

Abstract
The NF‐κB signal transduction pathway is a critical regulator of multiple cellular functions that ultimately shift the balance between cell survival and death. The cascade is activated by many intrinsic and extrinsic stimuli, which is transduced via adaptor proteins to phosphorylate the IκB kinase (IKK) complex, which in turn phosphorylates the inhibitory IκBα protein to undergo proteasomal degradation and sets in motion nuclear events in response to the initial stimulus. Viruses are important modulators of the NF‐κB cascade and have evolved multiple mechanisms to activate or inhibit this pathway in a manner conducive to viral multiplication and establishment of a productive infectious cycle. This is a subject of extensive research by multiple laboratories whereby unraveling the interactions between specific viral components and members of the NF‐κB signal transduction cascade can shed unique perspectives on infection associated pathogenesis and novel therapeutic targets. In this review, we highlight the interactions between components of the IKK complex and multiple RNA and DNA viruses with the emphasis on mechanisms by which the interaction feeds the infection. Understanding these interactions will shed light on the exploitative capabilities of viruses to maintain an environment favorable for a productive infection.
Keywords: IKK complex, host response, viral proteins
The IKK complex is targeted by multiple viruses in manners that cripple host responses and enhance infection.
Introduction
The NF‐κB cascade is a major signaling pathway that is vital to the functioning of a cell. A variety of stimuli can activate the NF‐κB signaling pathway. These include cytokine stimuli (TNF, IL‐1, IL‐6 and IL‐8), UV stress, DNA damage, double stranded (ds) RNA, and viruses (Hai et al., 2006; Solt & May, 2008; Israel, 2010; Gamble et al., 2012; Le Negrate, 2012; Lee et al., 2012; Liu et al., 2012; Diamant & Dikstein, 2013; Ersing et al., 2013; Hoesel & Schmid, 2013). Activation of the NF‐κB effector molecules regulates a number of genes by activating or repressing transcription (Hayden & Ghosh, 2004; Hai et al., 2006; Perkins, 2007; Solt & May, 2008; Lee et al., 2012; Diamant & Dikstein, 2013; Hoesel & Schmid, 2013). Such genes include those that control cellular stress response, apoptosis, cell proliferation, and cell adhesion as well as the innate and adaptive immune responses (Hayden & Ghosh, 2004; Perkins, 2007; Solt & May, 2008; Le Negrate, 2012; Ersing et al., 2013). In acute inflammation scenarios, NF‐κB levels return to normal; however, in cases of chronic inflammation, NF‐κB activity remains elevated, which can contribute to cancers and tumor progression (Diamant & Dikstein, 2013; Hoesel & Schmid, 2013).
The NF‐κB transcription factor complex comprises the REL‐homology domain (RHD) proteins p50, p52, RelA (also called p65), RelB, and cREL. In the cytoplasm of a resting cell, the NF‐κB complex is bound to inhibitory κB (IκB) proteins (Karin, 1999; Hayden & Ghosh, 2004; Hai et al., 2006; Perkins, 2007; Solt & May, 2008; Israel, 2010; Gamble et al., 2012; Le Negrate, 2012; Lee et al., 2012; Liu et al., 2012; Ersing et al., 2013; Hoesel & Schmid, 2013), which function in part by masking the nuclear localization sequence (NLS) found in a RHD of the NF‐κB subunits (Karin, 1999; Hayden & Ghosh, 2004; Hai et al., 2006; Perkins, 2007; Le Negrate, 2012; Hoesel & Schmid, 2013). Once stimulated, IκB kinase (IKK) complex phosphorylates IκBα, which is followed by subsequent ubiquitylation and proteasomal degradation of IκBα (Hayden & Ghosh, 2004; Perkins, 2007; Solt & May, 2008; Gamble et al., 2012; Lee et al., 2012; Liu et al., 2012; Diamant & Dikstein, 2013; Ersing et al., 2013; Hoesel & Schmid, 2013). Once released from IκBα, the NF‐κB transcription factors translocate to the nucleus where they regulate transcription (Solt & May, 2008; Gamble et al., 2012; Diamant & Dikstein, 2013; Ersing et al., 2013).
The IKK complex is comprised of IKKα, IKKβ, and IKKγ or NF‐κB essential modulator (NEMO) (Karin, 1999; Hayden & Ghosh, 2004; Hai et al., 2006; Solt & May, 2008; Israel, 2010; Shifera, 2010a, b, Le Negrate, 2012; Liu et al., 2012; Ersing et al., 2013; Hoesel & Schmid, 2013). This multi‐protein complex (c. 700–900 kDa) is deemed to be the ‘master coordinator of NF‐κB activation’ (Hayden & Ghosh, 2004; Israel, 2010; Gamble et al., 2012; Le Negrate, 2012; Liu et al., 2012; Ersing et al., 2013). This review will highlight the current understanding of the role that the IKK complex plays in the event of a viral infection and to discern how this impacts the viral life cycle. We will begin by describing the components of the NF‐κB cascade followed by an in‐depth account of viral interactions with each of the components of the IKK complex.
NF‐κB canonical and alternative pathways – differential involvement of IKK components with different outcomes
Depending on the type of stimuli a cell detects, the NF‐κB signaling cascade can diverge to the canonical pathway or an alternative noncanonical pathway, both of which have differences in the kinetics and molecular biology of the responses (Fig. 1) (Liu et al., 2012; Diamant & Dikstein, 2013; Ersing et al., 2013; Hoesel & Schmid, 2013). The canonical pathway is activated primarily by TNF‐α, IL‐1, lipopolysaccharide, and viruses (Karin, 1999; Perkins, 2007; Solt & May, 2008; Israel, 2010; Shifera, 2010a, b; Gamble et al., 2012; Le Negrate, 2012; Liu et al., 2012; Hoesel & Schmid, 2013). Activation of the canonical pathway, through multiple intermediate steps, ultimately phosphorylates IKKβ at S177 and S181 (Fig. 1) (Gamble et al., 2012). IKK activation is defined as the phosphorylation of S176 and S180 on IKKα and S177 and S181 on IKKβ, which leads to a conformational change in the activation loop of the complex, catalytically activating the kinase domain (Liu et al., 2012). Activation of the IKK complex via the canonical pathway also requires IKKγ (Perkins, 2007). The activated IKKβ rapidly phosphorylates IκBα on S32 and S36, resulting in subsequent ubiquitylation and proteasomal degradation of IκBα (Karin, 1999; Perkins, 2007; Solt & May, 2008; Israel, 2010; Gamble et al., 2012). A critical aspect of the canonical response that distinguishes it from the noncanonical pathway is that the former is a rapid response which is usually transient in nature. The noncanonical response, in contrast, usually takes longer to respond to stimuli and the response demonstrates increased longevity. The activated IKKβ also phosphorylates IκBβ on S19 and S23 (Gamble et al., 2012), albeit at a much slower rate and IκBβ and IκBε are ultimately degraded as well (Karin, 1999; Perkins, 2007). The literature alludes to the premise that in the classical pathway, IKKβ exclusively phosphorylates IκBα and ‐β (Israel, 2010). The assumption stems from the fact that the role for IKKα in the canonical pathway has not yet been fully determined. This could be attributed to cell‐specific phenomenon; IKKβ compensates for the lack of IKKα to activate the NF‐κB cascade in response to proinflammatory stimuli in the liver and keratinocytes (Senftleben et al., 2001).
Figure 1.
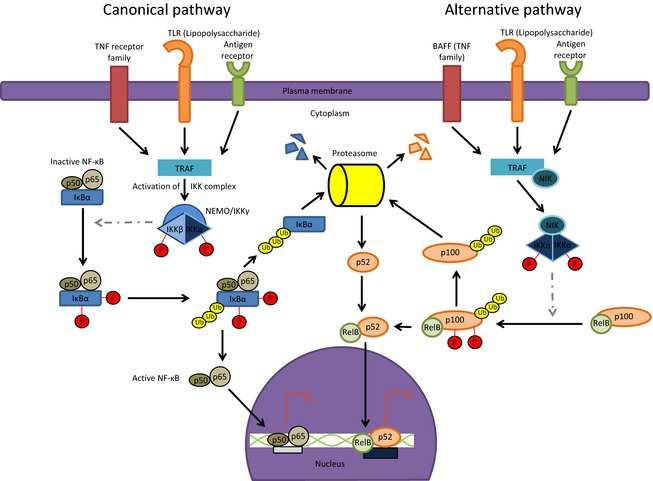
The canonical and noncanonical NF‐κB pathways. The canonical pathway is stimulated by a variety of signals that are detected by cell surface receptors such as TLRs, TNFRs, and antigen receptors. The signal is transduced to the adaptor protein TRAF such that TBK‐1 phosphorylates IKKβ and IKKα. IKK‐mediated IκBα phosphorylation is followed by proteasomal degradation resulting in nuclear translocation of the NF‐κB heterodimer p65/p50 to activate or repress target gene expression. The noncanonical NF‐κB pathway is triggered by stimuli such as lipopolysaccharide. In this pathway, NIK phosphorylates the homodimer IKKα which mediates the phosphorylation and ubiquitylation of p100/RelB. Consequently, p100 is processed to the smaller isoform p52 and the p52/RelB heterodimer translocates to the nucleus to activate or repress target gene expression.
The less characterized noncanonical pathway is stimulated by CD40, lymphotoxin‐B receptors, B‐cell activating factor of the TNF family (BAFF), lipopolysaccharide, and some viral proteins (Perkins, 2007; Israel, 2010; Ersing et al., 2013; Hoesel & Schmid, 2013). Additional factors that can activate this pathway include hydrogen peroxide and hypoxia (Perkins, 2007; Solt & May, 2008; Gamble et al., 2012). In the noncanonical pathway, the IKK complex comprises IKKα homodimers and IKKγ and is completely independent of the IKKβ subunit. Upon activation adaptor proteins, such as nuclear factor κB inducing kinase (NIK) phosphorylates IKKα, which in turn phosphorylates the NF‐κB precursor p100 on S99, S108, S115, S123, and S872, consequently resulting in ubiquitylation and proteasomal processing to the lower molecular weight isoform, p52. This isoform translocates to the nucleus to modulate transcription of its target genes (Fig. 1) (Hayden & Ghosh, 2004; Perkins, 2007; Solt & May, 2008; Israel, 2010; Gamble et al., 2012; Liu et al., 2012; Ersing et al., 2013). It has been reported that neither IKKβ nor IKKγ deficiency affects the noncanonical pathway (Liu et al., 2012; Hoesel & Schmid, 2013); therefore, IKKα homodimers plays a major role in regulating p100 processing (Senftleben et al., 2001; Perkins, 2007; Gamble et al., 2012; Hoesel & Schmid, 2013). Interestingly, IKKα does not always function as a kinase and was found to be essential for proper skeletal morphogenesis and differentiation of the epidermis in mice (Senftleben et al., 2001).
It is important to realize that while majority of the literature focuses on degradation of IκB and release of NF‐κB effector subunits as being primary functions associated with the IKK complex, it is also documented that IKKα and IKKβ can stimulate other signaling pathways such as pro‐ and antiapoptotic, pro‐inflammatory, and proliferative pathways independent of NF‐κB activation (Perkins, 2007). This idea that the IKK complex has additional roles in the cell outside of the traditional inflammatory response activation makes the IKK a complex regulator of cellular function and an excellent candidate for modulation by viral components.
Besides the classical IKKα, IKKβ, and IKKγ complex, there is another distinct IKK complex, which we will briefly introduce. This ‘nonclassical’ complex comprises the IKK‐related kinases, IKKε, and tank‐binding kinase 1 (TBK‐1). IKKε activation has been implicated in NF‐κB induction by stimuli such as lipopolysaccharide, cytokines, and phorbol 12‐myristate 13‐acetate (PMA) (Peters et al., 2000; Kawai & Akira, 2007; Möser et al., 2011). TBK‐1 was shown to activate the NF‐κB pathway by interacting and modulating the function of TANK (Kawai & Akira, 2007). In response to viral infections, IKKε and TBK‐1 have been shown to phosphorylate interferon regulatory factor (IRF)‐3 and IRF‐7 to activate the type I IFN response (Möser et al., 2011). Although IKKε shares similar sequence and functional domain organization to IKKα and IKKβ, the former kinase has been shown to only phosphorylate S36 on IκBα in a PMA‐mediated fashion (Peters et al., 2000). In this review, we will focus on the role that the classical IKK complex plays during viral infections and the impact these interactions have in the viral life cycle.
IKK complex components
The IKK complex comprises 3 components: IKKα, IKKβ, and IKKγ (Karin, 1999; Hayden & Ghosh, 2004; Hai et al., 2006; Solt & May, 2008; Israel, 2010; Shifera, 2010a, b; Gamble et al., 2012; Le Negrate, 2012; Liu et al., 2012; Ersing et al., 2013; Hoesel & Schmid, 2013). Biochemical studies demonstrate that the ratio of IKKα, IKKβ, and IKKγ subunits in the IKK complex is 1 : 1 : 2 (Solt & May, 2008; Gamble et al., 2012; Liu et al., 2012). This implies that the heterodimer IKKα and IKKβ are complexed with the homodimer IKKγ (Karin, 1999; Solt & May, 2008; Gamble et al., 2012; Liu et al., 2012). IKK subunits can exist in various orientations, indicated by the ability of canonical NF‐κB signaling to be activated despite the absence of IKKα in a TNF signaling event (Solt & May, 2008; Liu et al., 2012). Furthermore, an IL‐1‐mediated activation of canonical NF‐κB requires the interaction of IKKα and IKKγ in the absence of IKKβ (Solt & May, 2008).
IKKα (85 kDa) and IKKβ (87 kDa) are structurally similar proteins with enzymatic (phosphorylating) activity and differing only in the presence of a predicted NLS on IKKα (Liu et al., 2012). These 2 kinases share c. 51% overall sequence homology identity and 64% sequence identity across the catalytic domains (Karin, 1999; Solt & May, 2008; Israel, 2010; Gamble et al., 2012; Liu et al., 2012). Phosphorylation of S176 and S180 for IKKα and S177 and S181 for IKKβ is essential for the kinase functions (Liu et al., 2012). IKKα can shuttle from the cytoplasm to nucleus to phosphorylate histone H3 S10 upon TNF stimulation, or a variety of other targets. In contrast, targets of activated IKKβ are restricted to the cytosolic domain (Liu et al., 2012).
IKKγ is a 48 kDa protein with no known catalytic activity (Karin, 1999; Israel, 2010; Shifera, 2010a, b; Gamble et al., 2012) and can be found in the nucleus and the cytoplasm (Shifera, 2010a, b). It does have a role in regulation as a scaffold protein and is also required for kinase activity (Karin, 1999; Solt & May, 2008; Gamble et al., 2012). IKKγ has very different structural features compared to IKKα and IKKβ (Liu et al., 2012). The role of IKKγ in NF‐κB activation was demonstrated in site‐specific mutational studies, which showed that oligomeric IKKγ binding to K63‐linked polyubiquitin chains was necessary (Solt & May, 2008; Liu et al., 2012). Additionally, IKK activation involves the interaction between the N‐terminus of IKKγ and a NEMO binding domain (NBD) on the IKK complex (Solt & May, 2008; Liu et al., 2012). The regulatory role for IKKγ in NF‐κB activation is still not completely understood, but it is presumed to be both a positive and negative regulator of the IKK complex itself (Liu et al., 2012). Liu et al. (2012) have described IKKγ as a chaperone protein functioning as a positive regulator of the IKK complex. As a chaperone protein, IKKγ brings the IKK complex to upstream kinases to phosphorylate and activate IKKα and IKKβ (Liu et al., 2012). When IKKγ is phosphorylated on S68, it functions as a negative regulator of the IKK complex (Liu et al., 2012). Furthermore, IKKγ has been implicated in interactions with protein phosphatases PP2A and PP2C as well as deubiquitinases A20 and CYLD (Solt & May, 2008; Liu et al., 2012).
The mechanism of phosphorylation of the IKK complex has not been fully elucidated (Liu et al., 2012). To date, there are two schools of thought: (1) as a heterodimeric IKK complex, IKKα can phosphorylate IKKβ and vice versa; and (2) upstream IKK kinase, TGF‐β activated kinase 1 (TAK1) can phosphorylate and activate IKKβ (Solt & May, 2008; Liu et al., 2012). Crystal structures of IKKα and IKKβ show the presence of a large ‘space’ between these two subunits such that IKKα and IKKβ homodimers are less likely to phosphorylate each other. IKKγ has oligomerization properties that may bring IKKα and IKKβ closer to each other for phosphorylation to occur (Liu et al., 2012). The second school of thought emerged mainly due to identification of kinases such as TAK1, which is a mitogen‐activated protein kinase (MAPK) kinase involved in the Jun N‐terminal kinase (JNK) signaling pathway (Solt & May, 2008; Liu et al., 2012; Ersing et al., 2013). However, deletion mutant and RNAi studies demonstrate contradictory evidence on the dependence of TAK1 for IKKβ phosphorylation (Liu et al., 2012). Receptor‐interacting protein‐1 (RIP1) and MEKK3 are also potential candidates that can phosphorylate the IKKα, IKKβ, and IKKγ subunits, but this remains to be proven (Liu et al., 2012).
IKK regulation in viral infections
NF‐κB activation is a hallmark signaling pathway of most viral infections (DeLuca et al., 1999; Li et al., 1999; Xiao & Sun, 2000; Hiscott et al., 2001; Sun & Yamaoka, 2005; Amici et al., 2006; Victoriano et al., 2006; Fang et al., 2007; Harhaj et al., 2007; Shifera, 2010a, b; Jin et al., 2011; Ember et al., 2012; Gao et al., 2012; Le Negrate, 2012; Lee et al., 2012; Ersing et al., 2013; Li et al., 2013). As pro‐inflammatory molecules are the products of an activated NF‐κB cascade, many viruses have evolved to encode proteins to overcome this host strategy (Le Negrate, 2012). The constant battle between the better antagonist manufacturers has led to the assortment of effectors to regulate the NF‐κB pathway, such as secreted ligands or intracellular NF‐κB inhibitors (Le Negrate, 2012). Viruses such as Human immunodeficiency virus 1 (HIV‐1), Human T‐cell leukemia virus type 1 (HTLV‐1), Vaccinia virus (VACV), Kaposi's sarcoma‐associated herpesvirus (KSHV), Herpes simplex virus (HSV‐1), Hepatitis B virus (HBV), Hepatitis C virus (HCV), Epstein–Barr virus (EBV), and Influenza virus have been reported to activate the NF‐κB pathway (DeLuca et al., 1999; Li et al., 1999; Xiao & Sun, 2000; Hiscott et al., 2001; Gregory et al., 2004; Sun & Yamaoka, 2005; Amici et al., 2006; Victoriano et al., 2006; Harhaj et al., 2007; Shifera, 2010a, b; Jin et al., 2011; Ember et al., 2012; Le Negrate, 2012; Park et al., 2012; Ersing et al., 2013; Li et al., 2013). It has also been shown that viral proteins activate the NF‐κB response by targeting the IKK complex to increase viral replication (Hiscott et al., 2001; Wu et al., 2014). There are a number of possible reasons for a virus to modulate the NF‐κB pathway: promote viral replication, enhance infection, prevent viral detection and apoptosis (Hiscott et al., 2001). The IKK complex is a practical target for virus manipulation due to the IKK‐NF‐κB‐independent involvement in other biological functions such as cell proliferation, tumor suppression, and immune functions. To trigger these pathways, IKK will be activated by an independent set of stimuli that is unrelated to those that activate the NF‐κB cascade. Thus, a mechanistic understanding of viral interactions with the IKK complex is necessary to develop novel therapeutics that may modulate IKK to combat viral infections (Le Negrate, 2012). Here, we will discuss selected examples of viral manipulation of the IKK subunits to either activate or repress the host responses to enable a productive infection.
The IKKα subunit
Many viruses activate or repress the NF‐κB cascade to establish and maintain an infection. In this section of the review, we describe mechanisms by which viruses interact with and manipulate the IKKα subunit.
DNA viruses and IKKα
HBV is a DNA virus belonging to the Hepadnaviridae family and is known to manipulate the NF‐κB pathway to enhance viral replication and sustain a persistent infection (Huang et al., 2012). The 17 kDa polypeptide X protein of HBV (HBx) has been reported to be a potent activator of IKK (Hiscott et al., 2001; Huang et al., 2012). A study demonstrated that disrupting IKKα kinase activity resulted in inhibition of HBx‐mediated NF‐κB activation (Hiscott et al., 2001; Huang et al., 2012). Huang et al. (2012) showed that increased nuclear translocation of IKKα was observed in cells expressing stable and transient HBx. Furthermore, HBx‐mediated nuclear translocation of IKKα was facilitated by phosphorylation of IKKα T23 by the serine/threonine specific kinase, AKT followed by subsequent ubiquitylation (Huang et al., 2012). Moreover, mutating T23 and the NLS on IKKα not only inhibited nuclear transport of IKKα but also decreased migration and invasion of hepatocellular carcinoma cells (Huang et al., 2012). This suggests that hepatocellular carcinoma malignancy induced by HBx may be attributed to nuclear IKKα. Hence, HBV indirectly interacts with IKKα to activate the NF‐κB signaling pathway to induce a microenvironment to sustain a persistent infection.
The DNA dermatropic poxvirus, Molluscom contagiosum virus (MCV), is another example of a virus that inhibits the NF‐κB cascade. The viral protein MC160 prevented TNF‐α induced NF‐κB activation in 293T cells as a potential mechanism to inhibit the antiviral response (Nichols & Shisler, 2006; Le Negrate, 2012). The inhibition was attributed to MC160 destabilizing the IKK complex by degradation of IKKα, thus preventing phosphorylation of IκBα (Le Negrate, 2012). However, co‐immunoprecipitation (co‐IP) and in vitro kinase assay studies demonstrated that the observed NF‐κB inhibition was not a result of direct interaction of MC160 with the IKK subunits (Nichols & Shisler, 2006), but rather of an indirect inhibition. Two mechanisms of MC160 mediated NF‐κB inhibition have been reported (Le Negrate, 2012). First, the C‐terminus of MC160 can competitively bind to the heat‐shock protein 90 (HSP90) (Nichols & Shisler, 2009; Le Negrate, 2012). Heat‐shock proteins in general are produced in response to stress where they prevent protein denaturation and aggregation, assist in the refolding of damaged proteins, and facilitate their translocation to their correct intracellular locations (Salminen et al., 2008). In particular, HSP90 is a molecular chaperone which regulates biogenesis, stability, and enzymatic activity of protein kinases, thus affecting the function of several signal transduction and cell fate pathways (Salminen et al., 2008). IKK activation requires the formation of a heterocomplex with HSP90 and a co‐chaperone CDC37 (Salminen et al., 2008). Hence, MC160 binding to HSP90 prevents HSP90‐IKKα interaction, thus reducing IKKα stability and complex migration (Nichols & Shisler, 2009; Le Negrate, 2012). The second proposed mechanism is that binding of MC160 death effector domains to procaspase‐8 prevents procaspase‐8‐induced NF‐κB activation (Nichols & Shisler, 2009; Le Negrate, 2012). Hence, MCV inhibits the NF‐κB signaling pathway by indirect interaction with IKKα to establish and maintain an infection.
Viruses that utilize both IKKα and IKKβ to activate the NF‐κB pathway include the DNA viruses HSV and the gamma‐herpesvirus EBV. A member of Herpesviridae, HSV‐1, a large dsDNA virus that encodes c. 80 proteins, can establish a lytic and latent infection in infected cells (Amici et al., 2006; Jin et al., 2011; Xing et al., 2013). It was shown that efficient HSV replication was dependent on the activation of the NF‐κB cascade (Gregory et al., 2004). Employing the use of mouse embryonic fibroblasts (MEFs) deficient in IKKα (IKKα−/−) and IKKβ (IKKβ−/−), Gregory et al. (2004) showed that a 86% and 94% decrease in HSV replication was observed in IKKα−/− and IKKβ−/− MEFs, respectively. In addition, cells expressing the dominant negative form of IκBα resulted in a 98% reduction in viral protein production and replication. These results indicated that HSV‐1 activates NF‐κB by the IKK‐IκB‐p65 pathway to sustain productive viral replication cycles (Gregory et al., 2004). A possible reason for the higher dependence on IKKβ in HSV‐1 replication could likely be due to the kinase activity of the host protein. Perhaps, in the HSV life cycle, phosphorylation of (a) viral protein(s) is necessary to switch from a latent to lytic infection. Conversely, the requirement of IKKα could be to sustain a latent infection.
EBV infects c. 90% of people worldwide, targets B lymphocytes resulting in mononucleosis (a lymphoproliferative disease) that are associated with multiple human malignancies (Hiscott et al., 2001; Ersing et al., 2013). The EBV oncoprotein, latent membrane protein 1 (LMP1), lacks intrinsic enzymatic activity and therefore recruits and activates other enzymes to perform its functions (Ersing et al., 2013). LMP1 activates NF‐κB, MAPK, IRF7, and phosphatidylinositol 3‐kinase (PI3K) pathways to increase cell survival and growth (Hiscott et al., 2001; Ersing et al., 2013). The transformation effector site 2 (TES2) membrane distal signaling domain on LMP1 indirectly activates TRAF6 K63 ubiquitin ligase activity by binding to multiple adaptor proteins thus promoting TRAF6 auto‐K63‐ubiquitylation (Ersing et al., 2013). The newly formed ubiquitin chains recruit TGF‐β activated kinase 1/MAP3K7 binding protein 2 (TAB 2) and TAB 3 to activate TAK1 (Ersing et al., 2013). TES2 uses IKKα and IKKβ in partially redundant manners where depleting either one of these host kinases in an EBV‐infected system impairs TES2‐mediated IκBα phosphorylation. In addition, with a combined IKKα and IKKβ depletion, the TES2‐mediated IκBα phosphorylation and downstream effects were subdued (Ersing et al., 2013). A likely role for LMP1‐mediated canonical NF‐κB activation is in cell metabolism, glucose uptake, and the up‐regulation of cell microRNA, miR‐34A (Ersing et al., 2013). TES1, the proximal signaling domain, of LMP1 activates the noncanonical NF‐κB pathway via IKKα (Ersing et al., 2013). Mutational studies have shown that TES1 recruits TRAFs 1, 2, 3, and 5 through interaction by a specific motif (Ersing et al., 2013). The mechanism by which LMP1 activates the noncanonical NF‐κB pathway remains undetermined; however, studies are alluding to the likelihood that the activation is via NIK phosphorylation to allow LMP1 to trigger IKKα activation (Ersing et al., 2013).
RNA viruses and IKKα
HCV is an example of a single‐stranded (ss), positive‐sense RNA virus that inhibits TNF‐α‐induced NF‐κB activation to evade the host defense system (Park et al., 2012). The inflammatory mediator, TNF‐α, is a robust activator of the NF‐κB response. Stimuli that trigger inflammation including viral infection often result in increased levels of TNF‐α in circulation, which results in a controlled response process such as apoptosis. One can imagine that apoptosis of an infected cell would be deleterious to the virus from a microevolutionary perspective. As demonstrated by Western blot, HCV‐infected Huh‐7.5 cells treated with TNF‐α displayed decreased NF‐κB activity, which is in contrast to the expectation that TNF‐α stimulation would activate the NF‐κB cascade (Park et al., 2012). To identify HCV proteins responsible for the observed inhibition, cells were double transfected with plasmids encoding HCV proteins and a NF‐κB reporter plasmid encoding firefly luciferase under the control of NF‐κB‐responsive elements (Park et al., 2012). Data analyses demonstrated that IKK kinase activity was significantly reduced by HCV core, NS4B, and NS5B proteins (Park et al., 2012). Figure 2 illustrates the probable mechanism by which HCV inhibits TNF‐α‐induced NF‐κB activation. However, if the decrease in IKK activity was due to a direct interaction between HCV core protein, NS4B and NS5B with the IKK subunits remains to be determined. Li et al. (2013) identified IKKα as a host factor required for the later stages of HCV infection. HCV reliance on IKKα was found to be independent of NF‐κB activation, where IKKα is required for association of HCV core protein with lipid droplets during viral assembly (Li et al., 2013). Furthermore, Li et al. (2013) demonstrated that a DExD/H helicase, DDX3X (necessary for successful HCV infection), recognized and bound the HCV 3′ UTR region to activate IKKα. Activated IKKα then translocates to the nucleus to induce CBP/p300‐mediated lipogenic gene transcription (Li et al., 2013). In this fashion, core‐associated lipid droplets form to facilitate and promote HCV viral assembly (Li et al., 2013). Overall, HCV suppresses TNF‐α‐induced NF‐κB to initiate liver injury, which often leads to liver cirrhosis and hepatocellular carcinoma (Park et al., 2012).
Figure 2.
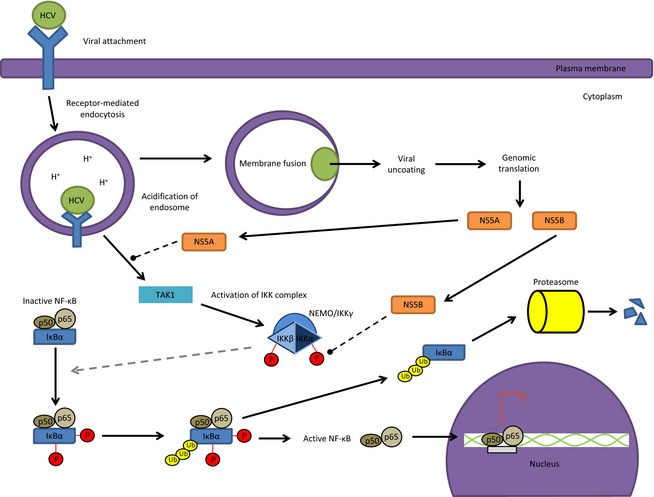
HCV infection inhibits the TNF‐α induced NF‐κB activation. In HCV infections, the nonstructural proteins NS5A and NS5B have been implicated in activating the NF‐κB pathway. The mechanism by which NS5A and NS5B inhibit NF‐κB has not yet been determined and as such we have denoted a probable mechanism by dashed lines. However, HCV utilizes IKKα independent of the NF‐κB pathway at later stages of infection. HCV core protein associates with lipid droplets during viral assembly.
A viral protein that indirectly acts on IKKα to inhibit NF‐κB‐induced IFN‐α production belongs to the negative sense ssRNA virus Human parainfluenza virus type 2 (HPIV2) – V protein. V protein has been reported to inhibit TLR7 and TLR9 dependent signaling for IFN‐α production (Kitagawa et al., 2013). Co‐IP studies revealed that in transfected HEK293T cells, V protein interacted with IRF7, TRAF6, IKKα, and MyD88 (Kitagawa et al., 2013). Knockdown studies demonstrated the requirement for TRAF6 for an indirect interaction between V protein with IRF7, IKKα, and MyD88 (Kitagawa et al., 2013). As a respiratory virus, the requirement here could be deemed to interactions with host proteins to have anti‐interferon properties in order for HPIV2 to sustain an infection.
The selectivity of utilizing IKKα in infection could be attributed to its nucleosomal function, which is not observed with IKKβ. With this function, activated IKKα contributes to NF‐κB‐mediated gene transcription, which ultimately results in termination and resolution of inflammatory responses (Liu & Malik, 2006). Nuclear IKKα utilizes its kinase activity to enhance transactivation and DNA binding of p65 and chromatin regulation through CREB‐binding protein (CBP) (Huang & Hung, 2013). The upstream kinase responsible for activation of and nuclear translocation of IKKα is NIK, which can be stimulated by lipopolysaccharide and TNF‐α (Huang & Hung, 2013). IKKα‐dependent CBP phosphorylation has been shown to enhance NF‐κB‐mediated gene expression and to suppress p53‐mediated gene expression by switching the binding preference of CBP from p53 to NF‐κB, to subsequently promote cell growth (Huang & Hung, 2013). Apart from NF‐κB modulation, nuclear IKKα plays a role in cell cycle, apoptosis, and tumor progression in cancers such as pancreatic cancer, breast cancer, and prostate cancer (Huang & Hung, 2013).
There may be differences in regulation of IKKα and IKKβ in the case of individual infections which may also be reflected in the fact that other transcription factors besides NF‐κB subunits may be regulated by IKK components and may play essential roles in viral replication. For example, an increase in phosphorylated IKKα was observed in HCV‐infected cells (Li et al., 2013). Phosphorylated IKKα is activated and hence can shuttle between the cytoplasmic and nuclear compartments (Li et al., 2013). Li et al. (2013) demonstrated that the 3′ UTR (a viral pathogen‐associated molecular pattern) of HCV specifically interacted with DDX3X and activated IKKα to mediate an NF‐κB‐independent function in the nucleus. Here, IKKα exerted a predominantly proviral effect to target the assembly stage of the HCV life cycle by activating sterol regulatory element‐binding proteins‐mediated lipogenesis and lipid droplet biogenesis (Li et al., 2013).
The IKKβ subunit
Only recently has the IKKβ subunit received attention in viral infection studies. Below, we provide a synopsis of the different viruses that directly or indirectly interact with IKKβ to activate or repress the NF‐κB response.
DNA viruses and IKKβ
KSHV is a dsDNA virus that both activates and inhibits NF‐κB signaling to establish and maintain an infection. KSHV is a gamma‐herpesvirus originally identified in Kaposi sarcoma lesions from HIV‐1‐infected individuals (Lee et al., 2012; Graham et al., 2013). KSHV is also associated with lymphoproliferative disorders, primary effusion lymphoma (PEL), and multicentric Castleman's disease (Graham et al., 2013). The KSHV protein expressed in latently infected cells, vFLIP, has been shown to activate NF‐κB, and as such promote survival, proliferation, differentiation, cytokine secretion, and oncogenic transformation, hence protecting cells from apoptosis (Lee et al., 2012; Graham et al., 2013). vFLIP activation of the NF‐κB pathway to award the host cell protection against B‐cell receptor‐induced apoptosis is a mechanism that not only requires initial activation of the pathway but maintenance of the signaling cascade as well (Graham et al., 2013). KSHV lymphomas survival was maintained by constitutive activation of the NF‐κB complex (Keller et al., 2000; Guasparri et al., 2004), whereby pharmacological inhibition of NF‐κB activation resulted in apoptosis of KSHV‐infected PEL cells (Keller et al., 2000). The constitutive activation of NF‐κB was achieved by a direct association of vFLIP with the IKK complex (Lee et al., 2012). In KSHV de novo infections, IKKβ and IKKε was found to be activated, resulting in subsequent NF‐κB signaling during latent infections, such that knockdown of these 2 kinases impaired NF‐κB activation and increased KSHV lytic gene expression (He et al., 2014). A second KSHV protein vIRF3 was shown to inhibit NF‐κB activity by exclusively inhibiting IKKβ‐mediated phosphorylation of IκBα (Seo et al., 2004; Le Negrate, 2012). GST‐binding assays revealed that vIRF3 physically associated with IKKβ to inhibit NF‐κB activation (Seo et al., 2004). vIRF3 and vFLIP function by selectively inhibiting and activating NF‐κB signaling in infected cells, which leads to the interesting possibility of distinctive regulation of individual components of the host responses that may be differentially modulated by viral proteins (Le Negrate, 2012).
VACV is a member of the Poxviridae family, genus Orthopoxvirus, and is commonly known as the live vaccine used to immunize against smallpox (Ember et al., 2012). These viruses have a dsDNA genome of c. 135 kb and replicate in the cytoplasm of infected cells (Ember et al., 2012). NF‐κB activation is inhibited by direct interaction of VACV viral proteins to the subunits of the IKK complex, such that these interactions will either inhibit IKK phosphorylation or interfere with upstream kinases that are required for phosphorylation of the IKK complex (Le Negrate, 2012). Specifically, B14 binds to IKKβ to inhibit activation and phosphorylation of IκBα (Ember et al., 2012), such that the B14‐IKKβ interaction inhibits phosphorylation of IKKβ (Le Negrate, 2012). Also, studies have shown that VACV N1 interacts with the IKK complex and TBK‐1 to inhibit NF‐κB activation (DiPerna et al., 2004; Le Negrate, 2012). Apart from IKKα modulation, HSV‐1 also affects IKKβ phosphorylation albeit through a viral protein. HSV‐1 encodes for γ134.5, a multifunctional virulence factor that promotes viral pathogenesis by preventing translational arrest by the protein kinase PKR. This action redirects protein phosphatase 1 (PP1) to dephosphorylate the α‐subunit of eukaryotic translation initiation factor 2 (Jin et al., 2011). Jin et al. (2011) demonstrated by co‐IP studies that in FLAG‐γ134.5‐transfected HeLa cells, NF‐κB activation was precluded. This was due to γ134.5 interacting with endogenous IKKα and/or IKKβ. A specific interaction with IKKβ and not with a control protein dN200 from a mutant Ebola virus VP35 determined the specificity of the interaction (Jin et al., 2011). The phosphorylation status of IKKβ was investigated to further dissect the mechanism by which γ134.5‐IKKβ interaction inhibits the NF‐κB cascade (Jin et al., 2011). FLAG‐PP1 or FLAG‐γ134.5 only slightly reduced phosphorylation of IKKβ. Interestingly, in combination, FLAG‐PP1 and FLAG‐γ134.5 completely reduced phosphorylation of IKKβ. These data demonstrate that γ134.5 interacts with PP1 to inhibit IKKβ phosphorylation thereby inhibiting NF‐κB activation (Jin et al., 2011). Furthermore, γ134.5 is the bridging protein via its N‐ and C‐terminal ends that form a complex with IKKβ and PP1, respectively, to mediate dephosphorylation of IKKβ to ultimately inhibit NF‐κB activation. The γ134.5 protein is expressed as early as 2–4 h and late in infection (Jin et al., 2009). The inhibition of IKKβ phosphorylation by HSV‐1 γ134.5 lends to the immediate response early on in an initial infection. This could be an evasive mechanism employed by the virus to establish an infection in the host cell.
RNA viruses and IKKβ
Rift valley fever virus (RVFV) is a negative sense ssRNA virus that belongs to the family Bunyaviridae. This arbovirus is a select agent and a known agricultural pathogen that infects livestock and humans (LaBeaud et al., 2010; Boshra et al., 2011; Narayanan et al., 2012). We have recently shown that the attenuated strain of RVFV, MP‐12, induced phosphorylation of p65 and IκBα via the canonical NF‐κB cascade (Narayanan et al., 2012). Size exclusion chromatography of MP‐12 infected human small airway epithelial cells revealed a molecular reorganization of the IKKβ subunit resulting in the formation of unique lower molecular weight protein complexes that are distinct from the traditional complexes that are known to exist in all cells. The smaller IKKβ complex was unique to virus‐infected cells and retained the kinase activity and ability to phosphorylate IκBα substrate as determined by in vitro kinase assays. Inhibitory assays using curcumin, an inhibitor of IKKβ, displayed a decrease in RVFV replication. An in vitro kinase assay demonstrated that the virulence factor NSs was indeed phosphorylated by the low molecular weight IKK complex 2 alluding to the possibility that RVFV requires a kinase‐active IKK complex for phosphorylation of NSs for efficient viral replication (Narayanan et al., 2012).
The positive‐sense ssRNA virus that belongs to the family Togaviridae, Venezuelan equine encephalitis virus (VEEV) produces a protein that interacts with IKKβ (Weaver et al., 2004; Garmashova et al., 2007; Lamb et al., 2010). We have recently reported an activation of the NF‐κB cascade in human astrocytes infected with the attenuated strain of VEEV, TC‐83. A molecular reorganization of IKKβ in TC‐83‐infected astrocytes was observed, which could be accredited to sensing of cytoplasmic dsRNA via the TLR3 pathway (Amaya et al., 2014). In vitro and in vivo inhibitor studies using specific inhibitors of the IKK complex affirmed that an active IKKβ was required for efficient VEEV replication. Liquid chromatography tandem MS of immunoprecipitated IKKβ from TC‐83‐infected cells determined an association between the viral nonstructural protein 3 (nsP3) with IKKβ. Confocal microscopy in combination with inhibitor treatments was utilized to validate the co‐localization and specificity between nsP3 and an active IKKβ (Amaya et al., 2014). Figure 3 models the interaction between nsP3 and IKKβ.
Figure 3.
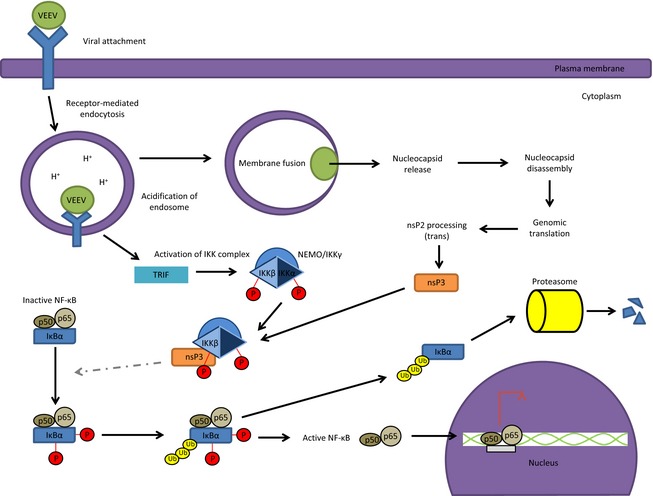
VEEV nsP3 interaction with IKKβ to activate the NF‐κB cascade. VEEV polyprotein processing results in nsP3 interacting with the IKKβ subunit of the IKK complex. It has yet to be determined whether this interaction is required for activation of the NF‐κB cascade and hence is denoted by dashed lines. The nsP3‐IKKβ interaction is required for VEEV replication in infected cells.
The ssRNA retrovirus, HIV‐1, is the causative agent of acquired immunodeficiency syndrome, has been shown to be positively regulated by NF‐κB‐induced cytokines (Victoriano et al., 2006), due to two NF‐κB binding sites located in the long terminal repeat enhancer region of HIV‐1 (Hiscott et al., 2001; Victoriano et al., 2006). DeLuca et al. (1999) demonstrated that in HIV‐1‐infected myeloid cells, IKKβ was constitutively active and maintained NF‐κB activity. The mechanism of HIV‐1 activation of IKK remains vague; however, there is the hypothesis that gp120, the HIV‐1 envelope glycoprotein, interacts with the CD4 receptor to activate NF‐κB (Hiscott et al., 2001).
The causative agent of severe acute respiratory syndrome (SARS), SARS‐associated coronavirus (SARS‐CoV), is an enveloped positive‐sense ssRNA virus (Fang et al., 2007). SARS‐CoV membrane (M) suppressed NF‐κB activity in HeLa and Vero E6 cells transfected with pcDNA3.1‐SARS‐CoV‐M and NF‐κB‐luciferase reporter plasmid. NF‐κB suppression was as a result of increased levels of cytoplasmic p50 and p65, indicating that nuclear translocation of p50 and p65 was affected (Fang et al., 2007). NF‐κB inhibition by M protein was a consequence of an interaction with IKKβ to inhibit phosphorylation and subsequent degradation of IκBα (Fang et al., 2007). M protein‐induced NF‐κB inhibition resulted in the downregulation of the inflammatory protein cyclooxygenase‐2 (COX‐2) at both the transcriptional and protein levels. Taken together, this offers a mechanistic approach as to SARS‐CoV evasion of the host immune response (Fang et al., 2007).
Influenza A virus, an Orthomyxovirus with eight segmented genes, is responsible for flu pandemics worldwide. The NS1 protein produced by Influenza A H5N1 was shown to inhibit IKK‐mediated NF‐κB activation and hence production of the induced antiviral genes. The mechanism behind the observed inhibition was determined by microarray analysis, where the C‐terminal effector domain of NS1 specifically interacted with IKKα and IKKβ (Gao et al., 2012). Hence, cytoplasmic NS1 blocks IKKβ‐mediated phosphorylation and degradation of IκBα (Gao et al., 2012). Cytoplasmic NS1 interferes with the noncanonical NF‐κB pathway by suppressing IKKα‐mediated processing of p100 to p52, thus inhibiting nuclear translocation of NF‐κB (Gao et al., 2012). Furthermore, nuclear NS1 prevents expression of NF‐κB target genes by inhibiting IKK‐mediated phosphorylation of S10 on histone H3 (Gao et al., 2012).
The IKKγ subunit
While the exact role of IKKγ within the IKK complex is not known, its interactions with viral proteins have become the subject of many recent studies. Studies implicate IKKγ in various cell types for the activation of NF‐κB via stimuli such as TNF‐α, IL‐1, HTLV‐1 Tax protein, lipopolysaccharide, and phorbol 12‐myristate 13‐acetate (PMA) (Shifera, 2010a, b).
DNA viruses and IKKγ
Lee et al. (2012) showed that over‐expression of the KSHV protein vFLIP, constitutively activated the NF‐κB cascade through the interaction and phosphorylation with IKKγ at S377. However, mutational studies to S377A in IKKγ indicated an increase in NF‐κB activity and IL‐6 production (Lee et al., 2012). Hence, IKKγ phosphorylation at S377 could be a feedback inhibitory mechanism to delay NF‐κB‐mediated immune responses (Lee et al., 2012).
RNA viruses and IKKγ
The retrovirus HTLV‐1 contains 2 copies of ssRNA and is the causative agent of adult T‐cell leukemia. HTLV‐1 encodes a viral oncoprotein Tax that is the major cause of adult T‐cell leukemia (Li et al., 1999; Harhaj et al., 2007; Shifera, 2010a, b). Protein–protein interactions that induce IKKγ phosphorylation can occur through interactions with HTLV‐1 Tax (Shifera, 2010a, b). HTLV‐1 Tax has been shown to interact with IKKγ and as a result is recruited to and activates the IKK complex resulting in constitutive activation of NF‐κB (Xiao & Sun, 2000; Sun & Yamaoka, 2005; Harhaj et al., 2007; Shifera, 2010a, b). Tax induces phosphorylation of IKKβ, which can then phosphorylate IKKγ (Li et al., 1999; Shifera, 2010a, b). The initial IKKβ phosphorylation event is required for Tax‐induced constitutive activation of IKK (Shifera, 2010a, b). Due to the constant activation of NF‐κB, host gene expression will be deregulated thus contributing to T‐cell transformation (Li et al., 1999; Sun & Yamaoka, 2005; Harhaj et al., 2007). Tax‐IKK complexes are capable of activating both the canonical and noncanonical pathways (Sun & Yamaoka, 2005; Harhaj et al., 2007). In a study conducted by Harhaj et al. (2007), Tax was shown to change the cellular localization of activated IKK complexes from the cytoplasm to concentrated perinuclear ‘hot spots’ in HTLV‐1‐transformed cell lines and in Tax‐expressing Jurkat cells. The relocalization of the IKK complex was likely due to Tax ubiquitylation (Harhaj et al., 2007). In another study conducted by Li et al. (1999) HTLV‐1‐infected T‐cells showed a significant increase in activity of the higher molecular weight IKK complex and an increase in IKKβ activity than in uninfected cells. These results suggested that Tax influences NF‐κB activity by modulating IKK factors.
Mazur et al. (2007) ascribed the IKKγ inhibitor activity of influenza infection to decreased expression of TRAIL and FasL reduced caspase activity that retained viral ribonucleoproteins in the nucleus. Furthermore, IKKγ protein expression was suppressed by influenza infection (Wang et al., 2012). The suppression of IKKγ enhanced replication of the virus to ultimately allow for an increase in NS1 protein expression, suggesting a requirement for NEMO to inhibit influenza viral replication in infected host cells (Wang et al., 2012).
The IKKε subunit
IKKε and TBK‐1 are viral targets used to antagonize IFN production during the course of infection (Ramanan et al., 2012). These targets are recent discoveries, whose exact interactions and mechanisms remain to be elucidated.
Wu et al. (2014) have shown that the phlebovirus Severe fever with thrombocytopenia syndrome virus (SFTSV), in the family Bunyaviridae, formed inclusion bodies that contain the viral protein NSs. The C‐terminal region of NSs was responsible for the induction of inclusion bodies (Wu et al., 2014). Wu et al. (2014) demonstrated that NSs inhibited IFN‐β and NF‐κB signaling. In addition, NSs co‐localized and interacted with TBK‐1 in the inclusion bodies that contained IKKε. The transcription factor, IRF‐3, is phosphorylated upon activation by TBK‐1. Interestingly, TBK‐1 and NSs confocal analysis indicated that phosphorylated IRF3 (p‐IRF3) was in the inclusion bodies, suggesting that NSs interacted indirectly with p‐IRF3 to sequester it in the inclusion bodies, preventing p‐IRF3 translocation to the nucleus, thereby suppressing IFN‐β signaling (Wu et al., 2014).
Marburgvirus (MARV), a filovirus, is the causative agent of lethal hemorrhagic fever in humans (Ramanan et al., 2012). MARV viral protein, VP35 (mVP35), is a multifunctional viral protein that has been shown to inhibit IRF3 phosphorylation by IFN kinases TANK, TBK‐1, and IKKε (Ramanan et al., 2012). mVP35 antagonizes IFN production through direct targeting of IKKε and the inhibition of retinoic‐acid inducible gene‐I (RIG‐I) like receptors (RLR) activation by sequestering dsRNA (Ramanan et al., 2012). IFN‐β promoter activity is inhibited by a dose‐dependent over‐expression of IKKε induced by mVP35 (Ramanan et al., 2012). Inhibition of IFN production, unlike inhibition of RLR activation, is not dsRNA‐dependent (Ramanan et al., 2012).
Targeting the IKK complex as a host‐based therapeutic against viral infections
The IKK complex is a highly attractive therapeutic target against viral infections because it has a diverse set of downstream effector molecules and is the center of many signaling pathways (Liu et al., 2012). Small molecule inhibitors that target the kinase activity of IKKα and/or IKKβ have shown promise (Liu et al., 2012) in viral infections. We have shown that the IKK inhibitors, BAY‐11‐7082, effectively inhibited VEEV replication in vitro and in vivo (Amaya et al., 2014) and that curcumin efficiently inhibited RVFV replication in vitro (Narayanan et al., 2012). Inhibitors of IKKα and IKKβ, wedelolactone, and IKK inhibitor XII were shown to reduce HCV core protein staining and infectious viral particle production in HCV‐infected Huh7.5.1 cells and primary human hepatocytes (Li et al., 2013). An IKKα and IKKβ inhibitor, 2‐amino‐6‐[2‐(cyclopropylmethoxy)‐6‐hydroxyphenyl]‐4‐piperidin‐4‐yl‐ nicotinonitrile (ACHP), was shown to reduce HIV‐1 production from latently infected cells (Victoriano et al., 2006). Despite the promise of IKK inhibitors in treating viral infections, there are considerations that need to be taken into account. The utilization of such inhibitors requires a comprehensive understanding of the IKK activated pathways such as downstream gene expression profiles, stimuli that trigger the activation of these pathways and upstream adaptor proteins. As the IKK complex is a critical component in host signaling, there is the concern of toxicity and side effects in all tissue types of an organism (Liu et al., 2012). However, the combination of small molecule IKK inhibitors with antiviral treatment as a host therapy to inhibit the deleterious consequences of enhanced inflammation that is often associated with many viral infections is highly plausible.
Conclusion
Viruses have developed unique mechanisms for efficient replication and infection of their target hosts. Similarly, hosts have developed innate cellular defenses to counter an infection and replication of these viruses. Productive and successful viral propagation depends on sequestering the host's cellular machinery concurrently evading the innate immune response. An infected host's first line of defense is the activation of the NF‐κB pathway to mount the antiviral response. However, from the above discussion, it seems that viruses can activate and/or repress the cascade to enhance their replication while avoiding the traditional survey mechanisms employed by the cell to detect viral infection. Although much has been elucidated about the mechanism of NF‐κB activation in particular via the IKK complex, a great deal still remains to be evaluated. In this review, we have determined the possible mechanisms by which both DNA and RNA viruses attack the NF‐κB cascade by either directly or indirectly interacting with the subunits of the IKK complex (IKKα, IKKβ, IKKγ, or IKKε). The selective interactions of viral proteins with the IKK subunits could be looked at from both perspectives: the function of the viral protein(s) and its interacting partner(s). When activated by viruses, IKKα relays a signaling cascade that is long lived. In this way, viruses could utilize the longevity of this type of response to maintain an environment in favor of latent infections. In contrast, IKKβ activation is a rapid response that is only transient in nature. Viral‐mediated activation of this kinase could be necessary for the virus to sequester cofactors that are already available to aid in viral replication or viral evasion. In this fashion, viral modulators may require these host molecules in the viral life cycle.
As discussed above activation of the NF‐κB cascade is helpful to the host cell when faced with an initial infection; however, viruses have utilized NF‐κB activation to enhance replication, to sustain a persistent infection and to evade the host defense system. For example, EBV, HCV and HBV have lytic and latent life cycles; hence it would be important to keep the infected cell alive to increase virion production and sustain a persistent infection.
In the canonical pathway, phosphorylation of IKKβ is required to phosphorylate IκBα to allow nuclear translocation of p65 to initiate transcription of target genes. Above, we have described mechanistic ways in which viral proteins prevent the phosphorylation of IκBα so as to inhibit NF‐κB activation: either by physically binding the IKK complex or by sequestering the kinase activities of the IKK subunits. In cases of VACV, HSV‐1 and KSHV infections viral proteins bind to IKKβ to inhibit subsequent phosphorylation of IκBα (Seo et al., 2004; Jin et al., 2011; Ember et al., 2012; Le Negrate, 2012). Interestingly, in RVFV and VEEV infections, viral proteins themselves maybe potentially phosphorylated by IKKβ to enhance viral replication and increase pathogenesis (Narayanan et al., 2012; Amaya et al., 2014). We and others have reported that by depleting IKKβ in the host cell viral replication is decreased (Gregory et al., 2004; Li et al., 2013; Amaya et al., 2014), which serves as an indicator that this host protein is required for viral replication. It would be interesting to determine whether other viral proteins that bind to IKKβ are phosphorylated to aid in viral replication or pathogenesis.
Studies in mice deficient of IKKβ resulted in reduced response to inflammatory cytokines accompanied by embryonal death (Huang & Hung, 2013). In IKKα deficient mice, the phenotype was one of the defective proliferation and differentiation of keratinocyte with limb and skeleton abnormalities (Huang & Hung, 2013). Under physiological conditions, IKKα is the regulatory component of the IKK heterodimeric complex to connect upstream activating signal to the IKKβ kinase activity (O'Mahony et al., 2000). IKKβ phosphorylation induced by a physiological agonist such as TNF‐α or by a pathological stimulus such as HTLV‐1 Tax proceeds directionally through IKKα then to IKKβ (O'Mahony et al., 2000). However, not all agonists will activate IKKβ through IKKα, such as with PKCθ that can activate IKKβ in the absence ofIKKα (O'Mahony et al., 2000). This implies that upstream kinases will target either IKK heterodimeric or homodimeric complexes differentially, thus increasing signal specificity (O'Mahony et al., 2000). This raises the possibility that distinct IKK complexes exist (O'Mahony et al., 2000), with even more variability in different cell types.
To summarize, viruses and viral products (proteins and nucleic acids) can either activate or inhibit the NF‐κB cascade by direct or indirect binding to the IKK subunits to enhance viral replication, evade the innate immune system, establish an infection, and develop pathogenicity. With the advancements in proteomics and in combination with bioinformatics, it would be beneficial to utilize the interactions between viral proteins and the IKK subunits as tools to identify biomarkers for diagnostic measures and host‐based therapeutics.
Acknowledgements
The authors would like to thank members of the Narayanan laboratory for help in manuscript preparation and revision. The authors declare that there are no conflict of interests relevant to this manuscript.
This minireview offers a perspective on the role of the interplay between host signaling pathways – specifically the IkB kinase complex pathway – and viral infections. Since this interplay is often necessary for viral replication and results in immune activation, inflammation and other negative consequences for the host, it becomes an attractive target for therapy.
References
- Amaya M, Voss K, Sampey G et al (2014) The role of IKKβ in Venezuelan equine encephalitis virus infection. PLoS One 9: e86745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amici C, Rossi A, Costanzo A, Ciafre S, Marinari B, Balsamo M, Levrero M & Santoro MG (2006) Herpes simplex virus disrupts NF‐kappaB regulation by blocking its recruitment on the IkappaBalpha promoter and directing the factor on viral genes. J Biol Chem 281: 7110–7117. [DOI] [PubMed] [Google Scholar]
- Boshra H, Lorenzo G, Busquets N & Brun A (2011) Rift valley fever: recent insights into pathogenesis and prevention. J Virol 85: 6098–6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca C, Petropoulos L, Zmeureanu D & Hiscott J (1999) Nuclear IkappaBbeta maintains persistent NF‐kappaB activation in HIV‐1‐infected myeloid cells. J Biol Chem 274: 13010–13016. [DOI] [PubMed] [Google Scholar]
- Diamant G & Dikstein R (2013) Transcriptional control by NF‐κB: elongation in focus. Biochim Biophys Acta 1829: 937–945. [DOI] [PubMed] [Google Scholar]
- DiPerna G, Stack J, Bowie AG, Boyd A, Kotwal G, Zhang Z, Arvikar S, Latz E, Fitzgerald KA & Marshall WL (2004) Poxvirus protein N1L targets the I‐kappaB kinase complex, inhibits signaling to NF‐kappaB by the tumor necrosis factor superfamily of receptors, and inhibits NF‐kappaB and IRF3 signaling by toll‐like receptors. J Biol Chem 279: 36570–36578. [DOI] [PubMed] [Google Scholar]
- Ember SW, Ren H, Ferguson BJ & Smith GL (2012) Vaccinia virus protein C4 inhibits NF‐κB activation and promotes virus virulence. J Gen Virol 93: 2098–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ersing I, Bernhardt K & Gewurz BE (2013) NF‐κB and IRF7 pathway activation by Epstein–Barr virus Latent Membrane Protein 1. Viruses 5: 1587–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Gao J, Zheng H, Li B, Kong L, Zhang Y, Wang W, Zeng Y & Ye L (2007) The membrane protein of SARS‐CoV suppresses NF‐kappaB activation. J Med Virol 79: 1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble C, McIntosh K, Scott R, Ho KH, Plevin R & Paul A (2012) Inhibitory kappa B Kinases as targets for pharmacological regulation. Br J Pharmacol 165: 802–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Song L, Li J, Zhang Z, Peng H, Jiang W, Wang Q, Kang T, Chen S & Huang W (2012) Influenza A virus‐encoded NS1 virulence factor protein inhibits innate immune response by targeting IKK. Cell Microbiol 14: 1849–1866. [DOI] [PubMed] [Google Scholar]
- Garmashova N, Atasheva S, Kang W, Weaver SC, Frolova E & Frolov I (2007) Analysis of Venezuelan equine encephalitis virus capsid protein function in the inhibition of cellular transcription. J Virol 81: 13552–13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham C, Matta H, Yang Y, Yi H, Suo Y, Tolani B & Chaudhary PM (2013) Kaposi's sarcoma‐associated herpesvirus oncoprotein K13 protects against B cell receptor‐induced growth arrest and apoptosis through NF‐κB activation. J Virol 87: 2242–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory D, Hargett D, Holmes D, Money E & Bachenheimer SL (2004) Efficient replication by herpes simplex virus type 1 involves activation of the IkappaB kinase‐IkappaB‐p65 pathway. J Virol 78: 13582–13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasparri I, Keller SA & Cesarman E (2004) KSHV vFLIP is essential for the survival of infected lymphoma cells. J Exp Med 199: 993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Hai T, Yeung ML, Wood TG, Wei Y, Yamaoka S, Gatalica Z, Jeang KT & Brasier AR (2006) An alternative splice product of IkappaB kinase (IKKgamma), IKKgamma‐delta, differentially mediates cytokine and human T‐cell leukemia virus type 1 tax‐induced NF‐kappaB activation. J Virol 80: 4227–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harhaj NS, Sun SC & Harhaj EW (2007) Activation of NF‐kappa B by the human T cell leukemia virus type I Tax oncoprotein is associated with ubiquitin‐dependent relocalization of I kappa B kinase. J Biol Chem 282: 4185–4192. [DOI] [PubMed] [Google Scholar]
- Hayden MS & Ghosh S (2004) Signaling to NF‐kappa B. Gene Dev 18: 2195–2224. [DOI] [PubMed] [Google Scholar]
- He Z, Zhao J, Zhang J, Jung JU & Feng P (2014) NF‐κB activation coordinated by IKKβ and IKKε enables latent infection of Kaposi's sarcoma‐associated herpesvirus. J Virol 88: 444–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiscott J, Kwon H & Genin P (2001) Hostile takeovers: viral appropriation of the NF‐kappaB pathway. J Clin Investig 107: 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoesel B & Schmid JA (2013) The complexity of NF‐κB signaling in inflammation and cancer. Mol Cancer 12: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WC & Hung MC (2013) Beyond NF‐κB activation: nuclear functions of IκB kinase α. J Biomed Sci 20: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WC, Chen WS, Chen YJ, Wang LY, Hsu SC, Chen CC & Hung MC (2012) Hepatitis B virus X protein induces IKKα nuclear translocation via Akt‐dependent phosphorylation to promote the motility of hepatocarcinoma cells. J Cell Physiol 227: 1446–1454. [DOI] [PubMed] [Google Scholar]
- Israel A (2010) The IKK complex, a central regulator of NF‐kappaB activation. Cold Spring Harb Perspect Biol 2: a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Ma Y, Prabhakar BS, Feng Z, Valyi‐Nagy T, Yan Z, Verpooten D, Zhang C, Cao Y & He B (2009) The gamma 1 34.5 protein of herpes simplex virus 1 is required to interfere with dendritic cell maturation during productive infection. J Virol 83: 4984–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Yan Z, Ma Y, Cao Y & He B (2011) A herpesvirus virulence factor inhibits dendritic cell maturation through protein phosphatase 1 and Ikappa B kinase. J Virol 85: 3397–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M (1999) The beginning of the end: IkappaB kinase (IKK) and NF‐kappaB activation. J Biol Chem 274: 27339–27342. [DOI] [PubMed] [Google Scholar]
- Kawai T & Akira S (2007) Signaling to NF‐kappaB by Toll‐like receptors. Trends Mol Med 13: 460–469. [DOI] [PubMed] [Google Scholar]
- Keller SA, Schattner EJ & Cesarman E (2000) Inhibition of NF‐kappaB induces apoptosis of KSHV‐infected primary effusion lymphoma cells. Blood 96: 2537–2542. [PubMed] [Google Scholar]
- Kitagawa Y, Yamaguchi M, Zhou M, Nishio M, Itoh M & Gotoh B (2013) Human parainfluenza virus type 2 V protein inhibits TRAF6‐mediated ubiquitination of IRF7 to prevent TLR7‐ and TLR9‐dependent interferon induction. J Virol 87: 7966–7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBeaud AD, Kazura JW & King CH (2010) Advances in Rift Valley fever research: insights for disease prevention. Curr Opin Infect Dis 23: 403–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb K, Lokesh GL, Sherman M & Watowich S (2010) Structure of a Venezuelan equine encephalitis virus assembly intermediate isolated from infected cells. Virology 406: 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Negrate G (2012) Viral interference with innate immunity by preventing NF‐kappaB activity. Cell Microbiol 14: 168–181. [DOI] [PubMed] [Google Scholar]
- Lee SH, Toth Z, Wong LY, Brulois K, Nguyen J, Lee JY, Zandi E & Jung JU (2012) Novel phosphorylations of IKKγ/NEMO. MBio 3: e00411–e00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XH, Murphy KM, Palka KT, Surabhi RM & Gaynor RB (1999) The human T‐cell leukemia virus type‐1 Tax protein regulates the activity of the IkappaB kinase complex. J Biol Chem 274: 34417–34424. [DOI] [PubMed] [Google Scholar]
- Li Q, Pène V, Krishnamurthy S, Cha H & Liang TJ (2013) Hepatitis C virus infection activates an innate pathway involving IKK‐α in lipogenesis and viral assembly. Nat Med 19: 722–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SF & Malik AB (2006) NF‐kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol 290: L622–L645. [DOI] [PubMed] [Google Scholar]
- Liu F, Xia Y, Parker AS & Verma IM (2012) IKK biology. Immunol Rev 246: 239–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur I, Wurzer WJ, Ehrhardt C, Pleschka S, Puthavathana P, Silberzahn T, Wolff T, Planz O & Ludwig S (2007) Acetylsalicylic acid (ASA) blocks influenza virus propagation via its NF‐kappaB‐inhibiting activity. Cell Microbiol 9: 1683–1694. [DOI] [PubMed] [Google Scholar]
- Möser CV, Kynast K, Baatz K et al (2011) The protein kinase IKKε is a potential target for the treatment of inflammatory hyperalgesia. J Immunol 187: 2617–2625. [DOI] [PubMed] [Google Scholar]
- Narayanan A, Kehn‐Hall K, Senina S et al (2012) Curcumin inhibits Rift Valley fever virus replication in human cells. J Biol Chem 287: 33198–33214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols DB & Shisler JL (2006) The MC160 protein expressed by the dermatotropic poxvirus molluscum contagiosum virus prevents tumor necrosis factor alpha‐induced NF‐kappaB activation via inhibition of I kappa kinase complex formation. J Virol 80: 578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols DB & Shisler JL (2009) Poxvirus MC160 protein utilizes multiple mechanisms to inhibit NF‐kappaB activation mediated via components of the tumor necrosis factor receptor 1 signal transduction pathway. J Virol 83: 3162–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Mahony A, Lin X, Geleziunas R & Greene WC (2000) Activation of the heterodimeric IkappaB kinase alpha (IKKalpha)‐IKKbeta complex is directional: IKKalpha regulates IKKbeta under both basal and stimulated conditions. Mol Cell Biol 20: 1170–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Kang W, Ryu SW et al (2012) Hepatitis C virus infection enhances TNFalpha‐induced cell death via suppression of NF‐kappaB. Hepatology 56: 831–840. [DOI] [PubMed] [Google Scholar]
- Perkins ND (2007) Integrating cell‐signalling pathways with NF‐kappa B and IKK function. Nat Rev Mol Cell Biol 8: 49–62. [DOI] [PubMed] [Google Scholar]
- Peters RT, Liao SM & Maniatis T (2000) IKKepsilon is part of a novel PMA‐inducible IkappaB kinase complex. Mol Cell 5: 513–522. [DOI] [PubMed] [Google Scholar]
- Ramanan P, Edwards MR, Shabman RS et al (2012) Structural basis for Marburg virus VP35‐mediated immune evasion mechanisms. P Natl Acad Sci USA 109: 20661–20666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Paimela T, Suuronen T & Kaarniranta K (2008) Innate immunity meets with cellular stress at the IKK complex: regulation of the IKK complex by HSP70 and HSP90. Immunol Lett 117: 9–15. [DOI] [PubMed] [Google Scholar]
- Senftleben U, Cao Y, Xiao G et al (2001) Activation by IKKalpha of a second, evolutionary conserved, NF‐kappa B signaling pathway. Science 293: 1495–1499. [DOI] [PubMed] [Google Scholar]
- Seo T, Park J, Lim C & Choe J (2004) Inhibition of nuclear factor kappaB activity by viral interferon regulatory factor 3 of Kaposi's sarcoma‐associated herpesvirus. Oncogene 23: 6146–6155. [DOI] [PubMed] [Google Scholar]
- Shifera AS (2010a) Proteins that bind to IKKgamma (NEMO) and down‐regulate the activation of NF‐kappaB. Biochem Biophys Res Commun 396: 585–589. [DOI] [PubMed] [Google Scholar]
- Shifera AS (2010b) Protein‐protein interactions involving IKKgamma (NEMO) that promote the activation of NF‐kappaB. J Cell Physiol 223: 558–561. [DOI] [PubMed] [Google Scholar]
- Solt LA & May MJ (2008) The IkappaB kinase complex: master regulator of NF‐kappaB signaling. Immunol Res 42: 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SC & Yamaoka S (2005) Activation of NF‐kappaB by HTLV‐I and implications for cell transformation. Oncogene 24: 5952–5964. [DOI] [PubMed] [Google Scholar]
- Victoriano AF, Asamitsu K, Hibi Y, Imai K, Barzaga NG & Okamoto T (2006) Inhibition of human immunodeficiency virus type 1 replication in latently infected cells by a novel IkappaB kinase inhibitor. Antimicrob Agents Chemother 50: 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhou J, Ruan C & Du Y (2012) Inhibition of type I interferon production via suppressing IKK‐gamma expression: a new strategy for counteracting host antiviral defense by influenza A viruses? J Proteome Res 11: 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver SC, Ferro C, Barrera R, Boshell J & Navarro JC (2004) Venezuelan equine encephalitis. Annu Rev Entomol 49: 141–174. [DOI] [PubMed] [Google Scholar]
- Wu X, Qi X, Qu B, Zhang Z, Liang M, Li C, Cardona CJ, Li D & Xing Z (2014) Evasion of antiviral immunity through sequestering of TBK1/IKKepsilon/IRF3 into viral inclusion bodies. J Virol 88: 3067–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao G & Sun SC (2000) Activation of IKKalpha and IKKbeta through their fusion with HTLV‐I tax protein. Oncogene 19: 5198–5203. [DOI] [PubMed] [Google Scholar]
- Xing J, Ni L, Wang S, Wang K, Lin R & Zheng C (2013) Herpes simplex virus 1‐encoded tegument protein VP16 abrogates the production of beta interferon (IFN) by inhibiting NF‐kappaB activation and blocking IFN regulatory factor 3 to recruit its coactivator CBP. J Virol 87: 9788–9801. [DOI] [PMC free article] [PubMed] [Google Scholar]