

Abstract
BACKGROUND
In 2015, the 68th World Health Assembly declared that effective, rapid, low-cost diagnostic tools were needed for guiding optimal use of antibiotics in medicine. This review is devoted to interferon-inducible myxovirus resistance proteins as potential biomarkers for differentiating viral from bacterial infections.
CONTENT
After viral infection, a branch of the interferon (IFN)-induced molecular reactions is triggered by the binding of IFNs with their receptors, a process leading to the activation of mx1 and mx2, which produce antiviral Mx proteins (MxA and MxB). We summarize current knowledge of the structures and functions of type I and III IFNs. Antiviral mechanisms of Mx proteins are discussed in reference to their structural and functional data to provide an in-depth picture of protection against viral attacks. Knowing such a mechanism may allow the development of countermeasures and the specific detection of any viral infection. Clinical research data indicate that Mx proteins are biomarkers for many virus infections, with some exceptions, whereas C-reactive protein (CRP) and procalcitonin have established positions as general biomarkers for bacterial infections.
SUMMARY
Mx genes are not directly induced by viruses and are not expressed constitutively; their expression strictly depends on IFN signaling. MxA protein production in peripheral blood cells has been shown to be a clinically sensitive and specific marker for viral infection. Viral infections specifically increase MxA concentrations, whereas viruses have only a modest increase in CRP or procalcitonin concentrations. Therefore, comparison of MxA and CRP and/or procalcitonin values can be used for the differentiation of infectious etiology.
The early observation that an animal infected with 1 virus acquires resistance to coinfection with another virus was observed in the 1930s and was termed as “viral interference.” A soluble factor with innate antiviral activity was later discovered and named “interferon” (IFN)4 (1). Subsequently, this factor was identified as belonging to a group of proteins; currently, 3 types of IFNs are known as types I, II, and III with distinct structures, biological properties, and activities (2). Type I IFNs contain 2 major subtypes: the fibroblast or IFN-β and the leukocyte or IFN-α. They are commonly produced by all nucleated cells exposed to a viral infection (3, 4). Although the type III IFNs (IFN-λ) are also produced by nucleated cells in response to an infectious virus (3, 4), they are primarily active on epithelial cells (5) because unlike the type I IFN receptors that are broadly produced on most cell types, type III IFN receptors are largely restricted to cells of epithelial origin (3).
IFNs induce expression of numerous IFN-stimulated genes (ISGs) involved in resistance to viral infections. Myxovirus resistance genes mx15 and mx2 also belong to the class of ISGs (6, 7); they were originally identified as factors conferring resistance to lethal influenza A virus infections in mice (8–12) and that encode the interferon-inducible guanine triphosphatases (GTPases) MxA and MxB in humans (9, 13). Their gene expression is strictly controlled by virus-induced type I and type III IFNs (14, 15).
In this review, we present the current state of knowledge about the structure and function of type I and III IFNs and their receptors and in addition trace the cascade of reactions that is switched on after binding of IFNs with their receptors and that leads to activation of mx1 and mx2. The structural and functional aspects of the molecules involved are described to illustrate on what confidence level the antiviral mechanisms are understood. However, the overall desire is to distinguish the bacterial infections from the viral and develop such distinguishing tools for the diagnostic purposes.
Type I IFNS And Their Receptors
In 2 decades after the discovery of IFNs, the genes for human (hu) IFN-α and huIFN-β were cloned and sequenced (16, 17). Sequence alignment of the interferons showed that 48 out of the 166 amino acid positions compared were identical (29% of identity) (17). On the basis of sequence identity, it was concluded that the 2 genes were derived from a common evolutionary ancestor.
Soon after decoding of the huIFN-α and huIFN-β sequences, molecular modeling of the secondary and three-dimensional structures was performed (18). On the basis of the sequences, it was predicted that huIFN-α and huIFN-β would contain 60%–70% α-helices. With the stereochemical method of packing the α-helices into globular proteins, 5 segments of α-helices, capable of forming tightly packed hydrophobic cores, were localized (18). Thus, the predicted globular structure of huIFN-α and huIFN-β was a 5-helix (named the A–E) bundle (19). Although the overall identity between the sequences of huIFN-α and huIFN-β was only 29%, the sequences of the AB loop were 46% identitical. Most of the conserved amino acid residues in the AB loop were hydrophilic and, therefore, exposed to the surface. There was also 70% identity in the region of α-helix E, and therefore it was suggested that the AB loop and the α-helix E together could form the active site (18). Later, the sequences of 28 IFN-α and IFN-β genes of different species were compared (19). Twenty-one conservative positions were identified in which there was no amino acid substitution or only 1 homologous replacement. A cluster of 7 conservative positions was found in the AB loop. Five of the 7 conservative positions composing this cluster, were hydrophilic and, therefore, very likely to reside on the surface of the molecule. The second cluster of 6 conservative positions was found in the α-helix E and in the DE loop (19). With the same predictive approach as for the huIFN-α and huIFN-β (18), it was found that many cytokines (IL2, IL3, IL4, IL5, IL6, IL7; macrophage colony-stimulating factor, granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, and chicken myelomonocytic growth factor) were related to α-helical proteins and contained 54% to 73% of α-helices (20). Helical segments (in main 5 segments) capable of forming tightly packed hydrophobic cores were localized (20). Hence, IFNs are in fact closely related to the family of α-helical cytokines (21).
At present, the type I IFNs comprise a family with 17 members: 13 subtypes of IFN-α and IFN-β, IFN-ε, IFN-κ, and IFN-ω (2, 22). They exhibit approximately 20%–60% sequence identity, and despite their differential activities, all type I IFNs initiate intracellular signaling by binding to a common receptor composed of 2 subunits, IFNAR1 and IFNAR2 (22). The IFNAR1 subunit has low affinity to huIFN-α2. Therefore, to get a high affinity for IFNAR1, a triple mutant (H57Y, G58N, Q61S) of huIFN-α2 was designed. It was designated as huIFN-α2 (YNS). The stable crystal structures of type I IFN ternary signaling complexes containing both receptor chains IFNAR1 and IFNAR2 in complex with huIFN-α2 (YNS) were solved (23). The crystallographic analysis confirmed the structure of huIFN-α to be a 5-helix bundle (23, 24). The major α-helices, composing a bundle with 5 α-helices, are labeled A–E, as shown in Fig. 1. The arrows indicate the direction of the corresponding α-helices from the N- to C-terminus.
Fig. 1. The crystal structure of huIFN-α2 (YNS) triple mutant, designed to get a high affinity for IFNAR1 (23), shown in huIFN-α2-IFNAR ternary complex (A) and separately in 2 different projections (B, C).
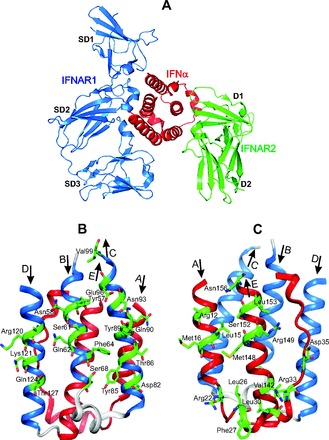
The colored blue B, C, and D α helices, which form the IFNAR1 binding site, and the side chains of residues, which contribute to the binding with IFNAR1 (23), are shown in front of B projection. The colored red A and E α helices and the AB loop, which form the IFNAR2 binding site (23), and the side chains of residues, which contribute to the binding with IFNAR2, are shown in front at C projection. The figure is reconstructed on the basis of the amino acid sequence, secondary structure, and coordinates of atoms from the Protein Data Bank under accession code 3SE3 (huIFN-α2-IFNAR ternary complex).
IFNAR1 and IFNAR2 bind to opposing surfaces of the huIFN-α2 molecule (Figs. 1 and 2A) (23). A high-affinity binding site for IFNAR2 in the huIFN-α2 molecule is mostly composed of the AB loop and the α-helix E (Fig. 1, A and C). Arg33 in the huIFN-α2 (YNS), corresponding to Arg35 in the huIFN-ω, is the most important residue for the binding of huIFN-α2 or huIFN-ω to IFNAR2 (23). The ligand forms a network of hydrogen bonds with the main chain carbonyl oxygen atoms of Ile45 and Glu50 of IFNAR2 and the side chain of Thr44 of IFNAR2. Replacing Arg33 by Ala in the huIFN-α2 (YNS) destabilizes the binding more than any other mutation in the huIFN-α2 (23). Previous attention was drawn to Leu30 in the huIFN-α2 (the equivalent of Leu32 in the huIFN-ω), which is conserved in all type I IFNs (19, 21). In a crystal structure of the ternary complex, both amino acids bind to similar hydrophobic clusters in IFNAR2 (23). Therefore, it was suggested that these residues are energetically critical for the shared anchor points mediating the IFN cross-reactivity. The α-helix E comprises Val142, Met148, Arg149, Ser152, Leu153, and Asn156, with side chains participating in binding of IFNAR2 (23). The binding site for IFNAR1 in the IFN-α2 molecule is formed by the α-helices B, C, and D (Fig. 1, A and B).
Fig. 2. The type I IFNs initiate viral signaling by binding to the common receptors called IFNAR1 and IFNAR2 (A), and the type III IFNs initiates signaling by binding to receptors IFNLR1 and IL10R2 (B).

The figure is redrawn on the basis of data presented in references (2, 23, 32, 34).
IFNAR2 is related to the class II receptors for α-helical cytokines (22, 25). The extracellular domain (ECD) of the class II receptors consists of 2 fibronectin III-like domains (D1 and D2) (Figs. 1A and 2A). The extracellular domain of IFNAR1 comprises 4 fibronectin III-like domains, labeled SD1 to SD4 (Figs. 1A and 2A), which have evolved from a gene duplication of a typical 2-domain structure (26). The huIFN-α2 binds to IFNAR1 at the level of a hinge between SD2 and SD3 domains and with the SD1 domain “capping” the top of the IFN molecule (Figs. 1A and 2A). The IFNAR1–huIFN-α2 interface is formed by residues of the SD1, SD2, and SD3 domains of IFNAR1 and by helices B, C, and D of the huIFN-α2 molecule (Figs. 1, A and B, and 2A).
The intracellular domains of IFNAR1 and IFNAR2 are associated with the Janus kinases (JAKs) Tyk2 and Jak1, respectively (27, 28). On the type I IFN binding by the IFNAR chains and the formation of the extracellular signaling complex, these JAKS initiate a phosphorylation cascade (28).
Type III IFNS and Their Receptors
In 2003, a novel type of IFN (named type III) was independently discovered by 2 research groups (3, 29). These IFNs were named IFN-λ1, IFN-λ2, and IFN-λ3 or IL29, IL28A, and IL28B, owing to their shared features with both type I IFN and the members of the IL10 family. The crystal structure of huIFN-λ3 was identified at the 2.8 Å level (30–32), and further evidence was obtained that the type III IFNs initiated signaling by binding to a common receptor composed of IFNLR1 and IL10R2 (33) (Fig. 2B). Furthermore, the crystal structures of huIFN-λ1 complex with its high-affinity receptor IFNLR1 (34) and huIL10R2 extracellular domain were also determined (35). The basic structure of the huIFN-λ1 (34) and huIFN-λ3 (30–32) is a 5-helix bundle (or elements A, C, D, E, F in the cited papers (26, 29, 30–32)). They are marked in Fig. 3 as A, B, C, D, E to facilitate comparison with type I IFNs. The topology of huIFN-λ1 and huIFN-λ3 is described by “up-up-down-up-down” 5-α-helix-bundle motif found previously for type I IFNs (20, 23).
Fig. 3. The crystal structure of huIFN-λ1 determined with the resolution 2.1 Å (34).
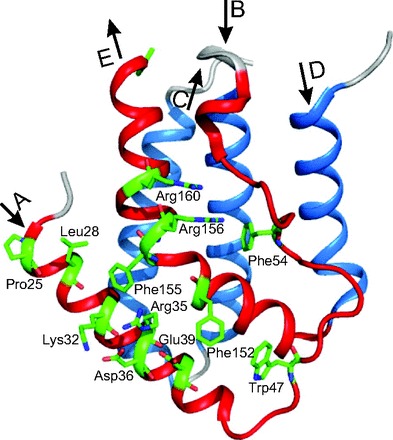
The figure is redrawn on the basis of the amino acid sequence, secondary structure, and coordinates of atoms taken from the PDB under accession code 3OG6 (huIFN-λ1-IFNLR1 binary complex).
The model of huIFN-λ3 contains residues 13–117 and 128–175 (Fig. 3). Residues 118–127 were not modeled owing to poor electron density, probably due to the flexibility of the CD loop or the DE loop (30–32), similar to the corresponding loop in the type I IFNs (20). The long AB loop or element B in the cited papers (30–32) in huIFN-λ3 is less rigid and adapts a flexible coil structure, whereas in huIFN-α2 the residues 26–29, 30–33, and 40–43 within the AB loop form 3 turns of 310 helix (310 A, 310 B, and 310 C) (20). Although the helix 310 A is linked to the N-terminus of α-helix E by a disulfide bond, it is flexible in solution. Out of 7 Cys residues in huIFN-λ3, 6 Cys residues form intramolecular disulfide bonds, whereas Cys48 is free in solution (30–32). Of the 3 intramolecular disulfide bonds, the first disulfide bond between Cys16 and Cys115 connects the N-terminus to the end of α-helix C, the second disulfide bond between Cys50 and Cys148 connects the AB loop to the beginning of helix E, and the third disulfide bond between Cys157 and Cys164 forms a small loop at the end of helix E.
The sequence of huIFN-λ3 is more similar to the type I IFNs than to the IL10 family. The sequence similarity between huIFN-λ3 and huIFN-α2 or huIFN-β is 33% and 31%, whereas the similarity between huIFN-λ3 and IL10, IL19, or IL22 is 23%, 22%, and 22%, respectively (30).
All type III IFNs signal through the IFNLR1/IL10R2 receptor complex (3, 29) (Fig. 2B), whereas all type I IFNs signal through the IFNAR2/IFNAR1 receptor complex (23) (Fig. 2A).
Type I and III IFNS Induce the Same Intracellular Signaling Pathway
Although the sequence of huIFN-λ3 is more similar to the type I IFNs than to the IL10 family, the receptor for type III IFNs shares the IL10R2 receptor chain, thus having a closer relationship with the IL10 family than with the type I IFNs. Nevertheless, binding of IFN-λ with IFNLR/IL10R2 receptor complex induces the same intracellular signaling pathway as the activation of the type I IFN receptor (2, 36) (Fig. 4).
Fig. 4. The main pathways of type I and type III IFNs induced gene expression.
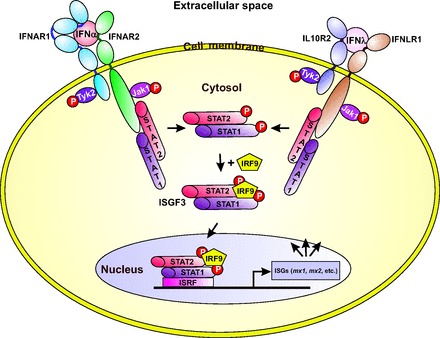
The figure is redrawn on the basis of data presented in references (2, 23, 32, 36).
Binding of the type I and III IFNs to the membrane-associated type I and III receptor complexes, respectively, leads to cross phosphorylation of the JAKs JAK1 and TYK2. The activated JAKs phosphorylate the signal transducer and activator of transcription 1 (STAT1) and STAT2 that induces their heterodimerization (STAT1-STAT2) (2, 3). The dimer recruits the interferon response factor 9 to form a trimeric transcription factor complex named as the interferon-stimulated gene factor 3 (ISGF3) that migrates to the nucleus, where it binds to the interferon-stimulated response element, thus promoting the transcription of the ISGs, including mx1 and mx2. The repertoire of genes induced by type III IFNs is essentially the same as the 1 induced by type I IFNs (37, 38), and as a consequence, type I and III IFNs display many of the same biological activities—including the antiviral activity—in a wide variety of target cells (2). Expression of the type III IFN genes and their corresponding proteins are inducible by infection with many types of viruses, and this process also applies to the genes induced by the type I IFNs. However, unlike the type I IFN receptors that are produced on most cell types, type III IFN receptors are mostly restricted to cells of epithelial origin (2).
Structure and Antiviral Activity of Mx Proteins
The gene for the mouse myxovirus resistance protein 1 (Mx1) was identified and cloned more than 3 decades ago (10, 11). Mx1 is the main IFN-induced intracellular restriction factor against influenza and influenza-like viruses in mice, and the Mx homologs in human serve similar functions (39). Their expression is strictly controlled by type I and III IFNs (7, 14, 15). In humans, MX1 (also known as MxA) and MX2 (also known as MxB) are encoded by closely linked genes on the long arm of chromosome 21 (map position 21q22.3) (10). Opposite to other IFN-stimulated genes, mx genes generally are not induced directly by viruses and are not expressed constitutively. In contrast, their expression is strictly dependent on IFN signaling (14, 15). Although some viruses like HIV may induce MxA without IFN type 1, in general mx genes are excellent markers for IFN action.
The 3-D structure of human MxA protein shows the elongated 3-domain architecture with N-terminal GTPase (G) domain, an antiparallel 3-helical bundle called the bundle-signaling element (BSE) and an antiparallel 4-helical bundle called the stalk (Fig. 5A) (40). The globular G domain comprises a central core consisting of 6 β-strands surrounded by α-helical segments. It is linked to a stalk that is composed of 4 long α-helices. The G domain binds and hydrolyzes guanosine triphosphate (GTP) and is located at the opposite site of the extended stalk in the MxA monomer (Fig. 5A). The connection between the G domain and the stalk is formed by the BSE. It is composed of 3 α-helices derived from the flanking regions of the G domain and the very C-terminal part of the molecule that folds back to the N-terminal G domain (Fig. 5A). The BSE is thought to function as a mediator of conformational coupling between the G domain and the stalk; it transmits a signal from the stalk to the G domain and vice versa transfers structural changes induced by GTP binding and hydrolysis to the stalk (41). The MxA molecule is flexible owing to the 2 hinge-like regions. Real-time domain dynamics of MxA were studied by single-molecule fluorescence resonance energy transfer (42). The G-domain-BSE region can adopt either an “open” or “closed” conformation in all nucleotide-loading conditions. Whereas the open conformation is preferred by the nucleotide-free, GDP · AlF4− -bound and GDP-bound forms, the loading of GTP activates the relative movement between the 2 domains and alters the conformational preference to a “closed” state. Moreover, frequent relative movement was observed to occur between the BSE and the stalk via hinge 1 (Fig. 5A). It was suggested that the MxA molecules within a helical polymer could collectively generate a stable torque through random cycles of GTP hydrolysis (42). The stalk mediates a ring formation by assembling the molecules in a zigzag fashion (Fig. 5B) (6). Cryotransmission electron microscopy shows that the G domains are directed to the outer side of the multimeric rings, whereas the stalks are directed to the opposite inner side (43) (Fig. 5B).
Fig. 5. The crystal structure of human MxA (A) and the schematic side view of an MxA oligomeric ring structure associated with lipid tubule, revealed by the cryotransmission electron microscopy (B).

(A) is redrawn on the basis of the MxA crystal structure data (39) from the PDB under accession code 3SZR, and (B) is redrawn on the basis of the cryotransmission electron microscopy data published in (43). The G domain is colored in orange, the BSE is colored in red, and the stalk domain is colored in green and blue. (B) shows a lipid tubule with inserted MxA molecules. The loop (residues 533–572), termed the L4, is shown black. It protrudes from the tip of the stalk and mediates membrane interaction and viral target recognition (43).
The 40 amino acids loop (residues 533–572), termed the L4, protrudes from the tip of the stalk (Fig. 5B, shown black) and mediates membrane interaction and viral target recognition (44). It was produced and characterized with a panel of G domain mutants and found that the residues in the catalytic center of MxA and the nucleotide itself were essential for the G domain dimerization (45). It was also demonstrated that the GTP binding contributes to the assembly of MxA in stable multimers associated with endoplasmic reticulum membranes, whereas the nucleotide hydrolysis facilitates the dynamic redistribution of MxA from the endoplasmic reticulum to the viral ribonucleoproteins or nucleocapsids, the actual targets of the MxA action.
Alignment of the human MxA and MxB amino acid sequences displays 63% identity (40), and the 3-dimensional structure shows identical folds and architecture of both GTPases (40). The main differences between the 2 proteins are observed within the relatively unstructured N-terminal sequences that extend from the BSE and in the unstructured L4 loop of the stalk. Each MxB stalk monomer contains a 4-helix bundle that forms a dimer (45). MxB dimer interfaces show that most of the residues forming the interface are different in MxB and MxA. As a result, the MxB dimer is more stable than that of the MxA. Although the MxA and MxB stalks share only 46.7% sequence identity, their overall structure of the stalk is conformationally similar. Although the human Mx proteins share common characteristics, their dimerization is unique, explaining their peculiar antiviral profile.
MxA protein is an important antiviral factor with broad activity against diverse RNA viruses. In particular, it has been demonstrated that MxA protein inhibits the multiplication of negative strand RNA viruses such as influenza virus, vesicular stomatitis virus, measles virus, and other viruses belonging to the family Bunyaviridae (46). In addition, it has been found that MxA-transfected cells were protected against Semliki Forest virus, a togavirus with a single-stranded RNA genome of positive polarity, whereas they were not protected against the multiplication of other positive-stranded RNA virus, mengovirus, and encephalomyocarditis virus belonging to the Picornaviridae family (46). The stably transfected Vero cells producing MxA in 98% of cells have been used to test the antiviral activity of MxA against coxsackievirus B4 (CVB4) (46). In Vero cells producing MxA, CVB4 yields were dramatically reduced. In addition, production of the VP1 CVB protein analyzed by immunofluorescence was highly restricted in comparison with control cells. Moreover, the accumulation of negative- and positive-strand CVB4 RNA was prevented, as shown by in situ hybridization and reverse transcription PCR. The obtained results evidently demonstrate the antiviral activity of MxA against CVB4 and indicate that its replication is inhibited at an early step in Vero cells producing MxA.
MxA protein also manifests antiviral activity to a few DNA viruses like the vaccinia virus (VACV), the monkey pox virus, and the African swine fever virus; for these viruses, MxA targets are unknown (47). Recently some well-studied DNA viruses representing the Poxviridae family were tested for their sensitivity to MxA (48). A cell line induced to produce inhibitory concentrations of MxA showed no antivaccinia virus activity, indicating either a lack of susceptibility of the virus or the existence of viral factors capable of counteracting the MxA inhibition. To determine if the VACV resistance to MxA was due to a virus-encoded anti-MxA activity, VACV and the MxA-sensitive vesicular stomatitis virus were coinfected (48). It was shown that VACV does not protect the vesicular stomatitis virus from the MxA inhibition at the protein level. Those results were extended to several additional VACV strains and 2 CPXV strains, thus confirming that these orthopoxviruses do not block the MxA action. Overall, these results point to a lack of susceptibility of the Poxviridae to MxA antiviral activity.
Recently, naturally occurring allelic variations of mx1 coding for the influenza restriction factor MxA were investigated (49). It was found that nonsynonymous variations in the GTPase domain cause a loss of both antiviral and enzymatic activities (49). Additionally, it was demonstrated that these amino acid substitutions disrupt the interface for the GTPase domain dimerization required for the stimulation of the GTP hydrolysis (49). The results obtained convincingly show that the naturally occurring mutations in the human mx1 gene can influence MxA function, which may also explain the individual variations in influenza virus susceptibility in the human population.
In addition, 2 isoforms of the MxB protein have been discovered: a long 78-kDa and a short 76-kDa molecule (50). The long 78-kDa isoform comprises a nuclear localization sequence-like stretch of basic amino acid residues that promote the protein localization preferentially to the nuclear pores (51). Translation of the short 76 kDa MxB form starts from an alternative methionine codon at position 26. Therefore, it is lacking the nuclear localization sequence and localized in cytoplasm (49).
MxA protein residing in the cytoplasm inhibits a broad set of viruses, independently of their replication site. In comparison, the long form of the human MxB is localized to the cytoplasmic side of the nuclear pores via its N-terminus and inhibits the import of the HIV type 1 preintegration complex into the nucleus (51). Additional evidence indicated that the N-terminus of the human MxB is responsible for binding with the HIV type 1 capsid (52).
The information obtained from the crystal structure studies of the MxA (Fig. 5A) (40), together with the mutational studies, led to the proposition of an antiviral mechanism that depends on the multimeric assembly of the Mx proteins. In this model, Mx forms tetramers that at higher Mx concentrations can further oligomerize into large Mx rings (Fig. 5B) (43). GTPase domains are situated on the outside of the ring, and stalks are directed inwards. Furthermore, the Mx rings can interact through their GTPase domains, increasing the GTPase activity. The C-termini of the Mx proteins are important for binding of the viral targets. Such a mode of assembly and oligomerization possibly leads to multiple Mx rings wrapping around the viral target structures (15). The arrangement of repetitive nucleoprotein or capsid protein molecules potentiates multiple bonds with the Mx oligomers. Therefore, even a low-affinity interaction could result in a high-avidity binding. Mx rings could prevent the assembly of nucleoprotein complexes with the viral polymerase, thereby suppressing virus transcription or replication (Fig. 6) (15). The Mx rings could also translocate viral nucleoproteins or capsid proteins to perinuclear complexes in a GTPase-dependent manner, leading to their degradation. However, the detailed molecular mechanisms by which Mx GTPases inhibit the susceptible viruses need further elucidation (39).
Fig. 6. The mechanisms of antiviral action of MxA or B proteins.
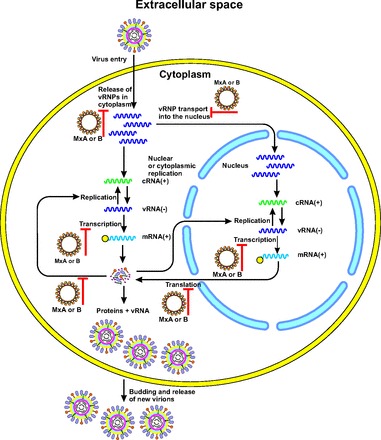
Milestones in the viral infection pathway in cell that are suppressed by Mx proteins are shown (15). MxA or B rings are assumed to prevent the assembly of nucleoprotein complexes with the viral polymerase, thereby suppressing virus transcription or replication (15). The figure is redrawn on the basis of the data presented in reference (15).
Mx Proteins as Biomarkers of Viral Infections
According to the Centers for Disease Control and Prevention, antibiotic resistance is one of the most serious health problems facing the US. Approximately 80% of all antibiotics are prescribed in primary care, and up to 80% of these are for respiratory tract infections (53). If an infection has a viral cause, antibiotics are useless and unlikely to provide clinical benefit to patients (53, 54). At the same time, the inappropriate therapy of using antibiotics for viral infections increases the risk of emergence of antibiotic-resistant bacterial strains. In May 2015, the 68th World Health Assembly endorsed a global action plan to tackle antibiotic resistance. One of the 5 main actions of the plan was to optimize the use of antibiotic medicines in human and animal health (55). The Assembly declared that “Decisions to prescribe antibiotics are rarely based on definitive diagnoses. Effective, rapid, low-cost diagnostic tools are needed for guiding optimal use of antibiotics in human and animal medicine, and such tools should be easily integrated into clinical, pharmacy and veterinary practices. Evidence-based prescribing and dispensing should be the standard of care.”
It is important to mention that MxA is an intracellular protein. Therefore, any method for its detection relies on the release of the MxA protein from cells. MxA can be detected in capillary blood; this characteristic is convenient in pediatrics (56) and could be beneficial for using MxA as biomarker.
The concentration of MxA protein in peripheral blood of healthy people is <50 ng/mL. It is induced <1.2 h after infection and has a half-life of 2.3 days (57, 58). MxA achieves peak concentration at 16 h and remains increased in the presence of increased IFN (59). MxA production in peripheral blood of patients has been shown to be a clinically sensitive and specific marker for viral infection (60–65). Viral infections increase MxA but only modestly increase C-reactive protein (CRP) concentrations, a protein marker suitable for bacterial infections (66–69).
The concentrations of MxA protein were investigated in healthy young children and in young children with symptoms of respiratory virus infection in an observational prospective cohort (70). Blood samples and nasal swabs were taken from 153 and 77 children with and without symptoms of respiratory infections, respectively, and tested with ELISA. The respiratory viruses in nasal swabs were tested with PCR-based detection. Respiratory viruses were detected in 81% of symptomatic children. These symptomatic children had significantly higher MxA (median [interquartile range]) concentrations in blood: 695 (345–1370) μg/L compared with control group 110 (55–170) μg/L; P < 0.001. In asymptomatic children, no significant differences were observed in MxA responses between the virus-positive and virus-negative groups. A cutoff value equal to 175 μg/L had a 92% clinical sensitivity and 77% clinical specificity for a symptomatic respiratory virus infection. Rhinovirus, respiratory syncytial virus, parainfluenza virus, influenza virus, coronavirus, and human metapneumovirus infections were also characterized by increased MxA concentrations. Asymptomatic virus-negative children vaccinated with a live virus vaccine had increased MxA protein concentrations of 240 (120–540) μg/L but had significantly lower concentrations than children with an acute respiratory infection who had not received vaccinations: 740 (350–1425) μg/L; P < 0.001. All in all, the concentrations of MxA protein in blood are increased in young children with symptomatic respiratory virus infections, including rhinovirus infections. Thus, MxA is an informative general marker for the most common acute virus infections.
A study of 533 children was performed in pediatric emergency hospitals in France (71). MxA in peripheral blood was measured in children with confirmed viral or bacterial infections, uninfected controls, and infections of unknown origin. Two monoclonal antibodies were used in the studies. One monoclonal antibody was directed to the C-terminal and the other to the N-terminal part of MxA. The antibodies were used respectively as detecting and capture antibodies in a sandwich-type immunoassay (71). This study showed MxA to be a valuable diagnostic marker for viral infections in children. In combination with a marker specific for bacterial infection, MxA could improve the management of children with signs of infection.
A prospective, single center, blinded, observational clinical trial was conducted at Beth Israel Deaconess Medical Center to determine the accuracy of a point-of-care immunoassay to identify a clinically significant immune response to viral and/or bacterial infection (72). Sixty patients with acute febrile respiratory infection were enrolled (19 pharyngitis and 41 lower respiratory tract infections). Participants provided fingerstick blood for immunoassay testing of MxA and CRP. The MxA ELISA Test Kit (Kyowa Medex Co., Ltd.) was used for the quantitative MxA testing. CRP was tested with the High Sensitivity CRP Enzyme Immunoassay Test Kit (Biocheck, Inc.). The immunoassay correctly categorized participants as 92% (22/24) negative, 80% (16/20) with bacterial infection, and 70% (7/10) with viral infection (72).
Throat swabs and blood samples taken from children (age, 1–16 years) with febrile pharyngitis were studied (73). A microbial cause was sought by bacterial culture, PCR, and serology. Blood MxA concentration was determined. A potential pathogen was detected in 88% of 83 patients: group A Streptococcus (GAS) alone in 10%, GAS and viruses in 13%, group C or G streptococci alone in 2% and together with viruses in 3%, and viruses alone in 59% of cases. Enteroviruses, rhinoviruses, and adenoviruses were the most frequently detected viruses. Blood MxA concentrations were higher in children with viral [median (IQR): 880 (245–1250) μg/L] or concomitant GAS-viral [340 (150–710) μg/L] than in those with sole GAS [105 (80–160) μg/L] infections. Thus, detection of respiratory viruses simultaneously with increased blood MxA concentrations supports the causative role of viruses in most children with pharyngitis.
Recently a new point-of-care rapid CRP/MxA immunoassay (FebriDx®, RPS Diagnostics) was used (74). A prospective, multicenter, cross-sectional study of adults and children with febrile upper respiratory tract infections was conducted to evaluate the diagnostic accuracy of the test to identify clinically significant bacterial infection with host response and acute pathogenic viral infection. Among 205 patients, 25 (12.2%) were classified as bacterial, 53 (25.9%) as viral, and 127 (62.0%) as negative by the reference standard. For bacterial detection, agreement between FebriDx and the reference standard was 91.7%, with FebriDx having a clinical sensitivity of 80% (95% CI, 59%–93%), clinical specificity of 93% (95% CI, 89%–97%), positive predictive value of 63% (95% CI, 45–79%), and a negative predictive value of 97% (95% CI, 94–99%). For viral detection, agreement was 84%, with a clinical sensitivity of 87% (95% CI, 75%–95%), clinical specificity of 83% (95% CI, 76%–89%), positive predictive value of 64% (95% CI, 63%–75%), and a negative predictive value of 95% (95% CI, 90%–98%).
mRNA indexes of MxA and 2 other interferon responsive genes (viperin and tripartite motif-containing protein 21) were investigated in nasal swabs as potential biomarkers of viral respiratory infection in children (75). Additionally, respiratory viruses in the same swabs were detected by PCR. Nasal MxA and viperin indexes were increased in symptomatic virus-positive children. The nasal viperin index was found to be a robust marker of viral respiratory tract infection with a clinical sensitivity of 80% and clinical specificity of 94% in distinguishing children with symptomatic virus infections from the asymptomatic virus-negative children.
Clinical studies show that the MxA protein is selectively increased in patients with viral infections and has the potential to greatly enhance the rapid distinction between viral and bacterial respiratory infections (76). Biomarkers such as CRP or procalcitonin independently may identify clinically significant infections, thereby reducing the risk of missing a clinically significant bacterial infection. However, these biomarkers lack adequate clinical specificity to differentiate a viral from a bacterial infection and ultimately lead to antibiotic overtreatment of viral infections. Moreover, the performance characteristics of CRP as a marker for bacterial infections vary considerable among published studies and are heavily influenced by the organ system involved and numerous comorbidities. Therefore, procalcitonin, which is gaining favor as a more specific biomarker of bacterial infection, should also be included in the diagnostics (76).
Conclusion
Viral infections in humans trigger a cascade of interferon-mediated signaling that leads to production of Mx proteins (MxA and MxB). Clinical studies show that the combined interpretation of MxA with either CRP and/or procalcitonin dramatically improves both clinical sensitivity and clinical specificity for differentiating infectious etiology and for excluding inappropriate therapy with antibiotics in viral infections.
4 Nonstandard abbreviations
- IFN
interferon
- ISGs
IFN-stimulated genes
- GTPases
guanine triphosphatases
- hu
human
- BSE
bundle-signaling element
- GTP
guanosine triphosphate
- GDP
guanosine diphosphate
- CVB4
coxsackievirus B4
- VACV
vaccinia virus
- CRP
C-reactive protein
- GAS
group A Streptococcus.
5 Genes
- mx1
myxovirus resistance protein 1
- mx2
myxovirus resistance protein 2
- MX1
dynamin-like GTPase 1
- MX2
dynamin-like GTPase 2.
Footnotes
Author Contributions: All authors confirmed they have contributed to the intellectual content of this paper and have met the following 4 requirements: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; (c) final approval of the published article; and (d) agreement to be accountable for all aspects of the article thus ensuring that questions related to the accuracy or integrity of any part of the article are appropriately investigated and resolved.
T. Wahlroos, administrative support.
Authors' Disclosures or Potential Conflicts of Interest: Upon manuscript submission, all authors completed the author disclosure form. Disclosures and/or potential conflicts of interest:
Employment or Leadership: T. Wahlroos, Labmaster ltd.
Consultant or Advisory Role: T.K. Korpela, Labmaster ltd.
Stock Ownership: T.K. Korpela, Labmaster ltd.
Honoraria: None declared.
Research Funding: None declared.
Expert Testimony: None declared.
Patents: None declared.
References
- 1. Isaacs A, Lindenmann J. Virus interference I: the interferon. Proc Roy Soc London Ser B 1957;147:258–67. [DOI] [PubMed] [Google Scholar]
- 2. Donnelly RP, Kotenko SV. Interferon-lambda: a new addition to an old family. J Interferon Cytokine Res 2010;30:555–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol 2003;4:69–77. [DOI] [PubMed] [Google Scholar]
- 4. Coccia EM, Severa M, Giacomini E, Monneron D, Remoli ME, Julkunen I. Viral infection and Toll-like receptor agonists induce a differential expression of type I and λ interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol 2004;34:796–805. [DOI] [PubMed] [Google Scholar]
- 5. Lasfar A, Lewis-Antes A, Smirnov SV, Anantha S, Abushahba W, Tian B. Characterization of the mouse IFN-lambda ligand–receptor system: IFN-lambdas exhibit antitumor activity against B16 melanoma. Cancer Res 2006;66:4468–77. [DOI] [PubMed] [Google Scholar]
- 6. Kochs G, Haener M, Aebi U, Haller O. Self-assembly of human MxA GTPase into highly ordered dynamin-like oligomers. J Biol Chem 2002;277:14172–6. [DOI] [PubMed] [Google Scholar]
- 7. Haller O, Kochs G. Human MxA protein: an interferon-induced dynamin-like GTPase with broad antiviral activity. J Interferon Cytokine Res 2011;31:79–87. [DOI] [PubMed] [Google Scholar]
- 8. Lindenmann J. Resistance of mouse to mice adapted influenza A virus. Virology 1962;16:203–4. [DOI] [PubMed] [Google Scholar]
- 9. Lindenmann J, Lane CA, Hobson D. The resistance of A2G mice to myxoviruses. J Immunol 1963;90:942–51. [PubMed] [Google Scholar]
- 10. Horisberger MA, Staeheli P, Haller O. Interferon induces a unique protein in mouse cells bearing a gene for resistance to influenza virus. Proc Natl Acad Sci USA 1983;80:1910–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Staeheli P, Haller O, Boll W, Lindenmann J, Weissmann C. Mx protein: constitutive expression in 3T3 cells transformed with cloned Mx cDNA confers selective resistance to influenza virus. Cell 1986;44:147–58. [DOI] [PubMed] [Google Scholar]
- 12. Haller O, Frese M, Kochs G. Mx proteins: mediators of innate resistance to RNA viruses. Rev Sci Tech 1998;17:220–30. [DOI] [PubMed] [Google Scholar]
- 13. Aebi M, Fah J, Hurt N, Samuel C, Thomis D, Bazzigher L. CDNA structures and regulation of two interferon-induced human Mx proteins. Mol Cell Biol 1989;9:5062–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haller O, Gao S, von der Malsburg A, Daumke O, Kochs G. Dynamin-like MxA GTPase: structural insights into oligomerization and implications for antiviral activity. J Biol Chem 2010;285:28419–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Verhelst J, Hulpiau P, Saelens X. Mx proteins: antiviral gatekeepers that restrain the uninvited. Microbiol Mol Biol Rev 2013;77:551–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nagata S, Taira H, Hall A, Johnsrud L, Streuli M, Ecsödi J. Synthesis in E. coli of a polypeptide with human leukocyte interferon activity. Nature 1980;284:316–20. [DOI] [PubMed] [Google Scholar]
- 17. Taniguchi T, Mantei N, Schwarzstein M, Nagata S, Muramatsu M, Weissmann C. Human leukocyte and fibroblast interferons are structurally related. Nature 1980;285:547–9. [DOI] [PubMed] [Google Scholar]
- 18. Zav'yalov VP, Denesyuk AI. Possible conformation of interferons: a prediction based on amino acid composition and sequence data. Immunol Lett 1982;4:7–14. [DOI] [PubMed] [Google Scholar]
- 19. Zav'yalov VP, Denesyuk AI, Zav'yalova GA. Theoretical analysis of conformation and active sites of interferons. Immunol Lett 1989;22:173–82. [DOI] [PubMed] [Google Scholar]
- 20. Zav'yalov VP, Denesyuk AI, Zav'yalova GA. Theoretical conformational analysis of a family of α-helical immunocytokines. Biochim Biophys Acta 1990;1041:178–85. [DOI] [PubMed] [Google Scholar]
- 21. Zav'yalov VP, Zav'yalova GA. Interferons alpha/beta and their receptors: place in the hierarchy of cytokines. Acta Pathol Microbiol Immunol Scand 1997;105:161–86. [DOI] [PubMed] [Google Scholar]
- 22. Uze′ G, Schreiber G, Piehler J, Pellegrini S. The receptor of the type I interferon family. Curr Top Microbiol Immunol 2007;316:71–95. [DOI] [PubMed] [Google Scholar]
- 23. Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell 2011;146:621–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Radhakrishnan R, Walter LJ, Hruza A, Reichert P, Trotta PP, Nagabhushan TL, Walter MR. Zinc mediated dimer of human interferon-alpha 2b revealed by X-ray crystallography. Structure 1996;4:1453–63. [DOI] [PubMed] [Google Scholar]
- 25. Uze G, Lutfcrllu G, Mogensen KE. α and β interferons and their receptor and their friends and relations. J Interferon Cytokine Res 1995;15:3–26. [DOI] [PubMed] [Google Scholar]
- 26. Gaboriaud C, Uze′ G, Lutfalla G, Mogensen K. Hydrophobic cluster analysis reveals duplication in the external structure of human alpha-interferon receptor and homology with gamma-interferon receptor external domain. FEBS Lett 1990;269:1–3. [DOI] [PubMed] [Google Scholar]
- 27. van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 2006;25:361–72. [DOI] [PubMed] [Google Scholar]
- 28. Schindler C, Plumlee C. Inteferons pen the JAK-STAT pathway. Semin Cell Dev Biol 2008;19:311–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol 2003;4:63–8. [DOI] [PubMed] [Google Scholar]
- 30. Gad HH, Dellgren C, Hamming OJ, Vends S, Paludan SR, Hartmann R. Interferon-lambda is functionally an interferon but structurally related to the interleukin-10 family. J Biol Chem 2009;284:20869–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gad HH, Hamming OJ, Hartmann R. The structure of human interferon lambda and what it has taught us. J Interferon Cytokine Res 2010;30:565–71. [DOI] [PubMed] [Google Scholar]
- 32. Hamming OJ, Gad HH, Paludan S, Hartmann R. Lambda interferons: new cytokines with old functions. Pharmaceuticals (Basel) 2010;3:795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zdanov A. Structural analysis of cytokines comprising the IL-10 family. Cytokine Growth Factor Rev 2010;21:325–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miknis ZJ, Magracheva E, Li W, Zdanov A, Kotenko SV, Wlodawer A. Crystal structure of human interferon-λ1 in complex with its high-affinity receptor interferon-λR1. J Mol Biol 2010;404:650–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yoon SI, Jones BC, Logsdon NJ, Harris BD, Deshpande A, Radaeva S. Structure and mechanism of receptor sharing by the IL-10R2 common chain. Structure 2010;18:638–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, Hartmann R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J Virol 2007;81:7749–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Doyle SE, Schreckhise H, Khuu-Duong K, Henderson K, Rosler R, Storey H. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006;44:896–906. [DOI] [PubMed] [Google Scholar]
- 38. Marcello T, Grakoui A, Barba-Spaeth G, Machlin ES, Kotenko SV, MacDonald MR, Rice CM. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006;131:1887–98. [DOI] [PubMed] [Google Scholar]
- 39. Haller O, Staeheli P, Schwemmle M, Kochs G. Mx GTPases: dynamin-like antiviral machines of innate immunity. Trends Microbiol 2015;23:154–63. [DOI] [PubMed] [Google Scholar]
- 40. Gao S, von der Malsburg A, Dick A, Faelber K, Schröder GF, Haller O. Structure of myxovirus resistance protein a reveals intra- and intermolecular domain interactions required for the antiviral function. Immunity 2011;35:514–25. [DOI] [PubMed] [Google Scholar]
- 41. Rennie ML, McKelvie SA, Bulloch EM, Kingston RL. Transient dimerization of human MxA promotes GTP hydrolysis, resulting in a mechanical power stroke. Structure 2014;22:1433–45. [DOI] [PubMed] [Google Scholar]
- 42. Chen Y, Zhang L, Graf L, Yu B, Liu Y, Kochs G. Conformational dynamics of dynamin-like MxA revealed by single-molecule FRET. Nat Commun 2017;8:15744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. von der Malsburg A, Abutbul-Ionita I, Haller O, Kochs G, Danino D. Stalk domain of the dynamin-like MxA GTPase protein mediates membrane binding and liposome tubulation via the unstructured L4 loop. J Biol Chem 2011;286:37858–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dick A, Graf L, Olal D, von der Malsburg A, Gao S, Kochs G, Daumke O. Role of nucleotide binding and GTPase domain dimerization in dynamin-like myxovirus resistance protein A for GTPase activation and antiviral activity. J Biol Chem 2015;290:12779–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xu B, Kong J, Wang X, Wei W, Xie W, Yu XF. Structural insight into the assembly of human anti-HIV dynamin-like protein MxB/Mx2. Biochem Biophys Res Commun 2015;456:197–201. [DOI] [PubMed] [Google Scholar]
- 46. Chieux V, Chehadeh W, Harvey J, Haller O, Wattré P, Hober D. Inhibition of coxsackievirus B4 replication in stably transfected cells expressing human MxA protein. Virology 2001;283:84–92. [DOI] [PubMed] [Google Scholar]
- 47. Patrick S. Mitchell PS, Emerman M, Malik HS. An evolutionary perspective on the broad antiviral specificity of MxA. Curr Opin Microbiol 2013;16:493–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lorenzo MM, Sanchez-Puig JM, Blasco R. Vaccinia virus and Cowpox virus are not susceptible to the interferon-induced antiviral protein MxA. PLoS One 2017;12:e0181459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Graf L, Dick A, Sendker F, Barth E, Marz M, Daumke O, Kochs G. Effects of allelic variations in the human myxovirus resistance protein A on its antiviral activity. J Biol Chem 2018;293:3056–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Melen K, Keskinen P, Ronni T, Sareneva T, Lounatmaa K, Julkunen I. Human MxB protein, an interferon-alpha-inducible GTPase, contains a nuclear targeting signal and is localized in the heterochromatin region beneath the nuclear envelope. J Biol Chem 1996;271:23478–86. [DOI] [PubMed] [Google Scholar]
- 51. King MC, Raposo G, Lemmon MA. Inhibition of nuclear import and cell-cycle progression by mutated forms of the dynamin-like GTPase MxB. Proc Natl Acad Sci USA 2004;101:8957–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kong J, Xu B, Wei W, Wang X, Xie W, Yu XF. Characterization of the amino-terminal domain of Mx2/MxB-dependent interaction with the HIV-1 capsid. Protein Cell 2014;5:954–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cals JW, Hopstaken RM, Butler CC, Hood K, Severens JL, Dinant GJ. Improving management of patients with acute cough by C-reactive protein point of care testing and communication training (IMPAC3T): study protocol of a cluster randomised controlled trial. BMC Fam Pract 2007;8:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gonzales R, Bartlett JG, Besser RE, Cooper RJ, Hickner JM, Hoffman JR, Sande MA. Principles of appropriate antibiotic use for treatment of nonspecific upper respiratory tract infections in adults: background, specific aims, and methods. Ann Emerg Med 2001;37:690–7. [PubMed] [Google Scholar]
- 55. WHO. Global action plan on antimicrobial resistance. http://www.wpro.who.int/entity/drug_resistance/resources/global_action_plan_eng.pdf (Accessed May 2015).
- 56. Chieux V, Hober D, Chehadeh W, Harvey J, Alm G, Cousin J. MxA protein in capillary blood of children with viral infections. J Med Virol 1999;59:547–51. [PubMed] [Google Scholar]
- 57. Horisberger MA. Interferon-induced human protein MxA is a GTPase which binds transiently to cellular proteins. J Virol 1992;66:4705–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ronni T, Melen K, Malygin A, Julkunen I. Control of IFN inducible MxA gene expression in human cells. J Immunol 1993;150:1715–26. [PubMed] [Google Scholar]
- 59. Goetschy JF, Zeller H, Content J, Horisberger MA. Regulation of the interferon-inducible IFI-78K gene, the human equivalent of the murine Mx gene, by interferons, double-stranded RNA, certain cytokines, and viruses. J Virol 1989;63:2616–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nakabayashi M, Adachi Y, Itazawa T, Okabe Y, Kanegane H, Kawamura M. MxA-based recognition of viral illness in febrile children by a whole blood assay. Pediatr Res 2006;60:770–4. [DOI] [PubMed] [Google Scholar]
- 61. Itazawa T, Adachi Y, Imamura H, Okabe Y, Yamamoto J, Onoue Y. Increased lymphoid MxA expression in acute asthma exacerbation in children. Allergy 2001;56:895–8. [DOI] [PubMed] [Google Scholar]
- 62. Itazawa T, Adachi Y, Nakabayashi M, Fuchizawa T, Murakami G, Miyawaki T. Theophylline metabolism in acute asthma with MxA-indicated viral infection. Pediatr Int 2006;48:54–7. [DOI] [PubMed] [Google Scholar]
- 63. Forster J, Schweizer M, Schumacher RF, Kaufmehl K, Lob S. MxA protein in infants and children with respiratory tract infection. Acta Paediatr 1996;85:163–7. [DOI] [PubMed] [Google Scholar]
- 64. Halminen M, Ilonen J, Julkunen I, Ruuskanen O, Simell O, Makela MJ. Expression of MxA protein in blood lymphocytes discriminates between viral and bacterial infections in febrile children. Pediatr Res 1997;41:647–50. [DOI] [PubMed] [Google Scholar]
- 65. Chieux V, Hober D, Harvey J, Lion G, Lucidarme D, Forzy G. The MxA protein levels in whole blood lysates of patients with various viral infections. J Virol Methods 1998;70:183–91. [DOI] [PubMed] [Google Scholar]
- 66. Makela MJ, Puhakka T, Ruuskanen O, Leinonen M, Saikku P, Kimpimaki M. Viruses and bacteria in the etiology of the common cold. J Clin Microbiol 1998;36:539–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Huang N, Morlock L, Lee CH, Chen LS, Chou YJ. Antibiotic prescribing for children with nasopharyngitis (common colds), upper respiratory infections, and bronchitis who have health professional parents. Pediatrics 2005;116:826–32. [DOI] [PubMed] [Google Scholar]
- 68. Baker MD, Bell LM, Avner JR. Outpatient management without antibiotics of fever in selected infants. N Engl J Med 1993;329:1437–41. [DOI] [PubMed] [Google Scholar]
- 69. Baskin MN, O'Rourke EJ, Fleisher GR. Outpatient treatment of febrile infants 28 to 89 days of age with intramuscular administration of ceftriaxone. J Pediatr 1992;120:22–7. [DOI] [PubMed] [Google Scholar]
- 70. Toivonen L, Schuez-Havupalo L, Rulli M, Ilonen J, Pelkonen J, Melen K. Blood MxA protein as a marker for respiratory virus infections in young children. J Clin Virol 2015;62:8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Engelmann I, Dubos F, Lobert PE, Houssin C, Degas V, Sardet A. Diagnosis of viral infections using myxovirus resistance protein A (MxA). Pediatrics 2015;135:e985–93. [DOI] [PubMed] [Google Scholar]
- 72. Towbin H, Schmitz A, Jakschies D, Von Wussow P, Horisberger MA. A whole blood immunoassay for the interferon inducible human Mx protein. J Interferon Res 1992;12:67–74. [DOI] [PubMed] [Google Scholar]
- 73. Sambursky R, Shapiro N. Evaluation of a combined MxA and CRP point-of-care immunoassay to identify viral and/or bacterial immune response in patients with acute febrile respiratory infection. Eur Clin Respir J 2015;2:28245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Self WH, Rosen J, Sharp SC, Filbin MR, Hou PC, Parekh AD. Diagnostic accuracy of FebriDx: a rapid test to detect immune responses to viral and bacterial upper respiratory infections. J Clin Med 2017;6:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yahya M, Rulli M, Toivonen L, Waris M, Peltola V. Detection of host response to viral respiratory infection by measurement of mRNA for MxA, TRIM21, and viperin in nasal swabs. J Infect Dis 2017;216:1099–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Joseph P, Godofsky E. Outpatient antibiotic stewardship: a growing frontier-combining myxovirus resistance protein A with other biomarkers to improve antibiotic use. Open Forum Infect Dis 2018;5:ofy024. [DOI] [PMC free article] [PubMed] [Google Scholar]