

Abstract
Friedreich's ataxia (FRDA) is the most common form of autosomal recessive ataxia caused by a deficit in the mitochondrial protein frataxin. Although demyelination is a common symptom in FRDA patients, no multicellular model has yet been developed to study the involvement of glial cells in FRDA. Using the recently established RNAi lines for targeted suppression of frataxin in Drosophila, we were able to study the effects of general versus glial-specific frataxin downregulation. In particular, we wanted to study the interplay between lowered frataxin content, lipid accumulation and peroxidation and the consequences of these effects on the sensitivity to oxidative stress and fly fitness. Interestingly, ubiquitous frataxin reduction leads to an increase in fatty acids catalyzing an enhancement of lipid peroxidation levels, elevating the intracellular toxic potential. Specific loss of frataxin in glial cells triggers a similar phenotype which can be visualized by accumulating lipid droplets in glial cells. This phenotype is associated with a reduced lifespan, an increased sensitivity to oxidative insult, neurodegenerative effects and a serious impairment of locomotor activity. These symptoms fit very well with our observation of an increase in intracellular toxicity by lipid peroxides. Interestingly, co-expression of a Drosophila apolipoprotein D ortholog (glial lazarillo) has a strong protective effect in our frataxin models, mainly by controlling the level of lipid peroxidation. Our results clearly support a strong involvement of glial cells and lipid peroxidation in the generation of FRDA-like symptoms.
INTRODUCTION
Friedreich's ataxia (FRDA) is an autosomal recessive neurodegenerative disease affecting the central (CNS) and peripheral (PNS) nervous systems (1,2). The FRDA neuropathology typically presents early degeneration of large sensory neurons in the dorsal root ganglia (DRG), followed by degeneration of sensory posterior columns, spinal-cerebellar tracts and cortical-spinal motor tracts together with atrophy of the large sensory fibers in peripheral nerves (reviewed in 3) and grumose degeneration in the dentate nucleus (4). FRDA is also characterized by hypertrophic cardiomyopathy that becomes the main cause of death among the patients (5) and by accumulation of labile iron in patient's mitochondria (6).
FRDA is caused by a decreased expression of the mitochondrial protein frataxin (7,8). Frataxin deficiency leads to a dysfunction of the respiratory chain complexes and Krebs cycle components mainly due to an inappropiate Fe–S cluster synthesis, therefore provoking a bioenergetic failure and the subsequent cell death (9–11).
Frataxin-deficient cells are hypersensitive to oxidative insult pointing to oxidative stress as a key factor in FRDA pathology (12–17). Furthermore, samples from FRDA patients normally show increased levels of oxidative stress markers such as malondialdehyde (MDA, a lipid peroxidation product), 8-hydroxy-2′-deoxyguanosine (an indicator of oxidized DNA) and higher glutathione transferase activity (18–20, respectively).
Dysregulated lipid metabolism and increased lipid peroxide levels are recurrent events in pathologies related to mitochondrial dysfunction (21–24). In addition, lipid imbalance has been reported to have a pivotal effect in the pathophysiology of multiple neurological disorders and neurodegenerative diseases, including bipolar disorders, schizophrenia, Alzheimer's, Parkinson's, Niemann–Pick and Huntington's diseases (reviewed in 25,26). Furthermore, as recently reviewed by Lessing and Bonini (27), Drosophila melanogaster has provided an impressive variety of examples which link mutations in lipid metabolism genes with neurodegenerative phenotypes such as bgm [bubblegum, encoding a fatty acid (FA) coenzyme A ligase], eas (ethanolamine kinase) or SNF4Aγ (encoding a subunit of AMP-activated protein kinase) or fumble (pantothenate kinase). Interestingly, iron metabolism is also disturbed in pantothenate kinase-associated neurodegeneration patients.
Very little is known about lipid homeostasis in FRDA. Although a typical symptom of FRDA is diabetes mellitus, several studies from patient autopsies or plasma samples showed undetectable changes in total cholesterol, triacylglycerides (TAG) and FA profiles compared with age-matched controls (28–30). However, levels of phosphatidylethanolamine, phosphatidylserine and linoleic acid were decreased (31,32). Nevertheless, electron microscopy analysis of the neuron/cardiac muscle frataxin-deficient mouse line revealed the presence of numerous lipid droplets in cardiac muscles (33).
Glial lazarillo (GLaz) is one of the Drosophila homologs to apolipoprotein D (ApoD), a human protein whose expression is increased in different neurological disorders such as Alzheimer's disease, schizophrenia and in the aging brain (reviewed in 34). Both, vertebrate ApoD and Drosophila GLaz, belong to the lipocalin family (35) and are predicted to carry small hydrophobic molecules such as steroids, bilins, retinoids and lipids (36). Both are expressed in glial cells in the developing and adult nervous system (37–39). Recent studies in plants, flies and mice indicate an important function for ApoD proteins as antioxidant defenses. Loss of function mutants showed increased sensitivity to oxidative stress (39–41). In contrast, overexpression enhanced resistance to oxidative stress and increased lifespan under normal conditions (41–43).
In this study, we address whether frataxin depletion in Drosophila results in an abnormal lipid metabolism and whether such metabolic dysfunction is an important element for the development of the disease. In addition, we hypothesized that GLaz might be an important candidate modulating the frataxin-deficient phenotype. We report that reduction of Drosophila frataxin (fh) expression either ubiquitously or in a pan-glial pattern produced a massive accumulation of FAs. In addition, loss of glial frataxin resulted in shortened lifespan, increased sensitivity to oxidative stress, impaired locomotor activity and progressive vacuolization of glial enriched regions. Interestingly, GLaz was able to significantly counteract some phenotypes detected in frataxin-deficient flies such as shortened lifespan, compromised locomotor performance and reduced aconitase activity. More importantly, this work provides in vivo evidence supporting a central role of lipid peroxides in the pathophysiology of FRDA.
RESULTS
Lack of frataxin in glial cells induces accumulation of lipid-like droplets
Although demyelination has been reported in post-mortem analysis of FRDA cases (44) and in two different FRDA mouse models (45,46), little is known about a possible glial dysfunction in FRDA neuropathology. Invertebrate glia do not generate myelin sheaths; however, they form multi-layered membrane protections around the neurons. To investigate these aspects in a Drosophila model system, expression of fh was downregulated in glial cells by means of the UAS-GAL4 system and the use of the pan-glial Repo-GAL4 driver. This driver was described by Sepp et al. (47) as a glial-specific GAL4 line expressed in all ectodermally derived glia within the PNS and CNS, reproducing the endogenous Repo expression (48).
Selective interference of frataxin in glial cells at 25°C using fhRNAi-1 produced viable offspring as well as some pre-adult lethality. The brains of these flies were examined in semi-thin plastic sections stained with toluidine blue by light microscopy. We found that when fh was specifically knocked-down in glial cells, frataxin-deficient flies showed abundant droplet-like structures throughout the entire brain cortex (Fig. 1B and C). The locations of these droplets correlate well with the staining pattern for anti-Repo observed in a wild-type brain (data not shown).
Figure 1.
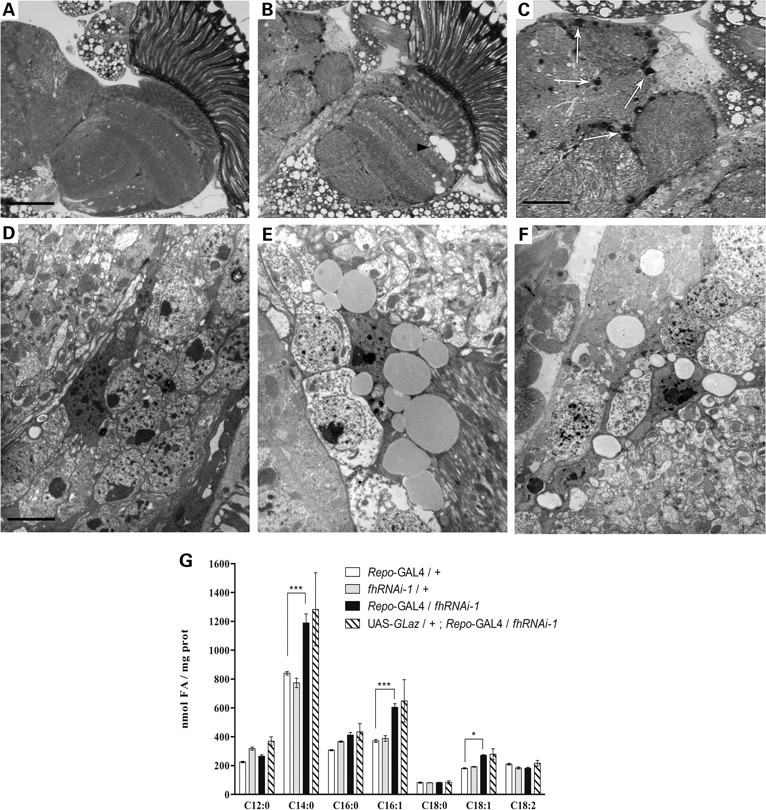
Lipid droplet accumulation in frataxin-deficient glial cells. (A and D) 35-day-old Repo-GAL4/+ controls. (B, C and E) 35-day-old Repo-GAL4/fhRNAi-1. (F) 35-day-old UAS-GLaz/+;Repo-GAL4/fhRNAi-1. (B) Frataxin-deficient brains displayed degenerative defects in the form of extensive outer chiasm vacuolization (arrowhead). (C) The appearance of lipid-like droplets is denoted with white arrows. (D and E) Ultrastructural analysis revealed accumulation of lipid bodies in glial cells of frataxin-deficient brains. (F) Altered properties of glial-lipid deposits after co-expression of GLaz. (G) GC/MS analysis of FAs from Drosophila heads. Frataxin deficiency results in a remarkable increase of C14:0, C16:1 and C18:1. GLaz is unsuccessful in restoring the amount of these three FAs to control levels. Significance in (G) was determined by one-way ANOVA with post hoc Newman–Keuls (***P < 0.001 and *P < 0.05). Error bar represents standard error. FA, fatty acid. The scale bar represents 50 μm (A and B); 25 μm (C) and 2.5 μm (D–F).
In order to analyze the structure of these droplets, the brains of fhRNAi-1 flies were further studied by electron microscopy. The ultrastructural examination revealed that the cell bodies of glial cells were densely packed with lipid-like vesicles (Fig. 1E) where the number and size progressively increased during aging (data not shown). To determine the lipidic composition of the droplets found in glia cells, we analyzed the FA content of Drosophila brains from 35-day-old flies by means of the gas chromatography-mass spectrometry (GC/MS) technique. We found that loss of frataxin in glial cells resulted in a robust accumulation of some FAs. Specifically, myristic (C14:0) and palmitoleic (C16:1) acids were highly increased, whereas oleic acid (C18:1) only showed a moderate increment (Fig. 1G).
These results suggest a disruption of lipid metabolism in glial cells triggered by loss of frataxin and the concomitant formation of multiple lipid droplets.
Frataxin knock-down increases free FA levels
Next, we wanted to characterize the lipid accumulation in a scenario that better resembles patient conditions (systemic frataxin deficiency). In order to elucidate this question, frataxin was ubiquitously reduced with daughterless-GAL4 (da-GAL4), and subsequently these flies were then subjected to a semi-quantitative analysis of lipid content using thin-layer chromatography.
In our hands, a strong systemic reduction of frataxin using fhRNAi-1 produced mainly long-lived L3 larvae, as described by Anderson et al. (49), and just a few of them were able to pupate. This second group of larvae was used to carry out the following experiments.
As shown in Figure 2, a 65% increase in the free FA content could be detected compared with control larvae (da-GAL4/+), whereas the TAG only showed a small 10% increase (Fig. 2A). The level of different phospholipids [phosphatidylcholine (PC), phosphatidylglycerin (PG) and phosphatidylinositol (PI)] did not show any relevant increase (Fig. 2B).
Figure 2.

Lipid analysis of frataxin interference lines detects a drastic increase in free FAs partially rescued by Drosophila GLaz. (A and B) Analysis of neutral and phospholipid levels by thin-layer chromatography in late L3 larvae. The values are expressed as a percentage of control (da-GAL4/+). Frataxin knock-down (da-GAL4/fhRNAi-1) mainly triggers accumulation of free FAs when compared with controls. These accumulations are partially rescued by GLaz co-expression (UAS-GLaz/+;da-GAL4/fhRNAi-1). (C and D) GC/MS analysis of FA content. (C) Comparison of the most abundant FA in our samples. (D) Comparison of low-level FA (<10%). Significance was determined by one-way ANOVA with post hoc Newman–Keuls (***P < 0.001; **P < 0.01 and *P < 0.05). Error bar represents standard error. FFA, free fatty acid; TAG, triacylglycerides; PC, phosphatidylcholine; PG, phosphatidylglycerin; PI, phosphatidylinositol; FA, fatty acid.
In order to determine whether such an accumulation was due to a general increase of FAs or only to a contribution of some specific species, we carried out a GC/MS analysis of FAs. In the L3 samples, we were able to detect a profile consisting of 19 different FAs. Eight of them were extremely low represented (<6‰) and were not considered in our study. Myristic acid (C14:0), palmitic acid (C16:0), palmitoleic acid (C16:1), oleic acid (C18:1) and linoleic acid (C18:2) were the most abundant acids (each of them representing >10% of total FA composition). In addition, FAs with chains containing more than 20 carbons were not detected. Figure 2C and D shows that frataxin deficiency induced a general accumulation of FAs independently of their relative abundance compared with control flies. In agreement with the glial results (Fig. 1G), the increase was higher in the saturated and monounsaturated FAs (from 65 up to 160%) in contrast to only slight increases detected in the polyunsaturated FAs (25%).
Taken together, these results suggest that loss of frataxin triggers a general accumulation of FAs either by an increased synthesis or by a defective catabolism. This finding is in agreement with our observation of accumulation of lipid-like droplets in glial cells of frataxin-deficient flies.
Overexpression of GLaz counteracts the accumulation of FAs in frataxin knock-down flies
GLaz is an important protein involved in antioxidant defense and lipid metabolism in glial cells and therefore a good candidate for attenuating the frataxin-dependent glial phenotype. A first hint for such an interaction came from the ultrastructural analysis, which shows a modification of the quality of lipid droplets in the glial cells of frataxin knock-down flies that co-express GLaz (compare Fig. 1E with F). In agreement with this observation, lipid analysis of L3 larvae that ubiquitously co-expressed GLaz showed a significant reduction of free FA content but no changes in TAG or phospholipids (Fig. 2A and B). To study any specific effects of GLaz on FA content, a GC/MS approach was performed identifying a biased action of GLaz with an exclusive and partial recovery of monounsaturated FAs, but not saturated or polyunsaturated FAs (Fig. 2C and D).
To exclude that the partial rescue of GLaz is due to a GAL4 dilution effect using multiple UAS constructs in one genotype, expression levels were compared by semi-quantitative real-time PCR. Both genotypes, single frataxin interference (da-GAL4/fhRNAi-1) and co-expression of GLaz (UAS-GLaz/+;da-GAL4/fhRNAi-1) showed a similar frataxin level of ∼10% relative to control values (data not shown).
Our results clearly indicate that GLaz is capable of partially rescuing the frataxin lipid phenotype by selectively reducing the increased amount of monounsaturated FAs.
Glial function of frataxin in lifespan and oxidative stress
Next, we investigated the impact of the glial function of frataxin on fly survival. Enhanced sensitivity to oxidative insult is a hallmark in FRDA models, hence we examined whether reducing frataxin expression in glial cells was sufficient to exacerbate the sensitivity to oxidative stress. Hyperoxia has been proven to be a relevant system to re-create an unbalanced oxidative status (50). As can be seen in Fig. 3A, frataxin interference (Repo-GAL4/fhRNAi-1) under oxygen-induced stress resulted in a drastic reduction of mean (73%) and maximum lifespan (55%) when compared with controls. Shortening of mean (25%) and maximum (32%) lifespan was also revealed by survival analysis under normal conditions (Fig. 3B).
Figure 3.

Physiological and behavioral effects of glial frataxin depletion. (A) Lifespan under hyperoxia (99.5% O2). Downregulation of frataxin in glia strongly enhances susceptibility to oxidative stress. (B) Lifespan under normoxia. Pan-glial reduction of fh expression shortens the mean and maximum lifespan compared with control flies. (C) Negative geotaxis experiment with 10-day-old individuals. Loss of frataxin in glia strongly reduced walking ability. (D–L) Autofluorescent paraffin brain sections. (D–F, Repo-GAL4/+controls); (G–I, Repo-GAL4/fhRNAi-1) and (J–L, fhRNAi-1/+controls). (D, G and J, 5-day-old flies); (E, H and K, 20-day-old flies) and (F, I and L, 35-day-old flies). Frataxin downregulation in glial cells induces age-dependent brain vacuolization focused on the outer chiasm, lamina and medulla (arrowheads). Statistical differences between survival curves in (A) and (B) were analyzed using the Kaplan–Meier test, and exclusively Repo-GAL4/fhRNAi-1 showed a statistical significant behavior (P < 0.001). Significance in (C) was determined by one-way ANOVA with post hoc Newman–Keuls (***P < 0.001). Error bar represents standard error. The scale bar represents 50 μm.
These results indicate that frataxin is essential for a correct function of glial cells. In particular, absence of glial frataxin increased sensitivity of this cell type towards reactive oxygen species (ROS), leading to premature death of the flies.
Depletion of frataxin expression in glial cells affects nervous system integrity
In order to study the effect of frataxin downregulation on the integrity and function of the Drosophila nervous system, locomotor activity and brain degeneration of Repo-GAL4/fhRNAi-1 flies were analyzed.
Loss of frataxin provoked a 55% reduction in the climbing ability of 10-day-old flies (Fig. 3C). Moreover, brain analysis showed an age-dependent degeneration that was first detected at day 20 and further increased at day 30 (Fig. 3G–I). The vacuolization is mainly focused on medulla, lamina and outer chiasm (Fig. 3I, arrowheads), which is consistent with a location of tissue enriched in glial cells. In addition, an ultrastructural analysis showed that the lipid droplets described in Figure 1 can be detected before the presence of vacuolization (data not shown).
Our results suggest that lack of frataxin initiates a glial dysfunction that correlates with the decrease in negative geotaxis performance concomitant with a lipid accumulation finally culminating in cell death.
GLaz overexpression partially rescues loss of frataxin phenotypes in glial cells
Next, we examined the rescue efficiency of GLaz in glia. As shown in Figure 4A and B, GLaz was able to clearly extend the lifespan of frataxin-deficient flies under oxidative stress as well as under normal conditions (50 and 25%, respectively). Furthermore, GLaz could significantly improve the climbing performance of the affected individuals at 5 and 10 days (Fig. 4C). The same behavioral experiments were conducted in fhRNAi-2 flies, which showed similar results, although with a stronger GLaz rescue effect (Fig. 4D–F).
Figure 4.

Lifespan and behavioral rescue of fh-deficient Drosophila by GLaz overexpression. (A and D) Lifespan experiments under hyperoxia conditions (99.5% O2) using either fhRNAi-1 (A) or fhRNAi-2 (D) flies. GLaz is able to partially protect frataxin-deficient Drosophila against oxidative insult although no benefit is detected in control flies. (B and E) Lifespan under standard conditions using fhRNAi-1 (B) or fhRNAi-2 (E) flies. Co-expression of GLaz shows an extension of mean and maximum lifespan of flies with reduced frataxin. (C and F) Negative geotaxis experiments of 5- and 10-day-old flies using fhRNAi-1 (C) or fhRNAi-2 (F) flies. GLaz alleviates the locomotor deficits in frataxin knock-down individuals. Survival curves were analyzed using the Kaplan–Meier test. Significance in (C) and (F) was determined by one-way ANOVA with post hoc Newman–Keuls (***P < 0.001). Error bar represents standard error.
In order to exclude possible GAL4 dilution artifacts due to the presence of a second UAS line, negative geotaxis experiments and survival under hyperoxia were also carried out with co-expression of a nuclear GFP construct (UAS-Stinger). No significant differences were observed in both experiments compared with frataxin interference (Fig. 4A and C).
Next, we asked whether GLaz, when overexpressed in glial cells, is also able to partially rescue the FA phenotype induced by a reduction of frataxin levels. By means of the GC/MS technique, we found that GLaz failed to reduce the amount of the monounsaturated FAs C16:1 and C18:1 in the head samples (Fig. 1G). As can be seen in Figure 1F, the ultrastructural analysis by electron microscopy of UAS-GLaz/+;Repo-GAL4/fhRNAi-1 brains also displayed the vacuolization and the lipid vesicles observed in Repo-GAL4/fhRNAi-1 individuals (Fig. 1E), with the peculiarity that the vesicles seemed empty after preparation. De Martino et al. (51) carefully studied the influence of distinct plastic embedding media and staining procedures on the lipid morphology in light and electron microscopy and they discovered that the binding affinity of osmium tetroxide varies proportionally with the oxidation status of the FAs. This would suggest that GLaz function has an influence on the nature of the lipids stored in the vesicles and that the oxidation state of these lipids might be altered by the co-expression of GLaz in glial cells.
These results show that GLaz is able to increase the lifespan and the locomotor ability of frataxin-deficient flies without directly reducing the excess of FAs in the glial cells and therefore indicating that there is not necessarily a direct link between lipid accumulation and behavioral phenotypes.
Reduction of frataxin expression promotes lipid peroxidation
Human ApoD and Drosophila GLaz have been reported to repress lipid peroxides (39,41,43). Moreover, lipid peroxidation products such as MDA have been regularly found in FRDA patients' samples (18). Therefore, we asked whether frataxin interference also increased lipid peroxidation levels in Drosophila. In order to elucidate the mechanism by which GLaz improved frataxin-deficient conditions, we studied the effect of systemic frataxin reduction on lipid peroxidation. As reported in Llorens et al. (15), a moderate ubiquitous reduction of frataxin (fhRNAi-2) was enough to bypass any pre-adult lethality and shows some typical features of FRDA, such as enhanced sensitivity to oxidative stress, together with reduced aconitase activity and shortened lifespan.
As shown in Figure 5A, the total amount of the MDA product was notably increased in 5- and 15-day-old frataxin-deficient flies. Nevertheless, neither young (7-day-old, Fig. 5B) nor older (30-day-old, data not shown) frataxin-deficient flies present significant alterations in the TAG or free FA content. Interestingly, co-expression of GLaz showed an almost complete rescue of this phenotype, with a strong reduction (30%) of lipid peroxide accumulation in young flies (5-day-old). A mild decrease (9%) was observed in older individuals (15-day-old).
Figure 5.

GLaz decreases lipid peroxidation levels and restores aconitase activity in frataxin-deficient flies. (A) Frataxin-deficient adults (fhRNAi-2/+;da-GAL4/+) display an increase in lipid peroxides that can be restored to control levels by GLaz in 5-day-old flies (5d) but not in 15-day-old flies (15d). (B) fhRNAi-2/+;da-GAL4/+ do not induce any significant changes in TAG or FFA content. (C and D) GLaz significantly protects aconitase from ROS-mediated inactivation in frataxin-deficient L3 (caused by fhRNAi-1 transgene) and adult flies (caused by fhRNAi-2 transgene). Significance was determined by one-way ANOVA with post hoc Newman–Keuls (***P < 0.001). Error bar represents standard error. FFA, free fatty acid; TAG, triacylglycerides.
These results suggest that lack of frataxin besides inducing hypersensitivity to oxidative insult also increases the production of oxidative radicals by itself. Our data also indicate that the abnormal peroxidation of lipids in frataxin knock-down flies is independent of lipid accumulation. In addition, GLaz is able to reduce the increased lipid peroxide generation, although in a time-restricted manner. Therefore, the increase in lipid peroxidation correlates well with the phenotypes described in glia, and GLaz was able to rescue the effects driven by the loss of frataxin.
GLaz restores aconitase activity in frataxin-deficient larvae and adult flies
One of the most characteristic biochemical defects associated with a loss of frataxin is the reduction of aconitase activity (9,15,33). Therefore, we asked whether co-expression of GLaz was able to restore the enzymatic activity in a frataxin-deficient environment. This hypothesis was tested in two different experimental scenarios: on the one hand, in L3 larvae with a strong reduction of frataxin produced by the fhRNAi-1 transgene (17); on the other hand, in young adult flies with a milder reduction of frataxin caused by the fhRNAi-2 transgene combined with oxidative stress (15). We used the da-GAL4 driver because it provides a more widespread expression facilitating the detection of changes in aconitase activity. Loss of frataxin provoked a 28% decrease in aconitase activity in hyperoxia-treated adult flies (Fig. 5C) and 46% in larvae (Fig. 5D). GLaz was able to restore aconitase activity effectively, up to levels comparable with control flies (Fig. 5C and D).
Taken together, these data indicate that preserving the redox balance is a major mechanism by which GLaz co-expression is able to counteract some of the effects caused by frataxin deficiency.
DISCUSSION
FRDA, the most frequent form of hereditary ataxia in the Caucasian population, is produced by a deficit in the mitochondrial protein frataxin. Although big efforts are being made using animal, cell culture and yeast models, the function of frataxin still remains elusive and controversial. Nevertheless, using microarray and proteomic approaches, new and interesting downstream consequences derived from frataxin depletion are currently being described. These pathways are suggesting novel therapeutic targets for future treatments designed to mitigate lack-of-frataxin phenotypes (52).
In this work, we could show that a reduction of frataxin expression compromised the lipid homeostasis at two different levels: increasing the total amount of lipids and enhancing the generation of peroxyl radicals. Our experiments revealed that strong ubiquitous fh interference with fhRNAi-1 triggered an accumulation of FAs in larvae. We also showed that a general reduction of frataxin to levels compatible with a normal development provoked an early overload of lipid peroxides without a detectable rise in the level of FAs. Thus, the increase in lipid peroxides is probably due to a higher ROS production rather than to an FA accumulation. However, we can speculate that the increase of FA mediated by a strong reduction of frataxin expression could exponentially increase the oxidation rate of FAs.
The FA accumulation observed in the Drosophila FRDA model is in good agreement with the lipid inclusions found previously in the cardiac muscle of FRDA conditional mouse models (33) and with the increased intracellular lipid content present in the striated muscle fibers and Schwann cells of inherited α-tocopherol deficiency patients (21), a disease that appears to be clinically indistinguishable from FRDA. Moreover, up to 40% of FRDA patients manifest different glucose metabolism problems (53) such as diabetes or insulin resistance, and remarkably type 2 diabetes patients also present an altered lipid status attributable to a mitochondrial dysfunction (54,55). However, the link between increased lipid amounts and FRDA is not completely clear. On the one hand, depletion of frataxin generates different scenarios that can lead to the inhibition of mitochondrial β-oxidation. For example, mitochondrial respiration defects induce a reduction of NAD+/NADH ratios compromising the β-oxidation pathway (22,56). Alternatively, impairment of aconitase activity increases citrate levels (57), and citrate is an allosteric activator of malonyl-CoA production, which in turn is a potent inhibitor of the mitochondrial β-oxidation (reviewed in 58). Furthermore, downregulation of peroxisome proliferator-activated receptor gamma (PPARγ) pathway has been observed in FRDA mice models, suggesting a reduction of lipid catabolism (59). Moreover, on the other hand, frataxin deficiency has been suggested to increase lipogenesis via hyperactivation of mitochondrial acyl-CoA thioesterase (60) or upregulating the expression of the sterol-responsive element-binding protein 1 (Srebp1) (59). Thus, derangement of lipid homeostasis in FRDA could be produced by either blocking the main degradation pathway of FAs or increasing their synthesis. In addition, lipid imbalance could be a critical event in FRDA progression since lipid metabolism is the main fuel source of the cell.
FAs are extremely sensitive to oxidative modification, resulting in the formation of lipid peroxides, which are highly cytotoxic and promote damage to other lipids, proteins and DNA (reviewed in 61). The link between oxidative stress and frataxin has been extensively analyzed. Indeed, loss of frataxin not only promotes hypersensitivity to oxidative insult in various experimental models (12–17), it also induces accumulation of oxidative stress markers such as anion superoxide (62–64), carbonylated proteins (16,65) and MDA or other lipoperoxidation products (46,66). Moreover, frataxin overexpression reduces endogenous levels of ROS in cell culture (67) and increases antioxidant activity in Drosophila (68). The age-dependent accumulation of lipid peroxides found in our Drosophila model corroborates the endogenous increase in ROS production led by loss of frataxin. Castilho et al. (69,70) reported that lipid peroxides trigger a cascade of events that includes a fall in mitochondrial membrane potential, mitochondrial swelling and loss of mitochondrial matrix components. More interestingly, lipid peroxidation is strongly catalyzed through the Fenton reaction by iron and iron complexes (71,72), being the iron–citrate complex one of the most toxic forms. Remarkably, iron is the transition metal that accumulates in frataxin-deficient yeast and in the mitochondria of different samples from FRDA patients (6,11,73). Thus, iron-mediated oxidative insult seems to develop a crucial role in the function of frataxin and in the progression of the disease. Since lipid peroxides are the most harmful oxidative stress mediators, strategies focused on quenching, recycling or protecting such products could be highly beneficial for the treatment of the disease.
The lipid metabolism failure is not restricted to a systemic reduction of frataxin. When fh expression is suppressed in glial cells, an accumulation of FAs was first detected with electron microscopy and confirmed by gas chromatography. Since neuron–glia interactions are crucial to ensure optimal brain function and glia are the principal neuron energy suppliers, our results may indicate a pivotal function of glial cells in the FRDA neuropathology. Indeed, posterior nerve roots and posterior sensory nerves in FRDA patients are characterized by an axonal atrophy as well as by a demyelination process, whereas the fine unmyelinated fibers are well preserved (44,74). Interestingly, Lu et al. (75) have recently reported that frataxin knock-down produced severe effects on Schwann cell cultures compared with DRG and with other neuronal cell lines. However, despite this pathological condition, the role of glial cells in the FRDA etiology has not yet been analyzed in detail. In this work, we report that the reduction of frataxin expression in Drosophila glial cells induces adverse effects such as a hypersensitive response against oxidative insult, a decrease in locomotor performance and shortened lifespan. Moreover, we present the first evidence of nervous system degeneration in an invertebrate model of FRDA. Interestingly, some phenotypes were detected prior to a clear lipid accumulation. Therefore, together with the results from the ubiquitous frataxin interference, we suggest that frataxin depletion in glial cells leads to increased lipid peroxidation that induced mitochondrial injury and dysfunction preceding the lipid accumulation and cell degeneration. Although we have not studied this aspect, alterations in glial cells might also occur in the PNS, contributing to the glial phenotypes described in this work.
If lipid dysregulation is a significant factor for FRDA pathology in Drosophila and lipid peroxides are a primary cause of the symptoms, we might be able to mitigate loss-of-frataxin effects by modulating the lipid content and/or the amount of lipoperoxides. GLaz, one of the Drosophila homologs of human ApoD, is expected to counteract both conditions (39,42,76). In frataxin-deficient flies, GLaz, indeed, exhibited cell-specific effects. First, general co-expression of GLaz specifically reduced the amount of monounsaturated FAs. In agreement with these results, ApoD and its homolog proteins have been shown to regulate systemic lipid homeostasis in mice (77) and Drosophila (78), as well as to control the brain lipid composition in mice (79). Our results would also suggest that oleic acid (C18:1), palmitoleic acid (C16:1) and myristoleic acid (C14:1) are reliable biological ligands of GLaz. These data correlate with the in vitro binding properties described for grasshopper Lazarillo (76). Second, ubiquitous co-expression of GLaz also decreased the level of lipid peroxides in frataxin knocked-down flies. Although levels of lipid peroxidation markers are highly increased in GLaz null mutants (39), it is not yet clear whether GLaz is able to bind lipid peroxidation products or, alternatively, is able to prevent lipid oxidation from occurring. However, all lipocalin family members are thought to bind small hydrophobic molecules due to their common tertiary structure (35) and some lipocalins such as human lcn-1 act directly on peroxidated lipids (80), pointing to a lipid-scavenging mechanism for GLaz. In addition, the plant ApoD ortholog knock-out also showed accumulation of other ROS (40). Third, restoration of lipid peroxides and other free radicals to normal levels by Glaz overexpression in frataxin-deficient flies appeared to be sufficient to prevent mitochondrial aconitase inactivation. Similarly, ectopic expression of hydrogen peroxide scavengers such as mitochondrial catalase or mitochondrial peroxiredoxin was able to extend lifespan of flies with reduced frataxin in the PNS. In addition, aconitase activity was restored using mitochondrial catalase in this Drosophila model of FRDA (17). Aconitase activity has also been rescued using the antioxidant compound idebenone in heart homogenates from FRDA patients (66) and by means of iron quelator molecules such as deferiprone (81) and mitochondrial ferritin (82) in frataxin-silenced mammalian cell cultures. These results would indicate that loss of aconitase activity in frataxin-deficient conditions is basically due to an imbalanced redox status in the cell and would suggest a role for frataxin in oxidative stress control. Some authors have proposed that frataxin carries out such tasks through a ferroxidase activity that detoxifies mitochondrial iron and prevents oxidative damage (83,84) or by acting as an iron sensor to negatively regulate the synthesis of Fe–S clusters (85), preventing the generation of oxidative damage from iron deposits and/or misfolded Fe–S clusters. Whereas, others have linked frataxin deficiency with impaired recruitment of antioxidant defenses (86–88).
Finally, co-expression of GLaz in the glia of frataxin-deficient flies is sufficient to produce at least a partial recovery of lifespan in both normoxic and hyperoxic conditions, as well as to cause significant improvements in locomotor activity, indicating that its modulation of lipid composition, lipid oxidation and aconitase activity do result in improvements in the physiological state of the whole organism. Human ApoD has been found to be highly upregulated in many neurological diseases or after nervous tissue damage (reviewed in 34,89); however, little is known about its abilities as a neuroprotective molecule. Beneficial effects of exogenous human ApoD have been reported after the induction of encephalitis in mice (90) and cytotoxic injury in hippocampal rat cultures (91). In both cases, the proposed mechanism implies the disposal of lipid peroxides and regulation of inflammatory responses. Remarkably, frataxin deficiency in Schwann cell cultures also triggered the upregulation of inflammatory genes (75). Therefore, we propose that, in our FRDA model, GLaz mainly exerts its protective function by ROS scavenging via quenching lipid peroxides.
Our results may suggest that initial aconitase failure is mostly due to the exposure to increased oxidative stress rather than to the lack of Fe–S clusters. We suggest that lack of frataxin alters the efficient production of these clusters. This dysregulation triggers a cascade of oxidative stress via iron-toxic precipitates, lipid peroxidation and H2O2 overproduction, that quickly would start to inactivate mitochondrial aconitase. This hypothesis would explain why antioxidant proteins are able to restore aconitase activity. However, reactivation of aconitase activity via mitochondrial catalase (17) or GLaz (this work) was not sufficient to bypass pre-adult lethality. This clearly indicates that other elements (for example, respiratory chain complexes) and/or other processes (namely, Fe–S production) are also affected and could not be rescued by these antioxidant mechanisms.
In the present study, we demonstrate for the first time that frataxin expression in glial cells is essential for normal development and adulthood in an in vivo animal model. Moreover, glia emerges as a crucial cell type for FRDA pathology. We also show that the control of lipid metabolism could represent a new strategy to improve some pathological conditions in patients. In addition, our results strongly support GLaz as a therapeutic agent in FRDA and in any neurodegenerative disorder in which redox imbalance is a major contributor to the pathophysiology.
Furthermore, our data promote the Drosophila glia model of FRDA as an excellent tool to study the mechanisms underlying nervous system degeneration in the disease as well as to test the beneficial effects of new drugs or genetic rescues for different aspects of the nervous system function.
MATERIALS AND METHODS
Drosophila stocks
The yw and w1118 strains were used as control lines in our experiments. P{da.G32-GAL4}, P{Repo-GAL4} and P{UAS-Stinger} were obtained from the Drosophila Bloomington Stock Center, and the line UDIR1 was kindly provided by John P. Phillips (University of Guelph). The stocks were routinely maintained on cornmeal agar medium at 25°C. The crosses of the GAL4 driver with the UAS responder lines were carried out at 25°C. UAS constructs were used in heterozygous configuration for all experiments. The lines UDIR1 (49) and UAS-fhIR (15) have been renamed in the text and figures as fhRNAi-1 and fhRNAi-2, respectively. fhRNAi-1 induced a strong interference with a 90% reduction of frataxin expression, whereas fhRNAi-2 produced a moderate 70% reduction compatible with a normal development. Rescue experiments were carried out first generating the stocks UAS-GLaz/CyO;fhRNAi-1/TM3 and fhRNAi-2 UAS-GLaz/CyO (in both lines, the presence of each construct was confirmed by PCR). UAS-Stinger/CyO;fhRNAi-1/TM3 was also created and used as control. A recent issue concerning the targeted gene silencing in Drosophila is the influence of off-target genes on the described phenotypes (92). Regarding FRDA fly models, the similar defects induced by both RNAi lines (fhRNAi-1 and fhRNAi-2) and the observed rescues with fh overexpression (data not shown) virtually excludes the possibility of a false-positive result.
Semi-quantitative real-time PCR
Total RNA was extracted from 75 L3 larvae or 7-day-old adult individuals, using TRIzol™ reagent (Invitrogen, Carlsbad, CA, USA), following manufacturer's instructions. Using QuantiTect Rev. Transcription Kit (Qiagen GmbH, Hilden, Germany), 1 μg RNA was converted into cDNA and then used for real-time PCR reactions with QuantiTect SYBR Green RT–PCR Kit (Qiagen GmgH) on Roche's lightcycler system. fh expression was detected using FHRTFw: 5′ACACCCTGGACGCACTGT3′ and FHRTRv: 5′CCAGGTTCACGGTTAGCAC3′ primers and GLaz expression with GLazExon1FOR: 5′ATTTGCTGGGACAGATGCCTAC3′ and GLazExon2REV: 5′TGTTGTACGGCTCAAACTGAAA3′ primers. The Ribosomal protein 49 (rp49) was used as internal control and amplified using RP49RTFw: 5′CCAAGCACTTCATCCGCCACC3′ and RP49RTRv: 5′GCGGGTGCGCTT GTTCGATCC3′ primers. The results were analyzed with the lightcycler software (version 3.5) from Roche Molecular Biochemical, gene expression levels were referred to the internal control and the relative quantification was carried out by means of the ΔCt method.
Lifespan and negative geotaxis assays
For lifespan assay, flies were collected within 24 h of eclosion and were raised at 25°C under a 12:12 h light/dark cycle and transferred to fresh food vials every 2–3 days. Hyperoxia treatment started 1 day post-eclosion and was performed by exposing flies in a glass container with a constant flux of 99.5% oxygen under a low positive pressure at 25°C. Flies were confined in groups of 25 and were transferred every day to new vials containing regular food. Climbing assays were performed as described in Botella et al. (93).
Brain histology
Adult flies were fixed with carnoy (ethanol:chloroform:acetic acid at a proportion of 6:3:1), dehydrated in ethanol and embedded in paraffin. Paraffin sections (7 μm) were analyzed under a fluorescence microscope.
For examination of adult fly brains with electron microscopy, ultrathin Epon plastic sections were post-stained with 2% uranyl acetate, followed by Reynolds' lead citrate and stabilized for transmission electron microscopy by carbon coating. Examination was done with a Zeiss EM10C/VR electron microscope at 80 kV. Glial cell material was identified by characteristically higher electron density.
Thin-layer and gas chromatography
Comparative measurement of neutral and phospholipid content was carried out using a thin-layer chromatography system from CAMAG Scientific, Inc.
Fifteen L3 male larvae or 80 male adult heads were placed in a Precellys cup containing 2.8 mm ceramic beads, frozen under liquid nitrogen and stored at −80°C until needed. Samples were then homogenized in 1 ml MQ water using a Precellys 24 homogenizer. Samples were cleaned from cellular debris by centrifugation at 2100g for 60 s. The protein content was determined from the cell homogenate by means of the Bradford assay. According to the procedure described by Bligh and Dyer (94), 500 μg of each sample were delipidated. Cellular lipids were separated on Silica Gel 60A Plates (Merck, Darmstadt, Germany) by high-performance thin-layer chromatography. Silica gel plates were run either with n-hexan:diethylether:acetic acid 80:20:1 (v/v) for neutral lipid quantification or with 2-propanol:methylacetat:chloroform:methanol:0.25% KCl 25:25:25:10:9 (v/v) for phospholipid evaluation, stained with 10% cupric sulfate (in 8% phosphoric acid) and heated to 170°C for 20 min. Quantitation was achieved by densitometric scanning with a CAMAG TLC scanner II at 366 nm.
For gas chromatography coupled with mass spectrometry (GC/MS), 20 μg from each sample were subjected to FA methyl ester derivatization (J. Ecker unpublished data). Peaks were assigned using both the retention time and fragmentation pattern. Introduction of internal standards C13:0 (tridecanoic acid) and C21:0 iso permitted quantification.
For both experiments, all individuals were grown under controlled conditions. Parental crosses were allowed to lay eggs for 12 h in order to avoid deleterious effects of limited nutritional conditions.
Enzymatic assays
Total aconitase activity was determined from whole individuals, using Bioxytech Aconitase-340™ Spectrophotometric Assay Kit (OXIS International, Inc., Portland, OR, USA).
Twenty larvae or 25 adults were placed in a tube containing 500 μl of ice-cold homogenization buffer (0.2 mm sodium citrate and 50 mm Tris–HCl, pH 7.4) and gently pounded. The resulting mashes were centrifuged twice at 800g for 10 min at 4°C, discarding the pellet every time. Supernatants were disrupted by sonication for 30 s, with 1 s interval between each pulse. To measure the aconitase activity, 100 μl of each extraction was used. The aconitase activity of each genotype was measured from four independent samples and each extraction was measured twice. The aconitase activity was finally referred to protein amount using the Bradford assay (Bio-Rad Laboratories, Inc).
Lipid peroxidation assay
The concentration of MDA was determined using the LPO-586 assay (OXIS International, Inc., Portland, OR, USA) from 60 male flies. Triplicates of each genotype and age were measured and each replica was measured twice. The procedure was conducted as described in Sanchez et al. (39), with minor modifications.
Statistical analysis
Survival data were analyzed using the Kaplan–Meier (semi-parametric log-rank) test. Locomotor, chromatographic, lipid peroxidation levels and aconitase activity data are presented as mean ± SEM of at least three replicates. Significance was determined by one-way ANOVA with post hoc Newman–Keuls multiple comparison test (***P < 0.001; **P < 0.01 and *P < 0.05). All statistical analyses were carried out with the GraphPad Prism 4.0 software.
FUNDING
This work was supported by Fondo Investigaciones Sanitarias (ISCIII06-PI060677 to M.D.M.), by the Elitenetzwerk Bayern (to J.A.N. and S.S.) and by the Ministerio de Ciencia e Innovación (BFU2008-01170 to M.D.G. and D.S.).
ACKNOWLEDGEMENTS
We would like to thank Uschi Roth, Judith R. Acebes and E. Martin-Tejedor for technical assistance and Alois Hofbauer for critically reading the manuscript. The authors thank John P. Phillips for the generous gift of the fhRNAi-1 line.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Harding A.E. Friedreich's ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain. 1981;104:589–620. doi: 10.1093/brain/104.3.589. [DOI] [PubMed] [Google Scholar]
- 2.Harding A.E. Clinical features and classification of inherited ataxias. Adv. Neurol. 1993;61:1–14. [PubMed] [Google Scholar]
- 3.Pandolfo M. Friedreich ataxia: the clinical picture. J. Neurol. 2009;256:3–8. doi: 10.1007/s00415-009-1002-3. [DOI] [PubMed] [Google Scholar]
- 4.Koeppen A.H. The hereditary ataxias. J. Neuropathol. Exp. Neurol. 1998;57:531–543. doi: 10.1097/00005072-199806000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Harding A.E., Hewer R.L. The heart disease of Friedreich's ataxia: a clinical and electrocardiographic study of 115 patients, with an analysis of serial electrocardiographic changes in 30 cases. Q. J. Med. 1983;52:489–502. [PubMed] [Google Scholar]
- 6.Delatycki M.B., Camakaris J., Brooks H., Evans-Whipp T., Thorburn D.R., Williamson R., Forrest S.M. Direct evidence that mitochondrial iron accumulation occurs in Friedreich ataxia. Ann. Neurol. 1999;45:673–675. [PubMed] [Google Scholar]
- 7.Campuzano V., Montermini L., Moltó M.D., Pianese L., Cossée M., Cavalcanti F., Monros E., Rodius F., Duclos F., Monticelli A., et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 8.Campuzano V., Montermini L., Lutz Y., Cova L., Hindelang C., Jiralerspong S., Trottier Y., Kish S.J., Faucheux B., Trouillas P., et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 1997;6:1771–1780. doi: 10.1093/hmg/6.11.1771. [DOI] [PubMed] [Google Scholar]
- 9.Rotig A., de Lonlay P., Chretien D., Foury F., Koenig M., Sidi D., Munnich A., Rustin P. Aconitase and mitochondrial iron–sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997;17:215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- 10.Lodi R., Cooper J.M., Bradley J.L., Manners D., Styles P., Taylor D.J., Schapira A.H. Deficit of in vivo mitochondrial ATP production in patients with Friedreich ataxia. Proc. Natl Acad. Sci. USA. 1999;96:11492–11495. doi: 10.1073/pnas.96.20.11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradley J.L., Blake J.C., Chamberlain S., Thomas P.K., Cooper J.M., Schapira A.H. Clinical, biochemical and molecular genetic correlations in Friedreich's ataxia. Hum. Mol. Genet. 2000;9:275–282. doi: 10.1093/hmg/9.2.275. [DOI] [PubMed] [Google Scholar]
- 12.Wong A., Yang J., Cavadini P., Gellera C., Lonnerdal B., Taroni F., Cortopassi G. The Friedreich's ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum. Mol. Genet. 1999;8:425–430. doi: 10.1093/hmg/8.3.425. [DOI] [PubMed] [Google Scholar]
- 13.Santos R., Buisson N., Knight S.A., Dancis A., Camadro J.M., Lesuisse E. Candida albicans lacking the frataxin homologue: a relevant yeast model for studying the role of frataxin. Mol. Microbiol. 2004;54:507–519. doi: 10.1111/j.1365-2958.2004.04281.x. [DOI] [PubMed] [Google Scholar]
- 14.Vázquez-Manrique R.P., González-Cabo P., Ros S., Aziz H., Baylis H.A., Palau F. Reduction of Caenorhabditis elegans frataxin increases sensitivity to oxidative stress, reduces lifespan, and causes lethality in a mitochondrial complex II mutant. FASEB J. 2006;20:172–174. doi: 10.1096/fj.05-4212fje. [DOI] [PubMed] [Google Scholar]
- 15.Llorens J.V., Navarro J.A., Martínez-Sebastián M.J., Baylies M.K., Schneuwly S., Botella J.A., Moltó M.D. Causative role of oxidative stress in a Drosophila model of Friedreich ataxia. FASEB J. 2007;21:333–344. doi: 10.1096/fj.05-5709com. [DOI] [PubMed] [Google Scholar]
- 16.Bulteau A.L., Dancis A., Gareil M., Montagne J.J., Camadro J.M., Lesuisse E. Oxidative stress and protease dysfunction in the yeast model of Friedreich ataxia. Free Radic. Biol. Med. 2007;42:1561–1570. doi: 10.1016/j.freeradbiomed.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 17.Anderson P.R., Kirby K., Orr W.C., Hilliker A.J., Phillips J.P. Hydrogen peroxide scavenging rescues frataxin deficiency in a Drosophila model of Friedreich's ataxia. Proc. Natl Acad. Sci. USA. 2008;105:611–616. doi: 10.1073/pnas.0709691105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emond M., Lepage G., Vanasse M., Pandolfo M. Increased levels of plasma malondialdehyde in Friedreich ataxia. Neurology. 2000;55:1752–1753. doi: 10.1212/wnl.55.11.1752. [DOI] [PubMed] [Google Scholar]
- 19.Schulz J.B., Dehmer T., Schöls L., Mende H., Hardt C., Vorgerd M., Bürk K., Matson W., Dichgans J., Beal M.F., Bogdanov M.B. Oxidative stress in patients with Friedreich ataxia. Neurology. 2000;55:1719–1721. doi: 10.1212/wnl.55.11.1719. [DOI] [PubMed] [Google Scholar]
- 20.Tozzi G., Nuccetelli M., Lo Bello M., Bernardini S., Bellincampi L., Ballerini S., Gaeta L.M., Casali C., Pastore A., Federici G., et al. Antioxidant enzymes in blood of patients with Friedreich's ataxia. Arch. Dis. Child. 2002;86:376–379. doi: 10.1136/adc.86.5.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burck U., Goebel H.H., Kuhlendahl H.D., Meier C., Goebel K.M. Neuromyopathy and vitamin E deficiency in man. Neuropediatrics. 1981;12:267–278. doi: 10.1055/s-2008-1059657. [DOI] [PubMed] [Google Scholar]
- 22.Watmough N.J., Bindoff L.A., Birch-Machin M.A., Jackson S., Bartlett K., Ragan C.I., Poulton J., Gardiner R.M., Sherratt H.S., Turnbull D.M. Impaired mitochondrial beta-oxidation in a patient with an abnormality of the respiratory chain. Studies in skeletal muscle mitochondria. J. Clin. Invest. 1990;85:177–184. doi: 10.1172/JCI114409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Y., Condell M., Plesken H., Edelman-Novemsky I., Ma J., Ren M., Schlame M. A Drosophila model of Barth syndrome. Proc. Natl Acad. Sci. USA. 2006;103:11584–11588. doi: 10.1073/pnas.0603242103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Picklo M.J., Montine T.J. Mitochondrial effects of lipid-derived neurotoxins. J. Alzheimers Dis. 2007;12:185–193. doi: 10.3233/jad-2007-12209. [DOI] [PubMed] [Google Scholar]
- 25.Adibhatla R.M., Hatcher J.F. Altered lipid metabolism in brain injury and disorders. Subcell. Biochem. 2008;49:241–268. doi: 10.1007/978-1-4020-8831-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adibhatla R.M., Hatcher J.F. Phospoholipase A(2), reactive oxygen species and lipid peroxidation in CNS pathologies. BMB Rep. 2008;41:560–567. doi: 10.5483/bmbrep.2008.41.8.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lessing D., Bonini N.M. Maintaining the brain: insight into human neurodegeneration from Drosophila melanogaster mutants. Nat. Rev. Genet. 2009;10:359–370. doi: 10.1038/nrg2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feki M., Belal S., Feki H., Souissi M., Frih-Ayed M., Kaabachi N., Hentati F., Ben Hamida M., Mebazaa A. Serum vitamin E and lipid-adjusted vitamin E assessment in Friedreich ataxia phenotype patients and unaffected family members. Clin. Chem. 2002;48:577–579. [PubMed] [Google Scholar]
- 29.Iorio L., De Michele G., Filla A., Di Martino L., Postiglione A., Patti L., Campanella G. Serum lipoprotein fatty acid profile in hereditary ataxias. Can. J. Neurol. Sci. 1993;20:206–209. doi: 10.1017/s0317167100047946. [DOI] [PubMed] [Google Scholar]
- 30.Walker J.L., Chamberlain S., Robinson N. Failure to detect abnormal fatty acid profiles in serum lipoproteins in Friedreich's ataxia. Ann. Neurol. 1980;8:74–76. doi: 10.1002/ana.410080113. [DOI] [PubMed] [Google Scholar]
- 31.Ross B.M., Eder K., Moszczynska A., Mamalias N., Lamarche J., Ang L., Pandolfo M., Rouleau G., Kirchgessner M., Kish S.J. Abnormal activity of membrane phospholipid synthetic enzymes in the brain of patients with Friedreich's ataxia and spinocerebellar atrophy type-1. Mov. Disord. 2000;15:294–300. doi: 10.1002/1531-8257(200003)15:2<294::aid-mds1013>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 32.Eder K., Kish S.J., Kirchgessner M., Ross B.M. Brain phospholipids and fatty acids in Friedreich's ataxia and spinocerebellar atrophy type-1. Mov. Disord. 1998;13:813–819. doi: 10.1002/mds.870130510. [DOI] [PubMed] [Google Scholar]
- 33.Puccio H., Simon D., Cossée M., Criqui-Filipe P., Tiziano F., Melki J., Hindelang C., Matyas R., Rustin P., Koenig M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe–S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001;27:181–186. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- 34.Rassart E., Bedirian A., Do Carmo S., Guinard O., Sirois J., Terrisse L., Milne R. Apolipoprotein D. Biochim. Biophys. Acta. 2000;1482:185–198. doi: 10.1016/s0167-4838(00)00162-x. [DOI] [PubMed] [Google Scholar]
- 35.Ganfornina M.D., Sanchez D., Greene L.H., Flower D.R. The lipocalin protein family: protein sequence, structure and relationships to the calycin superfamily. In: Akerström B., Borregaard N., Flower D.R., Salier J.P., editors. Lipocalins. Georgetown, TX: Landes Bioscience; 2006. pp. 17–27. [Google Scholar]
- 36.Vogt M., Skerra A. Bacterially produced apolipoprotein D binds progesterone and arachidonic acid, but not bilirubin or E-3M2H. J. Mol. Recognit. 2001;14:79–86. doi: 10.1002/1099-1352(200101/02)14:1<79::AID-JMR521>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 37.Sánchez D., Ganfornina M.D., Torres-Schumann S., Speese S.D., Lora J.M., Bastiani M.J. Characterization of two novel lipocalins expressed in the Drosophila embryonic nervous system. Int. J. Dev. Biol. 2000;44:349–359. [PubMed] [Google Scholar]
- 38.Navarro A., Del Valle E., Tolivia J. Differential expression of apolipoprotein D in human astroglial and oligodendroglial cells. J. Histochem. Cytochem. 2004;52:1031–1036. doi: 10.1369/jhc.3A6213.2004. [DOI] [PubMed] [Google Scholar]
- 39.Sanchez D., López-Arias B., Torroja L., Canal I., Wang X., Bastiani M.J., Ganfornina M.D. Loss of glial lazarillo, a homolog of apolipoprotein D, reduces lifespan and stress resistance in Drosophila. Curr. Biol. 2006;16:680–686. doi: 10.1016/j.cub.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 40.Charron J.B., Ouellet F., Houde M., Sarhan F. The plant apolipoprotein D ortholog protects Arabidopsis against oxidative stress. BMC Plant Biol. 2008;8:86–98. doi: 10.1186/1471-2229-8-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ganfornina M.D., Do Carmo S., Lora J.M., Torres-Schumann S., Vogel M., Allhorn M., González C., Bastiani M.J., Rassart E., Sanchez D. Apolipoprotein D is involved in the mechanisms regulating protection from oxidative stress. Aging Cell. 2008;7:506–515. doi: 10.1111/j.1474-9726.2008.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walker D.W., Muffat J., Rundel C., Benzer S. Overexpression of a Drosophila homolog of apolipoprotein D leads to increased stress resistance and extended lifespan. Curr. Biol. 2006;16:674–679. doi: 10.1016/j.cub.2006.01.057. [DOI] [PubMed] [Google Scholar]
- 43.Muffat J., Walker D.W., Benzer S. Human ApoD, an apolipoprotein up-regulated in neurodegenerative diseases, extends lifespan and increases stress resistance in Drosophila. Proc. Natl Acad. Sci. USA. 2008;105:7088–7093. doi: 10.1073/pnas.0800896105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hughes J.T., Brownell B., Hewer R.L. The peripheral sensory pathway in Friedreich's ataxia. An examination by light and electron microscopy of the posterior nerve roots, posterior root ganglia, and peripheral sensory nerves in cases of Friedreich's ataxia. Brain. 1968;91:803–818. doi: 10.1093/brain/91.4.803. [DOI] [PubMed] [Google Scholar]
- 45.Simon D., Seznec H., Gansmuller A., Carelle N., Weber P., Metzger D., Rustin P., Koenig M., Puccio H. Friedreich ataxia mouse models with progressive cerebellar and sensory ataxia reveal autophagic neurodegeneration in dorsal root ganglia. J. Neurosci. 2004;24:1987–1995. doi: 10.1523/JNEUROSCI.4549-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Al-Mahdawi S., Pinto R.M., Varshney D., Lawrence L., Lowrie M.B., Hughes S., Webster Z., Blake J., Cooper J.M., King R., Pook M.A. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics. 2006;88:580–590. doi: 10.1016/j.ygeno.2006.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sepp K.J., Schulte J., Auld V.J. Peripheral glia direct axon guidance across the CNS/PNS transition zone. Dev. Biol. 2001;238:47–63. doi: 10.1006/dbio.2001.0411. [DOI] [PubMed] [Google Scholar]
- 48.Campbell G., Göring H., Lin T., Spana E., Andersson S., Doe C.Q., Tomlinson A. RK2, a glial-specific homeodomain protein required for embryonic nerve cord condensation and viability in Drosophila. Development. 1994;120:2957–2966. doi: 10.1242/dev.120.10.2957. [DOI] [PubMed] [Google Scholar]
- 49.Anderson P.R., Kirby K., Hilliker A.J., Phillips J.P. RNAi-mediated suppression of the mitochondrial iron chaperone, frataxin, in Drosophila. Hum. Mol. Genet. 2005;14:3397–3405. doi: 10.1093/hmg/ddi367. [DOI] [PubMed] [Google Scholar]
- 50.Gruenewald C., Botella J.A., Bayersdorfer F., Navarro J.A., Schneuwly S. Hyperoxia-induced neurodegeneration as a tool to identify neuroprotective genes in Drosophila melanogaster. Free Radic. Biol. Med. 2009;46:1668–1676. doi: 10.1016/j.freeradbiomed.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 51.De Martino C., Natali P.G., Bruni C.B., Accinni L. Influence of plastic embedding media on staining and morphology of lipid bodies. A light and ultrastructural study. Histochemie. 1968;16:350–360. doi: 10.1007/BF00306358. [DOI] [PubMed] [Google Scholar]
- 52.Marmolino D., Acquaviva F., Pinelli M., Monticelli A., Castaldo I., Filla A., Cocozza S. PPAR-gamma agonist azelaoyl PAF increases frataxin protein and mRNA expression. New implications for the Friedreich's ataxia therapy. Cerebellum. 2009;8:98–103. doi: 10.1007/s12311-008-0087-z. [DOI] [PubMed] [Google Scholar]
- 53.Shapcott D., Melancon S., Butterworth R.F., Khoury K., Collu R., Breton G., Geoffroy G., Lemieux B., Barbeau A. Glucose and insulin metabolism in Friedreich's ataxia. Can. J. Neurol. Sci. 1976;3:361–364. doi: 10.1017/s0317167100025609. [DOI] [PubMed] [Google Scholar]
- 54.Petersen K.F., Dufour S., Befroy D., Garcia R., Shulman G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Befroy D.E., Petersen K.F., Dufour S., Mason G.F., de Graaf R.A., Rothman D.L., Shulman G.I. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes. 2007;56:1376–1381. doi: 10.2337/db06-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vankoningsloo S., Piens M., Lecocq C., Gilson A., De Pauw A., Renard P., Demazy C., Houbion A., Raes M., Arnould T.J. Mitochondrial dysfunction induces triglyceride accumulation in 3T3-L1 cells: role of fatty acid beta-oxidation and glucose. Lipid Res. 2005;46:1133–1149. doi: 10.1194/jlr.M400464-JLR200. [DOI] [PubMed] [Google Scholar]
- 57.Zahavi M., Tahori A.S. Citric acid accumulation with age in houseflies and other Diptera. J. Insect Physiol. 1965;11:811–816. doi: 10.1016/0022-1910(65)90160-5. [DOI] [PubMed] [Google Scholar]
- 58.Tong W.H., Rouault T.A. Metabolic regulation of citrate and iron by aconitases: role of iron–sulfur cluster biogenesis. Biometals. 2007;20:549–564. doi: 10.1007/s10534-006-9047-6. [DOI] [PubMed] [Google Scholar]
- 59.Coppola G., Marmolino D., Lu D., Wang Q., Cnop M., Rai M., Acquaviva F., Cocozza S., Pandolfo M., Geschwind D.H. Functional genomic analysis of frataxin deficiency reveals tissue-specific alterations and identifies the PPAR {gamma} pathway as a therapeutic target in Friedreich's ataxia. Hum. Mol. Genet. 2009;18:2452–2461. doi: 10.1093/hmg/ddp183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sutak R., Xu X., Whitnall M., Kashem M.A., Vyoral D., Richardson D.R. Proteomic analysis of hearts from frataxin knockout mice: marked rearrangement of energy metabolism, a response to cellular stress and altered expression of proteins involved in cell structure, motility and metabolism. Proteomics. 2008;8:1731–1741. doi: 10.1002/pmic.200701049. [DOI] [PubMed] [Google Scholar]
- 61.Nicolson G.L. Metabolic syndrome and mitochondrial function: molecular replacement and antioxidant supplements to prevent membrane peroxidation and restore mitochondrial function. J. Cell. Biochem. 2007;100:1352–1369. doi: 10.1002/jcb.21247. [DOI] [PubMed] [Google Scholar]
- 62.Busi M.V., Maliandi M.V., Valdez H., Clemente M., Zabaleta E.J., Araya A., Gomez-Casati D.F. Deficiency of Arabidopsis thaliana frataxin alters activity of mitochondrial Fe–S proteins and induces oxidative stress. Plant J. 2006;48:873–882. doi: 10.1111/j.1365-313X.2006.02923.x. [DOI] [PubMed] [Google Scholar]
- 63.Condò I., Ventura N., Malisan F., Tomassini B., Testi R. A pool of extramitochondrial frataxin that promotes cell survival. J. Biol. Chem. 2006;281:16750–16756. doi: 10.1074/jbc.M511960200. [DOI] [PubMed] [Google Scholar]
- 64.Napoli E., Taroni F., Cortopassi G.A. Frataxin, iron–sulfur clusters, heme, ROS, and aging. Antioxid. Redox Signal. 2006;8:506–516. doi: 10.1089/ars.2006.8.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Irazusta V., Moreno-Cermeño A., Cabiscol E., Ros J., Tamarit J. Major targets of iron-induced protein oxidative damage in frataxin-deficient yeasts are magnesium-binding proteins. Free Radic. Biol. Med. 2008;44:1712–1723. doi: 10.1016/j.freeradbiomed.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 66.Rustin P., Von Kleist-Retzow J.C., Chantrel-Groussard K., Sidi D., Munnich A., Rötig A. Effect of idebenone on cardiomyopathy in Friedreich's ataxia: a preliminary study. Lancet. 1999;354:477–479. doi: 10.1016/S0140-6736(99)01341-0. [DOI] [PubMed] [Google Scholar]
- 67.Shoichet S.A., Bäumer A.T., Stamenkovic D., Sauer H., Pfeiffer A.F., Kahn C.R., Müller-Wieland D., Richter C., Ristow M. Frataxin promotes antioxidant defense in a thiol-dependent manner resulting in diminished malignant transformation in vitro. Hum. Mol. Genet. 2002;11:815–821. doi: 10.1093/hmg/11.7.815. [DOI] [PubMed] [Google Scholar]
- 68.Runko A.P., Griswold A.J., Min K.T. Overexpression of frataxin in the mitochondria increases resistance to oxidative stress and extends lifespan in Drosophila. FEBS Lett. 2008;582:715–719. doi: 10.1016/j.febslet.2008.01.046. [DOI] [PubMed] [Google Scholar]
- 69.Castilho R.F., Meinicke A.R., Almeida A.M., Hermes-Lima M., Vercesi A.E. Oxidative damage of mitochondria induced by Fe(II)citrate is potentiated by Ca2+ and includes lipid peroxidation and alterations in membrane proteins. Arch. Biochem. Biophys. 1994;308:158–164. doi: 10.1006/abbi.1994.1022. [DOI] [PubMed] [Google Scholar]
- 70.Castilho R.F., Meinicke A.R., Vercesi A.E., Hermes-Lima M. The role of Fe(III) in Fe(II)-citrate mediated peroxidation of mitochondrial membrane lipids. Mol. Cell. Biochem. 1999;196:163–168. [PubMed] [Google Scholar]
- 71.Liochev S.I. The mechanism of ‘Fenton-like’ reactions and their importance for biological systems. A biologist's view. Met. Ions Biol. Syst. 1999;36:1–39. [PubMed] [Google Scholar]
- 72.Chen O.S., Hemenway S., Kaplan J. Genetic analysis of iron citrate toxicity in yeast: implications for mammalian iron homeostasis. Proc. Natl Acad. Sci. USA. 2002;99:16922–16927. doi: 10.1073/pnas.232392299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Babcock M., de Silva D., Oaks R., Davis-Kaplan S., Jiralerspong S., Montermini L., Pandolfo M., Kaplan J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science. 1997;276:1709–1712. doi: 10.1126/science.276.5319.1709. [DOI] [PubMed] [Google Scholar]
- 74.Pandolfo M., Koenig M. Friedreich's ataxia. In: Wells R.D., Warren S.T., editors. Genetic Instabilities and Hereditary Neurological Diseases. Texas A&M University, Houston, USA: Academic Press; 1998. pp. 371–398. [Google Scholar]
- 75.Lu C., Schoenfeld R., Shan Y., Tsai C., Hammock B., Cortopassi G. Frataxin deficiency induces Schwann cell inflammation and death. Biochim. Biophys. Acta. 2009;1792:1052–1061. doi: 10.1016/j.bbadis.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sanchez D., Ortega-Cubero S., Akerström B., Herrera M., Bastiani M.J., Ganfornina M.D. Molecular interactions of the neuronal GPI-anchored lipocalin Lazarillo. J. Mol. Recognit. 2008;21:313–323. doi: 10.1002/jmr.902. [DOI] [PubMed] [Google Scholar]
- 77.Do Carmo S., Fournier D., Mounier C., Rassart E. Human apolipoprotein D overexpression in transgenic mice induces insulin resistance and alters lipid metabolism. Am. J. Physiol. Endocrinol. Metab. 2009;296:E802–E811. doi: 10.1152/ajpendo.90725.2008. [DOI] [PubMed] [Google Scholar]
- 78.Hull-Thompson J., Muffat J., Sanchez D., Walker D.W., Benzer S., Ganfornina M.D., Jasper H. Control of metabolic homeostasis by stress signaling is mediated by the lipocalin NLaz. PLoS Genet. 2009;5:e1000460. doi: 10.1371/journal.pgen.1000460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thomas E.A., Yao J.K. Clozapine specifically alters the arachidonic acid pathway in mice lacking apolipoprotein D. Schizophr. Res. 2007;89:147–153. doi: 10.1016/j.schres.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 80.Lechner M., Wojnar P., Redl B. Human tear lipocalin acts as an oxidative-stress induced scavenger of potentially harmful lipid peroxidation products in a cell culture system. Biochem. J. 2001;356:129–135. doi: 10.1042/0264-6021:3560129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kakhlon O., Manning H., Breuer W., Melamed-Book N., Lu C., Cortopassi G., Munnich A., Cabantchik Z.I. Cell functions impaired by frataxin deficiency are restored by drug-mediated iron relocation. Blood. 2008;112:5219–5227. doi: 10.1182/blood-2008-06-161919. [DOI] [PubMed] [Google Scholar]
- 82.Zanella I., Derosas M., Corrado M., Cocco E., Cavadini P., Biasiotto G., Poli M., Verardi R., Arosio P. The effects of frataxin silencing in HeLa cells are rescued by the expression of human mitochondrial ferritin. Biochim. Biophys. Acta. 2008;1782:90–98. doi: 10.1016/j.bbadis.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 83.O'Neill H.A., Gakh O., Park S., Cui J., Mooney S.M., Sampson M., Ferreira G.C., Isaya G. Assembly of human frataxin is a mechanism for detoxifying redox-active iron. Biochemistry. 2005;44:537–545. doi: 10.1021/bi048459j. [DOI] [PubMed] [Google Scholar]
- 84.Gakh O., Park S., Liu G., Macomber L., Imlay J.A., Ferreira G.C., Isaya G. Mitochondrial iron detoxification is a primary function of frataxin that limits oxidative damage and preserves cell longevity. Hum. Mol. Genet. 2006;15:467–479. doi: 10.1093/hmg/ddi461. [DOI] [PubMed] [Google Scholar]
- 85.Adinolfi S., Iannuzzi C., Prischi F., Pastore C., Iametti S., Martin S.R., Bonomi F., Pastore A. Bacterial frataxin CyaY is the gatekeeper of iron–sulfur cluster formation catalyzed by IscS. Nat. Struct. Mol. Biol. 2009;16:390–396. doi: 10.1038/nsmb.1579. [DOI] [PubMed] [Google Scholar]
- 86.Chantrel-Groussard K., Geromel V., Puccio H., Koenig M., Munnich A., Rötig A., Rustin P. Disabled early recruitment of antioxidant defenses in Friedreich's ataxia. Hum. Mol. Genet. 2001;10:2061–2067. doi: 10.1093/hmg/10.19.2061. [DOI] [PubMed] [Google Scholar]
- 87.Jiralerspong S., Ge B., Hudson T.J., Pandolfo M. Manganese superoxide dismutase induction by iron is impaired in Friedreich ataxia cells. FEBS Lett. 2001;509:101–105. doi: 10.1016/s0014-5793(01)03140-4. [DOI] [PubMed] [Google Scholar]
- 88.Paupe V., Dassa E.P., Goncalves S., Auchère F., Lönn M., Holmgren A., Rustin P. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS ONE. 2009;4:e4253. doi: 10.1371/journal.pone.0004253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Van Dijk W., Do Carmo S., Rassart E., Dahlbäck B., Sodetz J.M. The plasma lipocalins a1-acid glycoprotein, apolipoprotein D, apolipoprotein M and complement protein C8g. In: Akerström B., Borregaard N., Flower D.R., Salier J.P., editors. Lipocalins. Georgetown, TX: Landes Bioscience; 2006. pp. 140–166. [Google Scholar]
- 90.Do Carmo S., Jacomy H., Talbot P.J., Rassart E. Neuroprotective effect of apolipoprotein D against human coronavirus OC43-induced encephalitis in mice. J. Neurosci. 2008;28:10330–10338. doi: 10.1523/JNEUROSCI.2644-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.He X., Jittiwat J., Kim J.H., Jenner A.M., Farooqui A.A., Patel S.C., Ong W.Y. Apolipoprotein D modulates F2-isoprostane and 7-ketocholesterol formation and has a neuroprotective effect on organotypic hippocampal cultures after kainate-induced excitotoxic injury. Neurosci. Lett. 2009;455:183–186. doi: 10.1016/j.neulet.2009.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Perrimon N., Mathey-Prevot B. Matter arising: off-targets and genome-scale RNAi screens in Drosophila. Fly (Austin) 2007;1:1–5. doi: 10.4161/fly.3601. [DOI] [PubMed] [Google Scholar]
- 93.Botella J.A., Ulschmid J.K., Gruenewald C., Moehle C., Kretzschmar D., Becker K., Schneuwly S. The Drosophila carbonyl reductase sniffer prevents oxidative stress-induced neurodegeneration. Curr. Biol. 2004;14:782–786. doi: 10.1016/j.cub.2004.04.036. [DOI] [PubMed] [Google Scholar]
- 94.Bligh E.G., Dyer W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]