

Abstract
BACKGROUND
First introduced in 2006, recombinase polymerase amplification (RPA) has stirred great interest, as evidenced by 75 publications as of October 2015, with 56 of them just in the last 2 years. The widespread adoption of this isothermal molecular tool in many diagnostic fields represents an affordable (approximately 4.3 USD per test), simple (few and easy hands-on steps), fast (results within 5–20 min), and sensitive (single target copy number detected) method for the identification of pathogens and the detection of single nucleotide polymorphisms in human cancers and genetically modified organisms.
CONTENT
This review summarizes the current knowledge on RPA. The molecular diagnostics of various RNA/DNA pathogens is discussed while highlighting recent applications in clinical settings with focus on point-of-care (POC) bioassays and on automated fluidic platforms. The strengths and limitations of this isothermal method are also addressed.
SUMMARY
RPA is becoming a molecular tool of choice for the rapid, specific, and cost-effective identification of pathogens. Owing to minimal sample-preparation requirements, low operation temperature (25–42 °C), and commercial availability of freeze-dried reagents, this method has been applied outside laboratory settings, in remote areas, and interestingly, onboard automated sample-to-answer microfluidic devices. RPA is undoubtedly a promising isothermal molecular technique for clinical microbiology laboratories and emergence response in clinical settings.
In the last decade, there was a remarkable increase in the development and adaptation of novel and existing isothermal amplification technologies for molecular diagnostics. More than a dozen isothermal technologies, differing in their respective enzymatic mechanisms, are known and have been recently reviewed elsewhere (1–3). In this review, we focus on recombinase polymerase amplification (RPA)3 because of its simplicity (few and easy hands-on manipulations), flexibility (different commercial kit formats offering various detection methods for both DNA and RNA), and speed (results in 5–20 min).
RPA entails 2 primers and 1 probe (optional) with simple design requirements. For DNA unwinding and primer annealing, RPA uses recombinase enzymes with accessory proteins. RPA has high specificity and efficiency (104-fold amplification in 10 min) (4) and displays a completely isothermal profile not requiring an additional temperature step for DNA denaturation. RPA reagents are available in dried formats, hence facilitating their application in diagnostics. Consequently, RPA technology has been increasingly used in different fields, resulting in a remarkable output of publications; 75 papers are now available on PubMed (with 56 of them published in the last 2 years; see Fig. 1 in the Data Supplement that accompanies the online version of this review at http://www.clinchem.org/content/vol62/issue7). We summarize and discuss here the current knowledge about this technology while focusing on recent diagnostic applications.
How Does RPA Work?
RPA comprises 2 key proteins to substitute for the usual heat denaturation step in PCR: the Escherichia coli RecA recombinase and single-strand DNA binding protein (SSB). The replication is performed by a DNA polymerase having strand-displacement activity necessary to extend the primer. Current commercial kits use the SauDNA polymerase from Staphylococcus aureus. Accessory proteins and cofactors also support the RPA reaction process, such as the T4 UvsY protein, a recombinase loading factor that assists RecA during the formation of the nucleoprotein filament. Polyethylene glycol is a macromolecule of high molecular weight shown to stimulate the interaction of RPA key proteins with DNA, by acting as a crowding agent. Creatine kinase uses phosphocreatine to generate ATP and fuel the enzymes of the system. This composition is used in typical RPA kits, but alternative components have been described; for instance, RecA could be replaced by T4 UvsX protein, SSB protein by T4 gp32, Sau polymerase by Bsu polymerase, and polyethylene glycol can be replaced by Carbowax20M (4).
In RPA, the recombinase, assisted by the loading factor, forms a nucleoprotein filament with single-stranded oligonucleotide primers and probes. This filament scans the double-stranded DNA (dsDNA) target searching for homologous sequences and, once homology is found, the filament invades the dsDNA, forming a D-loop structure that is a local separation of DNA strands in which the complementary strand is stabilized by SSBs and the target strand is hybridized with primer (Fig. 1). Recombinase disassembly from the nucleoprotein filament, induced by RecA protein hydrolyzing ATP, allows primer elongation by strand-displacing DNA polymerase. Newly generated DNA strands are used for another round of RPA. Consequently, an exponential amplification is accomplished by repeating the RPA cycle, which is described as self-perpetuating until exhaustion of the phosphocreatine pool (5).
Fig. 1. RPA cycle.
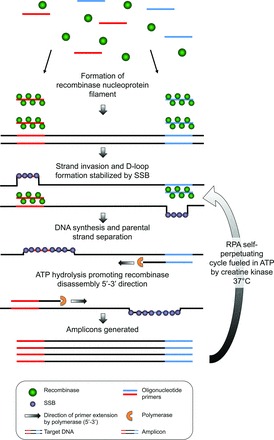
The 3 core proteins, recombinase, SSB, and strand-displacing polymerase enable exponential DNA amplification without the need for thermal cycling or an initial chemical or thermal melting step. The complete reaction is performed at a single temperature from 25–42 °C depending on RPA kit formulation.
Typically, RPA reactions are executed at a single temperature (37 °C) in 5–20 min depending on the starting template copy number and amplicon size (6). Additionally, it is possible to amplify RNA targets by incorporating a reverse transcriptase to RPA reagent components; 1-step reverse-transcriptase RPA (RT-RPA) operates at a single temperature (40–42 °C for exo RT or basic RT kits, respectively) while still enabling target detection in 20 min (7).
COMMERCIAL RPA KITS
RPA is currently commercialized for research use only (TwistDx) in kit configurations enabling DNA or RNA amplification, and detection of amplification products can be visualized by agarose gel electrophoresis, lateral flow strips, or real-time fluorescent probes (Table 1) (8). A typical RPA kit comprises individual tubes containing lyophilized RPA reagent pellets, 1× reaction buffer, magnesium acetate, and controls (positive control DNA with control primers and probe). It is possible to incorporate target-specific RPA primers/probe to the lyophilized RPA reagent pellet (9). RPA kits can be stored for up to 6 months at −20 °C before use.
Table 1.
RPA commercial kits.
| Kits (TwistAmp®)a | Target type | Incubation, ° C | Detection probe | Postamplification purification | Detection systemb | Specific application kits |
|---|---|---|---|---|---|---|
| Basic | DNA | 37–39 | No | Yes | AG | — |
| Basic RT | RNA | 40–42 | No | Yes | AG | — |
| nfo | DNA | 37–39 | Yes | Yes (only for AG detection) | LF/rt/AG | Salmonella, Red Snapperc |
| exo | DNA | 37–39 | Yes | No | rt | Salmonella, Listeria monocytogenes, Campylobacterd |
| exo RT | RNA | 40–42 | Yes | No | rt | — |
| fpg | DNA | 37–39 | Yes | Yes (only for AG detection) | rt/AG | — |
RT, reverse transcription; exo, exonuclease III real-time detection format; fpg, Fpg nuclease real-time detection format; nfo, Nfo nuclease lateral-flow detection format.
AG, agarose gel electrophoresis; rt, real-time; LF, lateral flow.
Kits for food safety and ID analysis: TwistFlow® Salmonella and TwistFlow® Red Snapper.
Kits for food safety and ID analysis: TwistGlow® Salmonella, TwistAmp® exo+ListeriaM, and TwistAmp® exo+Campylobacter.
The RPA experimental protocol necessitates simple hands-on manipulation. A 50-μL reaction solution is formed by mixing RPA primers and/or probe with a buffer solution, and target nucleic acids. This mixture is then added directly to 1 lyophilized RPA reagent pellet followed by brief mixing and centrifugation steps. The reaction is initiated by adding magnesium. Incubation is performed at a single temperature ranging between 37 and 42 °C depending on the kit used (Table 1). After incubation, the detection of RPA products can be either performed in real time or after the reaction. With real-time detection, results are obtained during the incubation stage, typically in <10 min. With post-RPA detection, when agarose gel electrophoresis is used, RPA amplicons must first be purified, and then separated on gel. With lateral-flow detection, RPA amplicons can be used directly without purification, generating results on the dipstick strip within 5 min post-RPA.
Design and Function of RPA Primers and Probes
According to the manufacturer's guidelines for RPA primer and probe design, RPA primers should be 30–35 bases long for the optimal formation of recombinase/primer filaments. Longer primers (>45 bases) are not recommended. Long tracks of one particular nucleotide or a large number of small repeats should be avoided. Low or high GC content (<30% or >70%) must also be avoided. RPA can amplify long sequences up to 1.5 kb; however, better results are obtained with shorter amplicons in the range of 80–400 bp (100–200 bp optimal) (4). In addition, there are no melting temperature requirements for RPA primers and probes because primer annealing and elongation are enzyme mediated and not temperature driven.
For real-time detection, 2 kinds of probes, RPA-exo or fpg, can be used. Conventional probes such as TaqMan cannot be used in RPA, and PCR Taq polymerases are not compatible with the amplification system either. The 5′→3′ exonuclease activity of Taq polymerase progressively digests the displaced strand during the strand displacement process, thus inhibiting DNA amplification. This explains the use of strand displacement polymerases in RPA that do not have the 5′→3′ exonuclease activity required for TaqMan probe detection (e.g., BsuDNA polymerase). The exo probe is a long oligonucleotide (46–52 bases) bearing an internal base analog [e.g., tetrahydrofuran (THF)] located between a fluorophore (e.g., FAM or TAMRA) and a quencher (e.g., Black Hole Quencher 1 or Black Hole Quencher 2) with a blocked 3′ end (e.g., 3′ phosphate group or dideoxynucleotide). This abasic residue serves as a substrate for E. coli exonuclease III that can cleave THF only after binding of the probe to the target sequence (10), thus separating the fluorophore from its quencher (Fig. 2A). Fluorescence generation typically yields a detectable signal within 5–10 min during the RPA reaction (6). It should be noted that exonuclease cleavage generates a free 3′-end of the exo probe, which is then extendable by polymerase enzyme serving as a forward primer (4). Compared to the exo probe, the fpg probe is shorter (32–35 bases) and cannot serve as a primer. It has a 5′ quencher and a fluorophore at 5–6 bases downstream attached to the ribose of an abasic nucleotide via a C-O-C linker, herein deoxyribose (dR)-fluorophore. Fpg, an 8-oxoguanine DNA glycosylase, has catalytic mode different from that of exonuclease III because it recognizes and cleaves the dR-fluorophore while leaving 2 nonextendable sequences (Fig. 2B) (10).
Fig. 2. RPA probes.
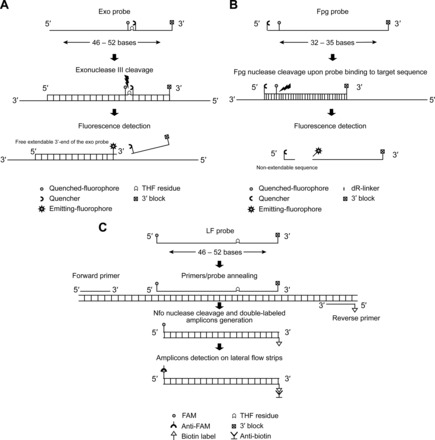
Real-time detection (A) RPA-exo probe; (B) RPA-fpg probe. Exonuclease III and Fpg nucleases recognize and cut the internal abasic residue, thus generating fluorescence. (C), Post-RPA detection with LF probe. Amplicon detection is accomplished by capture of tags with anti-FAM and anti-Biotin antibodies generating a visual colored line on LF strips.
For post-RPA lateral flow (LF) detection using the nfo kit, the design of an LF-probe requires a 5′-fluorophore tag (i.e., FAM) and a THF residue for E. coli endonuclease IV (Nfo) recognition and cleavage, thus serving as a primer similar to exo probe (Fig. 2B). Therefore, the Nfo endonuclease can replace exonuclease III because both recognize the same substrate (i.e., THF residue). However, Nfo generates a lower signal and incomplete cleavage; limiting the degradation of amplicons allows amplicon detection by gel electrophoresis, an option not possible when using an exo probe (10). For lateral flow assay, the reverse primer (opposing amplification primer) has a 5′-biotin label and forms with the labeled nfo probe a double labeled amplicon which is then captured with species-specific anti-FAM antibody coupled to gold nanoparticles. Another immobilized antibody captures biotin, thus forming the detection line on commercially available strips (MGHD 1, TwistDx). A control line with immobilized antispecies antibodies serves to monitor the lateral flow process (Fig. 2C).
By analyzing a total of 204 RPA primers and 64 probe [exo (75%), fpg (14%), and nfo (11%) probes] sequences originating from 40 of the 75 published RPA articles, we observed that some authors have succeeded in using RPA primers/probes presenting some differences from the manufacturer guidelines. For instance, 9% of RPA primers were below 30 bases and 7% were over 35 bases long (36–45 bases). Short sequences of 20–23 bases were used in studies for which primers also served as capture probes (11, 12). Ten percent of primers sequences had a G+C content <30% and 3% had a G+C content >65%. For published exo probes sequences, 17% of exo probes were shorter than the recommended length (34–44 bases instead of 46–52 bases) and 10% were of 53 and 58 bases long. A small proportion (6%) of exo probes had G+C content below 30%. Most of the probes that were considered in our review used the exo probe real-time detection method (75%) rather than fpg (14%) or nfo (11%) probes. Therefore, we considered that discrepant data for fpg and nfo probes were not as representative as those for exo probes (see online Supplemental Fig. 2).
The Diversity of RPA Applications
RPA was successfully used to detect major human pathogens including bacteria (6, 13–23), viruses (9, 24–32), fungi (33), and parasites (34, 35), as well as genetically modified organisms (36, 37) and genetic alterations observed in cancer cells (38, 39). RPA was also used for HIV diagnosis in low-resource settings (40, 41) (Tables 2 and 3). The analytical limit of detection and the turnaround time varied between assays: for RNA detection, 10–21 copies of target detected in 2–20 min and 100–5 × 104 copies in 4–20 min; for DNA detection, 1–50 copies detected in 7–35 min and 98–778 copies detected in 8–35 min, suggesting that RPA efficiency could be dependent on target sequence, amplicon size, and type of biological sample tested. It was demonstrated that fpg probes are less sensitive (104 target molecules detected) than exo probe assays (10 target molecules detected), suggesting that the Fpg nuclease has slower kinetics in real-time detection than that of exonuclease III (14). This explains the preferred use of real-time exo kit. The only advantage of fpg over exo kit is the fact that the former does allow detection by agarose gel electrophoresis.
Table 2.
Application of RPA for RNA pathogens.
| Pathogen | Biological samplesa | Analytical sensitivity, no. of copies | Time-to-result (min) | Detection methodb | Multiplex (no. primers; no. probes)c | Reference |
|---|---|---|---|---|---|---|
| Rift Valley fever virus | — | 19 | 8 | rt | — | Euler et al. (24) |
| Rift Valley fever virus | PlasmaS | 19 | 7 | rt | — | Euler et al. (14) |
| Ebola virus | PlasmaS | 21 | 7 | rt | — | Euler et al. (14) |
| Sudan virus | PlasmaS | 17 | 8 | rt | — | Euler et al. (14) |
| Marburg virus | PlasmaS | 21 | 8 | rt | — | Euler et al. (14) |
| Sigma virus | PlasmaS | 16 | 4 | rt | — | Euler et al. (14) |
| Bovine coronavirus | Fecal/nasal swabs | 19 | 10–20 | rt | — | Am et al. (32) |
| Middle East respiratory syndrome coronavirus | — | 21 | 3–7 | rt | — | Abd El Wahed et al. (26) |
| Foot-and-mouth disease virus | Vesicular material/saliva Serum/blood/nasal swabs | 1436 | 4–10 | rt | — | Abd El Wahed et al. (25) |
| Schmallenberg virus | Blood/serum | 6 × 103 | <10 | rt | — | Aebischer et al. (28) |
| Bovine viral diarrhea virus | Extracted RNA from blood/serum/tissue samples | 5 × 104 | <10 | rt | — | Aebischer et al. (28) |
| Yellow fever virus | RNA extracts/plasmaS | 21–44 | <10 | rt/LF | — | Escadafal et al. (42) |
| Dengue virus | Serum | 10 | <20 | rt | Yes (3;1) | Teoh et al. (31) |
| Dengue virus | RNA extracts/plasmaS | 14–241 | 3–7 | rt | — | Abd El Wahed et al. (43) |
| Little cherry virus 2 | Crude extracts | NDd | 25 | LF | — | Mekuria et al. (29) |
| Plum pox virus | Plant crude extracts | 104 | 15–20 | rt/LF | — | Zhang et al. (9) |
| Influenza A H7N9 | — | 10–100 | 2–7 | rt | — | Abd El Wahed et al. (7) |
S designates spiked biological sample.
rt, real-time, TwistAmp® exo kit; LF, lateral flow, TwistAmp® nfo kit or TwistAmp nfo® RT kit.
No value indicates simplex assay with only 2 primers and 1 probe.
ND, not determined; however, the reported sensitivity was of 0.1 ng of pure RNA (29).
Table 3.
Application of RPA for DNA pathogens.
| Pathogen | Biological samplesa | Analytical sensitivity, copy number | Time-to-result, min | Detection methodb | Multiplex (no. primers; no. probes)c | Reference |
|---|---|---|---|---|---|---|
| Methicillin-resistant Staphylococcus aureus | — | <10 | <30 | rt/LF/AG | Yes (4;3) | Piepenburg et al. (4) |
| Francisella tularensis | Tissue | 19 | 10 | rt | — | Euler et al. (13) |
| Francisella tularensis | PlasmaS | 19 | 10 | rt | — | Euler et al. (14) |
| Bacillus anthracis | PlasmaS | 16–778 | 7–8 | rt | — | Euler et al. (14) |
| Yersinia pestis | PlasmaS | 16 | 8 | rt | — | Euler et al. (14) |
| Variola virus | PlasmaS | 16 | 10 | rt | — | Euler et al. (14) |
| HIV-1 proviral | — | <10 | 20–30 | rt/LF | — | Boyle et al. (41) |
| Mycobacterium tuberculosis | Sputum/respiratory washes | ≅1*d | <20 | rt | — | Boyle et al. (16) |
| Leptospira | Serum/blood | <2 | ≅25 | rt | — | Ahmed et al. (15) |
| Chlamydia trachomatis | Urine | 5–12 | <20 | LF | — | Krolov et al. (21) |
| Shiga toxin-producing Escherichia coli | — | NDe | 5–10 | rt | — | Murinda et al. (22) |
| Streptococcus agalactiae | Vaginal/anal swabs | 98 | 9 | rt | Yes (4;2) | Daher et al. (17) |
| Plasmodium falciparum | Whole bloodS/serumS | ≅4**d | <20 | LF | — | Kersting et al. (35) |
| Giardia duodenalis | Extracted DNA from fresh stool | 50 | 35 | LF | — | Crannell et al. (69) |
| Fungi | Fungal cells suspension | ND | ≅60 | AG | — | Sakai et al. (33) |
| Shrimp white spot syndrome virus | Shrimp | 10 | 6.41 ± 0.17 | rt | — | Xia et al. (30) |
| Penaeus stylirostris densovirus | Shrimp | 100 | 35 | LF | — | Jaroenram et al. (70) |
| Infectious hypodermal and hematopoietic necrosis virus | Shrimp | 4 | 7 | rt | — | Xia et al. (71) |
S designates spiked biological sample.
rt, real-time, TwistAmp® exo kit; LF, lateral flow, TwistAmp® nfo kit; AG, agarose gel electrophoresis, TwistAmp® basic kit.
No value indicates simplex assay with only 2 primers and 1 probe.
*, the reported sensitivity was 6.5 fg, which was estimated to contain the equivalent of a single bacterial cell (16);
, the reported sensitivity was 100 fg of genomic DNA which was estimated to contain the equivalent of 4 parasites per reaction (35).
ND, not determined; however, the reported sensitivity was 5–7 CFU/mL (22).
For RNA detection, several groups (7, 9, 27, 29, 31) have used an exo RT kit that contained murine leukemia virus (MuLV) RT in the lyophilized pellet mixture (7, 25, 42, 43). RT-RPA assay sensitivities ranged between 10 and 104 target copies in 2–25 min (Table 2). We also designed an RNA detection assay using the same kit (TwistAmp® exo RT kit) for the detection of RNA transcript from influenza A virus in the presence of pooled nasopharyngeal swab samples (a combination of different nasopharyngeal samples negative for influenza A virus). We obtained a limit of detection of 8.77 ± 2.24 RNA copies (95% CI) vs 13.5 ± 6.6 RNA copies (95% CI) with and without the samples, respectively. In both cases, 5 copies of RNA transcript were detected in <20 min, thus correlating with results of other studies (see online Supplemental Table 1).
Other groups successfully added Transcriptor Reverse Transcriptase (Roche Molecular Diagnostics) to TwistAmp exo kit reagent mixture for 1-step RT-RPA (14, 24–26, 28, 29, 32). Euler et al. were the first to evaluate different RT enzymes with RPA reagents and demonstrated that Transcriptor RT performs better than MuLV RT (10 molecules detected with Transcriptor RT vs 102 molecules with MuLV RT) and Sensiscript RT (Qiagen; 103 molecules detected) when combined to exo kit in an assay for detecting the Rift Valley fever virus (24). Another study also reported a reduced performance of MuLV RT compared to Transcriptor in an assay detecting RNA extracts for foot-and-mouth disease virus from 45 samples (62% sensitivity for MuLV RT–based RT-RPA and 98% sensitivity for RT-RPA using Transcriptor RT) (25). When attempting to compare the performance of MuLV RT to Transcriptor RT, we obtained similar RT enzyme performances; 104 copies of Influenza A RNA transcript were detected in 3.5 min with MuLV RT compared to 5 min with Transcriptor RT with similar fluorescence signal intensity of 100 AFU (see online Supplemental Fig. 3). Therefore, it seems that RPA could be used with different RT enzymes. However, there is yet no publication on the use of RT-RPA for quantitative RNA measurements.
The Performance of RPA in the Presence of Contaminants
RPA has demonstrated a certain tolerance to common PCR inhibitors and it was shown to operate with nucleic acids extracted from various sample matrices such as blood (28), serum (31), fecal (32), nasal (16) and vaginal swabs (17), plasma (14), foodstuff (44), plants (9), animal tissues (30), milk (12), stool (34), and urine (21). For instance, RPA was able to function in the presence of 15%–25% of milk (6.3–7.2 mmol/L), which is the maximum concentration tolerated by PCR (12). The RPA method amplified targets even in presence of 50 g/L of hemoglobin, 4% vol/vol of ethanol, 0.5 U of heparin, or serum (35). However, RPA was inhibited by whole blood (35).
In a previous study, we evaluated the tolerance of RPA to different types of clinical samples including stool samples. Briefly, different volumes of a diluted stool sample [5 μL of stool diluted in 750 μL 1X TE buffer (10 mmol/L Tris, pH 8.0 with HCl and 1 mmol/L EDTA)] ranging from 0.5 to 5 μL were inoculated with 103 genome copies of Streptococcus agalactiae genomic DNA and amplified with the exo kit. RPA was still efficient with 5 μL of stool sample added (i.e., 1/10 of reaction volume) with no significant inhibition (results were generated in <15 min). Hence, this suggests that with appropriate sample preparation, direct RPA amplification and detection from crude samples may be feasible. For instance, by consolidating sample preparation with nucleic acid amplification/detection, Erh-Chia and colleagues were able to detect methicillin-resistant Staphylococcus aureus (MRSA) DNA directly from spiked blood samples. The process consisted of inertial plasma separation avoiding red blood cells lysis thereby eliminating a potential PCR-inhibitor, hemoglobin. This system proved to have >99% separation efficiency (45). Ultimately, the best sample preparation approach should be adjusted for each type of application and should consider many factors such as target concentration, presence of inhibitors, and lysis efficiency.
Alternative Post-RPA Detection Methods
RPA has been adapted into various end-point detection assays on the basis of immobilization of specific oligonucleotide sequence onto a solid-support and conducting either a pre-RPA amplification or surface RPA amplification. For example, hybridization assays have been performed on microtiter plates (i.e., RPA-ELISA) (44), in a sandwich assay using an aptamer (38), and on novel solid-phase platforms (e.g., on DVD, epoxy silane, or silicon microring surfaces) (11, 39, 46, 47).
RPA was combined with ELISA in a food safety analysis and in tests for detecting allergens, genetically modified organisms, bacteria, and fungi. RPA-ELISA performed as well as PCR-ELISA, showing similar sensitivities (1.29–19.74 μg of analyte/g of food for RPA-ELISA compared to 1.24–13.27 μg of analyte per gram of food for PCR-ELISA) and assay reproducibility (7.9%–11.3% for RPA-ELISA compared to 8.5%–14.5% for PCR-ELISA) (44). However, hybridization assays and ELISA tests are fastidious and require several optimizations (e.g., probes and antibody concentrations, hybridization temperature and time, or reaction volume). Furthermore, they typically involve multiple amplicon treatment steps taking at least 90 min/post RPA to yield signal detection (44).
RPA amplification was performed on a DVD surface for food analysis with an analytical sensitivity of 24–30 copies/mL (11) and 10–48 copies/mL (12). Several factors influence the efficiency and robustness of this amplification; for instance, evaporation problems must be reduced, and liquid volume, incubation temperature, concentration of bound and unbound primers, volume of the solution, and surface reaction products must be carefully determined (11).
These platforms are still for research use only and not yet commercially available. They also lack automation (i.e., several hands-on manipulations) and the on-chip sample preparation process carries the risk of cross-contamination due to the absence of closed-tube assay.
Automated Fluidic Platforms
Although extremely fast PCR (time-to-result <15 min) has been described (48), the procedure relies on a sophisticated instrument allowing fast PCR cycle (0.4–2.0 s/cycle instead of 10 s/cycle) through fast heat transfer, thus consuming high power. To match the fast instrument kinetics, primers and polymerase concentrations must be increased 10–20-fold, thus substantially raising assay costs.
The principal advantage behind microfluidic integration of nucleic acid amplification technologies is the possibility of automating the biological steps required for sample preparation, nucleic acid amplification, and detection. Automation results in a shortened turnaround time, reduced volume of sample and reagents, minimal handling steps, and enhanced assay throughput and cost-efficiency (49). Integrated systems may render the molecular assay amenable for point-of-care (POC) analysis, as close as possible to the patient. Several studies aiming to bring tests to POC have integrated RPA in miniaturized simple devices (11, 18–20, 23, 42, 46, 47, 49–54). Lutz et al. (49) were the first to integrate RPA exo in a centrifugal microfluidic cartridge containing prestored liquid and dry RPA reagents for the rapid detection of MRSA in 20 min. The cartridge allowed the conduction of 30 parallel RPA reactions and the disc contained 6 chambers having 5 cavities and each chamber was prestored with lyophilized RPA reagents. Real-time detection using exo probes was performed and fluorescence measurements were performed with an adjusted Rotor-Gene instrument. This system was a proof-of-principle not validated on clinical samples (49).
Other types of microfluidic platforms have also been used with RPA. A paper and plastic platform has been designed for HIV diagnosis (51). This platform differed from the previously mentioned microfluidic device in that it was made from simple, inexpensive, and user-friendly materials. No pumps were required, but the system was able to perform many functions of microfluidic devices. The paper and plastic device functioned on mixing reagents through simple diffusion by folding the platform. The detection of HIV DNA was made using lateral flow strips with a reported detection limit of 10 copies within 15 min (51). In another study, this platform served for the diagnosis of cryptosporidiosis from stool samples (8/10 infected samples were detected) (34). Despite its simplicity, however, there is a potential risk of contamination (34) because the handling is done in an open system; sample preparation and lateral flow detection are not integrated and reagents are manually dispensed.
To avoid nonspecific preamplification reaction and potential amplicon cross-contamination, RPA reagents were compartmentalized into separate wells on a SlipChip that could then be assembled by sliding the plates together. The digital RPA SlipChip platform allowed performing a quantitative analysis by endpoint fluorescence measurements (50) or real-time detection using exo probes (23). Quantification of RPA was also possible by compartmentalizing RPA mixture into individual aqueous microdroplets through a centrifugal step emulsification (55). Listeria monocytogenes DNA was detected (100 copies/μL) and quantified in <30 min. Automated droplet RPA was also applied for the real-time detection of antibiotic resistant carbapenemase producing β-lactamase E. coli, detecting a single copy within 15 min (56).
Most of the previously mentioned platforms, whether a portable disc, an origami paper, or a slipping chip, do not perform sample-to-answer analysis and lack an integrated sample preparation. However, Kim et al. have developed a sample-to-answer cartridge that is able to perform sample preparation via laser irradiation, and detection is achieved by an integrated lateral-flow strip (20). The cartridge is composed of 6 U simultaneously processing 6 independent RPA reactions. The workflow starts with an initial manual loading step of sample/RPA solution. The rest of the procedure is automated from sample preparation to detection. It was applied for the detection of Salmonella in spiked-milk samples and the full process was achieved in 30 min with a limit of detection of 100 cfu/mL (20). Although this completely enclosed microfluidic cartridge offers a rapid and simple sample-to-answer procedure for pathogen screening, it is not commercially available and still needs to be validated with more complex biological samples.
RPA was also applied in the field outside laboratory settings for RNA (7, 25, 42, 43, 57) and DNA (58) detection. A portable suitcase-sized laboratory containing all necessary equipment and reagents was developed to perform a normal bench-based RPA test in the field. Despite the advent of these novel platforms, none has been commercialized yet, because they require optimization in terms of instrument simplification, portability, and cost-effectiveness, as well as clinical validation to meet POC requirements.
RPA STRENGTHS AND WEAKNESSES
To better illustrate the strengths and weaknesses of RPA among other isothermal techniques, we made a concise comparison of RPA to a short list of novel or commonest isothermal technologies (see online Supplemental Table 2).
Specific criteria were taken into consideration: 1) turnaround time, 2) incubation temperature, 3) complexity of primers/probe design, 4) detection schemes, 5) multiplexing capability, 6) compatibility with miniaturized sample processing, 7) commercially available reagents, and 8) amplification of both DNA and RNA. The turnaround time reflects the speed of the isothermal technology for generating results; ideally, the speed of the test should be less than 1 h (59). The incubation temperature is an important element because it is associated with the complexity of instrumentation required for isothermal amplification. In general, isothermal technologies operate at lower temperatures (e.g., 30–42 °C), thereby needing less power, especially because they do not require initial denaturation and cycling steps at 95 °C. For instance, among the isothermal technologies listed in online Supplemental Table 2, only 5 of 10 are completely isothermal. These techniques (helicase-dependent amplification, RPA, nicking enzyme amplification reaction, ribonuclease-mediated amplification, and cross-priming amplification) enable nucleic acid amplification at a single temperature throughout the reaction, thus alleviating the need for temperature fluctuation and control and simplifying instrumentation. Several published studies have performed RPA using simple instruments such as ovens or heat blocks. Sodium sulfate was even used as an exothermic heat source for RPA reaction operation (52). This makes RPA suitable for noninstrumented nucleic acid amplification platforms, as demonstrated recently with loop-mediated isothermal amplification (LAMP) technology by a study from the Programs for Assessment of Technology in Health Research Institute (60). Moreover, the capacity of RPA to catalyze nucleic acid amplification using only body heat was also demonstrated (54). To our knowledge, RPA was the first isothermal technology to demonstrate noninstrumentation requirements (body heat), which is very important criteria for POC applications. In addition, the stability of RPA dried reagents allows transportation and storage without refrigeration (45). This means that a broad range of end users can operate RPA especially in low resource settings.
Furthermore, primer and probe design requirements play a key role in the specificity as well as the feasibility of multiplexing. Taking for example LAMP, 4–6 primers recognizing 6 different regions in the target sequence are required, rendering the amplification process highly specific. On the other hand, other isothermal technologies require certain particularities for primer and probe design, such as strand displacement amplification and nicking enzyme amplification reaction, which require short chimeric primers containing both DNA/RNA bases (61) or long (30–35/46–52 bases) RPA primers/probes. Despite the requirements for long primers/probes in RPA reactions, several multiplex assays have been reported (4, 11, 12, 17, 19, 31, 40).
Moreover, the detection strategies offered by isothermal technologies are associated with both speed of analysis and complexity of instrumentation; simple detection methods are usually fast. For instance, RPA offers several detection strategies either in real-time or post- amplification. Real-time RPA detection is performed with fluorescent probes generating results in 5–15 min in a closed-tube assay format avoiding downstream cross-contamination. For postamplification detection methods, RPA uses agarose gel electrophoresis (approximately 60 min for results) or lateral-flow strips (20–35 min) (Tables 2 and 3). By contrast, for LAMP the application of probe-based detection methods was not feasible because of the cauliflower-like structures of LAMP products (62). Therefore, real-time LAMP detection is restricted to fluorescent dyes for detection. Other postamplification LAMP detection methods could rely on the naked eye because turbidity of the reaction mixture increases because of pyrophosphate by-product formation (63). However, with end-point analysis (gel electrophoresis, lateral flow, or turbidimetry), as in the case of helicase-dependent amplification, nucleic acid sequence–based amplification, or LAMP, results required 60–180 min depending on the assay (61). Furthermore, LAMP products yield a ladder-like pattern during agarose gel electrophoresis, whereas RPA-purified amplicons can be directly identified by a specific band on agarose gel (35). RPA speed could be potentially exploited for NGS (next-generation sequencing) applications for presequencing amplification required for generating libraries (64).
RPA Validation and Costs
The evaluation of the RPA diagnostic performance for clinical testing involves comparison to a gold standard or a reference method. For instance, RPA was compared to PCR (17) and/or other widely used isothermal methods such as LAMP (31, 65). In these studies, the validation of RPA was made on clinical samples such as vaginal/anal lysates (n = 50, 96% clinical sensitivity and 100% clinical specificity) (17), surgical biopsy samples (n = 12, 100% sensitivity and 100% specificity) (65), or serum samples (n = 203, 77% sensitivity and 97.9% specificity) (31). RPA showed minimal requirements for sample preparation and multiplexing was feasible. Hence, integrating assay control was successfully applied to RPA tests (17, 66). Despite RPA showing comparable clinical performances to PCR and LAMP, it also shows some differences. Unlike LAMP (67), there is no software available to design RPA primers and probes. Consequently, screening of several RPA primer and probe sets is required to choose the optimal combination. Furthermore, the verification of RPA amplification by gel electrophoresis is hampered by the presence of high molecular weight proteins. A postamplification purification step is thus required and it can be achieved for amplicons generated using basic or fpg kits, but not for those from a real-time exo kit because of amplicons degradation by the exo nuclease.
Although PCR requires rapid and accurate temperature control during the amplification cycle, RPA tolerates temperatures ranging from 25 to 42 °C without losing reaction efficiency. This allows the simplification of instrumentation and the reduction of costs. Portable user-friendly instruments adapted for RPA reaction exists, the T-8 isothermal device (TwistDx) real-time fluorometer being 1 example. This portable (19 × 17.5–cm), battery-powered, $5494.5 USD device accommodates 8 RPA reactions per assay, monitors fluorescence in 2 channels, and offers magnetic mixing of RPA reactions (thus enhancing assay sensitivity). Similar to T-8, the T-16 instrument (TwistDx) supports 16 reactions/assay, offers magnetic mixing of RPA reactions as well, and monitors fluorescence in 3 different channels. While being portable, the price of this instrument is estimated to be $8048 USD (68), which is still affordable compared to more expensive thermocyclers (≥$30 000 USD). Currently, the cost of a RPA reaction is estimated to be $4.3 USD (51) and TwistDx offers a custom freeze-drying service for RPA reaction pellets, to facilitate the use and market penetration of RPA. This brings RPA a step closer to become a true POC isothermal molecular assay.
Conclusion
The RPA technology owes its speed, flexibility, and completely isothermal profile to the composition of a “protein soup” that mimics parts of the in vivo recombination process. It shows that exploiting a mixture of proteins (n ≥ 7) with known biological functions can reduce instrumentation costs and turnaround time for molecular diagnostics. Compared to the number of proteins involved in performing the thermal cycling of PCR, a savant mixture of 7 to 8 proteins enables the isothermal amplification of DNA or RNA in 5–20 min. The low operation temperature (near body temperature) of RPA and its minimal sample preparation requirements, known to tolerate a wide range of biological samples (e.g., serum, stool, urine, milk, nasal, vaginal, plasma, food, plants, and animal tissues) have led to the development of successful applications of the technology in different POC sample-to-answer systems for faster (bedside) diagnostics.
Although no RPA test or platform has yet been cleared by the FDA or CE marked, we think that it is the right time for RPA to emerge in clinical laboratories as a novel, cost-effective, and reliable isothermal molecular technique.
Supplementary Material
Acknowledgments
We thank Luc Bissonnette for reviewing this manuscript and Dominique K. Boudreau for providing useful technical insights to accomplish this work.
3 Nonstandard abbreviations
- RPA
recombinase polymerase amplification
- POC
point-of-care
- SSB
single-strand DNA binding protein
- dsDNA
double-stranded DNA
- RT-RPA
reverse transcriptase RPA
- THF
tetrahydrofuran
- dR
deoxyribose
- MuLV
murine leukemia virus
- LF
lateral flow
- MRSA
methicillin-resistant Staphylococcus aureus
- LAMP
loop-mediated amplification.
Footnotes
Author Contributions: All authors confirmed they have contributed to the intellectual content of this paper and have met the following 3 requirements: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; and (c) final approval of the published article.
Authors' Disclosures or Potential Conflicts of Interest: Upon manuscript submission, all authors completed the author disclosure form. Disclosures and/or potential conflicts of interest:
Employment or Leadership: None declared.
Consultant or Advisory Role: None declared.
Stock Ownership: None declared.
Honoraria: None declared.
Research Funding: M.G. Bergeron, Université Laval.
Expert Testimony: None declared.
Patents: None declared.
References
- 1. Yan L, Zhou J, Zheng Y, Gamson AS, Roembke BT, Nakayama S, Sintim HO. Isothermal amplified detection of DNA and RNA. Mol Biosyst 2014;10:970–1003. [DOI] [PubMed] [Google Scholar]
- 2. Li J, Macdonald J. Advances in isothermal amplification: novel strategies inspired by biological processes. Biosens Bioelectron 2015;64:196–211. [DOI] [PubMed] [Google Scholar]
- 3. de Paz HD, Brotons P, Munoz-Almagro C. Molecular isothermal techniques for combating infectious diseases: towards low-cost point-of-care diagnostics. Expert Rev Mol Diagn 2014;14:827–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Piepenburg O, Williams CH, Stemple DL, Armes NA. DNA detection using recombination proteins. PLoS Biol 2006;4:e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Piepenburg O, Williams CH, Armes NA. Methods for multiplexing recombinase polymerase amplification. 2011. https://www.google.com/patents/US8062850 (accessed February 2016).
- 6. Hill-Cawthorne GA, Hudson LO, El Ghany MF, Piepenburg O, Nair M, Dodgson A, et al. Recombinations in staphylococcal cassette chromosome mec elements compromise the molecular detection of methicillin resistance in Staphylococcus aureus. PLoS One 2014;9:e101419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Abd El Wahed A, Weidmann M, Hufert FT. Diagnostics-in-a-suitcase: development of a portable and rapid assay for the detection of the emerging avian influenza A (H7N9) virus. J Clin Virol 2015;69:16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Piepenburg O, Williams CH, Armes NA. Methods for multiplexing recombinase polymerase amplification October 02, 2014;2014:14/066,334. [Google Scholar]
- 9. Zhang S, Ravelonandro M, Russell P, McOwen N, Briard P, Bohannon S, Vrient A. Rapid diagnostic detection of plum pox virus in Prunus plants by isothermal AmplifyRP® using reverse transcription-recombinase polymerase amplification. J Virol Methods 2014;207C:114–20. [DOI] [PubMed] [Google Scholar]
- 10. Piepenburg O, Armes NA. DNA glycosylase/lyase and ap endonuclease substrates. 2011. http://www.google.com/patents/US20110053153 (Accessed February 2016).
- 11. Santiago-Felipe S, Tortajada-Genaro LA, Morais S, Puchades R, Maquieira Á. One-pot isothermal DNA amplification – hybridization and detection by a disc-based method. Sens Actuators B Chem 2014;204:273–81. [Google Scholar]
- 12. Santiago-Felipe S, Tortajada-Genaro LA, Morais S, Puchades R, Maquieira A. Isothermal DNA amplification strategies for duplex microorganism detection. Food Chem 2015;174:509–15. [DOI] [PubMed] [Google Scholar]
- 13. Euler M, Wang YJ, Otto P, Tomaso H, Escudero R, Anda P, et al. Recombinase polymerase amplification assay for rapid detection of Francisella tularensis. J Clin Microbiol 2012;50:2234–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Euler M, Wang Y, Heidenreich D, Patel P, Strohmeier O, Hakenberg S, et al. Development of a panel of recombinase polymerase amplification assays for the detection of biothreat agents. J Clin Microbiol 2013;51:1110–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ahmed A, van der Linden H, Hartskeerl RA. Development of a recombinase polymerase amplification assay for the detection of pathogenic leptospira. Int J Environ Res Public Health 2014;11:4953–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boyle DS, McNerney R, Teng Low H, Leader BT, Perez-Osorio AC, Meyer JC, et al. Rapid detection of Mycobacterium tuberculosis by recombinase polymerase amplification. PLoS One 2014;9:e103091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Daher RK, Stewart G, Boissinot M, Bergeron MG. Isothermal recombinase polymerase amplification assay applied to the detection of group B streptococci in vaginal/anal samples. Clin Chem 2014;60:660–6. [DOI] [PubMed] [Google Scholar]
- 18. del Río JS, Yehia Adly N, Acero-Sánchez JL, Henry OYF, O'Sullivan CK. Electrochemical detection of Francisella tularensis genomic DNA using solid-phase recombinase polymerase amplification. Biosens Bioelectron 2014;54:674–8. [DOI] [PubMed] [Google Scholar]
- 19. Kersting S, Rausch V, Bier F, von Nickisch-Rosenegk M. Multiplex isothermal solid-phase recombinase polymerase amplification for the specific and fast DNA-based detection of three bacterial pathogens. Microchim Acta 2014;1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim TH, Park J, Kim CJ, Cho YK. Fully integrated lab-on-a-disc for nucleic acid analysis of food-borne pathogens. Anal Chem 2014;86:3841–8. [DOI] [PubMed] [Google Scholar]
- 21. Krolov K, Frolova J, Tudoran O, Suhorutsenko J, Lehto T, Sibul H, et al. Sensitive and rapid detection of Chlamydia trachomatis by recombinase polymerase amplification directly from urine samples. J Mol Diagn 2014;16:127–35. [DOI] [PubMed] [Google Scholar]
- 22. Murinda SE, Ibekwe AM, Zulkaffly S, Cruz A, Park S, Razak N, et al. Real-time isothermal detection of shiga toxin-producing Escherichia coli using recombinase polymerase amplification. Foodborne Pathog Dis 2014;11:529–36. [DOI] [PubMed] [Google Scholar]
- 23. Tsaloglou MN, Watson RJ, Rushworth CM, Zhao Y, Niu X, Sutton JM, Morgan H. Real-time microfluidic recombinase polymerase amplification for the toxin B gene of Clostridium difficile on a SlipChip platform. Analyst 2015;140:258–64. [DOI] [PubMed] [Google Scholar]
- 24. Euler M, Wang YJ, Nentwich O, Piepenburg O, Hufert FT, Weidmann M. Recombinase polymerase amplification assay for rapid detection of Rift Valley fever virus. J Clin Virol 2012;54:308–12. [DOI] [PubMed] [Google Scholar]
- 25. Abd El Wahed A, El-Deeb A, El-Tholoth M, Abd El Kader H, Ahmed A, Hassan S, et al. A portable reverse transcription recombinase polymerase amplification assay for rapid detection of foot-and-mouth disease virus. PLoS One 2013;8:e71642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abd El Wahed A, Patel P, Heidenreich D, Hufert FT, Weidmann M. Reverse transcription recombinase polymerase amplification assay for the detection of Middle East respiratory syndrome coronavirus. PLoS Curr 2013;5:pii: ecurrents.outbreaks.62df1c7c75ffc96cd59034531e2e8364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Escadafal C, Paweska JT, Grobbelaar A, le Roux C, Bouloy M, Patel P, et al. International external quality assessment of molecular detection of Rift Valley fever virus. PLoS Negl Trop Dis 2013;7:e2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aebischer A, Wernike K, Hoffmann B, Beer M. Rapid genome detection of Schmallenberg virus and bovine viral diarrhea virus by use of isothermal amplification methods and high-speed real-time reverse transcriptase PCR. J Clin Microbiol 2014;52:1883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mekuria TA, Zhang S, Eastwell KC. Rapid and sensitive detection of little cherry virus 2 using isothermal reverse transcription-recombinase polymerase amplification. J Virol Methods 2014;205C:24–30. [DOI] [PubMed] [Google Scholar]
- 30. Xia X, Yu Y, Weidmann M, Pan Y, Yan S, Wang Y. Rapid detection of shrimp white spot syndrome virus by real time, isothermal recombinase polymerase amplification assay. PLoS One 2014;9:e104667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Teoh BT, Sam SS, Tan KK, Danlami MB, Shu MH, Johari J, et al. Early detection of dengue virus by use of reverse transcription-recombinase polymerase amplification. J Clin Microbiol 2015;53:830–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Am HM, Abd El Wahed A, Shalaby MA, Almajhdi FN, Hufert FT, Weidmann M. A new approach for diagnosis of bovine coronavirus using a reverse transcription recombinase polymerase amplification assay. J Virol Methods 2013;193:337–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sakai K, Trabasso P, Moretti ML, Mikami Y, Kamei K, Gonoi T. Identification of fungal pathogens by visible microarray system in combination with isothermal gene amplification. Mycopathologia 2014;178:11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Crannell ZA, Castellanos-Gonzalez A, Irani A, Rohrman B, White AC, Richards-Kortum R. Nucleic acid test to diagnose cryptosporidiosis: lab assessment in animal and patient specimens. Anal Chem 2014;86:2565–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kersting S, Rausch V, Bier FF, von Nickisch-Rosenegk M. Rapid detection of Plasmodium falciparum with isothermal recombinase polymerase amplification and lateral flow analysis. Malar J 2014;13:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu C, Li L, Jin W, Wan Y. Event-specific real-time RPA detection of transgenic rice kefeng 6. GMO Biosafety Res 2014;5:1–5. [Google Scholar]
- 37. Xu C, Li L, Jin W, Wan Y. Recombinase polymerase amplification (RPA) of CaMV-35S promoter and nos terminator for rapid detection of genetically modified crops. Int J Mol Sci 2014;15:18197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Loo JF, Lau PM, Ho HP, Kong SK. An aptamer-based bio-barcode assay with isothermal recombinase polymerase amplification for cytochrome-c detection and anti-cancer drug screening. Talanta 2013;115:159–65. [DOI] [PubMed] [Google Scholar]
- 39. Shin Y, Perera AP, Kim KW, Park MK. Real-time, label-free isothermal solid-phase amplification/detection (ISAD) device for rapid detection of genetic alteration in cancers. Lab Chip 2013;13:2106–14. [DOI] [PubMed] [Google Scholar]
- 40. Crannell ZA, Rohrman B, Richards-Kortum R. Quantification of HIV-1 DNA using real-time recombinase polymerase amplification. Anal Chem 2014;86:5615–9. [DOI] [PubMed] [Google Scholar]
- 41. Boyle DS, Lehman DA, Lillis L, Peterson D, Singhal M, Armes N, et al. Rapid detection of HIV-1 proviral DNA for early infant diagnosis using recombinase polymerase amplification. MBio 2013;4:pii:e00135–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Escadafal C, Faye O, Sall AA, Weidmann M, Strohmeier O, von Stetten F, et al. Rapid molecular assays for the detection of yellow fever virus in low-resource settings. PLoS Negl Trop Dis 2014;8:e2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Abd El Wahed A, Patel P, Faye O, Thaloengsok S, Heidenreich D, Matangkasombut P, et al. Recombinase polymerase amplification assay for rapid diagnostics of dengue infection. PLoS One 2015;10:e0129682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Santiago-Felipe S, Tortajada-Genaro LA, Puchades R, Maquieira A. Recombinase polymerase and enzyme-linked immunosorbent assay as a DNA amplification-detection strategy for food analysis. Anal Chim Acta 2014;811:81–7. [DOI] [PubMed] [Google Scholar]
- 45. Erh-Chia Yeh, Lee LP. One-step digital plasma separation for molecular diagnostics. 17th International Conference on Miniaturized Systems for Chemistry and Life Sci, Vol. CBMS-0001. Freiburg, Germany: μTAS 2013;1323–5. [Google Scholar]
- 46. Shin Y, Perera AP, Tang WY, Fu DL, Liu Q, Sheng JK, et al. A rapid amplification/detection assay for analysis of Mycobacterium tuberculosis using an isothermal and silicon bio-photonic sensor complex. Biosens Bioelectron 2015;68C:390–6. [DOI] [PubMed] [Google Scholar]
- 47. Lee TY, Shin Y, Park MK. A simple, low-cost, and rapid device for a DNA methylation-specific amplification/detection system using a flexible plastic and silicon complex. Lab Chip 2014;14:4220–9. [DOI] [PubMed] [Google Scholar]
- 48. Farrar JS, Wittwer CT. Extreme PCR: efficient and specific DNA amplification in 15–60 seconds. Clin Chem 2015;61:145–53. [DOI] [PubMed] [Google Scholar]
- 49. Lutz S, Weber P, Focke M, Faltin B, Hoffmann J, Muller C, et al. Microfluidic lab-on-a-foil for nucleic acid analysis based on isothermal recombinase polymerase amplification (RPA). Lab Chip 2010;10:887–93. [DOI] [PubMed] [Google Scholar]
- 50. Shen F, Davydova EK, Du W, Kreutz JE, Piepenburg O, Ismagilov RF. Digital isothermal quantification of nucleic acids via simultaneous chemical initiation of recombinase polymerase amplification reactions on SlipChip. Anal Chem 2011;83:3533–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rohrman BA, Richards-Kortum RR. A paper and plastic device for performing recombinase polymerase amplification of HIV DNA. Lab Chip 2012;12:3082–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lillis L, Lehman D, Singhal MC, Cantera J, Singleton J, Labarre P, et al. Non-instrumented incubation of a recombinase polymerase amplification assay for the rapid and sensitive detection of proviral HIV-1 DNA. PLoS One 9:e108189, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hakenberg S, Hugle M, Weidmann M, Hufert F, Dame G, Urban GA. A phaseguided passive batch microfluidic mixing chamber for isothermal amplification. Lab Chip 2012;12:4576–80. [DOI] [PubMed] [Google Scholar]
- 54. Crannell ZA, Rohrman B, Richards-Kortum R. Equipment-free incubation of recombinase polymerase amplification reactions using body heat. PLoS One 2014;9:e112146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schuler F, Schwemmer F, Trotter M, Wadle S, Zengerle R, von Stetten F, Paust N. Centrifugal step emulsification applied for absolute quantification of nucleic acids by digital droplet RPA. Lab Chip 2015;15:2759–66. [DOI] [PubMed] [Google Scholar]
- 56. Kalsi S, Valiadi M, Tsaloglou MN, Parry-Jones L, Jacobs A, Watson R, et al. Rapid and sensitive detection of antibiotic resistance on a programmable digital microfluidic platform. Lab Chip 2015;15:3065–75. [DOI] [PubMed] [Google Scholar]
- 57. TwistDx L. Resources/news. http://www.twistdx.co.uk/resources/news/ (Accessed March 2015).
- 58. Liljander A, Yu M, O'Brien E, Heller M, Nepper JF, Weibel DB, et al. A field-applicable recombinase polymerase amplification assay for rapid detection of Mycoplasma capricolum subsp. capripneumoniae. J Clin Microbiol 2015;53:2810–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bergeron MG. Revolutionizing the practice of medicine through rapid (< 1h) DNA-based diagnostics. Clin Invest Med 2008;31:E265–71. [DOI] [PubMed] [Google Scholar]
- 60. LaBarre P, Hawkins KR, Gerlach J, Wilmoth J, Beddoe A, Singleton J, et al. A simple, inexpensive device for nucleic acid amplification without electricity-toward instrument-free molecular diagnostics in low-resource settings. PLoS One 2011;6:e19738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Craw P, Balachandran W. Isothermal nucleic acid amplification technologies for point-of-care diagnostics: a critical review. Lab Chip 2012;12:2469–86. [DOI] [PubMed] [Google Scholar]
- 62. Dong H-J, Cho A-R, Hahn T-W, Cho S. Development of a multiplex loop-mediated isothermal amplification assay to detect shiga toxin-producing Escherichia coli in cattle. J Vet Sci 2014;15:317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res 2000;28:E63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Oyola SO, Otto TD, Gu Y, Maslen G, Manske M, Campino S, et al. Optimizing illumina next-generation sequencing library preparation for extremely at-biased genomes. BMC Genomics 2012;13:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ahmed SA, van de Sande WW, Desnos-Ollivier M, Fahal AH, Mhmoud NA, de Hoog GS. Application of isothermal amplification techniques for identification of Madurella mycetomatis, the prevalent agent of human mycetoma. J Clin Microbiol 2015;53:3280–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Crannell ZA, Rohrman B, Richards-Kortum R. Development of a quantitative recombinase polymerase amplification assay with an internal positive control. J Vis Exp 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Eiken GENOME. Lamp primer designing software primerexplorer. https://primerexplorer.jp/e/intro/index.html (Accessed February 2015).
- 68. TwistDx L. T-16 isothermal device. http://www.twistdx.co.uk/products/devices_and_accessories/t_16_device/ (Accessed June 2015).
- 69. Crannell ZA, Cabada MM, Castellanos-Gonzalez A, Irani A, White AC, Richards-Kortum R. Recombinase polymerase amplification-based assay to diagnose giardia in stool samples. Am J Trop Med Hyg 2014;92:583–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jaroenram W, Owens L. Recombinase polymerase amplification combined with a lateral flow dipstick for discriminating between infectious Penaeus stylirostris densovirus and virus-related sequences in shrimp genome. J Virol Methods 2014;208:144–51. [DOI] [PubMed] [Google Scholar]
- 71. Xia X, Yu Y, Hu L, Weidmann M, Pan Y, Yan S, Wang Y. Rapid detection of infectious hypodermal and hematopoietic necrosis virus (IHHNV) by real-time, isothermal recombinase polymerase amplification assay. Arch Virol 2015;160:987–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.