

Abstract
Background: Influenza A viruses are medically important viral pathogens that cause significant mortality and morbidity throughout the world. The recent emergence of a novel human influenza A virus (H1N1) poses a serious health threat. Molecular tests for rapid detection of this virus are urgently needed.
Methods: We developed a conventional 1-step RT-PCR assay and a 1-step quantitative real-time RT-PCR assay to detect the novel H1N1 virus, but not the seasonal H1N1 viruses. We also developed an additional real-time RT-PCR that can discriminate the novel H1N1 from other swine and human H1 subtype viruses.
Results: All of the assays had detection limits for the positive control in the range of 1.0 × 10−4 to 2.0 × 10−3 of the median tissue culture infective dose. Assay specificities were high, and for the conventional and real-time assays, all negative control samples were negative, including 7 human seasonal H1N1 viruses, 1 human H2N2 virus, 2 human seasonal H3N2 viruses, 1 human H5N1 virus, 7 avian influenza viruses (HA subtypes 4, 5, 7, 8, 9, and 10), and 48 nasopharyngeal aspirates (NPAs) from patients with noninfluenza respiratory diseases; for the assay that discriminates the novel H1N1 from other swine and human H1 subtype viruses, all negative controls were also negative, including 20 control NPAs, 2 seasonal human H1N1 viruses, 2 seasonal human H3N2 viruses, and 2 human H5N1 viruses.
Conclusions: These assays appear useful for the rapid diagnosis of cases with the novel H1N1 virus, thereby allowing better pandemic preparedness.
The emergence of a novel H1N1 influenza A virus of animal origin with transmissibility from human to human poses pandemic concern (1)(2). The virus was initially identified in people in Mexico and the US in March–April 2009. At the time of writing, there were 1490 confirmed human cases of this novel H1N1 infection in 21 cities across many continents. Hong Kong reported its first confirmed case of this novel H1N1 virus on May 1, 2009. The severity of the human disease remains unclear, with most cases outside of Mexico having a mild clinical course. However, the Spanish flu pandemic of 1918, the most severe influenza pandemic currently known, was also mild at the time of its initial emergence and only acquired virulence during the second wave of human disease in the winter season (3). Current mathematical model–based estimates predict that a new pandemic could result in 2–7.4 million deaths worldwide (4).
It is still unclear whether this novel H1N1 virus will be sustained in the human population. Some cases appear to have caused large outbreaks of local transmission (such as in New York), but many other cases have led to little or no secondary transmission. Early detection in suspected patients is crucial for containing the disease and preventing ongoing transmission. Few well-validated laboratory diagnostic methods exist for the diagnosis of this novel H1N1 infection. The WHO defines a probable clinical case as one that is confirmed by (1) specific RT-PCR–based detection methods, (2) isolation of the novel H1N1 influenza virus, or (3) detection of a 4-fold rise of neutralization antibodies to this virus (5). Biosafety level (BSL)1 2 laboratories with BSL3 practices are recommended for virus isolation and serology, whereas PCR detection requires only a BSL2 environment (6). Of the above diagnostic tests, only RT-PCR–based detection methods allow rapid detection of the swine-like H1N1 influenza virus within a few hours.
Based on the publicly released hemagglutinin (HA) sequence (A/California/04/2009, GenBank accession no. FJ966082), we developed and evaluated several RT-PCR assays for the detection of this virus. We used these assays to help confirm the first case found in Hong Kong (7). Here, we describe a conventional 1-step RT-PCR and a 1-step quantitative real-time RT-PCR that can detect this novel swine-like H1N1. Because many laboratories lack the novel A/California/04/2009-like H1N1 virus for use as a positive control for these tests, we used a swine H1 virus (A/SW/HK/PHK1578/03) previously isolated in our routine swine influenza surveillance program (unpublished results) as the positive control in this study. The primers and probes were designed to perfectly match both the swine-like H1N1 and A/SW/HK/PHK1578/03. We reasoned that regions of the genome that are conserved between swine H1 viruses in Asia and the swine-like novel H1N1 virus, but distinct from human seasonal H1N1 viruses, offer several advantages as targets for a molecular diagnostic test. More importantly, we reasoned that this approach might help to minimize the shipping and handling of A/California/04/2009-like H1N1 viruses to laboratories that do not have the recommended biosafety facilities. Accidental release of the human adapted A/California/04/2009-like viruses from the laboratory poses public health concerns. The positive control Asian swine H1N1 virus (A/SW/HK/PHK1578/03), which is not adapted to human transmission, is a much safer option and is available on request.
For performing conventional 1-step RT-PCR assays, we extracted RNA from 140 μL nasopharyngeal aspirate or viral culture with known titer using the QIAamp Virus RNA Mini Kit (Qiagen) as described (8). We performed the RT-PCR assays using a Qiagen OneStep RT-PCR kit as recommended by the manufacturer. Briefly, 2 μL purified RNA was reverse-transcribed and amplified in a 20-μL reaction containing 4 μL of 5× RT-PCR buffer (Qiagen), 0.4 mmol/L of each deoxynucleotide triphosphate, 4 U RNase inhibitor (Applied Biosystems), 0.8 μL RT-PCR enzyme mix (Qiagen), 0.6 μmol/L forward primer (HKU-SWF: 5′-GAGCTCAGTGTCATCATTTGAA-3′, corresponding to nucleotides 366–387 of the HA-encoding sequence), and 0.6 μmol/L reverse primer (HKU-SWR: 5′-TGCTGAGCTTTGGGTATGAA-3′, complementary to nucleotides 519–538). Reactions were first incubated at 50 °C for 30 min, followed by 95 °C for 15 min. Reactions were then thermal-cycled for 40 cycles (95 °C for 30 s, 57 °C for 30 s, and 72 °C for 20 s). The RT-PCR products were analyzed in 2% agarose gel using standard gel electrophoresis procedures.
To perform real-time 1-step RT-PCR assays, viral RNA was amplified by TaqMan EZ RT-PCR Core Reagents (Applied Biosystems) in a 7500 Sequence Detection System. Briefly, 4 μL purified RNA was amplified in a 25-μL reaction containing 2.5 U rTth DNA polymerase (Applied Biosystems), 5 μL of 5× EZ buffer A, 300 μmol/L of each dNTP (except 1200 μmol/L for dUTP), 3 mmol/L manganese acetate, 0.25 U AmpErase UNG, 800 nmol/L forward primer (HKU-qSWF: 5′-GGGTAGCCCCATTGCAT-3′, corresponding to nucleotides 188–204 of the HA-encoding sequence), 800 nmol/L reverse primer (HKU-qSWR: 5′-AGAGTGATTCACACTCTGGATTTC-3′, complementary to nucleotides 239–262), and 400 nmol/L probe [HKU-qSWP: 5′-(FAM)TGGGTAAATGTAACATTGCTGGCTGG(TAMRA)-3′, corresponding to nucleotides 206–231]. Reactions were first incubated at 50 °C for 2 min, followed by 60 °C for 40 min. After a 5-min denaturation at 95 °C, reactions were then thermal-cycled for 50 cycles (94 °C for 15 s, 55 °C for 1 min). Serially diluted positive viral RNA control samples were used as calibrators in each run.
With serially diluted positive control RNA, the detection limits of the conventional and quantitative RT-PCR assays were found to be 1.0 × 10−4 TCID50 (median tissue culture infective dose) per reaction and 2.0 × 10−3 TCID50 per reaction, respectively (Fig. 1A and B). In this study, 1 TCID50 of the positive sample was estimated to have 1000 copies of influenza matrix (M) gene (data not shown). We also determined the detection limit of our real-time assay that is specific for the A/California/4/2009-like viruses. Using RNA extracted from a A/California/4/2009 viral culture, the detection limit of the assay was found to be 8 × 10−3 TCID50 per reaction. The copy numbers of the M gene of the same RNA samples were also quantitated using a real-time RT-PCR assay, and 1 TCID50 of this virus was estimated to have approximately 5000 copies of M. All negative control samples, including 7 human seasonal H1N1 viruses, 1 human H2N2 virus, 2 human seasonal H3N2 viruses, 1 human H5N1 virus, 7 avian influenza viruses (HA subtypes 4, 5, 7, 8, 9, and 10), and 48 nasopharyngeal aspirate samples from patients with respiratory diseases other than influenza, were found to be negative in both assays (data not shown), indicating these assays were specific for the targeted sequences only. Both PCR assays were at least 500 times more sensitive than virus culture. The sensitivity of the real-time quantitative RT-PCR assays was less than that of the conventional RT-PCR, and further optimization of the real-time assay to enhance the sensitivity of that assay is in progress.
Figure 1.
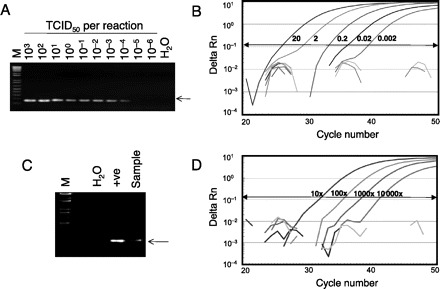
Molecular detection of the novel H1N1 influenza virus by conventional and real-time RT-PCR assays.
(A), Amplification of serially diluted positive control samples. The viral RNA inputs in these reactions are indicated in terms of TCID50. M, 1-kb DNA ladder markers (Invitrogen); H2O, water control; arrow, expected RT-PCR products (173 bp). (B), Amplification of serially diluted positive control samples. The amount of viral RNA (TCID50) used in each of the positive reactions is indicated. (C), Detection of the novel H1N1 from a clinical specimen by the conventional RT-PCR assay. +ve, positive control. (D), Detection of the novel H1N1 from a clinical specimen by the real-time RT-PCR assay. The clinical specimen was serially diluted and tested using the assay. The dilution factors used in each of these positive reactions are indicated. Delta Rn indicates the magnitude of the PCR signal.
As we were developing these assays, we received a respiratory specimen from a traveler from Mexico who was suspected to have infection with the novel H1N1 virus. A nasopharyngeal aspirate was collected within 24 h of clinical symptom onset. As indicated in Fig. 1C and D, RNA from this sample was positive in both conventional and real-time assays. The identity of the positive PCR product in the conventional RT-PCR was further confirmed by DNA sequencing. The RNA from the sample remained detectable in the real-time PCR assay when diluted 10 000-fold. The viral load in this respiratory sample determined by M gene real-time PCR was comparable to that from seasonal influenza virus infection (data not shown).
Such a RT-PCR assay for detection of the novel H1N1 virus should be part of a panel of molecular diagnostic tests for influenza type A including the M gene segment and human seasonal subtypes H1 and H3. A specimen that is positive for the type A M gene and negative for human seasonal H1 and H3, but positive for the test described here constitutes a provisional diagnosis of infection with the novel H1N1 virus. Such specimens need to be referred to a reference laboratory for further testing.
The conventional and real-time RT-PCR assays developed here detect the novel human adapted H1N1 and swine H1 viruses of the same viral linage, but not those of human seasonal influenza H1N1 or H3N2. Thus, a positive result in this assay clearly discriminates the novel H1N1 virus from seasonal H1N1 influenza viruses, but does not differentiate between the novel H1N1 and other swine H1 viruses. In the current context, however, a human specimen that has a H1 gene that is “swine-like” is sufficient to initiate a public health alert and referral to a reference laboratory for final confirmation. Although swine H1N1 or H1N2 viruses have infected humans in the past (9), they are exceedingly rare. A specimen found positive in the assay for the novel H1N1 virus we describe here can be confirmed by testing with other PCR methods specific for swine influenza A viruses (10) and by partial sequencing of one or more viral gene segments. For example, sequencing the RT-PCR amplicon generated from the M gene type A–specific assay (M30F: 5′-TTCTAACCGAGGTCGAAACG-3′ and M264R2: 5′-ACAAAGCGTCTACGCTGCAG-3′) (11) will allow differentiation of the swine H1N1 virus from the novel human-adapted H1N1 virus.
A real-time assay that is specific for the novel influenza A H1N1 and discriminates it from other swine (and human) H1 subtype viruses is under development. Preliminary reaction conditions are identical to the real-time assay as described above, with the following differences:
The primer and probe sequences are HKU-qABF1: 5′-GACAAGTTCATGGCCCAATCA-3′, HKU-qABR1: 5′-TTTGCTCCAGCATGAGGACAT-3′, and HKU-qABP1: 5′-(FAM)AACAAAGGTGTAACGGCAG(MGB)-3′. The PCR amplicon corresponds to nucleotides 408–476 of the HA-encoding sequence. Residues that differentiate swine influenza A/SW/HK/PHK1578/03 and human influenza A/California/04/2009-like viruses are underlined.
The annealing temperature is 60 °C instead of 55 °C.
In preliminary testing, the limit of detection determined by serial dilutions was similar to that of the other real-time PCR assay described here. Amplification of viral RNA from swine H1 virus (A/SW/HK/PHK1578/03) as well as other negative controls (20 control nasopharyngeal aspirates, 2 seasonal human H1N1 viruses, 2 seasonal human H3N2 viruses, and 2 human H5N1 viruses) was negative. Hence, this assay can be used to specifically confirm the presence of the novel H1N1 virus causing the current outbreaks. However, the A/California/4/2009-like virus is required as a positive control. Additional evaluation and optimization of this assay is in progress.
Acknowledgments
Author Contributions: All authors confirmed they have contributed to the intellectual content of this paper and have met the following 3 requirements: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data, (b) drafting or revising the article for intellectual content, and (c) final approval of the published article.
Authors’ Disclosures of Potential Conflicts of Interest: Upon manuscript submission, all authors completed the Disclosures of Potential Conflict of Interest form. Potential conflicts of interest:
Employment or Leadership: None declared.
Consultant or Advisory Role: None declared.
Stock Ownership: None declared.
Honoraria: None declared.
Research Funding: Area of Excellence Scheme of the University Grants Committee Hong Kong Grant AoE/M-12/06 and Research Grant Council of Hong Kong (HKU 773408M, LLM).
Expert Testimony: None declared.
Role of Sponsor: The funding organizations played no role in the design of study, choice of enrolled patients, review and interpretation of data, or preparation or approval of manuscript.
Acknowledgments: We thank Timothy Cheung, Edward Ma, Alex Chin, and Horasis Leung for their technical help. We also thank Dr. Wilina Lim for sharing clinical specimens and for advice.
Footnotes
Nonstandard abbreviations: BSL, biosafety level; HA, hemagglutinin; TCID50, median tissue culture infective dose; M, influenza matrix.
Contributor Information
Leo L M Poon, Email: llmpoon@hkucc.hku.hk.
J S M Peiris, Email: malik@hkucc.hku.hk.
References
- 1. Centers for Disease Control and Prevention. Swine influenza A (H1N1) infection in two children: Southern California, March–April 2009. MMWR Morb Mortal Wkly Rep 2009;58:400-402. [PubMed] [Google Scholar]
- 2. World Health Organization. Swine influenza. Wkly Epidemiol Rec 2009;84:149. [Google Scholar]
- 3.Taubenberger JK, Morens DM. Influenza: the mother of all pandemics. Emerg Infect Dis 2006;1918;12:15-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World Health Organization. Pandemic preparedness. http://www.who.int/csr/disease/influenza/pandemic/en/index.html (Accessed May 5, 2009). [Google Scholar]
- 5.World Health Organization. Interim WHO guidance for the surveillance of human infection with swine influenza A (H1N1) virus. http://www.who.int/csr/disease/swineflu/WHO_case_definition_swine_flu_2009_04_29.pdf (Accessed May 5, 2009). [Google Scholar]
- 6.Centers for Disease Control and Prevention. H1N1 influenza virus biosafety guidelines for laboratory workers. http://www.cdc.gov/h1n1flu/guidelines_labworkers.htm (Accessed May 5, 2009). [Google Scholar]
- 7.Centers for Disease Control and Prevention. Influenza A(H1N1): update 8.1. http://www.who.int/csr/don/2009_05_01a/en/index.html (Accessed May 5, 2009). [Google Scholar]
- 8.Poon LL, Wong OK, Chan KH, Luk W, Yuen KY, Peiris JS, Guan Y. Rapid diagnosis of a coronavirus associated with severe acute respiratory syndrome (SARS). Clin Chem 2003;49:953-955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Myers KP, Olsen CW, Gray GC. Cases of swine influenza in humans: a review of the literature. Clin Infect Dis 2007;44:1084-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.World Health Organization. CDC protocol of real-time RT-PCR for swine influenza A(H1N1). http://www.who.int/csr/resources/publications/swineflu/realtimeptpcr/en/index.html (Accessed May 5, 2009). [Google Scholar]
- 11.World Health Organization. Recommendations and laboratory procedures for detection of avian influenza A(H5N1) virus in specimens from suspected human cases. http://www.who.int/csr/disease/avian_influenza/guidelines/RecAIlabtestsAug07.pdf (Accessed May 5, 2009). [Google Scholar]