

Abstract
Ebola virus disease (EVD) is one of the most lethal transmissible infections characterized by a high fatality rate, and a treatment has not been developed yet. Recently, it has been shown that cationic amphiphiles, among them the antiarrhythmic drug amiodarone, inhibit filovirus infection. In the present work, we investigated how amiodarone interferes with Ebola virus infection. Wild-type Sudan ebolavirus and recombinant vesicular stomatitis virus, pseudotyped with the Zaire ebolavirus glycoprotein, were used to gain further insight into the ability of amiodarone to affect Ebola virus infection. We show that amiodarone decreases Ebola virus infection at concentrations close to those found in the sera of patients treated for arrhythmias. The drug acts by interfering with the fusion of the viral envelope with the endosomal membrane. We also show that MDEA, the main amiodarone metabolite, contributes to the antiviral activity. Finally, studies with amiodarone analogues indicate that the antiviral activity is correlated with drug ability to accumulate into and interfere with the endocytic pathway. Considering that it is well tolerated, especially in the acute setting, amiodarone appears to deserve consideration for clinical use in EVD.
Keywords: amiodarone, haemorrhagic fever, ebola, cationic amphiphiles, dronedarone, filovirus
The anti-arrhythmic drug amiodarone, and one of its active metabolites interfere with the early steps of Ebola virus life cycle by blocking the fusion step between the viral envelope and the endosomal membrane.
INTRODUCTION
Ebola virus disease (EVD) is a disease of human and non-human primates, caused by five genetically distinct members of the Ebolavirus genus, Filoviridae family. So far Zaire, Sudan and Bundibugyo viruses have been responsible for most of the human EVD outbreaks (Feldmann and Geisbert 2011). Ebola virus (EBOV) infects a broad range of cell types (Feldmann and Geisbert 2011). Binding and entry into target cells are mediated by the viral glycoprotein GP, a class I fusion protein composed of a receptor binding subunit (GP1) and a fusion subunit (GP2) (Lee et al.2008; Lee and Saphire 2009. After initial internalization via macropinocytosis, virus particle trafficking into the endolysosomal pathway ends up into late endosomes, where the low-pH-dependent cysteine proteases cathepsins B and L allow processing of the GP1 into a 19 kDa fusogenic form (Chandran et al.2005; Schornberg et al.2006; Dube et al.2009; Hood et al.2010; Miller et al.2012). Subsequently, the interaction between the processed GP1 and the late endosomal/lysosomal protein Neimann–Pick C1 (NPC-1) leads to GP2-dependent fusion of the viral envelope with the limiting membrane of the endosomes (Carette et al.2011; Cote et al.2011; Miller et al.2012). NPC-1 protein is necessary for virus infectivity, as cells from patients with Neimann–Pick disease, due to lack or malfunction of the NPC-1 protein, are resistant to EBOV infection (Carette et al.2011). In addition to the NPC-1, it has been recently demonstrated that EBOV entry into the target cells requires also the presence of the endosomal/lysosomal calcium channels called two-pore channels (Sakurai et al.2015).
Multiple cationic amphiphiles have been reported to inhibit EBOV infection (Carette et al.2011; Johansen et al.2013; Shoemaker et al.2013). In particular, it has been shown that the antiarrhythmic drugs amiodarone and dronedarone, multi-ion channel inhibitors, inhibit filovirus infection acting at an early step of viral life cycle (Gehring et al.2014). Interestingly, amiodarone and dronedarone induce an NPC-like phenotype in several cell types (Morissette et al.2009; Piccoli et al.2011).
Here we found that (i) amiodarone inhibits the fusion of the Zaire ebolavirus glycoprotein (ZEBOV GP) with the endosomal membrane; (ii) amiodarone and its main metabolite monodesethyl amiodarone (MDEA) potently inhibit Sudan ebolavirus (SUDV) entry and progeny release from infected cells; (iii) the antiviral activity correlates with amiodarone ability of inducing an NPC-like phenotype.
Since all these effects were observed at concentrations close to those found in the sera of patients treated for arrhythmias, it appears that amiodarone could be an attractive candidate for the treatment of EVD.
MATERIALS AND METHODS
Cells, plasmids and compounds
Human embryonic kidney (HEK293T) cells and African green monkey kidney Vero cells were grown at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium (Life Technologies) supplemented with 10% heat-inactivated foetal bovine serum (Life Technologies).
The plasmid expressing the ZEBOV GP, pVR-1012-ZEBOV-GP, was kindly provided by Gary Nabel (Vaccine Research Center, NIH, Bethesda, USA). The plasmid expressing the vesicular stomatitis virus (VSV) glycoprotein (pVSV-G) was previously described (Salata et al.2009).
Amiodarone was purchased from Sigma-Aldrich. Dronedarone was extracted from Multaq pills (Sanofi-Aventis) as previously described (Piccoli et al.2011). Synthesis and characteristics of amiodarone analogues (Fig. 1) have been previously reported (Quaglino et al.2004; Ha et al.2005).
Figure 1.
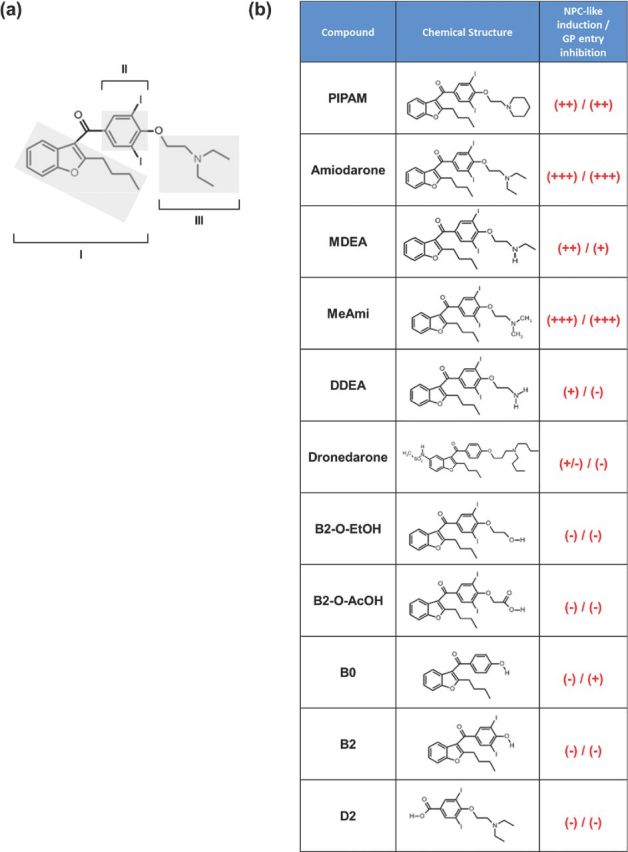
Amiodarone and analogues. (a) Amiodarone structural determinants: benzofuran moiety with butyl lateral group (I), diiodobenzoyl moiety (II) and diethylamino-β-ethoxy moiety with tertiary nitrogen atom (III). (b) Analogues listed in order of decreasing hydrophobicity; + or − indicate the ability of inducing an NPC-like phenotype and of inhibiting GP-mediated viral entry at the concentrations used in this work (0.3 μM dronedarone, 0.3 μM DDEA, 0.3 μM B2-O-EtOH, 2 μM MDEA, all other 10 μM).
Infection assays with the GP-pseudotyped virus and SUDV
The recombinant VSV containing the gene encoding the green fluorescent protein in place of the VSV-G gene (VSVΔG-GFP) was provided by Michael Whitt, University of Tennessee, USA. ZEBOV GP pseudotyped VSVΔG-GFP (GP-pseudotyped virus) was generated as previously described (Whitt 2010). Briefly, HEK293T cells were transfected by Lipofectamine 2000 (Life Technologies) with 16 μg of pVR-1012-ZEBOV-GP plasmid and infected with the recombinant VSVΔG-GFP virus at the multiplicity of infection (MOI) of 4 fluorescent focus-forming units (FFU)/cell. After 48 h, virus was harvested and stored at −80°C. Viral titer was evaluated by cytofluorimetric analysis (FACScalibur, Becton Dickinson) of Vero cells infected with limiting dilutions of viral stock, and data were analysed by CellQuest software.
In the antiviral activity assays, Vero cells were exposed to the appropriate compound for the time indicated. Next, cells were infected with recombinant viruses (MOI of 0.1 FFU/cell) and 24 h later GFP-positive cells were evaluated.
Infection with the SUDV was performed by adding the virus (MOI of 0.05 FFU/cell) to Vero cells. One hour later, unbound virus was removed and fresh medium with the appropriate compound was added. After 24 h, cells were fixed with ice-cold methanol/acetone at −20°C for 30 min. Cells were processed for immunofluorescence using an antibody targeting the viral nucleoprotein, as previously described (Weidmann et al.2011). Viral progeny release was evaluated on supernatants collected 48 h post-infection by immunostaining of infected Vero cells. All the experiments with SUDV were performed in the biosafety level 4 laboratory at the Public Health Agency of Sweden, Solna, Sweden.
Fusion assay
Vero cells grown on 13-mm coverslips were kept in control medium or pretreated for 16 h with either 10 μM amiodarone or 20 μM U18666A. Subsequently, cells were incubated for 3 h with GP-pseudotyped virus (MOI of 20 FFU/cell). Next, cells were fixed for 20 min with 2% paraformaldehyde (Sigma-Aldrich), and permeabilized for 30 min with 0.1% Triton X-100 (Sigma Aldrich). VSV matrix (VSV-M) protein was detected by incubations for 30 min at 37°C with 23H12 antibody (Kerafast Inc.), and then with Alexa 568-conjugated antibody (Life Technologies). Cells were counterstained with DRAQ5™ (Cell Signaling Technology) to visualize the nuclei. Images were acquired using a Nikon A1 confocal microscope (Nikon Instruments).
Cathepsin B and L activity assay
Cathepsin B substrate (Z-Arg-Arg-MCA) and Cathepsin L substrate (Z-Phe-Arg-MCA), both from Sigma-Aldrich, were stored at −80°C as 10 mM in DMSO stocks, and diluted to 100 μM with 0.1% Brij (Sigma-Aldrich) just before use. 10 mM in DMSO stocks of cathepsin B inhibitor CA-074Me (Calbiochem) was prepared and stored at −80°C. The concentration was brought to 25 μM with reaction buffer just before use. To measure the effects of amiodarone on cathepsin activity, Vero cells were grown to semi-confluence in six well plates. Three wells were incubated for 12 h with 10 μM amiodarone, while three wells served as controls. At the end of the incubation, cells were rinsed with cold PBS, pooled by scraping in two Eppendorf tubes, washed two more times with cold PBS and lysed in 200 μl of 100 mM sodium acetate, 1 mM EDTA, 0.5% Triton, pH 5.0 for 10 min in ice. Lysates were then centrifuged at 12 000 × g for 6 min and the supernatants were stored at −80°C. Cathepsin assay (Ebert et al.2002) was performed by using 0.5–5 μg of protein lysates and 50 μM of substrate in a total volume of 200 μl by combining (a) 80 μl of reaction buffer (100 mM sodium acetate, 1 mM EDTA, 4 mM dithiothreitol, pH 5.0) and 100 μl of Z-Arg-Arg-MCA solution (cathepsin B assay); (b) 80 μl of CA-074Me in reaction buffer (final concentration 10 μM) and 100 μl of Z-Phe-Arg-MCA (cathepsin L assay); (c) 80 μl of reaction buffer and 100 μl of Z-Phe-Arg-MCA (cathepsin B+L assay). The reaction, allowed to proceed for 1 h at 37°C in the dark, was stopped by the addition of 200 μl of cold reaction buffer. Fluorescence was measured by using a fluorometer with an excitation of 390 nm and an emission of 460 nm.
Statistical analysis
Data are expressed as mean ± standard deviation (SD). Differences were analysed by a Student's ‘t’ test. The level of accepted significance was P < 0.05.
RESULTS
Amiodarone and its metabolite MDEA inhibit EBOV infection in vitro
It has been recently reported that amiodarone inhibits filovirus replication in vitro, while the mechanism of inhibition remains unclear (Gehring et al.2014). In patients with arrhythmia chronically treated with 200–600 mg day−1 amiodarone, drug plasma concentration ranges from 1.7 ± 0.6 to 5.1 ± 0.2 μM, while after intravenous administration, plasma levels can increased up to 11 μM (Ha et al.2005; Breitenstein et al.2008). We have previously reported that 16 h of incubation with 10 μM amiodarone induces a full-fledged NPC-like phenotype (Piccoli et al.2011). In order to confirm the anti-ebolavirus activity of amiodarone and to analyse whether the NPC-like phenotype plays a role at this level, Vero cells were incubated for 16 h with 2, 5 or 10 μM amiodarone which previously displayed no appreciable cytotoxicity (Piccoli et al.2011). Cells were then infected with recombinant VSV encoding for the green fluorescent protein (GFP) in place of the glycoprotein G (VSVΔG-GFP), pseudotyped with either the envelope glycoprotein G of the VSV (VSV-recombinant virus) or the ZEBOV-envelope glycoprotein (GP-pseudotyped virus). A MOI of 0.1 FFU/cell was employed to achieve a percentage of infected cells which allows, with all the recombinant viruses tested, the evaluation of the antiviral effect without saturating the system. Two hours post-infection, cells were rinsed and incubated for additional 24 h in the presence of the appropriate drug concentration before evaluating GFP-positive cells by cytofluorimetry. The 2 h time frame was selected in order to ensure the adhesion to the target cells of all the used recombinant viruses, in the adopted experimental settings. Our results show that amiodarone inhibited GP-pseudotyped virus in a dose-dependent manner with an estimated IC50 (half maximal dose inhibitory concentration) of 5.6 μM (Fig. 2a). By contrast, the drug did not display such an effect on VSV-recombinant virus.
Figure 2.
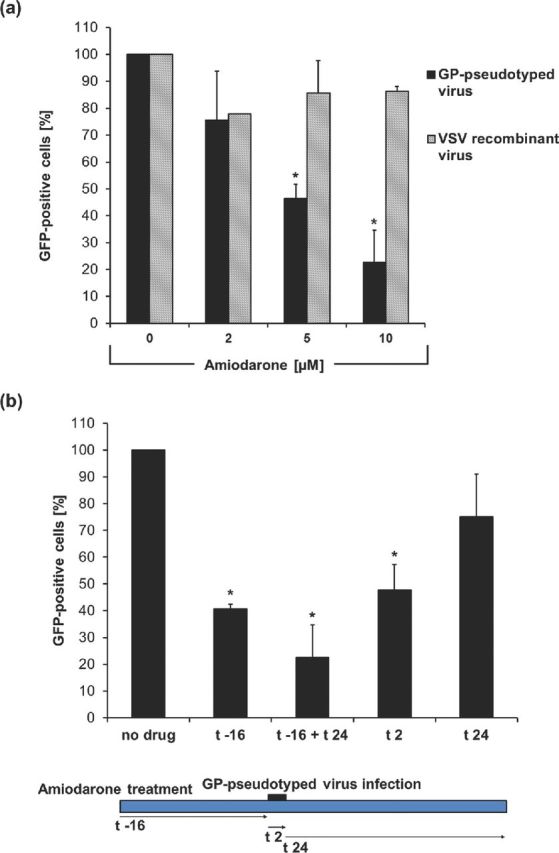
Amiodarone inhibits GP-mediated entry. (a) Vero cells treated for 16 h with amiodarone were infected for 2 h with the VSVΔG-GFP virus (MOI of 0.1 FFU/cell) bearing its native glycoprotein (VSV-recombinant virus) or the EBOV GP glycoprotein (GP-pseudotyped virus). After 24 h, GFP-positive cells were assayed by cytofluorimetry. (b) Vero cells treated with 10 μM amiodarone, as indicated, were infected for 2 h with GP-pseudotyped virus (MOI of 0.1 FFU/cell). After 24 h, GFP-positive cells were quantified by cytofluorimetry. No drug indicates no amiodarone treatment, (t –16) indicates 16 h of treatment before infection, (t + 24) stands for 24 h of treatment after infection, (t 2) indicates treatment during viral adhesion. Data (mean ± SD, N = 3 experiments in duplicate) are percentages of GFP-positive cells with respect to no drug, set as 100% (* = P < 0.05).
Next, to further dissect the mechanisms by which amiodarone inhibit the infection, GP-pseudotyped virus (MOI of 0.1 FFU/cell) was incubated with Vero cells untreated (no drug) or exposed to amiodarone for 16 h before infection (t −16), during viral adhesion (t 2), before and after (t −16 + t 24) or only after adhesion (t 24) (Fig. 2b). Cell fluorescence was then measured. We found a significant reduction in GFP-positive cells both with amiodarone pretreatment (t −16) and when amiodarone was present during viral adhesion (t 2). Only a limited effect was observed when amiodarone was added after viral adhesion (t 24). These results suggest that amiodarone could interfere with infection both by a direct effect during viral entry and through the induction of an NPC-like phenotype.
The above findings were confirmed in an experimental setting in which SUDV was used to infect Vero cells (MOI of 0.05 FFU/cell) either untreated (no drug) or treated with 10 μM amiodarone 16 h before and 24 h after infection (t −16 + t 24), during 1 h of viral adhesion (t 1), during and 24 h after adhesion (t 1 + t 24), or after adhesion, i.e. immediately (t 24) or 3 h post-viral adhesion (t 21) (Fig. 3). Cells were then analysed by immunofluorescence. SUDV infection was significantly inhibited (95.9 ± 0.2%) in the t −16 + t 24 condition (Fig. 3). Furthermore, 91.7 ± 2.9% of inhibition was also achieved with the t 1 + t 24 condition, while the treatment performed only during the adhesion (t 1) caused an inhibition of 81.3 ± 8.9% (Fig. 3). However, when the drug was added after the adhesion step, the effect of amiodarone was smaller, especially when the drug was added with 3 h delay (t 21) (Fig. 3). Furthermore, the amiodarone effect was stronger than the one observed in the presence of U18666A, a compound known to block the fusion of the EBOV envelope with the endosomal membrane (Carette et al.2011; Shoemaker et al.2013)
Figure 3.

Amiodarone interferes with early steps of SUDV infection. Vero cells, treated with 10 μM amiodarone, were infected for 1 h with 0.05 FFU/cell of SUDV. After 24 h, infected cells were identified by staining with an antibody anti-SUDV Nucleoprotein (NP). No drug indicates no amiodarone treatment, (t –16) stands for 16 h treatment before infection, (t 1) indicates treatment during infection, (t 24) indicates a treatment of 24 h following infection and (t 21) indicates 21 h treatment starting 3 h after infection. Data (mean ± SD, N = 2 experiments in duplicate) are percentages of NP-positive cells with respect to no drug, set as 100% (* = P < 0.05).
MDEA, the main metabolite of amiodarone, reaches plasma concentrations close to those of the parent drug and contributes both to antiarrhythmic activity and toxicity (Quaglino et al.2004; Ha et al.2005; Piccoli et al.2011). To investigate whether MDEA affects EBOV entry, Vero cells were incubated for 16 h with 2 or 10 μM amiodarone, 2 μM MDEA or with a combination of 2 μM amiodarone and 2 μM MDEA (a common condition in patients taking amiodarone chronically). Next, cells were infected with GP-pseudotyped virus (MOI of 0.1 FFU/cell), and 24 h later the infectivity was evaluated by cytofluorimetry. While amiodarone inhibits GP-pseudotyped virus infection more efficiently than MDEA, the combination of 2 μM amiodarone and 2 μM MDEA had a stronger inhibitory effect than the one achieved by treatment with each compound alone (Fig. 4a).
Figure 4.
Amiodarone and metabolite MDEA have additive antiviral activity. (a) Amiodarone and MDEA inhibit GP-dependent entry. Vero cells treated for 16 h with amiodarone or MDEA were infected for 2 h with GP-pseudotyped virus (MOI of 0.1 FFU/cell). Afterwards, cells were exposed to the same concentration of compounds for additional 24 h and then were evaluated by cytofluorimetry. Data (mean ± SD, N = 4 experiments conducted in duplicate) are percentages of GFP-positive cells with respect to no drug, set as 100%. (b) Effect of amiodarone, MDEA and dronedarone on SUDV entry. Vero cells treated for 16 h with amiodarone, amiodarone + MDEA, dronedarone or U18666A were infected with SUDV for 1 h (MOI of 0.05 FFU/cell). Afterwards, cells were exposed to the same concentration of each compound for further 24 h, and viral entry was evaluated by immunostaining of SUDV Nucleoprotein with a specific antibody. (c) Evaluation of infectious viral particles release. Vero cells treated for 16 h with amiodarone, amiodarone + MDEA, dronedarone or U18666A were infected with SUDV as reported above. Then, cells were exposed for 48 h to the same concentration of each compound. Next, viral particles released in the supernatants were titrated by infecting Vero cells and by counting SUDV Nucleoprotein-positive cells by fluorescence microscopy. Data in (b) and (c) (mean ± SD, N = 2 experiments in duplicate) are percentages of no drug, set as 100% (* = P < 0.05).
To further evaluate the effect of the amiodarone/MDEA combination on EBOV infection, we employed SUDV at the MOI of 0.05 FFU/cell. Twenty-four h later, viral entry was evaluated by indirect immunofluorescence, while progeny release was tested by viral titration at 48 h. U18666A and dronedarone were also included. In particular, dronedarone was used at a concentration of 0.3 μM, which is found in patients chronically treated by the oral route. Results showed that amiodarone and MDEA in combination inhibited viral entry and progeny release and that their effect was stronger than the one due to dronedarone and U18666A (Fig. 4b and c). We conclude that amiodarone and MDEA, at concentrations of clinical significance, inhibit EBOV infection.
Amiodarone inhibits EBOV fusion with the endosomal membrane
Since our data demonstrate that amiodarone has an effect on the early phases of ebolavirus replication, we focused our attention on the key step of viral envelope/endosomal membrane fusion. Vero cells, treated for 16 h with 10 μM amiodarone, were incubated for 3 h with GP-pseudotyped virus or with the VSV-recombinant virus as a control (MOI of 20 FFU/cell). Next, cells were fixed and stained with an anti-VSV-M antibody (Carette et al.2011). As shown in Fig. 5, untreated cells displayed a diffuse cytoplasmatic staining, with both GP-pseudotyped and VSV-recombinant viruses, indicating that the fusion of the viral envelope with the endosomal membrane occurred, allowing the release of VSV-M protein into the cytoplasm. By contrast, in the presence of amiodarone or U18666A, while in cells infected with VSV-recombinant virus a diffuse staining was observed, most of the cells infected by GP-pseudotyped virus displayed a punctate staining (Fig. 5). Similar results were also observed when cells were treated with amiodarone during the 3 h of viral incubation (data not shown).
Figure 5.

Amiodarone blocks fusion of GP-pseudotyped virus with the endosomal membranes. Vero cells treated for 16 h with 10 μM amiodarone or 20 μM U18666A were infected for 3 h with GP-pseudotyped virus (upper panels) or VSV-recombinant virus (lower panels) (MOI of 20 FFU/cell). Three hours post-infection, the VSV-M protein was stained with 23H12 specific antibody (red). Nuclei were stained with DRAQ5™ (blue). Scale bar of confocal images: 25 μm.
During viral entry, virion-associated GP is processed by cathepsins B and L into the 19 kDa fusogenic form (Martinez et al.2013). Although amiodarone did not modify the total cell content of cathepsins B and L (Fig. 6a), incubation of GP-pseudotyped virus with thermolysin before infection, a treatment that allows the in vitro processing of the GP1 into the 19 kDa form, clearly rescued viral infectivity (Fig. 6b). Overall these data suggest that amiodarone affects GP-driven fusion.
Figure 6.

Amiodarone antiviral effect is rescued by in vitro proteolysis of GP. (a) Amiodarone does not affect the cell content of cathepsins B and L. Vero cells treated for 12 h with 10 μM amiodarone were lysed and cathepsin activity was measured in the cell lysates. Data (mean ± SD) are presented as percentages of Cat B + Cat L measured in the absence of drug (no drug). (b) In vitro proteolysis of GP rescues viral infectivity. Vero cells, treated for 16 h with 10 μM amiodarone, were incubated for 2 h with GP-pseudotyped virus (MOI of 0.1 FFU/cell) treated for 1 h with either reaction buffer alone (mock pre-activated) or 0.2 mg ml–1 thermolysin (pre-activated). Cells were then incubated for 24 h in plain medium (no drug) or with 10 μM amiodarone, GFP-positive cells were estimated by cytofluorimetry. Data (mean ± SD, N = 3 experiments in duplicate) are percentages of GFP-positive cells with respect to cells exposed to mock pre-activated virus in the absence of amiodarone, set as 100% (* = P < 0.05).
Role of different structural groups of amiodarone on its antiviral activity
To clarify the structural basis of amiodarone ability to interfere with EBOV infection, we analysed the effect of amiodarone analogues (Fig. 1) on GP-pseudotyped virus infection. Analogues have already been tested for their toxicity and ability of inducing an NPC-like phenotype (Quaglino et al.2004; Bigler et al.2007; Piccoli et al.2011). Vero cells were treated for 16 h with amiodarone or its analogues (at concentrations of clinical relevance, when applicable, or at the ones sufficient to induce an NPC-like phenotype without being toxic to cells) and then incubated for 2 h with GP-pseudotyped virus (MOI of 0.1 FFU/cell). Infected cells were maintained in medium containing the appropriate compound for additional 24 h, and GFP-positive cells were evaluated by cytofluorimetry. As shown in Fig. 7, the diethylamino-β-ethoxy lateral group, which is a determinant for accumulation into acidic organelles, is required for antiviral activity. Furthermore, it appears that, among the compounds bearing this group, the antiviral activity correlates with the ability of inducing an NPC-like phenotype (MeAmi > Amiodarone > PIPAM > MDEA) (Piccoli et al.2011). In addition, a bulky benzofuran hydrophobic moiety is necessary for the antiviral activity, as D2, which has the diethylamino-β-ethoxy group but lacks a hydrophobic moiety, is inactive. Of note, the amiodarone metabolite didesethyl amiodarone (DDEA), usually found in traces in serum, is devoid of antiviral activity.
Figure 7.

Structural determinants of amiodarone antiviral activity. Vero cells were treated for 16 h with different compounds. Amiodarone, MDEA and dronedarone concentrations were close to maximum serum levels found in patients. The concentration of different analogues was the maximum achieved without cell toxicity. After 16 h of preincubation with amiodarone and its analogues, cells were infected for 2 h with GP-pseudotyped virus (MOI of 0.1 FFU/cell), and then exposed to the same concentration of each compound for additional 24 h. GFP-positive cells were then evaluated by cytofluorimetry. Data (mean ± SD, N = 4 experiments in duplicate) are percentages of no drug set as 100% (* = P < 0.05).
Overall, the antiviral activity of amiodarone seems to correlate with motifs which lead to an expanded lysosome phenotype (Logan et al.2014), namely the presence of an amine group, amphiphilicity, a bulky hydrophobic moiety and the ability to be incorporated into membranes (Mitterreiter et al.2010). It also appears that subtle changes of the alkyl group of the diethylamino-β-ethoxy chain (as in the MeAmi compound) might increase amiodarone antiviral effect.
DISCUSSION
EVD is one of the most lethal transmissible infections. Although in the past this disease appeared sporadically, the increase of human incursions into endemic regions and the higher mobility have turned EBOV into a high-priority public threat. This is sadly confirmed by the biggest epidemic outbreak ever, currently occurring in the Western Africa (CDC 2014). The fast track access to treatment options for EVD is a key priority for the World Health Organization (WHO). With worst-ever outbreak of Ebola raging in West Africa, WHO concluded that it is ethical to use unproven drugs and vaccines to try to combat the disease. However, no medicines so far have been approved, for routine use in EVD.
Currently, despite the fact that different anti-EBOV compounds have been reported in the literature, their antiviral efficacy has been evaluated only in vitro and in animal models (Basu et al.2011; Cote et al.2011; Iversen et al.2012; Johansen et al.2013; Madrid et al.2013; Shoemaker et al.2013; Elshabrawy et al.2014; Gehring et al.2014; Oestereich et al.2014; Picazo and Giordanetto 2014; Warren et al.2014). Even though some of these compounds have been already approved for treatment of human diseases (Johansen et al.2013; Shoemaker et al.2013; Gehring et al.2014), no clinical trials to evaluate their safety and efficacy in EVD have been completed so far. In this respect, the identification of commercially available drugs, already approved for the use in humans, and active against EBOV, might accelerate the organization of clinical trials and eventually clinical use.
Recently, Gehring et al. (2014) demonstrated that amiodarone and dronedarone, drugs approved for the treatment of cardiac arrhythmias, inhibit filovirus infection. The aim of the present work was to further characterize the effect of amiodarone on EBOV infection. Interestingly, we found that amiodarone inhibits EBOV infection in a dose-dependent manner and at concentrations comparable to plasma levels found in intravenously treated patients. The IC50 of amiodarone calculated by employing a recombinant GP-pseudotyped VSV on Vero cells was 5.6 μM. It has to be mentioned that a recent study (Gehring et al.2014) reported an IC50 of amiodarone around 2.2 μM, with a GP-pseudotyped lentivirus, and around 0.4 μM with the ZEBOV. Taking into account the different viral/cellular systems employed, overall these data indicate that amiodarone efficiently inhibits EBOV infection. The antiviral potency varies with the timing of cell exposure to the drug. Inhibition is maximum when amiodarone pretreatment induces an NPC-like phenotype, but a partial inhibition is also appreciable when amiodarone is added during very early steps of infection, suggesting that the anti-EBOV properties might derive on one side from cell structural modifications associated with amiodarone accumulation, and on the other side from a direct effect of the drug on specific steps of the viral life cycle. In this respect, amiodarone could act by modifying lipid fluidity (Chatelain, Laruel and Gillard 1985), by increasing membrane-proximal pH (Mitterreiter et al.2010), by competing for negatively charged groups on the surface or by affecting calcium channels, required for the maturation of the endosomes and/or viral entry (Kolter and Sandhoff 2010; Gehring et al.2014). Interestingly, we found that amiodarone affects viral fusion, as it has been previously reported for the U18666A inhibitor (Carette et al.2011). After viral internalization, GP cleavage proceeds through a 50 and a 20 kDa intermediate, ultimately generating the key 19 kDa core protein (Chandran et al.2005; Schornberg et al.2006; Dube et al.2009; Hood et al.2010; Miller et al.2012). Interestingly, the artificial activation of GP by thermolysin treatment of viral particles before infection rescued the effect of amiodarone. It has been demonstrated that amiodarone can affect the stability of specific proteins, such as SP-A (Baritussio et al.2001). Here we demonstrated that the cellular content of cathepsin B and L, required for the formation of the fusogenic form of GP, was not affected by drug treatment. However, we cannot exclude that amiodarone might influence the activity of these proteases, as it has been demonstrated for different proteolytic enzymes (Mitterreiter et al.2010; Piccoli et al.2011). Furthermore, specific effects on steps subsequent to the EBOV entry/fusion, as demonstrated in the case of the SARS coronavirus and HCV (Stadler et al.2008; Cheng et al.2013), cannot be ruled out.
Importantly, we found that there is an additive effect between amiodarone and its pharmacologically active metabolite MDEA on GP-mediated entry into target cells. Furthermore, our results show that, at concentrations of clinical interest, amiodarone and amiodarone/MDEA combination are more effective than dronedarone and U18666A in reducing SUDV infection.
Finally, we found that the presence of a tertiary amine in the compound plays a key role in the antiviral effect, as demonstrated by the use here of different amiodarone derivatives and by the anti-filovirus activity of additional amphiphilic cations (Johansen et al.2013; Shoemaker et al.2013). Moreover, a hydrophobic moiety is also critical, perhaps regulating the interactions with membranes. Overall, these observations support the idea that the antiviral activity of amiodarone relies on its ability to interact with membranes, to accumulate into the acidic organelles (Logan et al.2014) and to induce an NPC-like phenotype (Piccoli et al.2011; Shoemaker et al.2013; Gehring et al.2014).
In conclusion, here we demonstrate that the anti-arrhythmic drug amiodarone specifically interferes with the early steps of EBOV life cycle, thus blocking viral replication. Overall, our data provide evidence that amiodarone deserves serious consideration for the treatment of EVD.
Acknowledgments
The authors are grateful to Michael Whitt, University of Tennessee, Memphis, USA, for providing the recombinant VSV, to Gary Nabel, Vaccine Research Center, NIH, Bethesda, USA, for providing the ZEBOV GP plasmid. We would also like to thank Enzo Di Iorio's Group, Department of Molecular Medicine, University of Padova, Padova, Italy, for help with confocal microscopy, Nadia Inglese, Department of Molecular Medicine, University of Padova, for technical assistance with recombinant virus working stocks production, and Daniela Bruttomesso, Department of Medicine, University of Padova, for continuous encouragement.
FUNDING
This work was supported by University of Padova grants (ex60% to CS, AC, AB and Progetti di Ateneo 2008 to AB), and Regione Veneto grants to CP and GP.
Conflict of interest. None declared.
REFERENCES
- Baritussio A, Marzini S, Agostini M, et al. Amiodarone inhibits lung degradation of SP-A and perturbs the distribution of lysosomal enzymes. Am J Physiol-Lung C. 2001;281:L1189–99. doi: 10.1152/ajplung.2001.281.5.L1189. [DOI] [PubMed] [Google Scholar]
- Basu A, Li B, Mills DM, et al. Identification of a small-molecule entry inhibitor for filoviruses. J Virol. 2011;85:3106–19. doi: 10.1128/JVI.01456-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigler L, Spirli C, Fiorotto R, et al. Synthesis and cytotoxicity properties of amiodarone analogues. Eur J Med Chem. 2007;42:861–7. doi: 10.1016/j.ejmech.2006.12.031. [DOI] [PubMed] [Google Scholar]
- Breitenstein A, Stampfli SF, Camici GG, et al. Amiodarone inhibits arterial thrombus formation and tissue factor translation. Arterioscl Throm Vas. 2008;28:2231–8. doi: 10.1161/ATVBAHA.108.171272. [DOI] [PubMed] [Google Scholar]
- Carette JE, Raaben M, Wong AC, et al. Ebola virus entry requires the cholesterol transporter Niemann–Pick C1. Nature. 2011;477:340–3. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC. 2014 Ebola Outbreak in West Africa. 2014. http://www.cdc.gov/vhf/ebola/outbreaks/2014-west-africa/index.html (5 May 2015, date last accessed) [Google Scholar]
- Chandran K, Sullivan NJ, Felbor U, et al. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308:1643–5. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatelain P, Laruel R, Gillard M. Effect of amiodarone on membrane fluidity and Na+/K+ ATPase activity in rat-brain synaptic membranes. Biochem Bioph Res Co. 1985;129:148–54. doi: 10.1016/0006-291x(85)91415-9. [DOI] [PubMed] [Google Scholar]
- Cheng YL, Lan KH, Lee WP, et al. Amiodarone inhibits the entry and assembly steps of hepatitis C virus life cycle. Clin Sci. 2013;125:439–48. doi: 10.1042/CS20120594. [DOI] [PubMed] [Google Scholar]
- Cote M, Misasi J, Ren T, et al. Small molecule inhibitors reveal Niemann–Pick C1 is essential for Ebola virus infection. Nature. 2011;477:344–8. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube D, Brecher MB, Delos SE, et al. The primed ebolavirus glycoprotein (19-kilodalton GP1,2): sequence and residues critical for host cell binding. J Virol. 2009;83:2883–91. doi: 10.1128/JVI.01956-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, Deussing J, Peters C, et al. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J Biol Chem. 2002;277:24609–17. doi: 10.1074/jbc.M201107200. [DOI] [PubMed] [Google Scholar]
- Elshabrawy HA, Fan J, Haddad CS, et al. Identification of a broad-spectrum antiviral small molecule against severe acute respiratory syndrome coronavirus and Ebola, Hendra, and Nipah viruses by using a novel high-throughput screening assay. J Virol. 2014;88:4353–65. doi: 10.1128/JVI.03050-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann H, Geisbert TW. Ebola haemorrhagic fever. Lancet. 2011;377:849–62. doi: 10.1016/S0140-6736(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring G, Rohrmann K, Atenchong N, et al. The clinically approved drugs amiodarone, dronedarone and verapamil inhibit filovirus cell entry. J Antimicrob Chemoth. 2014;69:2123–31. doi: 10.1093/jac/dku091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha HR, Bigler L, Wendt B, et al. Identification and quantitation of novel metabolites of amiodarone in plasma of treated patients. Eur J Pharm Sci. 2005;24:271–9. doi: 10.1016/j.ejps.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Hood CL, Abraham J, Boyington JC, et al. Biochemical and structural characterization of cathepsin L-processed Ebola virus glycoprotein: implications for viral entry and immunogenicity. J Virol. 2010;84:2972–82. doi: 10.1128/JVI.02151-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iversen PL, Warren TK, Wells JB, et al. Discovery and early development of AVI-7537 and AVI-7288 for the treatment of Ebola virus and Marburg virus infections. Viruses. 2012;4:2806–30. doi: 10.3390/v4112806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen LM, Brannan JM, Delos SE, et al. FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci Transl Med. 2013;5:190–79. doi: 10.1126/scitranslmed.3005471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolter T, Sandhoff K. Lysosomal degradation of membrane lipids. FEBS Lett. 2010;584:1700–12. doi: 10.1016/j.febslet.2009.10.021. [DOI] [PubMed] [Google Scholar]
- Lee JE, Fusco ML, Hessell AJ, et al. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature. 2008;454:177–82. doi: 10.1038/nature07082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Saphire EO. Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 2009;4:621–35. doi: 10.2217/fvl.09.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan R, Kong AC, Axcell E, et al. Amine-containing molecules and the induction of an expanded lysosomal volume phenotype: a structure-activity relationship study. J Pharm Sci. 2014;103:1572–80. doi: 10.1002/jps.23949. [DOI] [PubMed] [Google Scholar]
- Madrid PB, Chopra S, Manger ID, et al. A systematic screen of FDA-approved drugs for inhibitors of biological threat agents. PLoS One. 2013;8:e60579. doi: 10.1371/journal.pone.0060579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez O, Ndungo E, Tantral L, et al. A mutation in the Ebola virus envelope glycoprotein restricts viral entry in a host species- and cell-type-specific manner. J Virol. 2013;87:3324–34. doi: 10.1128/JVI.01598-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EH, Obernosterer G, Raaben M, et al. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J. 2012;31:1947–60. doi: 10.1038/emboj.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitterreiter S, Page RM, Kamp F, et al. Bepridil and amiodarone simultaneously target the Alzheimer's disease beta- and gamma-secretase via distinct mechanisms. J Neurosci. 2010;30:8974–83. doi: 10.1523/JNEUROSCI.1199-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morissette G, Ammoury A, Rusu D, et al. Intracellular sequestration of amiodarone: role of vacuolar ATPase and macroautophagic transition of the resulting vacuolar cytopathology. Brit J Pharmacol. 2009;157:1531–40. doi: 10.1111/j.1476-5381.2009.00320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oestereich L, Ludtke A, Wurr S, et al. Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. Antiviral Res. 2014;105:17–21. doi: 10.1016/j.antiviral.2014.02.014. [DOI] [PubMed] [Google Scholar]
- Picazo E, Giordanetto F. Small molecule inhibitors of ebola virus infection. Drug Discov Today. 2014;20:277–86. doi: 10.1016/j.drudis.2014.12.010. [DOI] [PubMed] [Google Scholar]
- Piccoli E, Nadai M, Caretta CM, et al. Amiodarone impairs trafficking through late endosomes inducing a Niemann–Pick C-like phenotype. Biochem Pharmacol. 2011;82:1234–49. doi: 10.1016/j.bcp.2011.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaglino D, Ha HR, Duner E, et al. Effects of metabolites and analogs of amiodarone on alveolar macrophages: structure–activity relationship. Am J Physiol-Lung C. 2004;287:L438–47. doi: 10.1152/ajplung.00434.2003. [DOI] [PubMed] [Google Scholar]
- Sakurai Y, Kolokoltsov AA, Chen CC, et al. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science. 2015;347:995–8. doi: 10.1126/science.1258758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salata C, Curtarello M, Calistri A, et al. vOX2 glycoprotein of human herpesvirus 8 modulates human primary macrophages activity. J Cell Physiol. 2009;219:698–706. doi: 10.1002/jcp.21722. [DOI] [PubMed] [Google Scholar]
- Schornberg K, Matsuyama S, Kabsch K, et al. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J Virol. 2006;80:4174–8. doi: 10.1128/JVI.80.8.4174-4178.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CJ, Schornberg KL, Delos SE, et al. Multiple cationic amphiphiles induce a Niemann–Pick C phenotype and inhibit Ebola virus entry and infection. PLoS One. 2013;8:e56265. doi: 10.1371/journal.pone.0056265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler K, Ha HR, Ciminale V, et al. Amiodarone alters late endosomes and inhibits SARS coronavirus infection at a post-endosomal level. Am J Resp Cell Mol. 2008;39:142–9. doi: 10.1165/rcmb.2007-0217OC. [DOI] [PubMed] [Google Scholar]
- Warren TK, Wells J, Panchal RG, et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature. 2014;508:402–5. doi: 10.1038/nature13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidmann M, Sall AA, Manuguerra JC, et al. Quantitative analysis of particles, genomes and infectious particles in supernatants of haemorrhagic fever virus cell cultures. Virol J. 2011;8:81. doi: 10.1186/1743-422X-8-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitt MA. Generation of VSV pseudotypes using recombinant DeltaG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J Virol Methods. 2010;169:365–74. doi: 10.1016/j.jviromet.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]