

SUMMARY
Movement of chromosome sites within interphase cells is critical for numerous pathways including RNA transcription and genome organization. Yet, a mechanism for reorganizing chromatin in response to these events had not been reported. Here, we delineate a molecular chaperone dependent pathway for relocating activated gene loci in yeast. Our presented data support a model in which a two-authentication system mobilizes a gene promoter through a dynamic network of polymeric nuclear actin. Transcription factor-dependent nucleation of a myosin motor propels the gene locus through the actin matrix and fidelity of the actin association was ensured by ARP-containing chromatin remodelers. Motor activity of nuclear myosin was dependent on the Hsp90 chaperone. Hsp90 further contributed by biasing the remodeler-actin interaction towards nucleosomes with the non-canonical histone H2A.Z thereby focusing the pathway on select sites such as transcriptionally active genes. Together, the system provides a rapid and effective means to broadly yet selectively mobilize chromatin sites.
Graphical Abstract
eTOC
Chromosome reorganization is a central process critical for development and to maintain normal cell homeostasis. Here, Wang et al. report on a pathway able to direct the select movement of chromatin loci in interphase cells using the concerted activities of nuclear molecular chaperones, actin, myosin, and chromatin remodelers.
INTRODUCTION
Spatial genome organization is a conserved feature of cells (Cremer and Cremer, 2001; Misteli, 2007). While early light microscopy work led to the concept of “chromosome territories” (Boveri, 1909), electron microscopy as well as fluorescence in situ hybridization experiments validated the existence of structured chromatin fibers with non-random, higher-order chromosome organization (Comings, 1968; Wischnitzer, 1973; Lichter et al., 1988). More recent high-throughput molecular techniques (e.g., chromosome conformation capture assays) have shown that eukaryotic genomes are partitioned into distinct compartments that are further divided into topologically associated domains (TADs) as well as smaller self-associating regions (Lieberman-Aiden et al., 2009; Dixon et al., 2012; Nora et al., 2012). In Drosophila, genomes are largely unstructured prior to zygotic chromosome activation and although early expressed genes can serve as nucleation sites for TAD boundaries, boundary formation is not dependent upon transcription but rather relies upon a select transcription factor (Hug et al., 2017). Notably, the cell-type specific organization of a genome is mitigated during mitosis and is recurrently structured during the G1 phase in differentiated cells (Naumova et al., 2013; Dixon et al., 2015; Flyamer et al., 2017). In addition to transcription factors, epigenetic marks (i.e., mitotic bookmarking features) are believed to contribute to genome reorganization after cell division though the mechanistic contributions are unclear (Oomen and Dekker, 2017). Despite the prevalence and conservation of genome organization, the fundamental steps driving the process, including the directed motion of chromosome sites in interphase cells, had not been described.
Interphase chromatin displays two types of conserved movements—regular short-range fluctuations (<0.2 μm) and less frequent long-range transitions (>0.5 μm) (Heun et al., 2001; Levi et al., 2005). While constrained diffusion can explain the smaller steps (Marshall et al., 1997), a pathway mediating the longer movements under normal physiological conditions had yet to be reported. Prior studies showed it is an active process as the larger transitions are ATP-dependent and vary with the cell cycle (Dion and Gasser, 2013). In addition to genome organization, central pathways including DNA repair and transcription correlate with and depend on long-range movements (Soutoglou and Misteli, 2007). For instance, chromatin mobility increases near a double stranded break and repair efficiency by homologous recombination correlates with the motion (Neumann et al., 2012; Saad et al., 2014). Recent reports demonstrated that double-stranded DNA breaks within heterochromatin are moved long distances being clustered at the nuclear periphery utilizing Arp2/3-dependent actin filaments (Caridi et al., 2018; Schrank et al., 2018). Comparably, many inducible genes including INO1, GAL1, and HXK1 translocate from the inner nucleoplasm clustering at the nuclear envelope following activation (Brickner and Walter, 2004; Taddei et al., 2006). Nevertheless, how chromatin architecture is reconfigured during a transcriptional response is poorly understood.
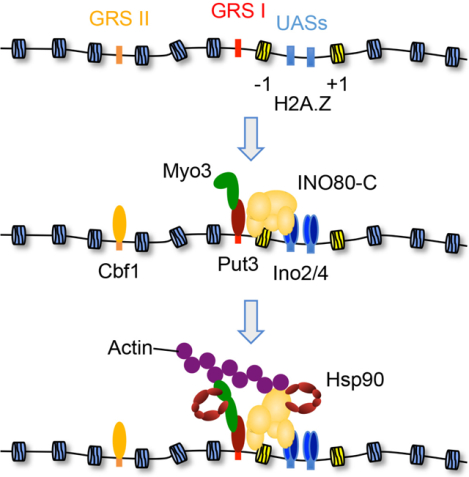
To investigate chromosome motion within interphase cells, we employed an experimentally tractable system in budding yeast where the INO1 locus has been tagged with nearby Lac operator repeats that are directly visualized by expressing a LacI-GFP fusion (Brickner and Walter, 2004). Following withdrawal of the carbocyclic sugar inositol, the inactive INO1 gene shifts from the inner nucleoplasm to the nuclear periphery where it is transcriptionally upregulated (Brickner and Walter, 2004). While a pathway mediating the movement had yet to be delineated, prior studies showed that INO1 mobility is reliant on one of two nearby DNA elements termed Gene Recruitment Sequences (GRSs) I and II (i.e., DNA zip codes) that are bound by the Put3 and Cbf1 transcription factors, respectively (Randise-Hinchliff and Brickner, 2016). The Put3-controlled GRS I is the dominant DNA zip code in the wild type promoter (GRS II is used if GRS I is mutated) and can direct clustering of INO1 at the nuclear periphery with other DNA loci having a GRS I element (Brickner et al., 2012). Yet, how a DNA zip code-bound transcription factor directly promotes chromosome motion was unclear.
Besides the DNA zip codes, the histone variant H2A.Z contributes to the nuclear positioning of INO1. H2A.Z, which is inserted by the SWR-C nucleosome remodeler, maintains INO1 at the nuclear envelope following removal of the stimuli (i.e., memory of nuclear position) (Brickner et al., 2007; Brickner et al., 2012). The INO80-C remodeler, on the other hand, catalyzes the reverse reaction by swapping in H2A, and also modulates INO1 transcription (Ebbert et al., 1999). While an impact of INO80-C in INO1 motion had yet to be reported, tethering INO80-C to an ectopic DNA site is sufficient to mobilize that region to the nuclear periphery (Neumann et al., 2012). Significantly, both INO80-C and SWR-C have been implicated in the movement of chromatin associated with a variety of nuclear pathways including transcription and DNA repair leading to the proposal that SWR-C and INO80-C foster chromosome mobility by enhancing the flexibility of the chromatin fiber (Neumann et al., 2012; Gerhold et al., 2015). Yet, increased chromatin elasticity alone would be insufficient for longer movements (>0.5 μm), as H2A.Z deposition by SWR-C or relocation of a few histone octamers by INO80-C would only provide very local changes to the fiber. If not chromatin flexibility, how might SWR-C and INO80-C contribute to long-range chromosome motion?
We were drawn to the chromosome motion process by our previous findings that the p23 (SBA1) molecular chaperone is genetically linked to genes encoding subunits of both INO80-C and SWR-C along with H2A.Z (Echtenkamp et al., 2011). Furthermore, cells with altered p23 levels have phenotypes similar to INO80-C and SWR-C mutants including sensitivities to DNA mutagens and chromosome instability, which led to an early naming of p23 as Cst18 (Chromosome stability 18) (Echtenkamp et al., 2011; Ouspenski et al., 1999). In addition to p23, its partner chaperone Hsp90 has been physically and genetically linked to these remodelers (Echtenkamp and Freeman, 2012). Hence, we checked whether p23 and Hsp90 influence chromosome motion using the INO1 GFP-marked system (Brickner and Walter, 2004).
RESULTS
Movement of INO1 is chaperone-dependent
In the presence of inositol the INO1 promoter is inactive and predominantly within the nucleoplasm but shifts to the periphery following activation triggered by inositol starvation (Figure 1A) (Brickner and Walter, 2004). While the onset of motion is stochastic during the first hour of inositol withdrawal (Figure S1A) (Brickner and Walter, 2004), once it begins it is rapid with a speed of ~100 nm/sec requiring ~5 sec to move half way across a yeast nuclei (Figure S1B and Videos S1 and S2). Perhaps notably, the rate of motion is consistent with motor-driven cargo transport in the cytosol (Ross et al., 2008; Agarwal and Zaidel-Bar, 2018). Yet in p23 null (p23Δ) cells or upon inhibition of Hsp90 with Radicicol no change in positioning was observed (Figure 1). If Radicicol was washed out, INO1 shifted to the periphery indicating an active chaperone role (Figure 1C). Furthermore, Radicicol also impaired the movement of the GAL1/GAL10 locus in reaction to galactose addition (Figure S2) thereby demonstrating a common chaperone-dependence to move an activated gene locus. Intriguingly, we also observed fast motion of the INO1 GFP-marked site in a newly formed daughter cell in the parental yeast but not in p23Δ yeast suggesting that a comparable pathway is used to reorganize the genome following cell division (Videos S3 and S4).
Figure 1. Long-range motion of the INO1 locus to the nuclear periphery is p23 and Hsp90 dependent.

(A) Representative images of the GFP-marked INO1 locus, which is visualized using an integrated array of lac binding sites near INO1 that are bound by GFP-LacI, in the presence of inositol (+) and absence (−). The outer and nuclear membranes are marked by ER05-mCherry (Randise-Hinchliff et al., 2016). (B) The reliance on p23 for inositol-dependent movement of INO1 was checked using p23 null (p23Δ) and parental yeast cells. (C) The impact of Hsp90 on INO1 motion was tested in the absence (DMSO), presence of an Hsp90 inhibitor (Radicicol), or after removal of the drug (Withdrawal). All INO1 motion data represent averages of 3 independent experiments with at least 50 counted cells in each trial and the error bars represent the SEM.
To identify chaperone targets within a particular pathway we typically focus on factors overlapping with our established p23 interactome (Echtenkamp et al., 2011). However, a path for moving an activated gene promoter was not known. Nevertheless, it is understood that INO1 is controlled by several transcription factors including Ino2 and Ino4 for promoter activity and Put3 and Cbf1 for nuclear positioning (Donahue and Henry, 1981; Randise-Hinchliff and Brickner, 2016). As p23 regulates the DNA occupancy of numerous transcription factors (Zelin et al., 2012), we checked whether this was occurring at INO1. Yet, under inducing conditions (i.e., inositol starvation), Ino2, Ino4, Put3, and Cbf1 associated equivalently with the INO1 locus in parental and p23Δ cells (Figure S3A). Besides these proteins, the noncanonical histone H2A.Z is involved with INO1.
H2A.Z-associated chromatin remodelers are required for INO1 motion
Minimally, H2A.Z retains INO1 at the periphery following deactivation of the promoter (Brickner et al., 2007). Curiously, H2A.Z is not needed to initially target INO1 to the periphery, yet the SWR-C motor protein Swr1 is necessary to move an ectopic chromatin site tagged with an INO1 DNA zip code (Brickner et al., 2007; Light et al., 2010). However, whether the entire SWR-C complex or H2A.Z deposition is needed was not clear since just tethering the SWR-C subunit Arp6 to DNA using LexA binding sites and LexA-Arp6 is sufficient to localize unrelated DNA to the periphery (Yoshida et al., 2010). Whether Arp6 has a role in INO1 nuclear localization had not been determined.
We checked the impact of SWR-C on INO1 nuclear positioning using swr1Δ cells along with isw1Δ, which encodes the motor for ISW1 remodelers. Loss of the SWR-C motor Swr1 blocked the transition of INO1 to the nuclear periphery (Figure 2A). The immobility did not result from a pleotropic disruption in the remodeling network since translocation was similar to WT in isw1Δ yeast (Figure 2A). While the role of SWR-C was tested using gene knockouts, we were unable to assess the other H2A.Z remodeler since INO80-C is essential in this yeast background. To check the influence of INO80-C on active chromosome motion, we engineered an INO80-C anchor-away strain to trigger the translocation of Ino80 to the cytosol (Xue et al., 2017). In brief, anchor-away exploits the Rapamycin-dependent high-affinity interaction between FKBP12 and the FKBP Rapamycin-Binding domain (FRB) of TOR1 along with the massive flow of ribosomal proteins from the nucleus to the cytoplasm (Haruki et al., 2008). To control the nuclear locale of INO80-C we expressed the motor protein Ino80 as an FRB fusion and found that in the presence of rapamycin INO1 was non-mobile (Figure 2B). Of note, the Ino80-FRB fusion did not cause fitness defects (Figure S3B). Overall, INO80-C and SWR-C are needed to mobilize INO1 in response to inositol levels.
Figure 2. Motion of INO1 to the nuclear periphery is dependent upon SWR-C and INO80-C but not the ISW1 remodeler.
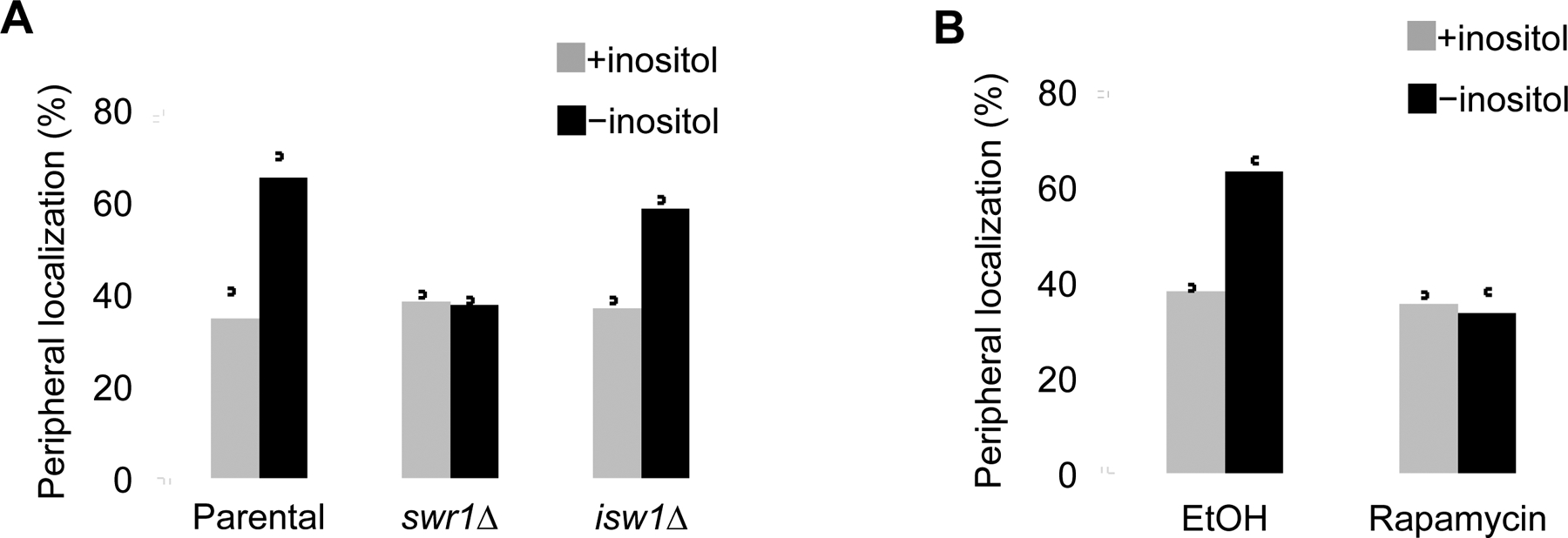
(A) INO1 position in parental, swr1Δ, or isw1Δ yeast was checked in the presence (+) and absence (−) of inositol (n = 3). (B) INO80-C was translocated to the cytosol utilizing the anchor away system (Rapamycin) or left unperturbed (EtOH) and the nuclear locale of INO1 was checked, as marked (n = 3).
Chaperones modulate the DNA binding activities of SWR-C and INO80-C
Previously we showed that p23 and Hsp90 regulate the RSC nucleosome remodeler (Echtenkamp et al., 2016). Interestingly, INO80-C and SWR-C occupancies at the INO1 promoter were enhanced in p23Δ cells (Figure 3A) suggesting that p23 affects these remodelers as it does RSC (Echtenkamp et al., 2016). Comparable to RSC the chaperones physically associated with the remodelers (Figure S3C), localized to the site of remodeler action at INO1 (Figure S3D), but did not modify the assembly of the complexes (Figures S3E). Next, we utilized biochemical tests to directly assess the impact of Hsp90 and p23 on the activities of INO80-C and SWR-C. Purified INO80-C and SWR-C bound DNA with high affinity (Kd ~8 nM) nonetheless both p23 and Hsp90 destabilized the assemblies (Figure 3B). In contrast to RSC, the chaperones cooperatively dissociated these complexes from DNA (Echtenkamp et al., 2016). Yet, Hsp90 did not significantly affect nucleosome remodeling by INO80-C or histone dimer exchange by SWR-C, p23 only had a mild impact, and the combination of both chaperones did not have a significant effect (Figure S3F and Table S1). Whether it is the capacity of the chaperones to dissociate the remodelers from DNA that regulates INO1 motion is not clear, since a means by which the remodelers contribute to INO1 movement had yet to be determined. Given the influence of the SWR-C subunit Arp6 in DNA localization, the general capacity of Actin Related Proteins (ARPs) to bind actin, and the established roles of actin in cellular transport events (Yoshida et al., 2010; Pollard, 2016), a relationship between the remodelers and actin was a plausible route.
Figure 3. Hsp90 and p23 modulate the DNA interactions of INO80-C and SWR-C in vivo and in vitro.
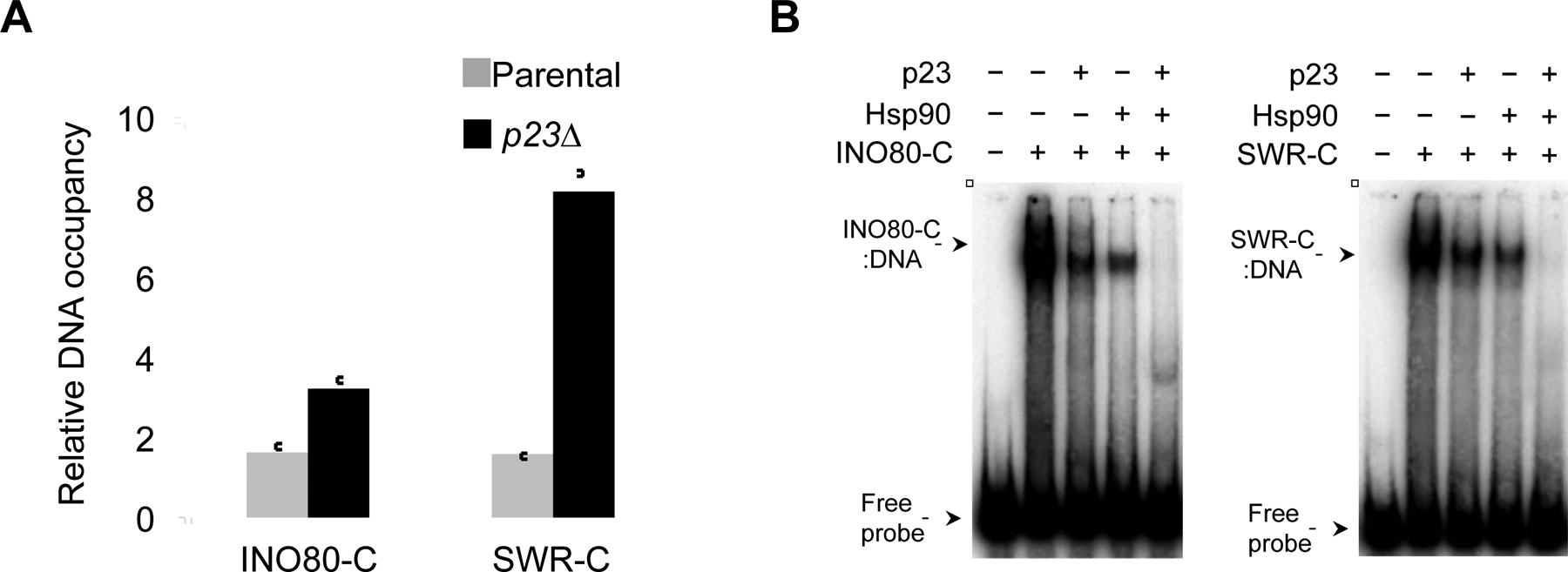
(A) ChIP was used to measure the inositol-dependent association of SWR-C (Swr1-TAP) or INO80-C (Ino80-TAP) at the INO1 promoter after 1 h of inositol withdrawal at 30°C in the presence (parent al) or absence of p23 (p23Δ). The data represent the fold enrichment of the indicated remodeler triggered by inositol withdrawal (n = 3). (B) INO80-C and SWR-C DNA binding activities were monitored by EMSA using purified complexes and radiolabeled INO1 promoter DNA, as indicated. The influence of p23 (1 μM) or Hsp90 (1 μM) on DNA binding was determined, as marked (n = 3). The nuclear concentration of Hsp90 is 1 μM and p23 is 4 μM (Echtenkamp et al., 2016).
Remodeler ARP-subunits are required for INO1 mobility
Since the discovery of actin in the nucleus, its role has been sought (Clark and Merriam, 1977). Actin trafficking is controlled by Importin-9 and Exportin-6 and blockage leads to general defects in transcription and chromosome organization (Stuven et al., 2003; Dopie et al., 2012). Indeed actin monomers are subunits of select chromatin modifiers including INO80-C and SWR-C but a role or even the existence of nuclear filamentous actin (f-actin) has been debated, since visualizing it within the nucleoplasm is difficult. Still, studies have shown that certain signals including DNA damage or serum-stimulation lead to polymerized actin in the nucleus (Plessner et al., 2015; Belin et al., 2015). Recent reports show that nuclear f-actin transiently forms at the mitotic exit to expand the nucleus and potentially promote chromatin reorganization and that Arp2/3-dependent f-actin is required for long-range clustering of a sub-set of double stranded DNA breaks (Caridi et al., 2018; Schrank et al., 2018; Belin et al., 2015; Baarlink et al., 2017). While it is becoming evident that f-actin exists in the nucleus (de Lanerolle, 2012; Belin and Mullins, 2013), how these polymers might physically connect to non-damaged chromosome sites was not understood.
As a starting point to investigate a potential connection between the remodelers and actin we checked the influence of the remodeler ARP subunits in INO1 motion. SWR-C has Arp4 and Arp6 while INO80-C has Arp4, Arp5, and Arp8 (Clapier and Cairns, 2009). The essential nature of ARP4 and ARP5 in our parental yeast precluded gene knockouts; however, deletion of ARP8 results in the loss of both Arp8 and Arp4 and deletion of IES6 triggers the loss of the Arp5 module (Watanabe et al., 2015). Thus, we tested arp6Δ, ies6Δ, and arp8Δ yeast and found all three to be required for INO1 motion (Figure 4A). Next, we inhibited actin polymerization using Latrunculin-A and observed that INO1 no longer moved to the periphery (Figure 4B). Our results show that actin polymerization is a conserved feature of chromosome mobility since it is needed for the directed movement of HSP70 to nuclear speckles in mammalian cells (Khanna et al., 2014). However, it’s been suggested that cytosolic f-actin modulates chromosome motion in conjunction with the monomeric actin subunits within remodelers (Spichal and Fabre, 2017). To assess whether yeast nuclei contain actin we developed a means to directly visualize it.
Figure 4. INO1 motion relies on remodeler ARP subunits and on actin polymerization.

(A) The position of INO1 in parental or cells deficient in an SWR-C ARP subunit (arp6Δ) or INO80-C ARP subunits (arp8Δ and ies6Δ as it results in loss of the Arp5-module) were monitored in the presence (+) and absence (−) of inositol (n = 3). (B) Parental yeast were treated with the carrier (DMSO) or Latrunculin-A (Lat-A) for 30 min then shifted to inositol free media. INO1 nuclear position was determined before (+) or after 30 min or 3 h of inositol starvation (−) (n = 3). (C) The INO1 GFP-marked yeast were transformed with an expression vector for an actin chromobody and examined using a DeltaVision OMX high-resolution microscope. The outer and nuclear membrane is visualized with ER05-mCherry and the bright green spot in the nucleus is the GFP-marked INO1 locus. (D) The nuclear actin signal was observed using an Airyscan confocal microscope. A still image captured from a high-speed Video (Video S7) is shown. (E) To assess the dynamic properties of the nuclear actin the images from 4 consecutive frames of an Airyscan video were averaged. A still image from Video S8 is shown. (F) The influence of expressing wild type actin fused to an NLS or the actin polymerization mutant R62D was checked in parental INO1-marked yeast in the presence (+) and absence (−) of inositol, as indicated (n = 3).
Nuclei contain a dynamic pool of short actin polymers
Filamentous actin is often detected with fluorophore-labeled phalloidin; yet, the limited recognition of f-actin conformations led to other methods including natural actin binding proteins (ABPs) fused to fluorescent proteins (FPs) (Riedl et al., 2008). We utilized a nanobody (i.e., single domain antibody) select for actin, which has been employed to visualize actin in mammalian and fly nuclei (Caridi et al., 2018; Schrank et al., 2018; Plessner et al., 2015) modified with yeast optimized GFP S65T and 3x NLS (chromobody) (Kaishima et al., 2016). The actin chromobody produced a punctate pattern within yeast nuclei visualized with a DeltaVision OMX super resolution microscope (Figure 4C; Video S5). Significantly, the nuclear signal was lost upon treatment with the actin polymerization inhibitor Cytochalasin D (Video S6) suggesting that short units of polymerized actin formed the puncta similar to what has been observed in unperturbed mammalian and fly nuclei (Caridi et al., 2018; Belin et al., 2015; Baarlink et al., 2017). Of note, the pattern produced by the actin chromobody was similar in yeast not expressing LacI-GFP (Figure S4A), expression of GFP as an NLS fusion protein without the actin nanobody only produced a diffuse nuclear staining (Figure S4B), and an actin chromobody without NLS tags visualized actin patches and cables as expected for a cytoplasmic actin probe (Figure S4C).
To check if the nuclear actin signal displayed any polymerization-type behavior we averaged consecutive exposures from high-speed movies captured with an Airyscan confocal microscope (Figure 4D and Video S7). This tactic has provided insights into the polymerization dynamics of both actin and tubulin (Kueh et al., 2010; Needleman et al., 2010). Averaging 4 contiguous frames revealed an actin flow tracking in a curvilinear fashion within the nucleoplasm implying that nuclear actin is constitutively and rapidly polymerizing/depolymerizing (Figure 4E and Video S8). Perhaps significantly, stable actin polymers were not observed to track with the GFP-marked INO1 locus as it moved within a yeast nucleus (Video S9), which is distinct from the long-range motion of heterochromatin double-strand DNA breaks (Caridi et al., 2018; Schrank et al., 2018). Importantly, expression of the nonpolymerisable actin mutant R62D as an NLS fusion protein blocked the inositol-dependent movement of INO1 thereby supporting a role for actin polymerization in the INO1 motion pathway (Figure 4F) (Posern et al., 2002). We believe INO1 is moved within a dynamic pool of short actin polymers (Figure 4, Videos S5 and S7), which is similar to cargo transport within the actin cortex of the cytoplasm (Ross et al., 2008; Agarwal and Zaidel-Bar, 2018).
Long-range motion of INO1 requires the actin regulatory protein formin
A key feature of actin-dependent cargo transport is dynamic actin filaments and actin polymerization is controlled by actin binding proteins (ABPs) in vivo (Moseley and Goode, 2006). Given the recent demonstration that the Arp2/3 inhibitor CK-666 blocked the nuclear periphery clustering of double-stranded heterochromatin DNA breaks (Caridi et al., 2018; Schrank et al., 2018), we tested whether Arp2/3 also modulates INO1 movement. While CK-666 was sufficient to disperse cytosolic actin patches (Figure S4D), CK-666 only had a mild influence on the translocation of INO1 to the nuclear periphery (Figure 5A). Hence, other ABPs likely control actin polymerization events mobilizing INO1.
Figure 5. Select actin binding proteins (ABPs) control INO1 movement including the Hsp90-dependent Myo3 myosin motor.

(A) The effect of the Arp2/3 inhibitor CK666 on INO1 motion was determined. Parental yeast were grown in the presence (+) or absence (−) of inositol along with carrier (Ethanol) or CK666 (50 μM) (n = 3). (B) Actin binding proteins (ABPs) influence the inositol-dependent transition of INO1 to the nuclear periphery. The genes encoding ABPs with established genetic interactions with either Hsp90 or p23 were knocked out and the nuclear location of INO1 was determined in the presence (+) and absence (−) of inositol (n = 3). The statistical significance of movement in each genetic background relative to WT was determined using non-paired t-tests: vrp1Δ (p=0.05), sla1Δ (p=0.87), myo3Δ (p=0.01), bnr1Δ (p=0.01), lsb1Δ (p=0.01), and tpm1Δ (p=0.003). (C) The inositol-dependent mobility of INO1 was checked in parental (WT) and she4Δ yeast (n = 3).
In budding yeast there are 27 ABPs (Moseley and Goode, 2006). As our chromosome motion pathway is chaperone-dependent, we narrowed our focus to the ABPs that share a genetic interaction with either chaperone leaving 6 ABPs. Loss of 4 of the ABPs (bnr1Δ, myo3Δ, lsb1Δ, or tpm1Δ) impaired the peripheral localization of INO1 (Figure 5B). Whereas vrp1Δ and sla1Δ, which both encode Arp2/3 modulators, had a weak to no apparent impact on INO1 motion (Figure 5B), respectively. The influential ABPs on INO1 included homologs of formin (Bnr1), type I myosin (Myo3), WASP inhibitor (Lsb1), and tropomyosin (Tpm1).
Prior reports showed that the transcription responses to serum stimulation or cell spreading in mammalian cells also involve formin-dependent nuclear actin filaments (Baarlink et al., 2013; Plessner and Grosse, 2015; Plessner et al., 2015). As yeast contains two formin homologs (Bnr1 and Bni1) and Bni1 is known to physically associate with Hsp90 (Miao et al., 2013), we checked whether loss of Bni1 also impacted INO1 motion and found that it was (Figure S4E). Yet, not all ABP homologs influenced chromatin motion since myo5Δ had no apparent affect (Figure S4E). Hence, the actin regulators mediating chromosome movement during gene activation (formin-dependent) appear distinct from those governing the response to double-stranded breaks within heterochromatin (Arp2/3-dependent) (Caridi et al., 2018; Schrank et al., 2018). Yet, other forms of DNA damage as well as DNA replication employ formin homologs (Belin et al., 2015; Parisis et al., 2017). Of note, deletion of any of the ABP genes declined the actin-chromobody signal (Figure S4F and S4G) suggesting that the maintenance of actin polymers in the nucleus is a highly cooperative process among the ABPs. Likely, all the ABPs contribute to the behavior of nuclear actin and select ABPs are called upon to direct specific actin structures in response to distinct cellular signals. Perhaps significantly, loss of chaperone, remodeler, or ABP genes resulted in a declined transcriptional induction of INO1 (Figure S5).
Hsp90 supports motor activity of Myo3
Actin-based transport typically depends on a motor protein to move cargo (Agarwal and Zaidel-Bar, 2018). Paralleling previous studies involving chromatin motion associated with either DNA repair or transcription, we found that translocation of the INO1 DNA locus was dependent on a type I myosin (Figure 5B). While the primary sequence of Myo3 is consistent with it being a single-headed (unconventional) motor, direct evidence for motor function had been lacking. Using a classic actin-gliding assay we found that Myo3 purified from yeast was inactive as a motor (Video S10). In fission yeast the type II myosins are known Hsp90 clients where the motor function is supported by the chaperone in conjunction with an UCS (Unc45-/Cro1-/She4) cochaperone (Barral et al., 2002; Lord and Pollard, 2004; Mishra et al., 2005). Intriguingly, the movement of double-stranded DNA breaks to the nuclear periphery is dependent upon Unc-45 (Caridi et al., 2018). Significantly, Myo3 gained motor function following the addition of Hsp90 and the budding yeast UCS homolog She4 in vitro (Video S11) and knockout of SHE4 impaired INO1 motion in vivo (Figure 5C). Hence, Hsp90, along with the She4 cochaperone, contribute to chromatin motion by supporting the nuclear motor activity of Myo3.
A DNA zip code-bound transcription factor connects INO1 to the myosin motor
In conjunction with recent DNA repair studies, it is evident that myosin motors are used to mobilize select chromatin sites in reaction to physiological cues (Caridi et al., 2018; Figure 5B). While a combination of Mre11, HP1a, and Smc5/6 are used to recruit myosins to double-stranded DNA breaks (Caridi et al., 2018), how a motor is targeted to a gene promoter was not evident. However, prior work had identified the Put3 and Cbf1 transcription factors as critical determinants in transitioning INO1 to the nuclear periphery, as these proteins bind to proximal DNA elements termed Gene Recruitment Sequences I and II, respectively, or “DNA zip codes” (D’Urso et al., 2016; Brickner et al., 2012; Ahmed et al., 2010). We speculated that these factors might direct Myo3 to the INO1 locus.
To assess whether Myo3 associated with INO1 in an inositol-dependent manner we employed ChIP and engineered yeast. In parental yeast Myo3 was nucleated at the Put3-controlled GRS I element at a time when INO1 is transitioning to the nuclear periphery following inositol withdrawal (Figure 6A and Figure S1A). In contrast to Myo3, neither the type II myosin Myo1 nor type V myosin Myo2 were found at either zip code yet the Myo3 paralog Myo5 was mildly recruited to GRS I (Figure 6A). Yet, deletion of the MYO5 gene had no apparent impact on INO1 motion (Figure S4E). Of note, the ChIP signal for Myo3 at GRS I was not evident if the crosslinker was added to whole extracts as opposed to live cells (data not shown). Significantly, in put3Δ cells Myo3 was no longer found at GRS I but rather was recruited to the Cbf1-bound GRS II site (Figure 6B), which agrees with prior studies showing that GRS I is the dominant DNA zip code in the normal promoter (Brickner et al., 2012). Thus, a critical role of the transcription factors binding to the DNA zip code elements is to nucleate a type I myosin motor at the chromatin target thereby actively engaging it with the nuclear actin network. In conjunction with recent DNA repair-linked chromosome motion (Caridi et al., 2018), it appears that myosin motors are commonly used to move targeted chromatin sites.
Figure 6. DNA zip code-bound transcription factors nucleate type I myosins at the INO1 locus.

(A) ChIP was used to measure the inositol-dependent association of Myo1, Myo2, Myo3, and Myo5 myosin motor proteins, as marked, at either GRS I or GRS II after inositol withdrawal (n = 3). (B) The nucleation of Myo3-TAP at either GRS I or GRS II after inositol withdrawal was determined in either parental, cbf1Δ, or put3Δ yeast, as indicated (n = 3). All ChIP data represent the fold enrichment of the indicated myosin protein following inositol withdrawal. (C) The influence of purified Myo2 or Myo3 on Put3 bound to either a UAS or GRS I oligonucleotide was determined by EMSA (n = 3). The position of free probe is marked. (D) The sufficiency of either full-length (FL) Myo3 or truncation proteins having the indicated combination of the amino-terminal motor (1), central TH (2), or carboxyl-terminal SH3 (3) domains to support inositol-dependent INO1 motion was determined, as indicated (n = 3).
Besides binding to the GRS I DNA zip code, Put3 also recognizes upstream activation sequences (UASs) to regulate the promoter activities of select genes (Siddiqui and Brandriss, 1989). Intriguingly, the sequences of the two elements are distinct (UAS consensus CGG(n10)CCG vs. GRS I GGGTTGGA) (Swaminathan et al., 1997; Ahmed et al., 2010), which might account for the ability of Put3 to yield distinct functional outcomes at the different sites. To test this concept, we checked if Put3 could directly associate with Myo3 on DNA using purified components in an electrophoresis mobility shift assay (EMSA). While recombinant Put3 bound to either a GRS I or UAS oligonucleotide, a supershifted complex was only apparent with Myo3 when Put3 was bound to a GRS I (Figure 6C). Thus, DNA allosteric changes in Put3 permit a Myo3-specific interaction thereby limiting the recruitment of a type I myosin motor to sites with a GRS I DNA zip code.
To further characterize the Myo3-Put3 interaction we determined the functional domains of the myosin motor required to support INO1 motion. The unconventional type I myosin motors are composed of three domains—an amino-terminal motor domain, a middle tail homology (TH) domain, and a carboxyl-terminal SH3 domain (Batters and Veigel, 2016). Expression of full length Myo3 in myo3Δ cells rescued the movement of INO1 to the nuclear periphery following inositol removal (Figure 6D). Comparably, a mutant missing the SH3-domain was sufficient; however, truncations lacking either the motor- or TH-domain were inadequate (Figure 6D). All the variants were expressed to levels at or above the full-length HA-tagged Myo3 protein (Figure S6).
INO80-C interacts with f-actin in an ARP- and chaperone-dependent manner
How might the chromatin remodelers, especially the more general INO80-C remodeler, contribute to the select motion of the INO1 locus? Although a prior report showed that mammalian BAF (SWI/SNF-like) remodeler associates with the ends and branch points of f-actin in a phosphatidylinositol (PIP2)-dependent manner (Rando et al., 2002), to our knowledge direct binding between a purified remodeler complex and f-actin had not been shown. To test whether the remodelers interact with f-actin we used a co-sedimentation assay in which f-actin is pelleted by centrifugation along with any associated proteins. In the absence of f-actin (−) SWR-C and INO80-C did not pellet (Figure 7A). In the presence of f-actin (+), INO80-C and SWR-C were found in the pellet albeit their interaction with f-actin was weaker relative to either Myo2 or Myo3 (Figure 7A). In contrast, the Put3 transcription factor did not associate with f-actin (Figure 7A). Of note, the remodeler interaction was dependent upon its ARP subunits since INO80-C isolated from either ies6Δ or arp8Δ yeast cells displayed reduced f-actin binding (Figures S7A).
Figure 7. The ARP-containing nucleosome remodelers interact directly with actin in a chaperone-dependent manner.

(A) Purified INO80-C, SWR-C, Myo2, and Myo3 bind directly to purified f-actin based on an actin spin-down assay. Dependence on actin to pellet each protein was checked using reactions without (−) or with (+) actin, as marked. The propensity of each protein to pellet with f-actin was quantified and the average ratio of the pellet to supernatant signal from 3 independent experiments is shown. The presence of Ino80-TAP, Swr1-TAP, Put3-TAP, Myo2-TAP, or Myo3-TAP in either the supernatant (S) or pellet (P) fractions was detected by immunoblot analysis using an anti-TAP antibody. (B) Hsp90 regulates the INO80-C/f-actin interaction in a nucleosome-dependent manner while INO80-C bound f-actin independent of nucleosome presence. The influence of Hsp90 (1 μM) and/or p23 (1 μM) on the INO80-C association with f-actin was determined in the absence or presence of canonical or H2A.Z-containing non-canonical nucleosomes, as marked. The presence of Ino80-TAP in either the supernatant (S) or pellet (P) fractions was detected by immunoblot analysis using an anti-TAP antibody (n = 3).
Significantly, we found that the Hsp90 and p23 chaperones regulated the association between the remodeling complexes and f-actin. Both SWR-C and INO80-C bound f-actin comparably in the absence or presence of canonical or non-canonical H2A.Z nucleosomes (Figure 7B and S7B). Hsp90 and p23 dissociated SWR-C from f-actin independent of nucleosomes (Figure S7B). Yet, the chaperones biased the interaction of INO80-C with f-actin in a nucleosome-dependent manner. In the absence of nucleosomes or in the presence of canonical nucleosomes Hsp90 disfavored INO80-C binding to f-actin yet promoted the association in the presence of H2A.Z-containing nucleosomes especially in the co-presence of p23 (Figure 7B). Thus, the chaperones differentially modulated the remodeler-actin interaction in a nucleosome-dependent manner. While the cellular role of SWR-C is H2A.Z-centric, INO80-C is a more general remodeler and therefore the ability of Hsp90 to restrict its actin-interactions to H2A.Z nucleosomes limits the chromatin sites targeted for motion. Overall, we believe the ARP-containing remodelers contribute to the process of chromosome motion by stabilizing the connection between Myo3 and the nuclear actin network thereby enhancing motion by the typically non-processive type I myosin motors (McIntosh and Ostap, 2016).
DISCUSSION
Central biological processes including genome organization, RNA transcription, and DNA repair rely on the select movement of chromosome sites within interphase cells (Soutoglou and Misteli, 2007). Here, we exploited an experimentally tractable system, the GFP-marked INO1 locus in budding yeast, to delineate a biological pathway capable of mediating the rapid and directed motion of a chromatin site within interphase cells. Our data support a model in which a dynamic pool of short actin polymers in the nucleus is engaged to move a gene locus through the concerted efforts of transcription factors, chromatin remodelers, molecular chaperones, and a myosin motor.
Prior work had established that the Put3 transcription factor bound to the GRS I DNA zip code element, which is near the INO1 promoter, is critical for mobilizing the locus to the nuclear periphery in response to inositol starvation (Brickner et al., 2012; Randise-Hinchliff and Brickner, 2016). Yet, the exact mechanistic contribution made by the Put3-GRS I complex was not evident. Our data show that a DNA zip code-associated transcription factor provides a means to selectively connect a target chromatin site to the nuclear actin network by tethering a myosin motor (Figure 6A). We propose that the nucleation of a myosin motor will be a common feature at DNA motifs sufficient for mobilizing chromosome sites within interphase cells (Randise-Hinchliff and Brickner, 2016; Brickner et al., 2019). Importantly, the sequence of the target DNA element is critical in determining whether a bound transcription factor mediates chromatin motion. For instance, binding sites used by Put3 to activate gene transcription (i.e., upstream activating sequences) are of a different sequence relative to the GRS I DNA zip code (Brickner et al., 2012). Hence, the GRS I DNA motif induces a conformation in the Put3 transcription factor (i.e., DNA allostery) that is favorable for myosin motor interactions (Figure 6C), which in turn fosters the translocation of the chromatin target to the nuclear periphery.
Likely, a single myosin motor is nucleated at a DNA zip code since a lone type I motor would be advantageous to quickly and efficiently move a chromatin site within the nuclear actin network. The speed of a moving INO1 locus (~100 nm/sec) was consistent with an effective mode of motor transport (Figure S1B and Videos S1 and S2). While further work is needed to fully characterize actin in the nucleus, minimally it appears to be comprised of short highly dynamic polymers and not long bundled filaments (Figure 4C–E; Videos S5–S7). As such, mobilized chromatin sites are not traversing along stable actin tracts to the nuclear periphery and our studies found no obvious occurrence of such actin filaments (Video S9). Rather, the nuclear actin network appears similar to the meshwork of the cytosolic actin cortex where actin polymers have a broad range of sizes and orientations (Ross et al., 2008; de Lanerolle, 2012; Belin and Mullins, 2013). Assessment of cargo transport within an actin meshwork shows that the engagement of multiple motors can create a “tug of war” in which motors can engage different actin filaments thereby pulling a cargo in various directions (Ross et al., 2008; Titus, 2018). Recruitment of only one myosin eliminates this complication, yet it would leave motion vulnerable to other potential inefficiencies inherent to a single motor.
A primary limitation of a employing only one myosin is the cycle of attachment and detachment of the motor head to the actin polymer since these events would slow the motion by minimally providing opportunities to diffuse away from the current actin polymer and/or engage a different actin filament (McIntosh and Ostap, 2016; Titus, 2018). We suggest that the ARP-containing chromatin remodelers circumvent this shortcoming by providing a secondary connection, which maintains a linkage to the actin polymer when the motor head detaches. Basically, the ARP-remodelers are serving as chromatin transport processivity factors. A similar strategy is used to move melanosomes by myosin Va with Melanophilin serving as the auxiliary protein stabilizing the motor interaction to the actin tract thereby fostering longer transport events by increasing the processivity of the myosin motors (Sckolnick et al., 2016).
While numerous studies have shown an influence of INO80-C and/or SWR-C on chromatin motion associated either with DNA repair or gene activation, most reports proposed that the remodelers contribute by decompacting the local chromatin structure (Seeber and Gasser, 2017). Yet, it is unclear how the assimilation of H2A.Z or the eviction of one or two nucleosomes enables the long-range movement of a chromosome section from the nuclear center to the periphery. Our model provides a rationale for why ARP-containing remodelers are commonly involved with long-distance chromatin movements—stabilization of the interaction between a type I myosin motor and the nuclear actin network. While both INO80-C and SWR-C have an innate ability to bind to f-actin (Figure 7A), in the presence of the chaperones the interaction is destabilized unless INO80-C is bound to an H2A.Z-nucleosome (Figure 7B). Hence, INO80-C is likely the remodeler directly contributing to the stability of the actin interaction and SWR-C adds to the pathway by depositing H2A.Z. Importantly, the chaperone-mediated restriction narrows the remodeler-actin association to chromatin loci linked to H2A.Z/H2A turnover (e.g., gene promoters). Overall, the directed movement of a chromosome site is dependent upon a two-factor authentication system that is comprised of a transcription factor-nucleated type I myosin motor and an ARP-containing chromatin modifier.
In contrast to recent studies outlining an Arp2/3-dependent actin pathway clustering heterochromatin double-stranded DNA breaks at the nuclear periphery (Caridi et al., 2018; Schrank et al., 2018), we found that transcription-associated chromatin motion was not reliant on Arp2/3 but rather required a Formin homolog (Figure 5B and S4E). Thus, the nucleoplasm actin system is following the basic principles used in the cytoplasm where select ABPs tune the behavior of actin for the task at hand (Pollard, 2016). In the case of DNA repair, Arp2/3 directs the formation of stable f-actin filaments to cluster numerous damaged heterochromatin sites at the periphery (Caridi et al., 2018; Schrank et al., 2018). Whereas the less intrusive transcription response does not seemingly use stable actin filaments but instead relies upon the constitutive pool of short actin polymers to mobilize the activated INO1 gene to the nuclear membrane in a formin-dependent manner. Likely, the capacity of Formins to promote the polymerization of unbranched actin polymers from actin monomers favors their use in directed chromatin movement events (Pollard, 2016).
Overall, our study identifies key determinants of a pathway capable of driving the long-range movement of a specific chromatin site in reaction to a physiological cue. In our model, the nucleus contains a dynamic network of short actin polymers that can be used to mobilize a target chromosome locus by engaging a myosin motor tethered to allosterically-regulated transcription factor. The actin-myosin interaction is stabilized by nearby ARP-containing chromatin remodelers and the activities of both the myosin and the remodelers are governed by the Hsp90 molecular chaperone. Working as a system these components provide a general means to mobilize chromosome regions that avoids spurious movements by employing a two-authentication mechanism (myosin and ARP-remodelers) to mediate the motion. We believe the described chromatin motion system represents a general mechanism for mobilizing genomic sites since chromosome motion in newly emerged daughter cells was chaperone-dependent (Videos S3 and S4).
It will be interesting to confirm whether the presented pathway is co-opted to drive other chromatin motion events, including genome reorganization following cell division. While prior studies have shown that targeting DNA loci to various nuclear landmarks including speckles, lamina, or general inter-chromosomal clustering relies on either select transcription factors or associated DNA elements (Spilianakis et al., 2005; Noma et al., 2006; Apostolou and Thanos, 2008; Haeusler et al., 2008; Hu et al., 2010; Schoenfelder et al., 2010; Zullo et al., 2012; Harr et al., 2015; Brickner et al., 2019), the means by which these chromatin sites are actively organized had not been resolved. Likely, the transcription factors provide specificity to the genome reorganization process through the recognition of select DNA elements (Fraser and Bickmore, 2007). We propose that the DNA-bound factors recruit myosin motors to drive the motion of the target chromatin loci and that the myosin-actin interaction is stabilized by ARP-containing chromatin modifiers, which can recognize epigenetic bookmarking features (Oomen and Dekker, 2017). Of note, chromatin positioning is not static, as it can vary in a cell-to-cell manner or with gene expression changes (Takizawa et al., 2008; Eskiw et al., 2010; Edelman and Fraser, 2012; Finn and Misteli, 2019). The continuous presence of a nuclear actin/myosin network would provide a means to mediate these dynamic genome-repositioning events. Notably, our work opens new avenues into how the molecular chaperone-, nucleoskeleton-, and chromatin remodeling-systems contribute to homeostasis as well providing important insights on how genomes are reconfigured in response to physiological cues within interphase cells.
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Brian Freeman (bfree@illinois.edu).
Plasmids and yeast strains generated in this study are listed in the Key Resources Table and are available at the laboratory upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti TAP | ThermoFisher | CAB1001 |
| Anti FLAG | Sigma-Aldrich | F3165 |
| Anti FLAG M2 Agarose beads | Sigma-Aldrich | A1205 |
| IgG Sepharose | GE Healthcare | 17-0969-01 |
| Calmodulin Affinity Resin | Agilent Technologies | 214303–52 |
| Bacterial and Virus Strains | ||
| DH5α | N/A | N/A |
| Rosetta | N/A | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| All chemicals unless otherwise noted | Sigma-Aldrich | N/A |
| Yeast Media Components | DIFCO | N/A |
| LB Media Components | DIFCO | N/A |
| Radicicol | AG Scientific | R-1130 |
| Rapamycin | GoldBio | R-101 |
| Latrunculin | AG Scientific | L-2471 |
| Cytochalasin D | EMD Millipore | 250255 |
| CK-666 | TOCRIS | 3950 |
| G418 | GoldBio | G-418 |
| 32-P γ-ATP | Perkin Elmer | BLU002Z250UC |
| SYPRO Ruby Protein Gel Stain | ThermoFisher | S12000 |
| Restriction Enzymes | New England Biolabs | N/A |
| Critical Commercial Assays | ||
| Pierce BCA Assay Kit | Fisher Scientific | PI23223 |
| Power SYBR Green RT-PCR Mix | Applied Biosystems | 4388869 |
| Experimental Models: Organisms/Strains | ||
| Saccharomyces cerevisiae | Strain Table S2 | N/A |
| TAP tag strain library | Open Biosystems | YSC1177 |
| Mat a Knockout library | Open Biosystems | YSC1053 |
| Oligonucleotides | ||
| Listed in methods | Eurofins | N/A |
| Recombinant DNA | ||
| Actin-Chromobody plasmid | Chromotek | acg |
| pRS315 tADH1 ActinVHH 3XNLS GFP | This manuscript | N/A |
| pRS315 tADH1 3XNLS GFP | This manuscript | N/A |
| pRS315 ADH1 ActinVHH GFP | This manuscript | N/A |
| pRS405 GAL1 3xNLS ACT1 | This manuscript | N/A |
| pRS405 GAL1 3xNLS Act1-R62D | This manuscript | N/A |
| pET28a SUMO Put3 | This manuscript | N/A |
| pRS405 ADH1 3xHA Myo3-FL | This manuscript | N/A |
| pRS405 ADH1 3xHA Myo3–1 | This manuscript | N/A |
| pRS405 ADH1 3xHA Myo3–2 | This manuscript | N/A |
| pRS405 ADH1 3xHA Myo3–3 | This manuscript | N/A |
| pRS405 ADH1 3xHA Myo3–12 | This manuscript | N/A |
| pRS405 ADH1 3xHA Myo3–23 | This manuscript | N/A |
| pRS405 ADH1 3xHA Myo3–13 | This manuscript | N/A |
| pRS405 ADH1 3xHA Myo3-FL | This manuscript | N/A |
| Software and Algorithms | ||
| Fiji | Free | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Saccharomyces cerevisiae cultures were grown in YPD or minimal media at 30°C unless otherwise noted. E. coli cultures were grown in LB media with the appropriate antibiotic at 37°C.
METHOD DETAILS
Chemicals, Media and Growth conditions
The Saccharomyces cerevisiae strains used in this study are described in Table S2. All gene deletion strains were created using a PCR-based method (Longtine et al., 1998). For experiments involving inositol starvation, cells were grown in synthetic complete medium lacking inositol or supplemented with 100 μM myo-inositol. Overnight cultures were used to inoculate corresponding medium to a final ODλ595 0.25. Cells were harvested for subsequent experiments at ODλ595 0.7–0.8. For Hsp82 inhibition, Radicicol (10 μM final concentration) or DMSO was added. For INO80 anchor-away, Rapamycin (1 μg/ml final concentration) or ethanol was added for 2 h followed by inositol withdrawal for 2 h. Latrunculin-A (Lat-A) (AG Scientific, L-2471) was added to the cultures to a final concentration of 100 μM and after 30 min of Lat-A treatment the cells were moved to inositol free media for 30 min or 3 h, as indicated. Cytochalasin D (EMD Millipore, 250255) was added to media to a final concentration of 20 μM for 2 h prior to visualizing the cells. CK-666 (TOCRIS, 3950) was added to media to a final concentration of 50 μM for 3 h.
Plasmid
The chromobody used to detect nuclear actin was constructed by replacing the TagGFP ORF from the Actin-Chromobody® plasmid (Chromotek) with yeast optimized GFP (S56T) and 3xNLS tag, then inserting the GFP (S56T)-actin chromobody fusion into the yeast expression vector pRS315 containing a truncated ADH1 promoter to drive low-level constitutive expression.
Protein purification
The yeast Hsp90 and p23 molecular chaperones were purified as described (Toogun et al., 2007). Hsp90 was expressed using a pET28 vector with a 6xHis-Tag in Rosetta cells and lysate was processed through a Cobalt column followed by a Resource Q column and a sizing column for purification. Similarly, p23 was expressed using a pET28 vector in Rosetta cells and purified by processing the lysate through a DEAE column, followed by a Resource Q, and sizing column. INO80-C WT and described subunit deletion mutants were purified from yeast strains expressing Ino80 as Tandem Affinity Purification (TAP)-tagged fusion. The purification process was previously described (Wu et al., 2008) with minor modifications. Briefly, 12 L cultures were grown in Yeast Extract/Peptone/Dextrose (YPD) media, and harvested cells were lysed in buffer E (20 mM HEPES, pH 7.5, 350 mM NaCl, 10% glycerol, 0.1% Tween and 1 mM dithiothreitol (DTT)) with 1x protease inhibitor cocktail (1 mM PMSF, 2 mM pepstatin A, 0.6 mM leupeptin, chymostatin (2 mg/mL), 2 mM benzamidine) by grinding the cell pellet with dry ice. Lysates were clarified at 17,000xg for 15 min at 4°C. Clarified lysates were incubated with IgG-Sepharose (GE Healthcare) and eluted by TEV protease cleavage. Eluted aliquots were incubated with calmodulin affinity resin (Agilent Technologies) in buffer E supplemented with 2 mM CaCl2 and eluted in buffer E supplemented with 10 mM EGTA. The proteins were concentrated using Amicon Ultra-15 Centrifugal Filter Units (Millipore). Purification of SWR-C was performed using a modified version of the protocol described (Mizuguchi et al., 2012). A SWR1–3xFlag htz1Δ yeast strain was grown in 24 L of YAP media with 3% glucose to an OD600 of 2.5–3.0 then pelleted at 4000 rpm for 10 min, washed with water, then subsequently pelleted at 4000 rpm for 10 min to remove remaining liquid. The yeast pellet was then noodled by using a 60 mL syringe to transfer the yeast into a beaker containing liquid N2. Yeast noodles were lysed using a Retsch Planetary Ball Mill PM 100 by chilling a 500 mL stainless steel jar in liquid N2 and grinding 6× 1.5 min cycles at 4000 rpm. Lysed cells were resuspended in an equal volume of 1x Lysis buffer [25 mM HEPES–KOH (pH 7.6), 1 mM EDTA, 20% glycerol, 10 mM β-glycerophosphate, 0.5 mM NaF, 1 mM Na-butylate, 0.5 mM DTT, 300 mM KCl, and 1x protease inhibitor cocktail (1 mM PMSF, 2 mM pepstatin A, 0.6 mM leupeptin, chymostatin (2 mg/mL), 2 mM benzamidine)]. Once lysed cells were fully dissolved in 1x Lysis buffer, cells were centrifuged at 35,000 rpm for 2 hours at 4°C. Whole cell extract was removed and incubated with 400 μL of anti-FLAG M2 agarose beads (A1205; Sigma-Aldrich) at 4°C for 3 h. Whole cell extract was removed by a 1000 rpm spin at 4°C for 2 min and the supernatant was aspirated. FLAG-resin was washed with 100 mL B0.5 [25 mM HEPES–KOH (pH 7.6), 1 mM EDTA, 2 mM MgCl2, 10% glycerol, 0.01% NP-40, 10 mM β-glycerophosphate, 0.5 mM NaF, 1 mM Na-butylate, 0.5 mM DTT, 1x protease inhibitor cocktail, and 0.5 M KCl] followed by a 20 mL wash with B0.1 (same as B0.5 but with 100 mM KCl). SWR-C was eluted from the resin 2x by 30 min incubations at 4°C with equal bed volume of 1 mg/mL 3xFlag peptide (F4799; Sigma-Aldrich) in buffer B0.1. The elutions were pooled and flash frozen in aliquots (20 μL). The isolated complexes were resolved on 10% SDS-PAGE and stained with SYPRO Ruby Protein Gel Stain (Thermo Fisher Scientific) to assess the purity and concentrations of the protein preparations.
Preparing fluorescent nucleosomal DNA
Nucleosomal DNA was generated from PCR amplification of the 601 positioning sequence on the pGEM-3Z-LowerStrand plasmid using Taq polymerase (M0273S, NEB) and the forward primer GTACCCGGGGATCCTCTAGAGT and reverse primer 5’/Cy3/CTGGAGAATCCCGGTGCC. The PCR product was purified using a DNA Clean & Concentrator™−500 (D4032, Zymo research) followed by ethanol precipitation and resuspending in TE (10 mM Tris pH 7.4, 1 mM EDTA).
Recombinant histones, octamer assembly, and nucleosome reconstitution
Recombinant histones were expressed independently in BL21 cells using pET vectors for each histone. Lysates were processes to generate inclusion bodies containing the histone proteins that were resuspended in unfolding buffer [7 M guanidinium hydrochloride, 20 mM Tris-HCl, pH 7.5, 10 mM DTT] and applied through a gel filtration column. Peak fractions were analyzed by 18% SDS-PAGE and fractions containing histones were pooled and dialyzed against distilled water containing 2 mM 2-mercaptoethanol. Samples were lyophilized, dissolved in SAU-200 [7 M deionized urea, 20 mM sodium acetate, pH 5.3, 0.2 M NaCl, 5 mL 2-mercaptoethanol, 1 mM Na-EDTA] and resolved through an HPLC column; fractions containing pure histone proteins were dialyzed against water and lyophilized. Aliquots of the four histones were dissolved in unfolding buffer, mixed to equimolar ratios, and adjusted to a total final protein concentration of 1 mg/ml. Refolding was carried out by dialysis against water and octamers and dimers were purified using a final resolution through a gel filtration column (Luger et al., 1999). The H2A derivative K119C was unfolded and labeled with Cyanine 5 maleimide (43080, Lumiprobe), as described (Zhou and Narlikar, 2016) before refolding with the other core histones to generate fluorescent yeast octamers. 77N0 FRET nucleosomes were generated as described (Luger et al., 1999) using the Cy3 DNA template and Cy5 octamer. Briefly, 400 nM Cy3 DNA and 400 nM octamers were mixed in Hi buffer (2 M KCl, 10 mM Tris pH 7.4, 1 mM EDTA, 1 mM DTT) and placed in Slide-A-Lyzer MINI dialysis Units 10,000MWCO (69572, Thermo Fisher). The dialysis units were placed on the surface of a beaker with 600 mL Hi buffer and 3 L of Lo buffer (50 mM KCl, 10 mM Tris pH 7.4, 1mM EDTA, 1mM DTT) was exchanged via a peristaltic pump. This process was performed in the dark and set to take 18–20 hours for complete transfer of Lo buffer. Nucleosome quality was gauged by electrophoresis on a 4.5% Native PAGE and by emission scan on a Tecan M1000 Infinite Pro by exciting at 530 nm and scanning from 550 nm to 700 nm.
FRET-based dimer exchange
FRET dimer exchange assays were performed using a Tecan M1000 Infinite Pro at 25–26°C with an excitation of 530 nm and measuring the emission at 670 nm. The reactions (50 μL) were performed in 96 well half area black clear bottom plates (3880, Corning) containing 25 nM SWR-C (B0.1 for controls), 10 nM labeled nucleosomes, 70 nM H2A.Z/H2B dimers, and chaperone proteins mixed in a reaction buffer (25 mM HEPES pH 7.6, 0.2 mM EDTA, 5 mM MgCl2, and 70 mM KCl. Remodeling was initiated by adding 500 μM ATP (A6559–25UMO, Sigma-Aldrich). All dimer exchange assay conditions were all performed in duplicate and normalized to no enzyme controls.
Nucleosome sliding assay
Nucleosome sliding assays were performed as previously described (Sinha and Peterson, 2009). Briefly, end positioned mononucleosomes were reconstituted onto a 255 bp 32P-labelled 601 DNA fragment. Mononuleosomes (5 nM) were incubated with purified INO80-C and 2 μM ATP in buffer A (10 mM Tris-HCl pH 8.0, 70 mM NaCl, 5 mM MgCl2, 0.1 mg/mL 1 BSA and 1 mM DTT) for 15 min at 30°C. The reactions were quenched with 5% glycerol and 1 mg/mL salmon sperm DNA, followed by 5 min incubation at 30°C. The products were resolved by Native-PAGE on 5% polyacrylamide 1xTBE gels. Gels were dried and products visualized using PhosphorImager (Molecular Dynamics).
Electro-Mobility Shift Assays (EMSAs) for DNA binding
A 32P-labelled INO1 promoter DNA probe (100 nM) was incubated with purified INO80-C or SWR-C at 30°C for 30 min. Reactions were resolved on pre-chilled (4°C) 3.2% native polyacrylamide 1x GTG (glycerol tolerant gel-90 mM Tris, 28.5 mM Taurine, and 0.5 mM EDTA) gels, dried, and imaged using a PhosphorImager (Molecular Dynamics). Probes were generated using the following primers and their reverse complements for Put3 UAS binding and GRSI binding: UAS: TCTCGGGAAGCCAACTCCGAAGC; GRSI: GTGTTCCGGGGTTGGATGCGGAA.
Chromatin Immunoprecipitation
Chromatin Immunoprecipitation (ChIP) assays were performed as previously described (Kuras and Struhl, 1999). Briefly, the indicated parental TAP strains and where noted the corresponding sba1, put3, or cbf1 null strains were grown logarithmically in synthetic complete medium with or without inositol. Isolated DNA was analyzed by qPCR using primers selected to the INO1 promoter. The relative DNA occupancies were calculated using the normalized PCR values obtained in the presence and absence of inositol. The primers for INO80 and SWR1 binding corresponded to the +1 nucleosome of the INO1 locus (Forward: TTCGATGTAACGCCCACTG; Reverse: AGCCATTGTTGCCACCTAA). Negative control primers were designed from a nucleosome depleted region (NDR) on the INO1 locus (Forward: TGGAGCTTTCGTCACCTTT; Reverse: GGGAATGAAACAGAATAAATCAAGG). Primers for detecting Ino2/4, Put3, and Cbf1 binding were the following: INO2/4 Forward: ATGCGGAATCGAAAGTGTTGAATGT; INO2/4 Reverse: TCTCAAATTAACATTGCCGCCAACG; CBF1 Forward: ACGTGATGAAGGCTCGTTTT; CBF1 Reverse: TGGTTGTTTGCTTTCTGCTG; PUT3 Forward: CTTCGTTCCTTTTGTTCTTCACGTC; PUT3 Reverse: AGACAATACTTTTCACATGCCGCAT.
Actin spin-down assay
The actin spin-down assays were performed as described previously (Normoyle and Brieher, 2012). In brief, polymerization of purified g-actin to f-actin was done by diluting the g-actin to a final concentration of 5 μM at room temperature in KMEI polymerization buffer (50 mM KCl, 1 mM MgCl2, 50 mM EGTA and 10 mM imidazole pH 7.0). After 1 h, the indicated INO80-C or SWR-C preparations (pre-cleared by centrifugation for 30 min at 60,000 rpm) were added to the polymerized actin (2 μM based on initial g-actin) with or without Hsp90, p23, or nucleosomes, as marked. The reactions were incubated at 30°C for 30 min and clarified at 60,000 rpm for 30 min at 4°C in a TLA100 rotor (Beckman Instruments, Fullerton, CA). Supernatants were mixed with 5x SDS loading buffer and pellets were resuspended in 1x SDS loading buffer prepared in KMEI buffer. Equal proportions of the pellets and supernatants were resolved on 8% SDS–PAGE and subjected to immunoblot analysis using an anti-TAP antibody (CAB1001, Thermo Fisher Scientific) or anti-FLAG antibody.
Phalloidin Staining of Actin Cytoskeleton
Phalloidin staining was carried out as per Higuchi et al. (2013). Briefly, cells were fixed by addition of 3.7% paraformaldehyde directly to the growth medium at 30°C with shaking for 50 min. Cells were then collected by centrifugation and washed three times with wash solution (0.025M KPi pH 7.5, 0.8M KCl), followed by one wash with PBT (PBS, 1% w/v BSA, 0.1% v/v Triton X-100, 0.1% NaN3). Staining was carried out with 1.65 μM FITC-phalloidin for 35 min at RT in the dark. After three washed in PBS, cells were mounted onto slides and visualized immediately.
Myo3 Domain Mapping
Myo3 domains were identified using Saccharomyces Genome Database. Primer pairs were generated flanking each domain – the amino terminal motor domain 1–716 (1), the IQ/TH domain 760–961 (2) and the SH3 domain 1107–1184 (3). The domains and their combinations (1, 2, 3, 1/2, 1/3, 2/3, Full Length) were amplified by PCR using yeast genomic DNA and cloned into the XbaI and SalI sites of pRS405 ADH1 3xHA. Primer pairs were as follows: 1 Forward: TTGTTGGCTAGCATGGCTGTCATAAAAAAGGGAGC; 1 Reverse: TTGTTGGTCGACCTATGCTCTCTGAATTCTAGCAGCCATGT; 2 Forward: TTGTTGGCTAGCTGGAGAAGGTTTCTTCAAAGGC; 2 Reverse: TTGTTGGTCGACCTAGCTATGTGTATGCTTATGAGTAG; 3 Forward: TTGTTGGCTAGCATGCATAGCCATAGAATTCATAGGGATGCTGCA; 3 Reverse: TTGTTGGTCGACTTACCAGTCATCATCATCATCGC.
Actin Gliding Assay
Purified TAP-Myo3 (0.2 mg/mL) was introduced into flow cell made from a microscope slide and coverslip by capillary action and was allowed to adhere to the coverslip surface for 10 min. The chamber was washed with running buffer (25 mM imidazole pH 7.4, 25 mM KCl, 4 mM MgCl2, 1 mM ATP, and 1 mM DTT) followed by a wash with running buffer with 1 mg/mL BSA for 2 min. The contents of the chamber were then replaced with She4 (0.2 mg/mL) and HSP90, (1.0 mg/mL) in running buffer for 10 minutes. A solution of actin (2 μM) in running buffer was then introduced into the chamber and allowed to polymerize for 2–3 min. The chamber was washed twice with running buffer before imaging. Filaments were observed by epi-fluorescence illumination with a 63× 1.4 N.A. objective and recorded at intervals of 5 s (Lord and Pollard, 2004).
Microscopy
INO1 nuclear positioning was determined by analyzing z-stack images acquired with a DeltaVision OMX microscope (GE Life Science) with 100x/1.40 NA oil immersion objective (Olympus). INO1 nuclear periphery localization was determined by stringent criteria as previously described (Brickner and Walter, 2004). All INO1 motion data represent averages of 3 independent trials with 50 counted cells in each and the error bars represent the SEM. Live cell microscopy was done according to a described protocol (Rines et al., 2011). Time lapse movies were acquired at indicated time intervals. Airyscan microscopy was done using Zeiss LSM880 confocal laser scanning equipped with an Airyscan detection unit. All imaging was performed with 63x/1.46 oil immersion objective with Immersol 518 F immersion media (ne = 1.518 (23°C); Carl Zeiss) using 2% of the maximum 488 nm laser power, a gain setting of 700, a pixel dwell time of ranging from 6–8 μs and averaging 2 frames were used to acquire the images. The intensity of fluorescence signal quantification and walking average were generated using Fiji.
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were performed in a minimum of 3 biological replicates as indicated in the figure legends with (n = 3). Additionally, in the INO1 motion experiments, at least 50 cells were counted in each condition. The error bars represent the SEM of the 3 biological replicates. Student’s t-test (unpaired or paired when appropriate) was used to compare with WT and the resulting p-values are presented in the figure legend.
DATA AND CODE AVAILABILITY
This study did not generate any datasets. Image analysis was done using Fiji, a free software available online.
Supplementary Material
Movie S1. Related to Figure 1.
The behavior of the control GFP-marked INO1 locus in the presence of inositol was monitored using a DeltaVision OMX microscope with an InsightSSI solid state illumination system and 100x 1.40 NA oil immersion objective collected at 1 frame per second.
Movie S2. Related to Figure 1.
To tract the motion of INO1 in the absence of inositol 5 min movies were taken 20 min after transferring cells to inositol free media and the acquired movies were checked for cells displaying a clear shift of INO1 from the inner nucleoplasm to the periphery. A typical cell displaying motion of INO1 from the inner nucleoplasm to the periphery is shown. The images were acquired using a DeltaVision OMX microscope.
Movie S3. Related to Figure 2.
The behavior of the GFP-marked INO1 locus was visualized in mother and a newly emerged daughter cell (bud neck apparent) in parental yeast. The images were acquired using a DeltaVision OMX microscope.
Movie S4. Related to Figure 2.
The behavior of the GFP-marked INO1 locus was visualized in mother and a newly emerged daughter cell (bud neck apparent) in p23 null yeast. The images were acquired using a DeltaVision OMX microscope.
Movie S5. Related to Figure 4.
Nuclear actin was visualized using an actin chromobody. Yeast with the GFP-marked INO1 locus were transformed with an expression vector for an actin chromobody, which is an actin nanobody (single domain antibody) fused to GFP (S65T) and 3x NLS tags. A representative image acquired with a DeltaVision OMX microscope is shown.
Movie S6. Related to Figure 4.
Transformants expressing the actin chromobody were exposed to the actin polymerization inhibitor Cytochalasin D (20 ?M) for 1 h prior to image collection with the DeltaVision OMX. The signal intensity of the GFP-mark near INO1 was used to normalize the exposures shown in Movie S5 and S6.
Movie S7. Related to Figure 4.
Images of parental yeast expressing the actin chromobody were acquired with a Zeiss LSM 880 Airyscan microscope using the 488 and 561 excitation channels with an oil immersion 63x objective collected at 1 frame per 100 msec.
Movie S8. Related to Figure 4.
To gauge whether the actin signal in the Airyscan images (Movie S7) displayed polymerization dynamics a movie was prepared by averaging 4 consecutive exposures using the data collected in the 488 channel (Needleman et al., 2010).
Movie S9. Related to Figure 4.
A mobile INO1 locus does not track along stable, extended actin fibers. A cell displaying multiple long-range motions of the GFP-mark within parental yeast grown under inositol starvation conditions is provided. The images were acquired using a DeltaVision OMX microscope.
Movie S10. Related to Figure 5.
Myo3-TAP purified from yeast is an inactive motor based upon an actin-gliding assay.
Movie S11. Related to Figure 5.
Co-incubation of Myo3-TAP with purified Hsp82 and TAP-She4 activates myosin motor activity in an actin-gliding assay.
Highlights.
Chromatin motion is dependent upon nuclear chaperones, remodelers, myosin, and actin
Allosterically-regulated transcription factors nucleate myosin to select chromatin loci
Motor activity of nuclear myosin relies on molecular chaperones
ARP-containing remodelers act as chromatin transport processivity factors
Acknowledgements
We thank Lindsey Bendix, Donna Brickner, Jason Brickner, Melinda Lynch-Day, Lisa Schuster, and Brian Nguyen for their helpful contributions to the work. Support by the Public Service grant GM118306. Authors declare no competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
REFERENCES
- Agarwal P and Zaidel-Bar R (2019). Principles of Actomyosin Regulation In Vivo. Trends in Cell Biology 29, 150–163 [DOI] [PubMed] [Google Scholar]
- Ahmed S, Brickner D, Light W, Cajigas I, McDonough M, Froyshteter A, Volpe T and Brickner J (2010). Erratum: DNA zip codes control an ancient mechanism for gene targeting to the nuclear periphery. Nature Cell Biology 12, 306–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolou E and Thanos D (2008). Virus Infection Induces NF-κB-Dependent Interchromosomal Associations Mediating Monoallelic IFN-β Gene Expression. Cell 134(10), 85–96. [DOI] [PubMed] [Google Scholar]
- Baarlink C, Plessner M, Sherrard A, Morita K, Misu S, Virant D, Kleinschnitz E, Harniman R, Alibhai D, Baumeister S, Miyamoto K, Endesfelder U, Kaidi A and Grosse R (2017). A transient pool of nuclear F-actin at mitotic exit controls chromatin organization. Nature Cell Biology 19, 1389–1399. [DOI] [PubMed] [Google Scholar]
- Baarlink C, Wang H and Grosse R (2013). Nuclear Actin Network Assembly by Formins Regulates the SRF Coactivator MAL. Science 340, 864–867. [DOI] [PubMed] [Google Scholar]
- Barral J, Hutagalung A, Brinker A, Hartl F and Epstein H (2002). Role of the Myosin Assembly Protein UNC-45 as a Molecular Chaperone for Myosin. Science 295, 669–671. [DOI] [PubMed] [Google Scholar]
- Batters C and Veigel C (2016). Mechanics and Activation of Unconventional Myosins. Traffic 17, 860–871. [DOI] [PubMed] [Google Scholar]
- Belin B and Mullins R (2013). What we talk about when we talk about nuclear actin. Nucleus 4, 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin B, Lee T and Mullins R (2015). Correction: DNA damage induces nuclear actin filament assembly by Formin-2 and Spire-1/2 that promotes efficient DNA repair. eLife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickner D, Cajigas I, Fondufe-Mittendorf Y, Ahmed S, Lee P, Widom J and Brickner J (2007). H2A.Z-Mediated Localization of Genes at the Nuclear Periphery Confers Epigenetic Memory of Previous Transcriptional State. PLoS Biology 5, p.e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickner D, Ahmed S, Meldi L, Thompson A, Light W, Young M, Hickman T, Chu F, Fabre E and Brickner J (2012). Transcription Factor Binding to a DNA Zip Code Controls Interchromosomal Clustering at the Nuclear Periphery. Developmental Cell 22, 1234–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickner D, Randise-Hinchliff C, Lebrun Corbin M, Liang J, Kim S, Sump B, D’Urso A, Kim S, Satomura A, Schmit H, Coukos R, Hwang S, Watson R and Brickner J (2019). The Role of Transcription Factors and Nuclear Pore Proteins in Controlling the Spatial Organization of the Yeast Genome. Developmental Cell 49(6), 936–947.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickner D, Sood V, Tutucci E, Coukos R, Viets K, Singer R and Brickner J (2016). Subnuclear positioning and interchromosomal clustering of the GAL1–10 locus are controlled by separable, interdependent mechanisms. Molecular Biology of the Cell 27, 2980–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickner J and Walter P (2004). Gene Recruitment of the Activated INO1 Locus to the Nuclear Membrane. PLoS Biology 2, e342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveri T (1909). Die Blastomerenkerne von Ascaris megalocephala und die Theorie der Chromosomenindividualität. Arch Zellforsch 3,181–268. [Google Scholar]
- Brown JM, Green J, das Neves RP, Wallace HA, Smith A,J, Hughes J, Gray N, Taylor S, Wood WG, Higgs DR, et al. (2008) Association between active genes occurs at nuclear speckles and is modulated by chromatin environment. J Cell Biol 182, 1083–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Leach J, Reittie JE, Atzberger A, Lee-Prudhoe J, Wood WG, Higgs DR, Iborra FJ and Buckle VJ (2006) Coregulated human globin genes are frequently in spatial proximity when active. J Cell Biol 172, 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K, Landry C, Hartl D and Cavalieri D (2008). Cascading transcriptional effects of a naturally occurring frameshift mutation in Saccharomyces cerevisiae. Molecular Ecology 17, 2985–2997. [DOI] [PubMed] [Google Scholar]
- Caridi C, D’Agostino C, Ryu T, Zapotoczny G, Delabaere L, Li X, Khodaverdian V, Amaral N, Lin E, Rau A and Chiolo I (2018). Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature 559, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang CH, Carpenter AE, Fuchsova B, Johnson T, de Lanerolle P and Belmont AS (2006) Long-range directional movement of an interphase chromosome site. Current Biology 16, 825–831. [DOI] [PubMed] [Google Scholar]
- Clapier C and Cairns B (2009). The Biology of Chromatin Remodeling Complexes. Annual Review of Biochemistry 78, 273–304. [DOI] [PubMed] [Google Scholar]
- Clark T and Merriam R (1977). Diffusible and bound actin in nuclei of xenopus laevis oocytes. Cell 12, 883–891. [DOI] [PubMed] [Google Scholar]
- Comings DE (1968). The rationale for an ordered arrangement of chromatin in the interphase nucleus. Am J Hum Genet. 20(5), 440–460. [PMC free article] [PubMed] [Google Scholar]
- Cremer T and Cremer C (2001). Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nature Reviews Genetics 2(4), 292–301. [DOI] [PubMed] [Google Scholar]
- de Lanerolle P (2012). Nuclear actin and myosins at a glance. Journal of Cell Science 125, 4945–4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion V and Gasser S (2013). Chromatin Movement in the Maintenance of Genome Stability. Cell 152, 1355–1364. [DOI] [PubMed] [Google Scholar]
- Dixon J, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu J and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485(7398), 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget J, Lee A, Ye Z, Kim A, Rajagopal N, Xie W, Diao Y, Liang J, Zhao H, Lobanenkov V, Ecker J, Thomson J and Ren B (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518(7539), 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue TF, & Henry SA (1981). Inositol Mutants of SACCHAROMYCES CEREVISIAE: Mapping the ino1 Locus and Characterizing Alleles of the ino1, ino2 and ino4 Loci. Genetics 98, 491–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dopie J, Skarp K, Kaisa Rajakyla E, Tanhuanpaa K and Vartiainen M (2012). Active maintenance of nuclear actin by importin 9 supports transcription. Proceedings of the National Academy of Sciences, 109, E544–E552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Urso A, Takahashi Y, Xiong B, Marone J, Coukos R, Randise-Hinchliff C, Wang J, Shilatifard A and Brickner J (2016). Set1/COMPASS and Mediator are repurposed to promote epigenetic transcriptional memory. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbert R, Birkmann A and Schuller H (1999). The product of the SNF2/SWI2 paralogue INO80 of Saccharomyces cerevisiae required for efficient expression of various yeast structural genes is part of a high-molecular-weight protein complex. Molecular Microbiology 32, 741–751. [DOI] [PubMed] [Google Scholar]
- Echtenkamp F and Freeman B (2012). Expanding the cellular molecular chaperone network through the ubiquitous cochaperones. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1823, 668–673. [DOI] [PubMed] [Google Scholar]
- Echtenkamp F, Gvozdenov Z, Adkins N, Zhang Y, Lynch-Day M, Watanabe S, Peterson C and Freeman B (2016). Hsp90 and p23 Molecular Chaperones Control Chromatin Architecture by Maintaining the Functional Pool of the RSC Chromatin Remodeler. Molecular Cell 64, 888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echtenkamp F, Zelin E, Oxelmark E, Woo J, Andrews B, Garabedian M and Freeman B (2011). Global Functional Map of the p23 Molecular Chaperone Reveals an Extensive Cellular Network. Molecular Cell 43, 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelman L and Fraser P (2012). Transcription factories: genetic programming in three dimensions. Current Opinion in Genetics & Development 22(2), 110–114. [DOI] [PubMed] [Google Scholar]
- Eskiw C, Cope N, Clay I, Schoenfelder S, Nagano T and Fraser P (2010). Transcription Factories and Nuclear Organization of the Genome. Cold Spring Harbor Symposia on Quantitative Biology 75(0), 501–506. [DOI] [PubMed] [Google Scholar]
- Finn E and Misteli T (2019). Molecular basis and biological function of variability in spatial genome organization. Science 365(6457), p.eaaw9498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyamer I, Gassler J, Imakaev M, Brandão H, Ulianov S, Abdennur N, Razin S, Mirny L and Tachibana-Konwalski K (2017). Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 544(7648), 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford J, Odeyale O and Shen C (2008). Activator-dependent recruitment of SWI/SNF and INO80 during INO1 activation. Biochemical and Biophysical Research Communications 373, 602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser P and Bickmore W (2007). Nuclear organization of the genome and the potential for gene regulation. Nature 447(7143), 413–417. [DOI] [PubMed] [Google Scholar]
- Gerhold C, Hauer M and Gasser S (2015). INO80-C and SWR-C: Guardians of the Genome. Journal of Molecular Biology 427, 637–651. [DOI] [PubMed] [Google Scholar]
- Haeusler R, Pratt-Hyatt M, Good P, Gipson T and Engelke D (2008). Clustering of yeast tRNA genes is mediated by specific association of condensin with tRNA gene transcription complexes. Genes & Development 22(16)_, 2204–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr J, Luperchio T, Wong X, Cohen E, Wheelan S and Reddy K (2015). Directed targeting of chromatin to the nuclear lamina is mediated by chromatin state and A-type lamins. Journal of Cell Biol. 208(1), 33–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J and Laemmli U (2008). The Anchor-Away Technique: Rapid, Conditional Establishment of Yeast Mutant Phenotypes. Mol. Cell 31, 925–932. [DOI] [PubMed] [Google Scholar]
- Heun P, Laroche T, Shimada K, Furrer P and Gasser S (2001). Chromosome Dynamics in the Yeast Interphase Nucleus. Science 294, 2181–2186. [DOI] [PubMed] [Google Scholar]
- Higuchi R, Vevea JD, Swayne TC, Chojnowski R, Hill V, Boldogh IR and Pon LA (2013). Actin Dynamics Affect Mitochondrial Quality Control and Aging in Budding Yeast. Current Biology 23, 2417–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Li H and Zhang J (2010). Analysis of transcriptional synergy between upstream regions and introns in ribosomal protein genes of yeast. Computational Biology and Chemistry 34(2), 106–114. [DOI] [PubMed] [Google Scholar]
- Hug C, Grimaldi A, Kruse K and Vaquerizas J (2017). Chromatin Architecture Emerges during Zygotic Genome Activation Independent of Transcription. Cell 169(2), 216–228.e19. [DOI] [PubMed] [Google Scholar]
- Kaishima M, Ishii J, Matsuno T, Fukuda N and Kondo A (2016). Expression of varied GFPs in Saccharomyces cerevisiae: codon optimization yields stronger than expected expression and fluorescence intensity. Scientific Reports 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna N, Hu Y and Belmont A (2014). HSP70 Transgene Directed Motion to Nuclear Speckles Facilitates Heat Shock Activation. Current Biology 24, 1138–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kueh H, Brieher W and Mitchison T (2010). Quantitative Analysis of Actin Turnover in Listeria Comet Tails: Evidence for Catastrophic Filament Turnover. Biophysical Journal 99, 2153–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuras L and Struhl K (1999). Binding of TBP to promoters in vivo is stimulated by activators and requires PolII holoenzyme. Nature 399, 609–613. [DOI] [PubMed] [Google Scholar]
- Levi V, Ruan Q, Plutz M, Belmont A and Gratton E (2005). Chromatin Dynamics in Interphase Cells Revealed by Tracking in a Two-Photon Excitation Microscope. Biophysical Journal 89, 4275–4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichter P, Cremer T, Borden J, Manuelidis L and Ward D (1988). Delineation of individual human chromosomes in metaphase and interphase cells by in situ suppression hybridization using recombinant DNA libraries. Human Genetics 80(3), 224–234. [DOI] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum N, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie B, Sabo P, Dorschner M, Sandstrom R, Bernstein B, Bender M, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny L, Lander E and Dekker J (2009). Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 326(5950), 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light W, Brickner D, Brand V and Brickner J (2010). Interaction of a DNA Zip Code with the Nuclear Pore Complex Promotes H2A.Z Incorporation and INO1 Transcriptional Memory. Molecular Cell 40, 112–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine M, Mckenzie III A, Demarini D, Shah N, Wach A, Brachat A, Philippsen P and Pringle J (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961. [DOI] [PubMed] [Google Scholar]
- Lord M and Pollard T (2004). UCS protein Rng3p activates actin filament gliding by fission yeast myosin-II. The Journal of Cell Biology 167, 315–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Rechsteiner T and Richmond T (1999). Expression and Purification of Recombinant Histones and Nucleosome Reconstitution. Chromatin Protocols 119, 1–16. [DOI] [PubMed] [Google Scholar]
- Marshall W, Straight A, Marko J, Swedlow J, Dernburg A, Belmont A, Murray A, Agard D and Sedat J (1997). Interphase chromosomes undergo constrained diffusional motion in living cells. Current Biology 7, 930–939. [DOI] [PubMed] [Google Scholar]
- McIntosh B and Ostap E (2016). Myosin-I molecular motors at a glance. Journal of Cell Science 129, 2689–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra M, D’souza V, Chang K, Huang Y and Balasubramanian M (2005). Hsp90 Protein in Fission Yeast Swo1p and UCS Protein Rng3p Facilitate Myosin II Assembly and Function. Eukaryotic Cell 4, 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T (2007). Beyond the Sequence: Cellular Organization of Genome Function. Cell 128(4), 787–800. [DOI] [PubMed] [Google Scholar]
- Mizuguchi G, Wu W, Alami S and Luk E (2012). Biochemical Assay for Histone H2A.Z Replacement by the Yeast SWR1 Chromatin Remodeling Complex. Methods in Enzymology, 512, 275–291. [DOI] [PubMed] [Google Scholar]
- Moseley J and Goode B (2006). The Yeast Actin Cytoskeleton: from Cellular Function to Biochemical Mechanism. Microbiology and Molecular Biology Reviews 70, 605–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumova N, Imakaev M, Fudenberg G, Zhan Y, Lajoie B, Mirny L and Dekker J (2013). Organization of the Mitotic Chromosome. Science 342(6161), 948–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman D, Groen A, Ohi R, Maresca T, Mirny L and Mitchison T (2010). Fast Microtubule Dynamics in Meiotic Spindles Measured by Single Molecule Imaging: Evidence That the Spindle Environment Does Not Stabilize Microtubules. Molecular Biology of the Cell 21, 323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann F, Dion V, Gehlen L, Tsai-Pflugfelder M, Schmid R, Taddei A and Gasser S (2012). Targeted INO80 enhances subnuclear chromatin movement and ectopic homologous recombination. Genes & Development 26, 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma K, Cam H, Maraia R and Grewal S (2006). A Role for TFIIIC Transcription Factor Complex in Genome Organization. Cell 125)5), 859–872. [DOI] [PubMed] [Google Scholar]
- Nora E, Lajoie B, Schulz E, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum N, Meisig J, Sedat J, Gribnau J, Barillot E, Blüthgen N, Dekker J and Heard E (2012). Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485(7398), 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normoyle K and Brieher W (2012). Cyclase-associated Protein (CAP) Acts Directly on F-actin to Accelerate Cofilin-mediated Actin Severing across the Range of Physiological pH. Journal of Biological Chemistry 287, 35722–35732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oomen M and Dekker J (2017). Epigenetic characteristics of the mitotic chromosome in 1D and 3D. Critical Reviews in Biochemistry and Molecular Biology 52(2), 185–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouspenski I, Elledge S and Brinkley B (1999). New yeast genes important for chromosome integrity and segregation identified by dosage effects on genome stability. Nucleic Acids Research 27, 3001–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papamichos-Chronakis M and Peterson C (2013). Chromatin and the genome integrity network. Nature Reviews Genetics 14, 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisis N, Krasinska L, Harker B, Urbach S, Rossignol M, Camasses A, Dewar J, Morin N and Fisher D (2017). Initiation of DNA replication requires actin dynamics and formin activity. The EMBO Journal 36, 3212–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plessner M and Grosse R (2015). Extracellular signaling cues for nuclear actin polymerization. European Journal of Cell Biology 94, 359–362. [DOI] [PubMed] [Google Scholar]
- Plessner M, Melak M, Chinchilla P, Baarlink C and Grosse R (2015). Nuclear F-actin Formation and Reorganization upon Cell Spreading. Journal of Biological Chemistry 290, 11209–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard T (2016). Actin and Actin-Binding Proteins. Cold Spring Harbor Perspectives in Biology 8, p.a018226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posern G, Sotiropoulos A, and Treisman R (2002). Mutant Actins Demonstrate a Role for Unpolymerized Actin in Control of Transcription by Serum Response Factor. Molecular Biology Of The Cell 13, 4167–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randise-Hinchliff C and Brickner J (2016). Transcription factors dynamically control the spatial organization of the yeast genome. Nucleus 7, 369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randise-Hinchliff C, Coukos R, Sood V, Sumner M, Zdraljevic S, Meldi Sholl L, Garvey Brickner D, Ahmed S, Watchmaker L and Brickner J (2016). Strategies to regulate transcription factor–mediated gene positioning and interchromosomal clustering at the nuclear periphery. The Journal of Cell Biology 212, 633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando O, Zhao K, Janmey P and Crabtree G (2002). Phosphatidylinositol-dependent actin filament binding by the SWI/SNF-like BAF chromatin remodeling complex. Proceedings of the National Academy of Sciences 99, 2824–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J, Crevenna A, Kessenbrock K, Yu J, Neukirchen D, Bista M, Bradke F, Jenne D, Holak T, Werb Z, Sixt M and Wedlich-Soldner R (2008). Lifeact: a versatile marker to visualize F-actin. Nature Methods 5, 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rines D, Thomann D, Dorn J, Goodwin P and Sorger P (2011). Live Cell Imaging of Yeast. Cold Spring Harbor Protocols 2011, pdb.top065482–pdb.top065482. [DOI] [PubMed] [Google Scholar]
- Ross J, Ali M and Warshaw D (2008). Cargo transport: molecular motors navigate a complex cytoskeleton. Current Opinion in Cell Biology 20, 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad H, Gallardo F, Dalvai M, Tanguy-le-Gac N, Lane D and Bystricky K (2014). DNA Dynamics during Early Double-Strand Break Processing Revealed by Non-Intrusive Imaging of Living Cells. PLoS Genetics 10, p.e1004187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawarkar R,, Sievers C and Paro R (2012) Hsp90 globally targets paused RNA polymerase to regulate gene expression in response to environmental stimuli. Cell 149, 807–818. [DOI] [PubMed] [Google Scholar]
- Schoenfelder S, Sexton T, Chakalova L, Cope N, Horton A, Andrews S, Kurukuti S, Mitchell J, Umlauf D, Dimitrova D, Eskiw C, Luo Y, Wei C, Ruan Y, Bieker J and Fraser P (2010). Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nature Genetics 42, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrank B, Aparicio T, Li Y, Chang W, Chait B, Gundersen G, Gottesman M and Gautier J (2018). Nuclear ARP2/3 drives DNA break clustering for homology-directed repair. Nature 559, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sckolnick M, Krementsova E, Warshaw D and Trybus K (2016). Tropomyosin isoforms bias actin track selection by vertebrate myosin Va. Molecular Biology of the Cell 27, 2889–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeber A and Gasser S (2017). Chromatin organization and dynamics in double-strand break repair. Current Opinion in Genetics & Development 43, 9–16. [DOI] [PubMed] [Google Scholar]
- Seeber A, Hauer M and Gasser S (2018). Chromosome Dynamics in Response to DNA Damage. Annual Review of Genetics 52, 295–319. [DOI] [PubMed] [Google Scholar]
- Siddiqui A and Brandriss M (1989). The Saccharomyces cerevisiae PUT3 activator protein associates with proline-specific upstream activation sequences. Molecular and Cellular Biology 9, 4706–4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha M and Peterson C (2009). Chromatin dynamics during repair of chromosomal DNA double-strand breaks. Epigenomics 1, 371–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutoglou E and Misteli T (2007). Mobility and immobility of chromatin in transcription and genome stability. Current Opinion in Genetics & Development 17, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spichal M and Fabre E (2017). The Emerging Role of the Cytoskeleton in Chromosome Dynamics. Frontiers in Genetics, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilianakis C, Lalioti M, Town T, Lee G and Flavell R (2005). Interchromosomal associations between alternatively expressed loci. Nature 435(7042), 637–645. [DOI] [PubMed] [Google Scholar]
- Stuven T, Hartmann E and Görlich D (2003). Exportin 6: a novel nuclear export receptor that is specific for profilin{middle dot}actin complexes. The EMBO Journal 22, 5928–5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan K, Flynn P, Reece R and Marmorstein R (1997). Crystal structure of a PUT3–DNA complex reveals a novel mechanism for DMA recognition by a protein containing a Zn2Cys6 binuclear cluster. Nature Structural Biology 4, 751–759. [DOI] [PubMed] [Google Scholar]
- Taddei A, Van Houwe G, Hediger F, Kalck V, Cubizolles F, Schober H and Gasser S (2006). Nuclear pore association confers optimal expression levels for an inducible yeast gene. Nature 441, 774–778. [DOI] [PubMed] [Google Scholar]
- Takizawa T, Gudla P, Guo L, Lockett S and Misteli T (2008). Allele-specific nuclear positioning of the monoallelically expressed astrocyte marker GFAP. Genes & Development 22(4), 489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanizawa H, Iwasaki O, Tanaka A, Capizzi J, Wickramasinghe P, Lee M, Fu Z and Noma K (2013). Mapping of Long-Range Associations throughout the Fission Yeast Genome Reveals Global Genome Organization Linked to Transcriptional Regulation. Biophysical Journal 104, 425a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus M (2018). Myosin-Driven Intracellular Transport. Cold Spring Harbor Perspectives in Biology 10, p.a021972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toogun O, Zeiger W and Freeman B (2007). The p23 molecular chaperone promotes functional telomerase complexes through DNA dissociation. Proceedings of the National Academy of Sciences 104, 5765–5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S and Workman J (2015). Histone exchange, chromatin structure and the regulation of transcription. Nature Reviews Molecular Cell Biology 16, 178–189. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Tan D, Lakshminarasimhan M, Washburn M, Erica Hong E, Walz T and Peterson C (2015). Structural analyses of the chromatin remodelling enzymes INO80-C and SWR-C. Nature Communications 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischnitzer S (1973). The Submicroscopic Morphology of the Interphase Nucleus. International Review of Cytology, 1–48. [DOI] [PubMed] [Google Scholar]
- Wu W, Wu C, Ladurner A, Mizuguchi G, Wei D, Xiao H, Luk E, Ranjan A and Wu C (2008). N Terminus of Swr1 Binds to Histone H2AZ and Provides a Platform for Subunit Assembly in the Chromatin Remodeling Complex. Journal of Biological Chemistry 284, 6200–6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M and Cook P (2008). Similar active genes cluster in specialized transcription factories. The Journal of Cell Biology 181, 615–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Pradhan S, Sun F, Chronis C, Tran N, Su T, Van C, Vashisht A, Wohlschlegel J, Peterson C, Timmers H, Kurdistani S and Carey M (2017). Mot1, Ino80C, and NC2 Function Coordinately to Regulate Pervasive Transcription in Yeast and Mammals. Molecular Cell 67, 594–607.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Vinayachandran V and Pugh B (2013). SWR-C and INO80 Chromatin Remodelers Recognize Nucleosome-free Regions Near +1 Nucleosomes. Cell 154, 1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Shimada K, Oma Y, Kalck V, Akimura K, Taddei A, Iwahashi H, Kugou K, Ohta K, Gasser S and Harata M (2010). Actin-Related Protein Arp6 Influences H2A.Z-Dependent and -Independent Gene Expression and Links Ribosomal Protein Genes to Nuclear Pores. PLoS Genetics 6, p.e1000910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelin E, Zhang Y, Toogun O, Zhong S and Freeman B (2012). The p23 Molecular Chaperone and GCN5 Acetylase Jointly Modulate Protein-DNA Dynamics and Open Chromatin Status. Molecular Cell 48, 459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C and Narlikar G (2016). Analysis of Nucleosome Sliding by ATP-Dependent Chromatin Remodeling Enzymes. Methods in Enzymology 573, 119–135. [DOI] [PubMed] [Google Scholar]
- Zullo J, Demarco I, Piqué-Regi R, Gaffney D, Epstein C, Spooner C, Luperchio T, Bernstein B, Pritchard J, Reddy K and Singh H (2012). DNA Sequence-Dependent Compartmentalization and Silencing of Chromatin at the Nuclear Lamina. Cell 149(7), 1474–1487. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Related to Figure 1.
The behavior of the control GFP-marked INO1 locus in the presence of inositol was monitored using a DeltaVision OMX microscope with an InsightSSI solid state illumination system and 100x 1.40 NA oil immersion objective collected at 1 frame per second.
Movie S2. Related to Figure 1.
To tract the motion of INO1 in the absence of inositol 5 min movies were taken 20 min after transferring cells to inositol free media and the acquired movies were checked for cells displaying a clear shift of INO1 from the inner nucleoplasm to the periphery. A typical cell displaying motion of INO1 from the inner nucleoplasm to the periphery is shown. The images were acquired using a DeltaVision OMX microscope.
Movie S3. Related to Figure 2.
The behavior of the GFP-marked INO1 locus was visualized in mother and a newly emerged daughter cell (bud neck apparent) in parental yeast. The images were acquired using a DeltaVision OMX microscope.
Movie S4. Related to Figure 2.
The behavior of the GFP-marked INO1 locus was visualized in mother and a newly emerged daughter cell (bud neck apparent) in p23 null yeast. The images were acquired using a DeltaVision OMX microscope.
Movie S5. Related to Figure 4.
Nuclear actin was visualized using an actin chromobody. Yeast with the GFP-marked INO1 locus were transformed with an expression vector for an actin chromobody, which is an actin nanobody (single domain antibody) fused to GFP (S65T) and 3x NLS tags. A representative image acquired with a DeltaVision OMX microscope is shown.
Movie S6. Related to Figure 4.
Transformants expressing the actin chromobody were exposed to the actin polymerization inhibitor Cytochalasin D (20 ?M) for 1 h prior to image collection with the DeltaVision OMX. The signal intensity of the GFP-mark near INO1 was used to normalize the exposures shown in Movie S5 and S6.
Movie S7. Related to Figure 4.
Images of parental yeast expressing the actin chromobody were acquired with a Zeiss LSM 880 Airyscan microscope using the 488 and 561 excitation channels with an oil immersion 63x objective collected at 1 frame per 100 msec.
Movie S8. Related to Figure 4.
To gauge whether the actin signal in the Airyscan images (Movie S7) displayed polymerization dynamics a movie was prepared by averaging 4 consecutive exposures using the data collected in the 488 channel (Needleman et al., 2010).
Movie S9. Related to Figure 4.
A mobile INO1 locus does not track along stable, extended actin fibers. A cell displaying multiple long-range motions of the GFP-mark within parental yeast grown under inositol starvation conditions is provided. The images were acquired using a DeltaVision OMX microscope.
Movie S10. Related to Figure 5.
Myo3-TAP purified from yeast is an inactive motor based upon an actin-gliding assay.
Movie S11. Related to Figure 5.
Co-incubation of Myo3-TAP with purified Hsp82 and TAP-She4 activates myosin motor activity in an actin-gliding assay.
Data Availability Statement
This study did not generate any datasets. Image analysis was done using Fiji, a free software available online.