Science Foundation and the European Commission (SARS-DTV SP22-CT-2004-511064).
Biographies

Volker Thiel received his M.Sc. in 1993 and his Ph.D. in 1998 from the Institute of Virology at the University of Würzburg, Germany. He has studied the genome expression of human and animal coronaviruses for over 15 years and has pioneered the development of coronavirus reverse genetic systems. His research has contributed to the elucidation of coronavirus polyprotein processing pathways and the identification of gene products involved in coronavirus replication and discontinuous transcription. His group was involved in sequencing the SARS coronavirus Frankfurt-1 isolate and provided a first molecular analysis on mechanisms and enzymes involved in SARS coronavirus genome expression. In 2003 he served as a Temporary Advisor for the WHO on “Needs and Opportunities for SARS Vaccine Research and Development”. In 2003 he joined the Research Department of the Kantonal Hospital in St. Gallen, Switzerland, where he continued his research with a particular focus on the analysis of coronavirus genome expression, coronavirus–host interactions and the development of coronavirus vaccine vectors. He has published over 30 research papers, review articles, book chapters and has recently edited a book on coronavirus molecular and cellular biology.

Friedemann Weber received his M.Sc. in 1993 from the Department of Microbiology and his Ph.D. in 1997 from the Department of Virology at the University of Freiburg, Germany. He was an EMBO Long Term Postdoctoral Fellow at the Institute of Virology in Glasgow, UK, and is currently a research group leader in the Department of Virology, Freiburg, Germany. In 2003, he has received the Milstein Young Investigator Award from the International Society for Interferon and Cytokine Research (ISICR) and in 2004 the Heine-Medin Medal of the European Society for Clinical Virology. In 2007 he received the “Löffler-Frosch-Preis” of the Gesellschaft für Virologie. He is interested in the innate immune responses to highly pathogenic RNA viruses such as SARS-Coronavirus and bunyaviruses. The particular focus is on interferon-inducing viral structures, their intracellular receptors, and the viral escape strategies. He has published over 50 research papers, review articles and book chapters.
References
- 1.Siddell S.G., Ziebuhr J., Snijder E.J. Coronaviruses, toroviruses, and arteriviruses. In: Mahy B.W.J., ter Meulen V., editors. Topley and Wilson's microbiology and microbial infections. Hodder Arnold; London: 2005. pp. 823–856. [Google Scholar]
- 2.Weiss S.R., Navas-Martin S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev. 2005;69:635–664. doi: 10.1128/MMBR.69.4.635-664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fouchier R.A., Kuiken T., Schutten M., van Amerongen G., van Doornum G.J., van den Hoogen B.G. Aetiology: Koch's postulates fulfilled for SARS virus. Nature. 2003;423:240. doi: 10.1038/423240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S. The Genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 5.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 6.Li W., Shi Z., Yu M., Ren W., Smith C., Epstein J.H. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 7.Perlman S., Dandekar A.A. Immunopathogenesis of coronavirus infections: implications for SARS. Nat Rev Immunol. 2005;5:917–927. doi: 10.1038/nri1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J Mol Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S. Mechanisms and enzymes involved in SARS coronavirus genome expression. J Gen Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 10.Gorbalenya A.E., Enjuanes L., Ziebuhr J., Snijder E.J. Nidovirales: evolving the largest RNA virus genome. Virus Res. 2006;117:17–37. doi: 10.1016/j.virusres.2006.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziebuhr J. The coronavirus replicase. Curr Top Microbiol Immunol. 2005;287:57–94. doi: 10.1007/3-540-26765-4_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sawicki S.G., Sawicki D.L., Siddell S.G. A contemporary view of coronavirus transcription. J Virol. 2006;81:20–29. doi: 10.1128/JVI.01358-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Almazan F., Galan C., Enjuanes L. The nucleoprotein is required for efficient coronavirus genome replication. J Virol. 2004;78:12683–12688. doi: 10.1128/JVI.78.22.12683-12688.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schelle B., Karl N., Ludewig B., Siddell S.G., Thiel V. Selective replication of coronavirus genomes that express nucleocapsid protein. J Virol. 2005;79:6620–6630. doi: 10.1128/JVI.79.11.6620-6630.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ziebuhr J., Snijder E. The coronavirus replicase gene: special enzymes for special viruses. In: Thiel V., editor. Coronaviruses: molecular and cellular biology. Caister scientific press; Norwich, UK: 2007. [Google Scholar]
- 16.Gorbalenya A.E., Snijder E.J., Spaan W.J. Severe acute respiratory syndrome coronavirus phylogeny: toward consensus. J Virol. 2004;78:7863–7866. doi: 10.1128/JVI.78.15.7863-7866.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bergmann C.C., Lane T.E., Stohlman S.A. Coronavirus infection of the central nervous system: host–virus stand-off. Nat Rev Microbiol. 2006;4:121–132. doi: 10.1038/nrmicro1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hemmila E., Turbide C., Olson M., Jothy S., Holmes K.V., Beauchemin N. Ceacam1a−/− mice are completely resistant to infection by murine coronavirus mouse hepatitis virus A59. J Virol. 2004;78:10156–10165. doi: 10.1128/JVI.78.18.10156-10165.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barthold S.W., Beck D.S., Smith A.L. Enterotropic coronavirus (mouse hepatitis virus) in mice: influence of host age and strain on infection and disease. Lab Anim Sci. 1993;43:276–284. [PubMed] [Google Scholar]
- 20.De Albuquerque N., Baig E., Ma X., Zhang J., He W., Rowe A. Murine hepatitis virus strain 1 produces a clinically relevant model of severe acute respiratory syndrome in A/J mice. J Virol. 2006;80:10382–10394. doi: 10.1128/JVI.00747-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Homberger F.R. Enterotropic mouse hepatitis virus. Lab Anim. 1997;31:97–115. doi: 10.1258/002367797780600189. [DOI] [PubMed] [Google Scholar]
- 22.Homberger F.R., Zhang L., Barthold S.W. Prevalence of enterotropic and polytropic mouse hepatitis virus in enzootically infected mouse colonies. Lab Anim Sci. 1998;48:50–54. [PubMed] [Google Scholar]
- 23.Sarma J.D., Fu L., Hingley S.T., Lavi E. Mouse hepatitis virus type-2 infection in mice: an experimental model system of acute meningitis and hepatitis. Exp Mol Pathol. 2001;71:1–12. doi: 10.1006/exmp.2001.2378. [DOI] [PubMed] [Google Scholar]
- 24.Falsey A.R., McCann R.M., Hall W.J., Criddle M.M., Formica M.A., Wycoff D. The “common cold” in frail older persons: impact of rhinovirus and coronavirus in a senior daycare center. J Am Geriatr Soc. 1997;45:706–711. doi: 10.1111/j.1532-5415.1997.tb01474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Hoek L., Pyrc K., Berkhout B. Human coronavirus NL63, a new respiratory virus. FEMS Microbiol Rev. 2006;30:760–773. doi: 10.1111/j.1574-6976.2006.00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Hoek L., Sure K., Ihorst G., Stang A., Pyrc K., Jebbink M.F. Croup is associated with the novel coronavirus NL63. PLoS Med. 2005;2:e240. doi: 10.1371/journal.pmed.0020240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haller O., Kochs G., Weber F. The interferon response circuit: Induction and suppression by pathogenic viruses. Virology. 2006;344:119–130. doi: 10.1016/j.virol.2005.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Samuel C.E. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weber F., Kochs G., Haller O. Inverse interference: how viruses fight the interferon system. Viral Immunol. 2004;17:498–515. doi: 10.1089/vim.2004.17.498. [DOI] [PubMed] [Google Scholar]
- 30.Hertzog P.J., O’Neill L.A., Hamilton J.A. The interferon in TLR signaling: more than just antiviral. Trends Immunol. 2003;24:534–539. doi: 10.1016/j.it.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 31.Le Bon A., Tough D.F. Links between innate and adaptive immunity via type I interferon. Curr Opin Immunol. 2002;14:432–436. doi: 10.1016/s0952-7915(02)00354-0. [DOI] [PubMed] [Google Scholar]
- 32.Stark G.R., Kerr I.M., Williams B.R., Silverman R.H., Schreiber R.D. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 33.Roberts R.M., Ezashi T., Rosenfeld C.S., Ealy A.D., Kubisch H.M. Evolution of the interferon tau genes and their promoters, and maternal-trophoblast interactions in control of their expression. Reprod Suppl. 2003;61:239–251. [PubMed] [Google Scholar]
- 34.van Pesch V., Lanaya H., Renauld J.C., Michiels T. Characterization of the murine alpha interferon gene family. J Virol. 2004;78:8219–8228. doi: 10.1128/JVI.78.15.8219-8228.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Pesch V., Michiels T. Characterization of interferon-alpha 13, a novel constitutive murine interferon-alpha subtype. J Biol Chem. 2003;278:46321–46328. doi: 10.1074/jbc.M302554200. [DOI] [PubMed] [Google Scholar]
- 36.Delhaye S., Paul S., Blakqori G., Minet M., Weber F., Staeheli P. Neurons produce type I interferon during viral encephalitis. Proc Natl Acad Sci USA. 2006;103:7835–7840. doi: 10.1073/pnas.0602460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marie I., Durbin J.E., Levy D.E. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Colonna M., Krug A., Cella M. Interferon-producing cells: on the front line in immune responses against pathogens. Curr Opin Immunol. 2002;14:373–379. doi: 10.1016/s0952-7915(02)00349-7. [DOI] [PubMed] [Google Scholar]
- 39.Diebold S.S., Montoya M., Unger H., Alexopoulou L., Roy P., Haswell L.E. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 2003;424:324–328. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- 40.Hornung V., Ellegast J., Kim S., Brzozka K., Jung A., Kato H. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 41.Pichlmair A., Schulz O., Tan C.P., Naslund T.I., Liljestrom P., Weber F. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′ phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 42.Plumet S., Herschke F., Bourhis J.M., Valentin H., Longhi S., Gerlier D. Cytosolic 5′-triphosphate ended viral leader transcript of measles virus as activator of the RIG I-mediated interferon response. PLoS ONE. 2007;2:e279. doi: 10.1371/journal.pone.0000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weber F., Wagner V., Rasmussen S.B., Hartmann R., Paludan S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol. 2006;80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andrejeva J., Childs K.S., Young D.F., Carlos T.S., Stock N., Goodbourn S. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci USA. 2004;101:17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 46.Yoneyama M., Kikuchi M., Natsukawa T., Shinobu N., Imaizumi T., Miyagishi M. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 47.Yoneyama M., Kikuchi M., Matsumoto K., Imaizumi T., Miyagishi M., Taira K. Shared and unique functions of the DExD/H-Box Helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175:2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 48.Gitlin L., Barchet W., Gilfillan S., Cella M., Beutler B., Flavell R.A. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA. 2006;103:8459–8464. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fitzgerald K.A., McWhirter S.M., Faia K.L., Rowe D.C., Latz E., Golenbock D.T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 50.Sharma S., TenOever B.R., Grandvaux N., Zhou G.P., Lin R., Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 51.Ozato K., Tailor P., Kubota T. The interferon regulatory factor family in host defense: the mechanism of action. J Biol Chem. 2007;282:20065–20069. doi: 10.1074/jbc.R700003200. [DOI] [PubMed] [Google Scholar]
- 52.Paun A., Pitha P.M. The IRF family, revisited. Biochimie. 2007;89:744–753. doi: 10.1016/j.biochi.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J Biol Chem. 2007;282:15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- 54.Sato M., Suemori H., Hata N., Asagiri M., Ogasawara K., Nakao K. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 55.Iwamura T., Yoneyama M., Yamaguchi K., Suhara W., Mori W., Shiota K. Induction of IRF-3/-7 kinase and NF-kappaB in response to double- stranded RNA and virus infection: common and unique pathways. Genes Cells. 2001;6:375–388. doi: 10.1046/j.1365-2443.2001.00426.x. [DOI] [PubMed] [Google Scholar]
- 56.tenOever B.R., Sharma S., Zou W., Sun Q., Grandvaux N., Julkunen I. Activation of TBK1 and IKK epsilon kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J Virol. 2004;78:10636–10649. doi: 10.1128/JVI.78.19.10636-10649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chu W.M., Ostertag D., Li Z.W., Chang L., Chen Y., Hu Y. JNK2 and IKKbeta are required for activating the innate response to viral infection. Immunity. 1999;11:721–731. doi: 10.1016/s1074-7613(00)80146-6. [DOI] [PubMed] [Google Scholar]
- 58.Paladino P., Cummings D.T., Noyce R.S., Mossman K.L. The IFN-independent response to virus particle entry provides a first line of antiviral defense that is independent of TLRs and retinoic acid-inducible gene I. J Immunol. 2006;177:8008–8016. doi: 10.4049/jimmunol.177.11.8008. [DOI] [PubMed] [Google Scholar]
- 59.Alexopoulou L., Holt A.C., Medzhitov R., Flavell R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 60.Iwasaki A., Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 61.Uematsu S., Akira S. Toll-like receptors and type I interferons. J Biol Chem. 2007;282:15319–15323. doi: 10.1074/jbc.R700009200. [DOI] [PubMed] [Google Scholar]
- 62.Kerkmann M., Rothenfusser S., Hornung V., Towarowski A., Wagner M., Sarris A. Activation with CpG-A and CpG-B oligonucleotides reveals two distinct regulatory pathways of type I IFN synthesis in human plasmacytoid dendritic cells. J Immunol. 2003;170:4465–4474. doi: 10.4049/jimmunol.170.9.4465. [DOI] [PubMed] [Google Scholar]
- 63.Prakash A., Smith E., Lee C.K., Levy D.E. Tissue-specific positive feedback requirements for production of type I interferon following virus infection. J Biol Chem. 2005;280:18651–18657. doi: 10.1074/jbc.M501289200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Asselin-Paturel C., Trinchieri G. Production of type I interferons: plasmacytoid dendritic cells and beyond. J Exp Med. 2005;202:461–465. doi: 10.1084/jem.20051395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tsujimura H., Tamura T., Ozato K. Cutting edge: IFN consensus sequence binding protein/IFN regulatory factor 8 drives the development of type I IFN-producing plasmacytoid dendritic cells. J Immunol. 2003;170:1131–1135. doi: 10.4049/jimmunol.170.3.1131. [DOI] [PubMed] [Google Scholar]
- 66.Honda K., Ohba Y., Yanai H., Negishi H., Mizutani T., Takaoka A. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434:1035–1040. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- 67.de Weerd N.A., Samarajiwa S.A., Hertzog P.J. Type I interferon receptors: biochemistry and biological functions. J Biol Chem. 2007;282:20053–20057. doi: 10.1074/jbc.R700006200. [DOI] [PubMed] [Google Scholar]
- 68.Levy D.E., Darnell J.E., Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 69.Schindler C., Levy D.E., Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282:20059–20063. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- 70.Der S.D., Zhou A., Williams B.R., Silverman R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci USA. 1998;95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haller O., Kochs G. Interferon-induced mx proteins: dynamin-like GTPases with antiviral activity. Traffic. 2002;3:710–717. doi: 10.1034/j.1600-0854.2002.31003.x. [DOI] [PubMed] [Google Scholar]
- 72.Haller O., Staeheli P., Kochs G. Interferon-induced Mx proteins in antiviral host defense. Biochimie. 2007;89:812–818. doi: 10.1016/j.biochi.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 73.Kochs G., Haller O. Interferon-induced human MxA GTPase blocks nuclear import of Thogoto virus nucleocapsids. Proc Natl Acad Sci USA. 1999;96:2082–2086. doi: 10.1073/pnas.96.5.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reichelt M., Stertz S., Krijnse-Locker J., Haller O., Kochs G. Missorting of LaCrosse virus nucleocapsid protein by the interferon-induced MxA GTPase involves smooth ER membranes. Traffic. 2004;5:772–784. doi: 10.1111/j.1600-0854.2004.00219.x. [DOI] [PubMed] [Google Scholar]
- 75.Garcia M.A., Meurs E.F., Esteban M. The dsRNA protein kinase PKR: virus and cell control. Biochimie. 2007;89:799–811. doi: 10.1016/j.biochi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 76.Williams B.R. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18:6112–6120. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- 77.Hovanessian A.G., Justesen J. The human 2′-5′oligoadenylate synthetase family: unique interferon-inducible enzymes catalyzing 2′-5′ instead of 3′-5′ phosphodiester bond formation. Biochimie. 2007;89:779–788. doi: 10.1016/j.biochi.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 78.Bisbal C., Silverman R.H. Diverse functions of RNase L and implications in pathology. Biochimie. 2007;89:789–798. doi: 10.1016/j.biochi.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sarkar S.N., Sen G.C. Novel functions of proteins encoded by viral stress-inducible genes. Pharmacol Ther. 2004;103:245–259. doi: 10.1016/j.pharmthera.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 80.Terenzi F., Pal S., Sen G.C. Induction and mode of action of the viral stress-inducible murine proteins, P56 and P54. Virology. 2005;340:116–124. doi: 10.1016/j.virol.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 81.Degols G., Eldin P., Mechti N. ISG20, an actor of the innate immune response. Biochimie. 2007;89:831–835. doi: 10.1016/j.biochi.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 82.Espert L., Degols G., Gongora C., Blondel D., Williams B.R., Silverman R.H. ISG20, a new interferon-induced RNase specific for single-stranded RNA, defines an alternative antiviral pathway against RNA genomic viruses. J Biol Chem. 2003;278:16151–16158. doi: 10.1074/jbc.M209628200. [DOI] [PubMed] [Google Scholar]
- 83.Espert L., Degols G., Lin Y.L., Vincent T., Benkirane M., Mechti N. Interferon-induced exonuclease ISG20 exhibits an antiviral activity against human immunodeficiency virus type 1. J Gen Virol. 2005;86:2221–2229. doi: 10.1099/vir.0.81074-0. [DOI] [PubMed] [Google Scholar]
- 84.Aurisicchio L., Delmastro P., Salucci V., Paz O.G., Rovere P., Ciliberto G. Liver-specific alpha 2 interferon gene expression results in protection from induced hepatitis. J Virol. 2000;74:4816–4823. doi: 10.1128/jvi.74.10.4816-4823.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fuchizaki U., Kaneko S., Nakamoto Y., Sugiyama Y., Imagawa K., Kikuchi M. Synergistic antiviral effect of a combination of mouse interferon-alpha and interferon-gamma on mouse hepatitis virus. J Med Virol. 2003;69:188–194. doi: 10.1002/jmv.10286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pei J., Sekellick M.J., Marcus P.I., Choi I.S., Collisson E.W. Chicken interferon type I inhibits infectious bronchitis virus replication and associated respiratory illness. J Interferon Cytokine Res. 2001;21:1071–1077. doi: 10.1089/107999001317205204. [DOI] [PubMed] [Google Scholar]
- 87.Cinatl J., Morgenstern B., Bauer G., Chandra P., Rabenau H., Doerr H.W. Treatment of SARS with human interferons. Lancet. 2003;362:293–294. doi: 10.1016/S0140-6736(03)13973-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hensley L.E., Fritz L.E., Jahrling P.B., Karp C.L., Huggins J.W., Geisbert T.W. Interferon-beta 1a and SARS coronavirus replication. Emerg Infect Dis. 2004;10:317–319. doi: 10.3201/eid1002.030482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Spiegel M., Pichlmair A., Mühlberger E., Haller O., Weber F. The antiviral effect of interferon-beta against SARS-coronavirus is not mediated by MxA. J Clinical Virol. 2004;30:211–213. doi: 10.1016/j.jcv.2003.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stroher U., DiCaro A., Li Y., Strong J.E., Aoki F., Plummer F. Severe acute respiratory syndrome-related coronavirus is inhibited by interferon-alpha. J Infect Dis. 2004;189:1164–1167. doi: 10.1086/382597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zheng B., He M.L., Wong K.L., Lum C.T., Poon L.L., Peng Y. Potent inhibition of SARS-associated coronavirus (SCOV) infection and replication by type I interferons (IFN-alpha/beta) but not by type II interferon (IFN-gamma) J Interferon Cytokine Res. 2004;24:388–390. doi: 10.1089/1079990041535610. [DOI] [PubMed] [Google Scholar]
- 92.Cervantes-Barragan L., Zust R., Weber F., Spiegel M., Lang K.S., Akira S. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood. 2007;109:1131–1137. doi: 10.1182/blood-2006-05-023770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hogan R.J., Gao G., Rowe T., Bell P., Flieder D., Paragas J. Resolution of primary severe acute respiratory syndrome-associated coronavirus infection requires Stat1. J Virol. 2004;78:11416–11421. doi: 10.1128/JVI.78.20.11416-11421.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Haagmans B.L., Kuiken T., Martina B.E., Fouchier R.A., Rimmelzwaan G.F., Van Amerongen G. Pegylated interferon-alpha protects type 1 pneumocytes against SARS coronavirus infection in macaques. Nat Med. 2004;10:290–293. doi: 10.1038/nm1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhou H., Perlman S. Preferential infection of mature dendritic cells by the JHM strain of mouse hepatitis virus. Adv Exp Med Biol. 2006;581:411–414. doi: 10.1007/978-0-387-33012-9_74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stockman L.J., Bellamy R., Garner P. SARS: systematic review of treatment effects. PLoS Med. 2006;3:e343. doi: 10.1371/journal.pmed.0030343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Loutfy M.R., Blatt L.M., Siminovitch K.A., Ward S., Wolff B., Lho H. Interferon alfacon-1 plus corticosteroids in severe acute respiratory syndrome: a preliminary study. JAMA. 2003;290:3222–3228. doi: 10.1001/jama.290.24.3222. [DOI] [PubMed] [Google Scholar]
- 98.Pestka S. The interferons: 50 years after their discovery, there is much more to learn. J Biol Chem. 2007;282:20047–20051. doi: 10.1074/jbc.R700004200. [DOI] [PubMed] [Google Scholar]
- 99.Akerstrom S., Mousavi-Jazi M., Klingstrom J., Leijon M., Lundkvist A., Mirazimi A. Nitric oxide inhibits the replication cycle of severe acute respiratory syndrome coronavirus. J Virol. 2005;79:1966–1969. doi: 10.1128/JVI.79.3.1966-1969.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Versteeg G.A., Bredenbeek P.J., van den Worm S.H., Spaan W.J. Group 2 coronaviruses prevent immediate early interferon induction by protection of viral RNA from host cell recognition. Virology. 2007;361:18–26. doi: 10.1016/j.virol.2007.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Garcia-Sastre A., Biron C.A. Type 1 interferons and the virus–host relationship: a lesson in detente. Science. 2006;312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 102.Spiegel M., Pichlmair A., Martinez-Sobrido L., Cros J., Garcia-Sastre A., Haller O. Inhibition of beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J Virol. 2005;79:2079–2086. doi: 10.1128/JVI.79.4.2079-2086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Versteeg G.A., Slobodskaya O., Spaan W.J. Transcriptional profiling of acute cytopathic murine hepatitis virus infection in fibroblast-like cells. J Gen Virol. 2006;87:1961–1975. doi: 10.1099/vir.0.81756-0. [DOI] [PubMed] [Google Scholar]
- 104.Zhou H., Perlman S. Mouse hepatitis virus does not induce beta interferon synthesis and does not inhibit its induction by double-stranded RNA. J Virol. 2007;81:568–574. doi: 10.1128/JVI.01512-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cheung C.Y., Poon L.L., Ng I.H., Luk W., Sia S.F., Wu M.H. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J Virol. 2005;79:7819–7826. doi: 10.1128/JVI.79.12.7819-7826.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ziegler T., Matikainen S., Ronkko E., Osterlund P., Sillanpaa M., Siren J. Severe acute respiratory syndrome coronavirus fails to activate cytokine-mediated innate immune responses in cultured human monocyte-derived dendritic cells. J Virol. 2005;79:13800–13805. doi: 10.1128/JVI.79.21.13800-13805.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Spiegel M., Weber F. Inhibition of cytokine gene expression and induction of chemokine genes in non-lymphatic cells infected with SARS coronavirus. Virol J. 2006;3:17. doi: 10.1186/1743-422X-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gosert R., Kanjanahaluethai A., Egger D., Bienz K., Baker S.C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J Virol. 2002;76:3697–3708. doi: 10.1128/JVI.76.8.3697-3708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Prentice E., Jerome W.G., Yoshimori T., Mizushima N., Denison M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem. 2004;279:10136–10141. doi: 10.1074/jbc.M306124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Snijder E.J., van der Meer Y., Zevenhoven-Dobbe J., Onderwater J.J., van der Meulen J., Koerten H.K. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J Virol. 2006;80:5927–5940. doi: 10.1128/JVI.02501-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stertz S., Reichelt M., Spiegel M., Kuri T., Martinez-Sobrido L., Garcia-Sastre A. The intracellular sites of early replication and budding of SARS-coronavirus. Virology. 2007;361:304–315. doi: 10.1016/j.virol.2006.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Roth-Cross J.K., Martinez-Sobrido L., Scott E.P., Garcia-Sastre A., Weiss S.R. Inhibition of the alpha/beta interferon response by mouse hepatitis virus at multiple levels. J Virol. 2007;81:7189–7199. doi: 10.1128/JVI.00013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kopecky-Bromberg S.A., Martinez-Sobrido L., Frieman M., Baric R.A., Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol. 2007;81:548–557. doi: 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Frieman M., Yount B., Heise M., Kopecky-Bromberg S.A., Palese P., Baric R.S. SARSCoV ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rER/golgi membrane. J Virol. 2007;81:9812–9824. doi: 10.1128/JVI.01012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kopecky-Bromberg S.A., Martinez-Sobrido L., Palese P. 7a protein of severe acute respiratory syndrome coronavirus inhibits cellular protein synthesis and activates p38 mitogen-activated protein kinase. J Virol. 2006;80:785–793. doi: 10.1128/JVI.80.2.785-793.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kamitani W., Narayanan K., Huang C., Lokugamage K., Ikegami T., Ito N. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc Natl Acad Sci USA. 2006;103:12885–12890. doi: 10.1073/pnas.0603144103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Züst R., Cervantes-Barragan L., Kuri T., Blakqori G., Weber F., Ludewig B. Identification of coronavirus non-structural protein 1 as a major pathogenicity factor—implications for the rational design of live attenuated coronavirus vaccines. PlOS Pathogens. 2007;3:e109. doi: 10.1371/journal.ppat.0030109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Weber F., Haller O. Viral suppression of the interferon system. Biochimie. 2007;89:836–842. doi: 10.1016/j.biochi.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ye Y., Hauns K., Langland J.O., Jacobs B.L., Hogue B.G. Mouse hepatitis coronavirus A59 nucleocapsid protein is a type I interferon antagonist. J Virol. 2007;81:2554–2563. doi: 10.1128/JVI.01634-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lee H.K., Lund J.M., Ramanathan B., Mizushima N., Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 121.Taguchi F., Siddell S.G. Difference in sensitivity to interferon among mouse hepatitis viruses with high and low virulence for mice. Virology. 1985;147:41–48. doi: 10.1016/0042-6822(85)90225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cameron M.J., Ran L., Xu L., Danesh A., Bermejo-Martin J.F., Cameron C.M. Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in severe acute respiratory syndrome (SARS) patients. J Virol. 2007;81:8692–8706. doi: 10.1128/JVI.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ebihara H., Takada A., Kobasa D., Jones S., Neumann G., Theriault S. Molecular determinants of Ebola virus virulence in mice. PLoS Pathog. 2006;2:e73. doi: 10.1371/journal.ppat.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kash J.C., Mühlberger E., Carter V., Grosch M., Perwitasari O., Proll S.C. Global suppression of the host anti-viral response by Ebola and Marburg viruses: increased antagonism of the type I IFN response is associated with enhanced virulence. J Virol. 2006;80:3009–3020. doi: 10.1128/JVI.80.6.3009-3020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lin R., Heylbroeck C., Genin P., Pitha P.M., Hiscott J. Essential role of interferon regulatory factor 3 in direct activation of RANTES chemokine transcription. Mol Cell Biol. 1999;19:959–966. doi: 10.1128/mcb.19.2.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Leung T.H., Hoffmann A., Baltimore D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell. 2004;118:453–464. doi: 10.1016/j.cell.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 127.Cinatl J., Jr., Hoever G., Morgenstern B., Preiser W., Vogel J.U., Hofmann W.K. Infection of cultured intestinal epithelial cells with severe acute respiratory syndrome coronavirus. Cell Mol Life Sci. 2004;61:2100–2112. doi: 10.1007/s00018-004-4222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Stiles L.N., Hardison J.L., Schaumburg C.S., Whitman L.M., Lane T.E. T cell antiviral effector function is not dependent on CXCL10 following murine coronavirus infection. J Immunol. 2006;177:8372–8380. doi: 10.4049/jimmunol.177.12.8372. [DOI] [PubMed] [Google Scholar]
- 129.Cinatl J., Jr., Michaelis M., Morgenstern B., Doerr H.W. High-dose hydrocortisone reduces expression of the pro-inflammatory chemokines CXCL8 and CXCL10 in SARS coronavirus-infected intestinal cells. Int J Mol Med. 2005;15:323–327. [PubMed] [Google Scholar]
- 130.Rempel J.D., Quina L.A., Blakely-Gonzales P.K., Buchmeier M.J., Gruol D.L. Viral induction of central nervous system innate immune responses. J Virol. 2005;79:4369–4381. doi: 10.1128/JVI.79.7.4369-4381.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Frieman M., Heise M., Baric R. SARS coronavirus and innate immunity. Virus Res. 2007 doi: 10.1016/j.virusres.2007.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Barnes B.J., Moore P.A., Pitha P.M. Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon alpha genes. J Biol Chem. 2001;276:23382–23390. doi: 10.1074/jbc.M101216200. [DOI] [PubMed] [Google Scholar]
- 133.Takaoka A., Yanai H., Kondo S., Duncan G., Negishi H., Mizutani T. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434:243–249. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 134.Chang Y.J., Liu C.Y., Chiang B.L., Chao Y.C., Chen C.C. Induction of IL-8 release in lung cells via activator protein-1 by recombinant baculovirus displaying severe acute respiratory syndrome-coronavirus spike proteins: identification of two functional regions. J Immunol. 2004;173:7602–7614. doi: 10.4049/jimmunol.173.12.7602. [DOI] [PubMed] [Google Scholar]
- 135.He R., Leeson A., Andonov A., Li Y., Bastien N., Cao J. Activation of AP-1 signal transduction pathway by SARS coronavirus nucleocapsid protein. Biochem Biophys Res Commun. 2003;311:870–876. doi: 10.1016/j.bbrc.2003.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Polyak S.J., Khabar K.S., Paschal D.M., Ezelle H.J., Duverlie G., Barber G.N. Hepatitis C virus nonstructural 5A protein induces interleukin-8, leading to partial inhibition of the interferon-induced antiviral response. J Virol. 2001;75:6095–6106. doi: 10.1128/JVI.75.13.6095-6106.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Stiles L.N., Hosking M.P., Edwards R.A., Strieter R.M., Lane T.E. Differential roles for CXCR3 in CD4+ and CD8+ T cell trafficking following viral infection of the CNS. Eur J Immunol. 2006;36:613–622. doi: 10.1002/eji.200535509. [DOI] [PubMed] [Google Scholar]
- 138.Sanda C., Weitzel P., Tsukahara T., Schaley J., Edenberg H.J., Stephens M.A. Differential gene induction by type I and type II interferons and their combination. J Interferon Cytokine Res. 2006;26:462–472. doi: 10.1089/jir.2006.26.462. [DOI] [PubMed] [Google Scholar]
- 139.Yu S.Y., Hu Y.W., Liu X.Y., Xiong W., Zhou Z.T., Yuan Z.H. Gene expression profiles in peripheral blood mononuclear cells of SARS patients. World J Gastroenterol. 2005;11:5037–5043. doi: 10.3748/wjg.v11.i32.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Huang K.J., Su I.J., Theron M., Wu Y.C., Lai S.K., Liu C.C. An interferon-gamma-related cytokine storm in SARS patients. J Med Virol. 2005;75:185–194. doi: 10.1002/jmv.20255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jiang Y., Xu J., Zhou C., Wu Z., Zhong S., Liu J. Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome. Am J Respir Crit Care Med. 2005;171:850–857. doi: 10.1164/rccm.200407-857OC. [DOI] [PubMed] [Google Scholar]
- 142.Tang N.L., Chan P.K., Wong C.K., To K.F., Wu A.K., Sung Y.M. Early enhanced expression of interferon-inducible protein-10 (CXCL-10) and other chemokines predicts adverse outcome in severe acute respiratory syndrome. Clin Chem. 2005;51:2333–2340. doi: 10.1373/clinchem.2005.054460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Christensen J.E., de Lemos C., Moos T., Christensen J.P., Thomsen A.R. CXCL10 is the key ligand for CXCR3 on CD8+ effector T cells involved in immune surveillance of the lymphocytic choriomeningitis virus-infected central nervous system. J Immunol. 2006;176:4235–4243. doi: 10.4049/jimmunol.176.7.4235. [DOI] [PubMed] [Google Scholar]
- 144.Jones B.M., Ma E.S., Peiris J.S., Wong P.C., Ho J.C., Lam B. Prolonged disturbances of in vitro cytokine production in patients with severe acute respiratory syndrome (SARS) treated with ribavirin and steroids. Clin Exp Immunol. 2004;135:467–473. doi: 10.1111/j.1365-2249.2003.02391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Reghunathan R., Jayapal M., Hsu L.Y., Chng H.H., Tai D., Leung B.P. Expression profile of immune response genes in patients with severe acute respiratory syndrome. BMC Immunol. 2005;6:2. doi: 10.1186/1471-2172-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.SARS BGoNRPf Dynamic changes in blood cytokine levels as clinical indicators in severe acute respiratory syndrome. Chin Med J (Engl) 2003;116:1283–1287. [PubMed] [Google Scholar]
- 147.Ward S.E., Loutfy M.R., Blatt L.M., Siminovitch K.A., Chen J., Hinek A. Dynamic changes in clinical features and cytokine/chemokine responses in SARS patients treated with interferon alfacon-1 plus corticosteroids. Antivir Ther. 2005;10:263–275. [PubMed] [Google Scholar]
- 148.Wong C.K., Lam C.W., Wu A.K., Ip W.K., Lee N.L., Chan I.H. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol. 2004;136:95–103. doi: 10.1111/j.1365-2249.2004.02415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hsueh P.R., Chen P.J., Hsiao C.H., Yeh S.H., Cheng W.C., Wang J.L. Patient data, early SARS epidemic. Taiwan. Emerg Infect Dis. 2004;10:489–493. doi: 10.3201/eid1003.030571. [DOI] [PubMed] [Google Scholar]
- 150.Wang W.K., Chen S.Y., Liu I.J., Kao C.L., Chen H.L., Chiang B.L. Temporal relationship of viral load, ribavirin, interleukin (IL)-6, IL-8, and clinical progression in patients with severe acute respiratory syndrome. Clin Infect Dis. 2004;39:1071–1075. doi: 10.1086/423808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Xu L., Ran L., Cameron M.J., Persad D., Danesh A., Gold W. Proinflammatory gene expression profiles and severity of disease course in SARS patients. International Conference on SARS, One Year After the (first) Outbreak; Luebeck, Germany; 2004. [Google Scholar]
- 152.Yen Y.T., Liao F., Hsiao C.H., Kao C.L., Chen Y.C., Wu-Hsieh B.A. Modeling the early events of severe acute respiratory syndrome coronavirus infection in vitro. J Virol. 2006;80:2684–2693. doi: 10.1128/JVI.80.6.2684-2693.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhang Y., Li J., Zhan Y., Wu L., Yu X., Zhang W. Analysis of serum cytokines in patients with severe acute respiratory syndrome. Infect Immun. 2004;72:4410–4415. doi: 10.1128/IAI.72.8.4410-4415.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ng L.F., Hibberd M.L., Ooi E.E., Tang K.F., Neo S.Y., Tan J. A human in vitro model system for investigating genome-wide host responses to SARS coronavirus infection. BMC Infect Dis. 2004;4:34. doi: 10.1186/1471-2334-4-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tang B.S., Chan K.H., Cheng V.C., Woo P.C., Lau S.K., Lam C.C. Comparative host gene transcription by microarray analysis early after infection of the Huh7 cell line by severe acute respiratory syndrome coronavirus and human coronavirus 229E. J Virol. 2005;79:6180–6193. doi: 10.1128/JVI.79.10.6180-6193.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Law H.K., Cheung C.Y., Ng H.Y., Sia S.F., Chan Y.O., Luk W. Chemokine upregulation in SARS coronavirus infected human monocyte derived dendritic cells. Blood. 2005;106:2366–2374. doi: 10.1182/blood-2004-10-4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tseng C.T., Perrone L.A., Zhu H., Makino S., Peters C.J. Severe acute respiratory syndrome and the innate immune responses: modulation of effector cell function without productive infection. J Immunol. 2005;174:7977–7985. doi: 10.4049/jimmunol.174.12.7977. [DOI] [PubMed] [Google Scholar]
- 158.Ding Y., He L., Zhang Q., Huang Z., Che X., Hou J. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: implications for pathogenesis and virus transmission pathways. J Pathol. 2004;203:622–630. doi: 10.1002/path.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 160.Farcas G.A., Poutanen S.M., Mazzulli T., Willey B.M., Butany J., Asa S.L. Fatal severe acute respiratory syndrome is associated with multiorgan involvement by coronavirus. J Infect Dis. 2005;191:193–197. doi: 10.1086/426870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gu J., Gong E., Zhang B., Zheng J., Gao Z., Zhong Y. Multiple organ infection and the pathogenesis of SARS. J Exp Med. 2005;202:415–424. doi: 10.1084/jem.20050828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lau Y.L., Peiris J.S. Pathogenesis of severe acute respiratory syndrome. Curr Opin Immunol. 2005;17:404–410. doi: 10.1016/j.coi.2005.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Li T., Qiu Z., Zhang L., Han Y., He W., Liu Z. Significant changes of peripheral T lymphocyte subsets in patients with severe acute respiratory syndrome. J Infect Dis. 2004;189:648–651. doi: 10.1086/381535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Peiris J.S., Yuen K.Y., Osterhaus A.D., Stohr K. The severe acute respiratory syndrome. N Engl J Med. 2003;349:2431–2441. doi: 10.1056/NEJMra032498. [DOI] [PubMed] [Google Scholar]
- 165.Tsui P.T., Kwok M.L., Yuen H., Lai S.T. Severe acute respiratory syndrome: clinical outcome and prognostic correlates. Emerg Infect Dis. 2003;9:1064–1069. doi: 10.3201/eid0909.030362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wong R.S., Wu A., To K.F., Lee N., Lam C.W., Wong C.K. Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. BMJ. 2003;326:1358–1362. doi: 10.1136/bmj.326.7403.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chan K.H., Poon L.L., Cheng V.C., Guan Y., Hung I.F., Kong J. Detection of SARS coronavirus in patients with suspected SARS. Emerg Infect Dis. 2004;10:294–299. doi: 10.3201/eid1002.030610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ding Y., Wang H., Shen H., Li Z., Geng J., Han H. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J Pathol. 2003;200:282–289. doi: 10.1002/path.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lang Z.W., Zhang L.J., Zhang S.J., Meng X., Li J.Q., Song C.Z. A clinicopathological study of three cases of severe acute respiratory syndrome (SARS) Pathology. 2003;35:526–531. doi: 10.1080/00313020310001619118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Nicholls J.M., Poon L.L., Lee K.C., Ng W.F., Lai S.T., Leung C.Y. Lung pathology of fatal severe acute respiratory syndrome. Lancet. 2003;361:1773–1778. doi: 10.1016/S0140-6736(03)13413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bermejo J.F., Munoz-Fernandez M.A. Severe acute respiratory syndrome, a pathological immune response to the new coronavirus—implications for understanding of pathogenesis, therapy, design of vaccines, and epidemiology. Viral Immunol. 2004;17:535–544. doi: 10.1089/vim.2004.17.535. [DOI] [PubMed] [Google Scholar]
- 172.Ng M.L., Tan S.H., See E.E., Ooi E.E., Ling A.E. Proliferative growth of SARS coronavirus in Vero E6 cells. J Gen Virol. 2003;84:3291–3303. doi: 10.1099/vir.0.19505-0. [DOI] [PubMed] [Google Scholar]
- 173.Mazzulli T., Farcas G.A., Poutanen S.M., Willey B.M., Low D.E., Butany J. Severe acute respiratory syndrome-associated coronavirus in lung tissue. Emerg Infect Dis. 2004;10:20–24. doi: 10.3201/eid1001.030404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hung I.F., Cheng V.C., Wu A.K., Tang B.S., Chan K.H., Chu C.M. Viral loads in clinical specimens and SARS manifestations. Emerg Infect Dis. 2004;10:1550–1557. doi: 10.3201/eid1009.040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Peiris J.S., Guan Y., Yuen K.Y. Severe acute respiratory syndrome. Nat Med. 2004;10:S88–S97. doi: 10.1038/nm1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lee C.H., Chen R.F., Liu J.W., Yeh W.T., Chang J.C., Liu P.M. Altered p38 mitogen-activated protein kinase expression in different leukocytes with increment of immunosuppressive mediators in patients with severe acute respiratory syndrome. J Immunol. 2004;172:7841–7847. doi: 10.4049/jimmunol.172.12.7841. [DOI] [PubMed] [Google Scholar]
- 177.de Lang A., Baas T., Teal T., Leijten L.M., Rain B., Osterhaus A.D. Functional genomics highlights differential induction of antiviral pathways in the lungs of SARS-CoV infected macaques. PlOS Pathogens. 2007;3:e112. doi: 10.1371/journal.ppat.0030112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Roberts A., Deming D., Paddock C.D., Cheng A., Yount B., Vogel L. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 2007;3:e5. doi: 10.1371/journal.ppat.0030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tseng C.T., Huang C., Newman P., Wang N., Narayanan K., Watts D.M. Severe acute respiratory syndrome coronavirus infection of mice transgenic for the human angiotensin-converting enzyme 2 virus receptor. J Virol. 2007;81:1162–1173. doi: 10.1128/JVI.01702-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.McCray P.B., Jr., Pewe L., Wohlford-Lenane C., Hickey M., Manzel L., Shi L. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J Virol. 2007;81:813–821. doi: 10.1128/JVI.02012-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yoneyama M., Fujita T. Function of RIG-I-like receptors in antiviral innate immunity. J Biol Chem. 2007;282:15315–15318. doi: 10.1074/jbc.R700007200. [DOI] [PubMed] [Google Scholar]