

SUMMARY
Genome editing technologies have transformed our ability to engineer desired genomic changes within living systems. However, detecting precise genomic modifications often requires sophisticated, expensive, and time-consuming experimental approaches. Here, we describe DTECT (Dinucleotide signaTurE CapTure), a rapid and versatile detection method that relies on the capture of targeted dinucleotide signatures resulting from the digestion of genomic DNA amplicons by the type IIS restriction enzyme AcuI. DTECT enables the accurate quantification of marker-free precision genome editing events introduced by CRISPR-dependent homology-directed repair, base editing, or prime editing in various biological systems, such as mammalian cell lines, organoids, and tissues. Furthermore, DTECT allows the identification of oncogenic mutations in cancer mouse models, patient-derived xenografts, and human cancer patient samples. The ease, speed, and cost efficiency by which DTECT identifies genomic signatures should facilitate the generation of marker-free cellular and animal models of human disease and expedite the detection of human pathogenic variants.
Graphical Abstract
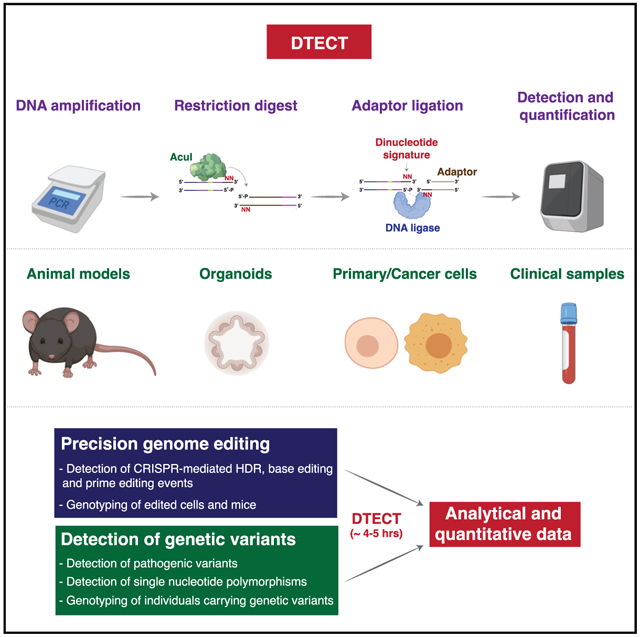
In Brief
Billon et al. report the development of a versatile detection method based on the capture of targeted genomic signatures. This method allows the detection and quantification of genomic signatures introduced by marker-free precision genome editing or resulting from genetic variation.
INTRODUCTION
Precision genome editing allows the modeling and correction of desired genomic variants containing insertions or deletions of specific nucleotide sequences or changes in single DNA bases (Anzalone et al., 2019; Barbieri et al., 2017; Cong et al., 2013; Dow, 2015; Guo et al., 2018; Liu et al., 2018; Mali et al., 2013; Roy et al., 2018). Precise editing of the genome can be obtained by CRISPR-dependent homology-directed repair (HDR) of Cas9-induced DNA double-stranded breaks (DSBs) (Jasin and Haber, 2016). Alternatively, precision genome editing can result from the use of DSB-free methods, such as CRISPR-dependent base editing, which uses cytidine or adenosine deaminases fused to a nickase Cas9 (nCas9) mutant to generate base transitions (Gaudelli et al., 2017; Komor et al., 2016), or prime editing, which employs a reverse transcriptase-nCas9 fusion and a template prime editing guide RNA (pegRNA) to install into the genome a large variety of genomic changes, including transversions, transitions, and small insertions and deletions (indels) (Anzalone et al., 2019).
Genome editing has been facilitated by the development of accessible and cost-effective methods for the detection of small indels resulting from the repair of Cas9-induced DSBs, such as the T7E1 and Surveyor nuclease assays (Mashal et al., 1995; Qiu et al., 2004; Ran et al., 2013). However, because these methods do not determine the identity of DNA bases, they are ill suited for the detection of genomic changes introduced by precision genome editing (Germini et al., 2018). Precision genome editing events can be detected by the addition of genomic markers by CRISPR-dependent HDR or prime editing, such as silent mutations that create or disrupt restriction sites, or selectable reporters encoding for antibiotic resistance or fluorescent proteins. However, the use of genomic markers entails an elaborate experimental design that is unique for each targeted site and can result in unintended perturbations of coding or non-coding genomic elements. Moreover, marker-based detection methods are not compatible with CRISPR-dependent base editing strategies, which induce single DNA base changes (Rees and Liu, 2018). Alternative methods that employ Sanger sequencing or next-generation sequencing (NGS) enable the detection of precise genomic changes without the use of genomic markers (Brinkman et al., 2014; Pinello et al., 2016). However, Sanger-sequencing-based approaches suffer from low sensitivity and precision because of the variable quality of the sequencing reactions and background signals that often affect the sequencing reads (Brinkman et al., 2014, 2018). Furthermore, NGS-dependent detection strategies, while highly sensitive (Clement et al., 2019; Lindsay et al., 2016; Pinello et al., 2016), remain expensive and time consuming, which limits their value for the development of mutant cell lines and animal models and for applications that require a rapid turnaround time, such as the identification of pathogenic variants in certain clinical settings. Therefore, a simple, efficient, inexpensive, and rapid method that enables quantitative detection of genetic variants in complex biological systems is needed.
In this study, we describe a versatile method that uses standard molecular biology techniques to detect variants introduced by precision genome editing or resulting from genetic variation. We show that this detection method, designated Dinucleotide signaTurE CapTure (DTECT), enables accurate and sensitive quantification of marker-free precision genome editing events induced by CRISPR-dependent HDR, base editing, and prime editing. In addition, we show that DTECT can readily identify oncogenic mutations in cancer mouse models, patient-derived xenografts (PDXs), and cancer patient samples. These studies establish a cost-effective method for the rapid detection of genetic variants, which will aid the generation of marker-free cellular and animal models of human disease and expedite the detection of pathogenic variants for clinical applications.
RESULTS
Design of DTECT, a Detection Method Based on the Capture of Dinucleotide Signatures
In our detection method, we take advantage of the property of type IIS restriction enzymes to generate single-stranded DNA overhangs at a specific distance from their recognition motif. Based on the preceding property, we hypothesized that single-stranded DNA overhangs generated by digestion of genomic DNA (gDNA) sequences with type IIS restriction enzymes could be captured and identified using DNA adaptors containing overhangs complementary to the exposed DNA signatures (Figure 1A). To identify type IIS enzymes with efficient and accurate endonuclease activity, we analyzed the properties of known type IIS enzymes. Restriction enzymes optimal for our method exhibit the following characteristics: (1) they cleave far from their recognition motif, enabling the incorporation of non-complementary type IIS recognition motifs into PCR primers without disrupting gDNA amplification (Figures 1A and S1A); (2) they bind a single recognition motif (Bath et al., 2002; Figure S1A); and (3) they possess highly specific endonuclease activity, generating a limited number of cleavage byproducts due to slippage activity (Lundin et al., 2015; Figure S1B). Among the >40 known types IIS endonucleases, only 6 enzymes cleave at a distance of ≥14 bases from their recognition motif (AcuI, BpmI, BpuEI, BsgI, MmeI, and NmeAIII) (Figure S1C). Of those enzymes, only AcuI and BpuEI have a single recognition motif, and AcuI exhibits the lowest slippage activity of the two enzymes (slippage byproducts: AcuI, 1.1%; BpuEI, 41.4%) (Lundin et al., 2015). In particular, upon DNA cleavage, AcuI exposes a dinucleotide signature located 15/16 nucleotides from its recognition site (Figure S1D). Based on the preceding considerations, AcuI is the most suitable restriction enzyme for our detection method.
Figure 1. Identification of Targeted Dinucleotide Signatures Using DTECT.
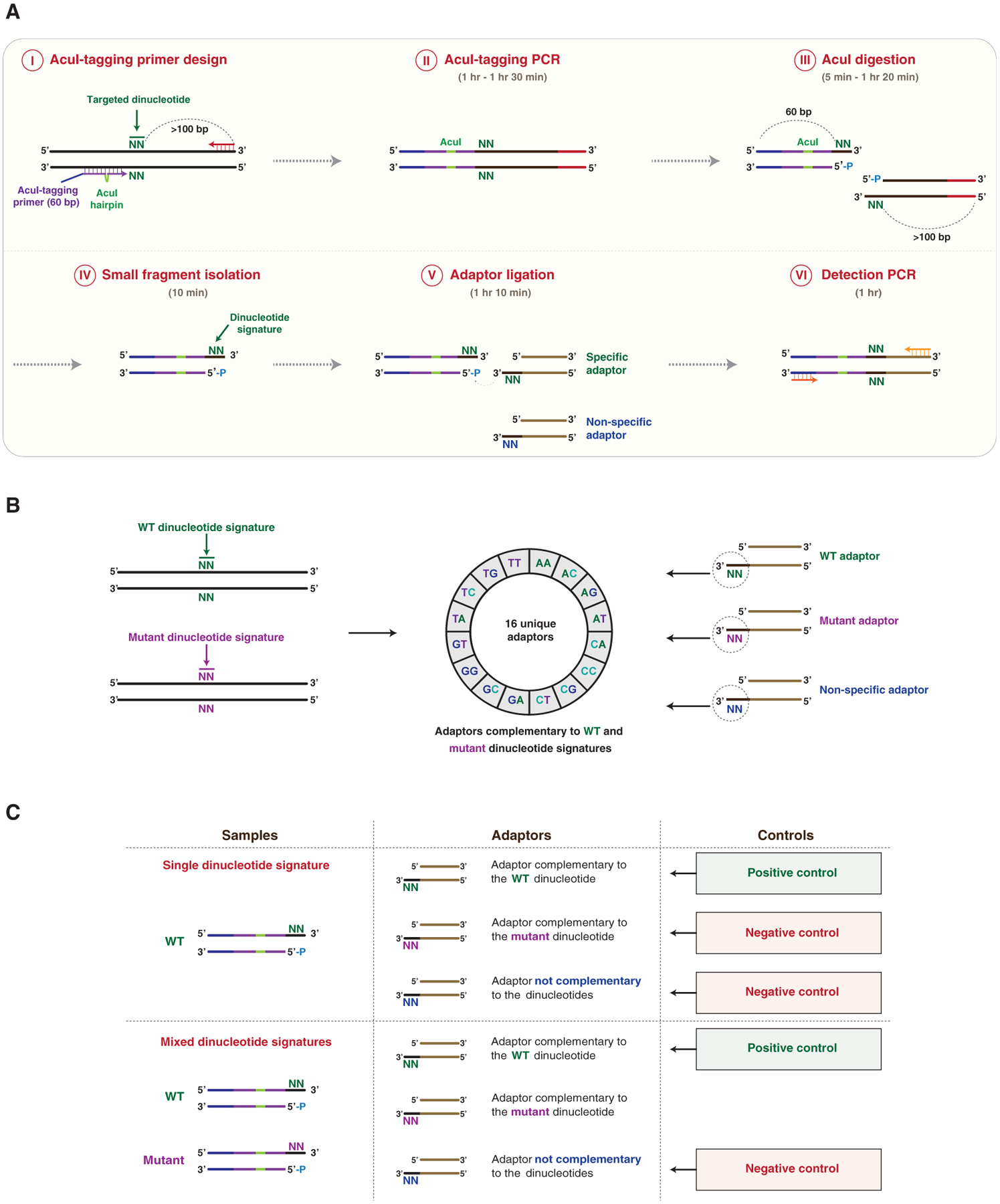
(A) Schematic representation of DTECT. The targeted genomic locus containing a hypothetical targeted dinucleotide (N = A, C, G, or T; green) is PCR amplified using a forward AcuI-tagging primer juxtaposed to the targeted dinucleotide and a locus-specific DNA primer (AcuI-tagging primer design and PCR, steps I and II). The AcuI-tagging primer (60 nt) consists of DNA sequences complementary to the genomic locus (purple) interrupted by a hairpin containing an AcuI recognition site (green), and a non-complementary DNA sequence (blue). The locus-specific reverse primer (red) is located >100 bp from the targeted dinucleotide. The obtained PCR product is subsequently cleaved by the AcuI restriction enzyme in a position adjacent to the targeted dinucleotide, resulting in the generation of two DNA fragments of 60 and >100 bp (AcuI digestion, step III). The 60 bp fragment containing the exposed signature of the targeted dinucleotide is then isolated using SPRI beads, with higher affinity toward >100 bp DNA products (small fragment isolation, step IV). The 60 bp fragment is then ligated to DNA adaptors containing 3′ overhangs of two bases complementary (specific) or not (non-specific) to the dinucleotide signature (adaptor ligation, step V). The ligated product is then subjected to PCR amplification for analytical or quantitative detection (detection PCR, step VI). The approximate time required for each step is indicated.
(B) Schematics of the DTECT adaptor library. Control (green) and mutant (purple) dinucleotide signatures (left panel) are detected using a library of 16 unique adaptors (middle panel). The library contains adaptors with dinucleotides complementary to the control (green) or mutant (purple) signature, as well as non-specific adaptors (blue) (right panel).
(C) Schematics of the positive and negative controls used in DTECT experiments to identify signatures of interest (e.g., mutant allele) in allele populations. In gDNA samples containing only the WT dinucleotide signature, the adaptor complementary to the WT dinucleotide signature (green) serves as a positive control, while the adaptor complementary to the mutant signature of interest (purple) and a non-specific adaptor (blue) are used as negative controls. In gDNA samples containing a mixture of the WT and the mutant dinucleotide signature, the adaptor complementary to the WT dinucleotide signature (green) is used as a positive control and a non-specific adaptor (blue) serves as a negative control. The adaptor complementary to the mutant dinucleotide signature (purple) is used to detect the presence of the variant of interest and quantify its frequency.
See also Figure S1.
In our approach, the genomic locus of interest is PCR amplified using a locus-specific DNA primer (red) and a DNA oligonucleotide (AcuI-tagging primer) containing two regions of complementarity to the genomic locus (purple) interrupted by an AcuI recognition site (AcuI hairpin, green) positioned 14 bp upstream of a dinucleotide of interest (Figure 1A, steps I and II). Tagging the genomic amplicon with an AcuI motif allows AcuI-mediated digestion of the sequence of interest on the 3′ side of the targeted dinucleotide. Upon AcuI-mediated digestion, the signature of the targeted dinucleotide becomes exposed (Figure 1A, step III). To proceed with a single DNA fragment containing the targeted dinucleotide, the larger DNA fragment (>100 bp) resulting from AcuI-mediated digestion is removed using solid-phase reversible immobilization (SPRI) beads (Figure 1A, step IV) and the smaller DNA fragment (60 bp) containing the targeted dinucleotide is ligated to an adaptor with a 3′ overhang complementary to the exposed signature (Figure 1A, step V). The ligated DNA products are subsequently detected by analytical or quantitative PCR (qPCR) (Figure 1A, step VI). This method, which we named DTECT, can be completed within 4–5 h (Figure 1A). A common set of DNA primers that anneal to constant regions in the AcuI-digested fragments (blue) and the ligated adaptors (brown) is used in all DTECT experiments (Figure 1A, step VI), avoiding locus-specific amplification bias and variability in qPCR efficiency among distinct sets of samples. Considering the total number of 16 unique dinucleotides (24), a library of 16 distinct adaptors is sufficient to capture all dinucleotide signatures that can be generated by AcuI (Figure 1B). Positive and negative controls are used in DTECT assays to determine the efficiency and specificity of dinucleotide capture (Figure 1C), providing a highly controlled assessment of successful and specific capture of dinucleotide signatures.
DTECT Efficiently Captures Dinucleotide Signatures Generated by AcuI-Mediated Digestion
To demonstrate the feasibility of DTECT, we designed two AcuI-tagging DNA primers flanking four adjacent bases (5′-TTGG-3′) on opposite DNA strands (TT and CC signatures, blue) (Figure 2A). Upon PCR amplification using AcuI-tagging primers and locus-specific DNA primers, the PCR amplicons were digested and ligated to adaptors with either complementary or non-specific 3′ overhangs (GG or AA). Detection of the ligated products by PCR, as described earlier, revealed that the GG and AA adaptors specifically captured the DNA fragments containing the CC and TT dinucleotides, respectively (Figure 2B). Sanger sequencing confirmed that the amplicons of the ligated DNA products had the expected genomic sequence (purple) adjacent to the AcuI motif (green) and the GG or AA adaptors (brown) (Figures S2A and S2B). Importantly, robust amplification of captured DNA products was observed only upon (1) capture of the AcuI-digested products with complementary adaptors (Figure 2B), (2) AcuI-mediated cutting and generation of 5′-phosphorylated DNA fragments (Figures 2C and 2D), and (3) DNA ligation by the T4 DNA ligase (Figure 2D). We additionally showed that each DNA base can be identified by designing 4 independent AcuI-tagging primers (2 on each DNA strand), enabling the capture of 4 distinct signatures per gDNA base (Figures 2E and 2F). This DTECT feature allows flexible AcuI-mediated cleavage of gDNA amplicons containing targeted DNA sequences. In additional studies, we confirmed that each of the 16 possible dinucleotide signatures generated by AcuI at two independent target sites can be efficiently captured using DNA adaptors containing complementary DNA overhangs (Figure S2C). Altogether, these studies establish DTECT as a rapid and efficient method to identify DNA bases through the capture of AcuI-induced dinucleotide signatures using a common and unique set of adaptors.
Figure 2. Detection and Quantification of Dinucleotide Signatures Using DTECT.
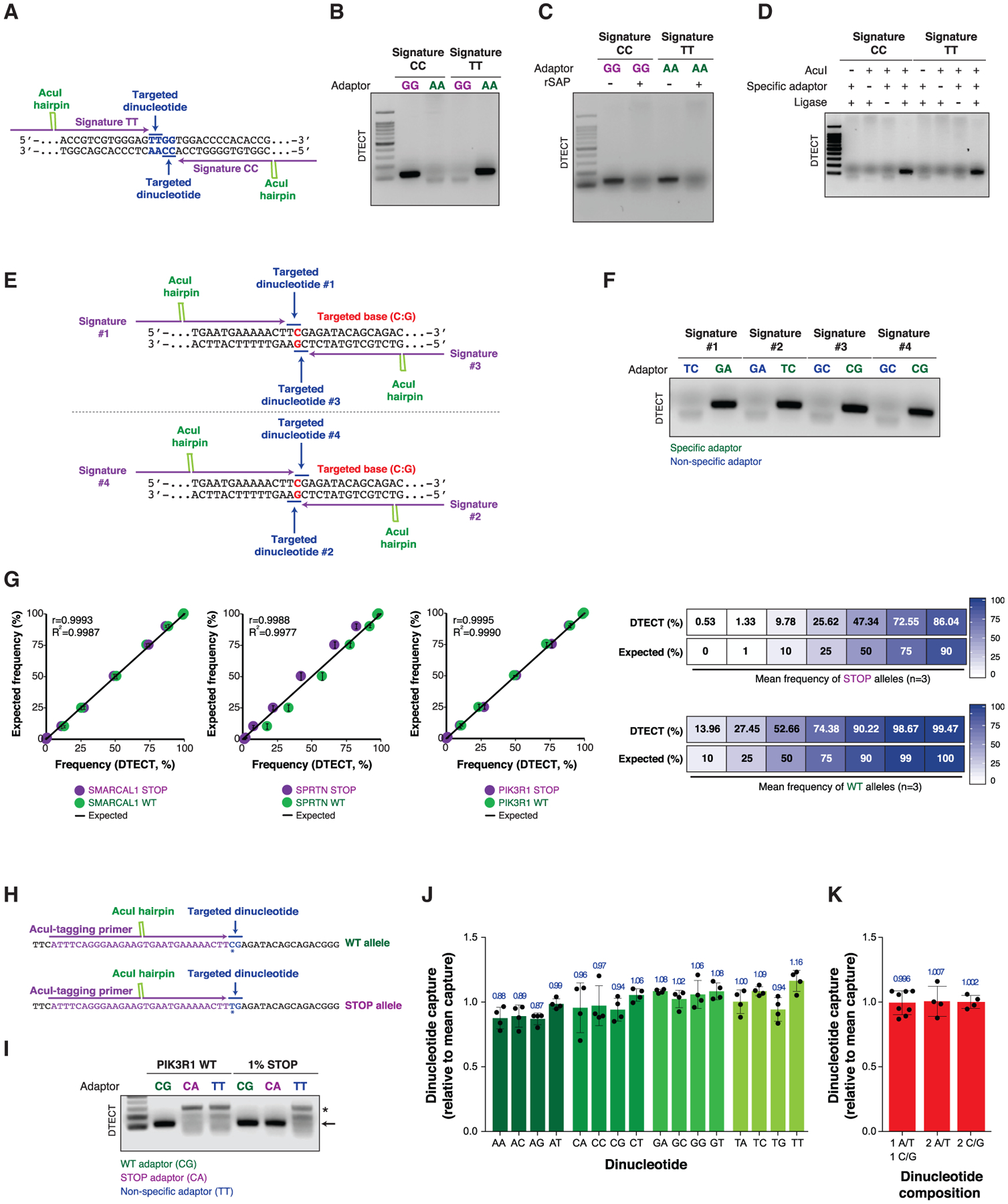
(A) Design of AcuI-tagging primers that allow the capture of two dinucleotide signatures (CC and TT, blue) on opposite DNA strands.
(B) PCR amplification (22 cycles) of the AcuI-digested DNA products containing the CC and TT signatures shown in (A), which have been captured using GG or AA adaptors.
(C) PCR amplification (22 cycles) of DNA fragments captured as in (B) with or without dephosphorylation of the AcuI-digested products by the shrimp alkaline phosphatase (rSAP).
(D) PCR amplification (22 cycles) of DNA fragments captured as in (B) in the absence or presence of AcuI, DNA adaptors (GG adaptor for signature CC and AA adaptor for signature TT), or T4 DNA ligase.
(E) Schematic representation of the AcuI-tagging primer design for detecting four possible dinucleotide signatures (1–4) containing the same targeted base (C:G, red) in the PIK3R1 gene.
(F) Detection of the four dinucleotide signatures shown in (E) by DTECT (18 PCR cycles) using specific (green) and non-specific (blue) adaptors.
(G) Quantification by DTECT of the relative abundance of SMARCAL1, SPRTN, and PIK3R1 WT (green) and STOP (purple) dinucleotide signatures in mixtures of WT and STOP alleles at predefined ratios. Graphs (left) represent the correlation between the frequency of WT and STOP variants determined by DTECT and the expected frequency of the same variants in the mixed populations for each of the preceding 3 genes. Error bars represent the SD of independent experiments (n = 2). Pearson correlation (r) was determined by comparing expected and DTECT-based frequency. Comparison of the mean frequency of STOP and WT signatures determined by DTECT and their expected frequency is shown in the right panel (n = 3 independent genes, SMARCAL1, SPRTN, and PIK3R1).
(H) Representation of the AcuI-tagging primers used to detect the WT and STOP alleles of the PIK3R1 gene. The targeted dinucleotides are shown in blue, the edited base is indicated with an asterisk, and part of the AcuI-tagging primer sequence is shown in purple.
(I) PCR amplification (25 cycles) of WT and STOP PIK3R1 alleles (arrow) captured using DTECT from WT:STOP allele mixtures (i.e., 100:0 and 99:1). An adaptor (CG) specific for the WT allele is used as a positive control, and a non-specific adaptor (TT) is used as a negative control. An adaptor that captures the STOP PIK3R1 allele (CA) serves as an additional negative control in the reaction containing only the WT allele. Background non-specific PCR products are indicated with an asterisk.
(J) Fold change variation in the frequency of capture of each of the 16 dinucleotide signatures relative to the mean dinucleotide capture frequency. Oligonucleotides containing distinct dinucleotide signatures are captured using specific adaptors. The fraction of captured material is then quantified by qPCR and normalized to the mean value obtained from the capture of all 16 dinucleotide signatures. Error bars indicate the SD of 4 independent experiments. Dots represent individual data points.
(K) Fold change variation in the frequency of capture of dinucleotide signatures with 1 A/T + 1 C/G, 2 A/T, or 2 C/G bases relative to the mean dinucleotide capture frequency, determined as described in (J). Error bars represent the SD of 8 mean values for dinucleotides with 1 A/T + 1 C/G and 4 mean values for dinucleotides with 2 A/T and 2 C/G, as determined in (J).
See also Figures S2, S3, and S10.
Figure 3. Detection and Quantification of Precision Genome Editing by CRISPR-Mediated HDR, Base Editing, and Prime Editing Using DTECT.
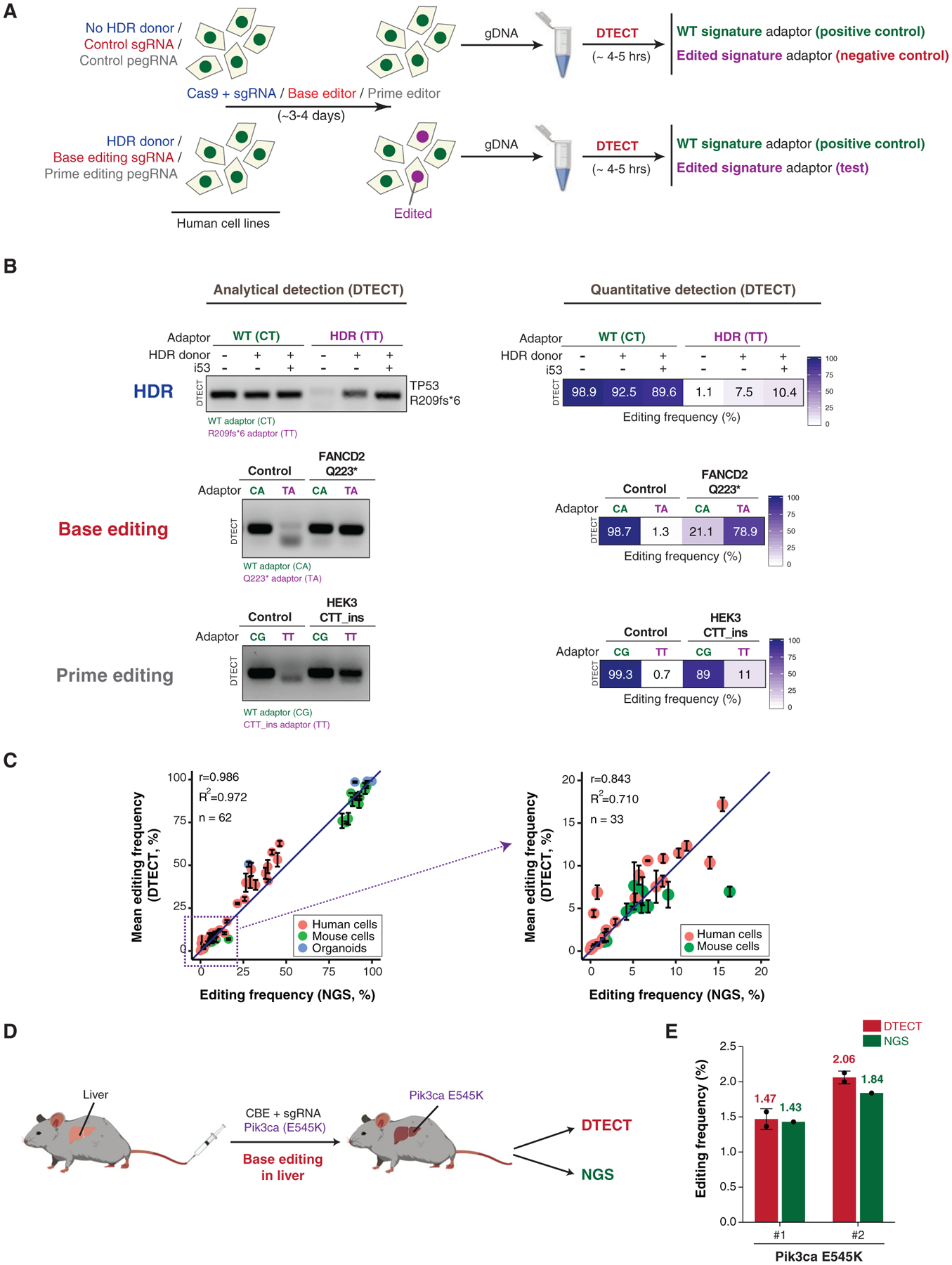
(A) Schematics of the protocol used to identify genomic changes introduced by CRISPR-dependent HDR, base editing, or prime editing. In HDR experiments (blue), HEK293T cells were transfected with Cas9 and an sgRNA targeting a gene of interest with or without donor DNA molecules. In base editing experiments (red), HEK293T cells were transfected with BE3 base editors with either control or base editing sgRNAs. Base editing experiments were also conducted in cells stably expressing FNLS-BE3. In prime editing experiments (gray), HEK293T cells were transfected with PE2 with or without pegRNA. gDNA was then extracted from cell populations and subjected to DTECT using adaptors specific for WT (green) or edited (purple) variants.
(B) Identification by DTECT of WT and HDR-edited (R209fs*6) TP53 alleles (top), WT and base-edited (Q223*) FANCD2 alleles (middle), and WT and prime-edited (CTT_ins) HEK3 alleles (bottom). Adaptors specific for the WT (CT, CA, and CG; green) or edited (TT and TA; purple) signatures were used in DTECT experiments. Captured samples were subjected to analytical PCR (left, 21 cycles) or qPCR (right). In the HDR experiment, cells were transfected with Cas9, sgRNA, and an ssODN specific for the TP53 locus with or without the HDR stimulatory factor i53. The ssODN was omitted in control reactions. In the base editing experiment, cells were transfected with BE3 and an sgRNA to induce Q223* in FANCD2. In prime editing experiments, cells were transfected with PE2 and pegRNA to introduce a CTT insertion in the HEK3 locus.
(C) Graphical representation of the correlation of DTECT- and NGS-based estimations of the frequency of genetic variants introduced by precision genome editing in human and mouse cells and mouse intestinal organoids (n = 62). Data points in the dashed box (frequency < 20%) of the left panel are enlarged in the right panel (n = 33). Error bars indicate the SEM of 2–5 independent replicates. The sources of the edited samples are indicated by distinct colors.
(D) Schematic representation of the experiments conducted to measure the efficiency of precision genome editing in vivo using DTECT. Editing of the mouse liver was performed by hydrodynamic injection of the cytidine base editor (CBE) FNLS-BE3 and an sgRNA to introduce the Pik3ca E545K variant. DTECT (red) and NGS (green) were used to determine the efficiency of editing in the mouse liver sample.
(E) Quantification by DTECT (red) and NGS (green) of the Pik3ca E545K variant introduced by CRISPR-mediated base editing in the mouse liver, as shown in (D). Error bars indicate the SD of 2 independent experiment. Dots represent individual data points.
See also Figures S4, S5, and S11.
DTECT Enables Specific and Sensitive Quantification of DNA Variants
Next, we examined whether DTECT can determine the relative abundance of DNA variants with distinct DNA signatures, including low-abundance DNA variants. To this end, we transfected HEK293T cells with single guide RNAs (sgRNAs) that introduce nonsense mutations into the SPRTN, PIK3R1, and SMARCAL1 genes using iSTOP, a CRISPR-mediated base editing approach that creates STOP codons within genes of interest (Billon et al., 2017; Figure S3A). We then cloned both wild-type (WT) and mutant alleles, which differ by a single base change (C→T) (Figure S3B), and subjected them to PCR amplification using a locus-specific DNA primer and an AcuI-tagging primer flanking the iSTOP-targeted DNA base (Figure S3C). The WT and edited PCR products were then mixed at different ratios (WT:STOP allele = 100:0, 99:1, 90:10, 75:25, 50:50, 25:75, or 10:90) and digested with AcuI. The resulting DNA fragments were then captured using adaptors complementary to WT (green) and STOP (purple) dinucleotide signatures (Figure S3A). Remarkably, qPCR analysis of the captured DNA fragments accurately determined the relative abundance of the WT and STOP alleles at the three loci indicated earlier (Figure 2G), demonstrating that DTECT can estimate the frequency of dinucleotide signatures in a mixed population with high precision, including variants with low abundance (1%) (Figure 2G). Low-abundance STOP variants in SPRTN and PIK3R1 were also detectable by analytical PCR (Figures 2H, 2I, S3C and S3D), confirming the high sensitivity and accuracy of DTECT. Direct comparison of the 16 DTECT adaptors revealed comparable efficiency in the capture of oligonucleotides containing complementary dinucleotide signatures (Figures 2J and 2K). In addition, all adaptors exhibited low levels of non-specific capture background (mean = 0.325%, ranging from 0.16% to 0.876%) (Figure S3E). The preceding observations indicate that the adaptor ligation is conducted under optimal conditions, as confirmed by kinetic analysis of the adaptor ligation reaction (Figure S3F). Altogether, these findings demonstrate that DTECT captures dinucleotide variants and quantifies their relative abundance with high specificity and sensitivity.
DTECT Accurately Identifies Genomic Changes Introduced by CRISPR-Dependent HDR, Base Editing, and Prime Editing in Mammalian Cells
To examine the ability of DTECT to identify precise genomic changes introduced into mammalian cell populations, we used CRISPR-mediated HDR for generating various types of disease-related mutations using single-stranded oligodeoxynucleotides (ssODNs), including a cancer-associated frameshift mutation in TP53 (i.e., R209fs*6), a missense mutation in HBB (i.e., G6V) that causes sickle cell anemia, a small tandem duplication in BRCA2 (dupAGAAGAT) identified in breast cancer, and small insertions into JAK2 and EMX1 (Paulsen et al., 2017), two genes associated with myeloproliferative disorders and Kallmann syndrome, respectively. Three days after co-transfection of Cas9 with site-specific sgRNAs and ssODNs into HEK293T cells, we harvested the cellular gDNA and used DTECT to determine by analytical and qPCR whether the desired changes were incorporated into the targeted chromosomal loci (Figure 3A). For comparison, a restriction fragment-length polymorphism (RFLP) assay that monitors restriction sites disrupted or created by the preceding mutations in the targeted genomic loci was conducted in parallel. In these experiments, DTECT readily captured the specific signature of the mutant variants (Figures 3B and S4A–S4C), while the RFLP assay either failed to detect or weakly detected the same mutant variants (Figures S4F–S4H). In addition, DTECT was able to discern the HDR stimulatory effect induced by i53 (Figures 3B, S4A, and S4B), a genetically encoded 53BP1 inhibitor that was previously shown to increase the frequency of HDR events (Canny et al., 2018). DTECT also clearly determined which mutations failed to be incorporated by the HDR machinery (e.g., BRCA2 dupAGAAGAT), as confirmed by NGS analysis (Figures S4D and S4E).
Next, to determine whether DTECT can identify precise genomic changes introduced by CRISPR-mediated base editing in mammalian cell populations, we used a cytidine base editor to install nonsense mutations into the Fanconi-anemia-associated genes FANCD2, FANCM, and SLX4; the DNA replication and circadian clock gene TIMELESS; and the Treacher Collins syndrome gene TCOF1. These experiments showed that DTECT was able to capture the signatures of the newly introduced variants in all preceding genes (Figures 3B, S4I, and S4J). To test whether DTECT is also able to identify genomic signatures generated by prime editing, we transiently transfected a prime editor and a pegRNA into HEK293T cells to introduce a 3-bp insertion (CTT_ins) in the HEK3 locus (Anzalone et al., 2019). As shown in Figure 3B, DTECT specifically identified the newly created signature and quantified its frequency in the transfected cell population, indicating that DTECT is also suitable to identify prime editing events. The specificity and accuracy of the preceding DTECT studies were confirmed by both positive and negative controls (e.g., CG and TT adaptors in the control unedited sample of Figure 3B).
To confirm the accuracy of DTECT in quantifying precision genome editing, we compared the frequency of editing events determined by either DTECT or NGS across 62 samples derived from human cells, mouse cells, and intestinal organoids, which were modified using CRISPR-mediated HDR or base editing (Zafra et al., 2018). As shown in Figures 3C (left panel) and S5A, the frequencies of editing events obtained by DTECT and NGS were comparable (mean frequency: DTECT, 35.43%; NGS, 33.47%; r = 0.9857, n = 62), indicating that the quantification of precision genome editing by DTECT is accurate. Similar to NGS, DTECT is also accurate in the detection of less abundant (<20% frequency) variants (mean frequency: DTECT, 5.41%; NGS, 5.06%, r = 0.843, n = 33) (Figure 3C, right panel). Altogether, these experiments demonstrate that DTECT precisely identifies and quantifies genetic variants introduced by precision genome editing in various biological systems.
Recent studies led to the development of Sanger-sequencing-based methods, such as Interference of CRISPR Editing (ICE, Synthego; https://ice.synthego.com/#/) or EditR (Kluesner et al., 2018), that enable the detection of genomic variants based on the deconvolution of chromatogram peaks. To compare DTECT with the preceding methods, we subjected to Sanger sequencing the genomic amplicons of 23 samples edited by precision genome editing. In these experiments, we used two primers annealing to opposite DNA strands to obtain independent sequencing duplicates of the same amplicons and analyzed the Sanger-sequencing reads using either ICE or EditR. Notably, ~10% of the sequencing reactions failed to generate high-quality reads required for ICE or EditR, despite using high-quality amplicons for sequencing (Mendeley Data; Key Resources Table). Independent repeats using new genomic amplicons did not improve the sequencing outcome (Mendeley Data; Key Resources Table). In addition, we noted that technical duplicates of Sanger-sequencing reactions analyzed by ICE or EditR displayed lower levels of consistency relative to technical replicates of DTECT assays (Figure S5B). These studies indicate that DTECT displays greater robustness and reliability compared with Sanger-based detection methods, which heavily rely on the quality of Sanger-sequencing reactions.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | ||
|---|---|---|---|---|
| Bacterial and Virus Strains | ||||
| Subcloning Efficiency DH5α | ThermoFisher Scientific | 18265017 | ||
| Chemicals, Peptides, and Recombinant Proteins | ||||
| Q5 High-Fidelity DNA polymerase | NEB | M0491L | ||
| T4 DNA ligase | ThermoFisher Scientific | 15224017 | ||
| AcuI | NEB | R0641L | ||
| rSAP | NEB | M0371L | ||
| SybrGold (for gel staining) | ThermoFisher Scientific | S-11494 | ||
| SybrGold (for qPCR) | ThermoFisher Scientific | 4367659 | ||
| BamHI-HF | NEB | R3136S | ||
| dNTPs | NEB | N0447L | ||
| T4 Polynucleotide Kinase | NEB | M0201S | ||
| Critical Commercial Assays | ||||
| Agencourt AMPure XP magnetic beads | Beckman Coulter | A63881 | ||
| Zymoclean gel DNA recovery kit | Zymo Research | D4008 | ||
| Quick Extract DNA Extraction Solution | Epicenter | QE09050 | ||
| Zero BLUNT II TOPO PCR Cloning kit | ThermoFisher Scientific | 450245 | ||
| Deposited Data | ||||
| Unprocessed images of gels | This paper, Mendeley Data | Raw gel images | ||
| Raw Sanger sequencing files | This paper, Mendeley Data | Sequences of BRCA1–2 edited cells; Repeated sequences | ||
| Raw NGS sequencing files | This paper; Zafra et al., 2018 | NCBI BioProject # PRJNA603357; Sequence Read Archive # SRP151111 | ||
| Raw and processed qPCR data | This paper, Mendeley Data | Raw and processed qPCR data | ||
| Raw and processed DTECT, ICE, EditR and NGS data | This paper, Mendeley Data | DTECT_ICE_EditR_NGS data | ||
| Experimental Models: Cell Lines | ||||
| Human: HEK293T | ATCC | CCL-11268 | ||
| Human: DLD1 | ATCC | CRL-221 | ||
| Mouse: NIH/3T3 | ATCC | CRL-1658 | ||
| Experimental Models: Organisms/Strains | ||||
| Mouse: C57BL/6N | Charles River | C57BL/6NCrl | ||
| Mouse: Brca1S1598F/+ | Shakya et al., 2011 | N/A | ||
| Mouse: Bard1S563F/+ | Billing et al., 2018 | N/A | ||
| Mouse: Mx1Cre+;CD45.1 | Mullally et al., 2010 | N/A | ||
| Mouse: Mx1-Cre+;CD45.2 Jak2V617F/+ | Mullally et al., 2010 | N/A | ||
| Mouse: NRG | The Jackson Laboratory | 007799 | ||
| Oligonucleotides | ||||
| Primers for PCR | This paper | Table S1 | ||
| Oligonucleotides for sgRNA cloning | This paper | Table S1 | ||
| Oligonucleotides for adaptors | This paper | Table S1 | ||
| ssODNs (for HDR) | This paper | Table S1 | ||
| Recombinant DNA | ||||
| Plasmid: B52 (containing 2 empty sgRNA-expressing cassettes) | Addgene | 100708 | ||
| pCMV-PE2 | Addgene | 132775 | ||
| pCMV-BE3 | Addgene | 73021 | ||
| DTECT - Plasmid for standard curve | This paper, Addgene | 139333 | ||
| pTOPO-SPRTN WT | This paper | N/A | ||
| pTOPO-SPRTN STOP | This paper | N/A | ||
| pTOPO-SMARCAL1 WT | This paper | N/A | ||
| pTOPO-SMARCAL1 STOP | This paper | N/A | ||
| pTOPO-PIK3R1 WT | This paper | N/A | ||
| pTOPO-PIK3R1 STOP | This paper | N/A | ||
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | Addgene | 42230 | ||
| pCDNA3-Flag::UbvG08 I44A, deltaGG | Addgene | 74939 | ||
| pU6-Sp-pegRNA-HEK3-CTT_ins | Addgene | 132778 | ||
| Plasmids expressing sgRNAs for base editing of FANCD2, BRCA1 and BRCA2 | This paper, Addgene | 139321–139332, and 139511 | ||
| Software and Algorithms | ||||
| R Studio Desktop IDE 1.0.143 | RStudio | https://rstudio.com/ | ||
| Bioconductor R packages | Bioconductor | https://www.bioconductor.org | ||
| R 3.4.1 | The R project for statistical computing | https://www.r-project.org | ||
| Other | ||||
| ClinVar database | NCBI | https://www.ncbi.nlm.nih.gov/clinvar/ | ||
| LI-COR Odyssey | LI-COR | https://www.licor.com/bio/products/imaging_systems/odyssey | ||
| q-PCR QuantStudio 3 | Applied Biosystems | N/A | ||
DTECT Enables the Identification of Precision Genome Editing Events In Vivo
The modeling and correction of pathogenic mutations in adult mice is critical for the development of novel approaches to therapeutic intervention against cancer and other diseases (Chadwick et al., 2017; Gao et al., 2018; Levy et al., 2020; Ryu et al., 2018; Song et al., 2020; Villiger et al., 2018; Yin et al., 2016; Yin et al., 2014). To examine whether DTECT can determine the frequency of precision genome editing in adult mouse tissue, we hydrodynamically delivered into the mouse liver (Tschaharganeh et al., 2014) a cytidine base editor and an sgRNA introducing the oncogenic Pik3ca E545K mutation (Zafra et al., 2018; Figure 3D). We then used both DTECT and NGS to quantify the oncogenic Pik3ca signature in DNA samples derived from the edited livers of two mice. DTECT analysis identified base editing events in the mouse liver at a frequency of ~1%–2%, comparable to the editing rates obtained by NGS (Figure 3E). This study revealed that DTECT can accurately quantify low-abundance genetic variants introduced by precision genome editing in vivo.
DTECT Is Capable of Identifying Multiple Genome Editing Events Occurring within a Single Locus or Distinct Loci
The preceding studies indicate that DTECT can determine the identity of individual genomic changes. To examine whether DTECT can also identify complex sets of mutations, we employed CRISPR-dependent base editing to target two adjacent cytosines in the EMX1 locus that had previously been converted into four distinct dinucleotide combinations (i.e., CC, CT, TC, or TT) by base editing (Komor et al., 2016; Figure 4A). As shown in Figure 4A, DTECT readily distinguished each of the four combinations in an sgRNA-dependent manner. Furthermore, DTECT was able to identify base editing byproducts (Figure S6A; Komor et al., 2017; Wang et al., 2017), demonstrating that it can detect a complex mixture of allelic variants. In addition, to determine whether DTECT can be employed to monitor genomic changes at multiple loci, we simultaneously introduced two clinically relevant point mutations into two distinct genes (i.e., BRCA1 and BRCA2) (Figure 4B). As shown in Figure 4C, DTECT correctly identified these genomic changes. These studies indicate that DTECT can readily identify complex genome editing events occurring within single or multiple genomic loci.
Figure 4. Identification of Multiple Genome Editing Events in a Single Locus or Distinct Loci by DTECT.
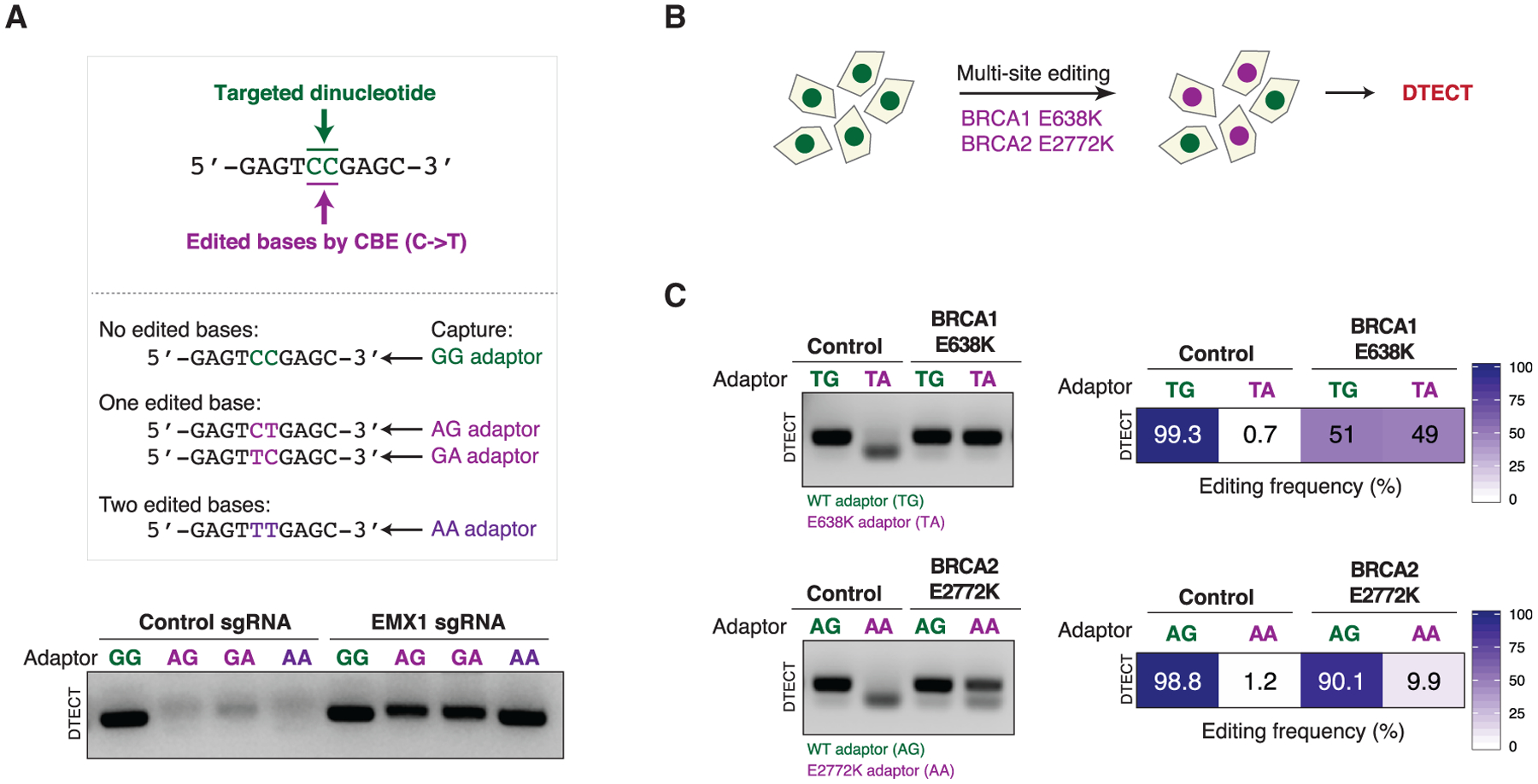
(A) Detection by PCR (21 cycles) of allelic mixtures induced by CRISPR-mediated base editing events occurring at a CC sequence (green) in the EMX1 gene. The sequences of the EMX1 alleles resulting from four possible C→T base transitions (CC, CT, TC, and TT) induced by CRISPR-mediated base editing and the adaptors to capture them (GG, AG, GA, and AA) are shown. In these experiments, HEK293T cells constitutively expressing the cytidine base editor (CBE) FNLS-BE3 were transfected with an sgRNA targeting the EMX1 locus.
(B) Schematics of the experiments conducted to detect multiple simultaneously induced variants using DTECT. HEK293T cells constitutively expressing the base editor FNLS-BE3 were transfected with two sgRNAs to introduce simultaneously the BRCA1 E638K and the BRCA2 E2772K mutations by CRISPR-mediated base editing.
(C) Detection of multiple precision genome editing events introduced by CRISPR-mediated base editing in HEK293T cell populations, as illustrated in (B). WT and edited BRCA1 and BRCA2 alleles captured using adaptors specific for the WT (TG and AG, green) or edited (TA and AA, purple) alleles were subjected to analytical PCR (left, 21 cycles) or qPCR (right).
See also Figure S6.
DTECT Expedites the Derivation of Marker-free Cell Lines Carrying Clinically Relevant Mutations and Facilitates the Genotyping of Cellular and Animal Disease Models
Precision genome editing allows the modeling of clinically relevant gene variants. Given that DTECT enables the identification of newly created DNA signatures without requiring the insertion of markers or elaborate experimental designs specific for each edited site, we tested whether DTECT could facilitate the generation of multiple cell lines harboring clinically relevant mutations. In particular, we focused our attention on mutations in the BRCA1 and BRCA2 genes, which in heterozygosity can predispose women to the development of breast and/or ovarian cancer (Apostolou and Fostira, 2013) and in homozygosity can cause Fanconi anemia (Ceccaldi et al., 2016). More than 7,000 clinically associated single nucleotide variants (SNVs) have been identified in BRCA1/2, according to the ClinVar database, but efforts to characterize their functional impact and pathogenic potential have been limited partly by the challenge of generating cell lines that carry such a large number of individual homozygous and heterozygous variants. To determine whether DTECT can facilitate the production of cell lines harboring clinically relevant BRCA1/2 SNVs, we expressed a cytidine base editor in HEK293T cells, along with individual sgRNAs, to generate 23 different BRCA1/2 mutations identified in patients with ovarian and breast cancers, as reported in ClinVar (Figures 5A and 5D). We then used DTECT to determine by analytical PCR which variants were introduced in the transfected cell populations and to quantify the editing efficiency for each variant by qPCR (Figures 5B, 5C, 5E, 5F, S6B, and S6C). The accuracy of DTECT in the quantification of the editing events was confirmed by NGS (Figures 5B and 5E). The preceding approach proved effective for rapidly identifying cell populations with high levels of editing. Upon isolation of single clones from edited cell populations (e.g., BRCA1 E638K mutant cells), we tested whether DTECT could be used for clone genotyping. Importantly, DTECT allowed rapid genotyping of multiple clones (Figure S7A) and accurately determined the genotype of each clone, including WT and homozygous and heterozygous mutant clones (Figures 5G and 5H).
Figure 5. DTECT-Mediated Identification of Clinically Relevant BRCA1/2 Mutations Generated by Precision Genome Editing and Genotyping of Cell Lines and Animal Models Carrying BRCA1 or BARD1 Mutations.
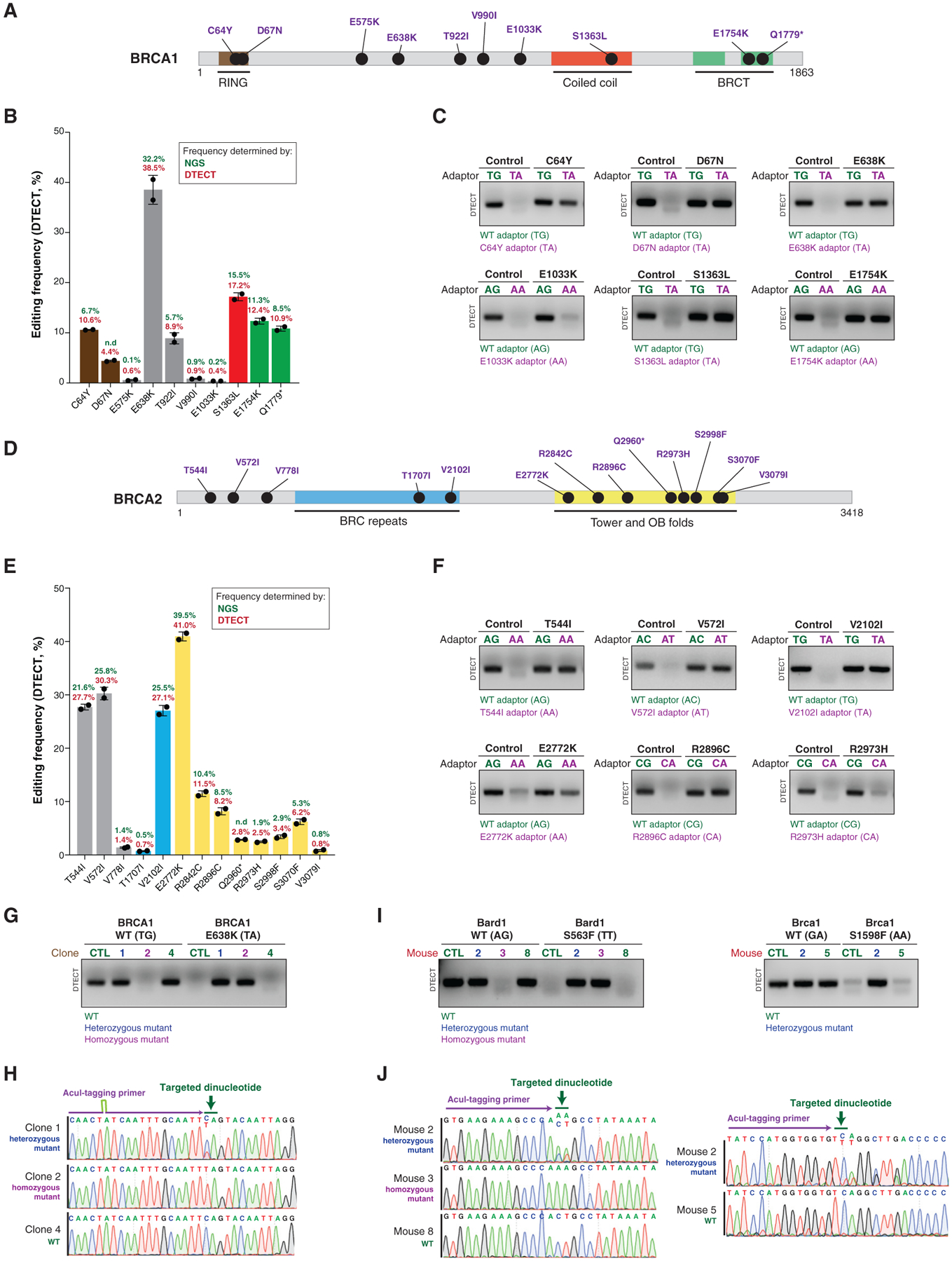
(A) Schematic representation of the human BRCA1 protein. BRCA1 domains and ClinVar BRCA1 mutations generated in this study are indicated.
(B) Quantification using DTECT (red) and NGS (green) of the editing efficiency by which 10 BRCA1 mutations are introduced into HEK293T cells by CRISPR-mediated base editing. Experiments were conducted in cells expressing the base editor FNLS-BE3 upon transfection of sgRNAs to introduce the indicated mutations. Histograms show the mean frequency of the indicated variants estimated by DTECT, and error bars represent the SD from 2 independent DTECT assays for the same AcuI-tagged amplicon. ND, not determined due to sequencing failure.
(C) Analytical detection of the indicated BRCA1 mutations in HEK293T cell populations by DTECT (21 PCR cycles) using adaptors specific for WT (green) or mutant (purple) alleles.
(D) Schematic representation of the human BRCA2 protein. BRCA2 domains and ClinVar BRCA2 mutations generated in this study are indicated.
(E) Quantification using DTECT (red) and NGS (green) of the editing efficiency by which 13 BRCA2 mutations are introduced into HEK293T cells by CRISPR-mediated base editing, as described in (B).
(F) Analytical detection of the indicated BRCA2 mutations in HEK293T cell populations by DTECT (21 PCR cycles) using adaptors specific for WT (green) or mutant (purple) alleles. Experiments were conducted as in (C).
(G) Genotyping by DTECT-based analytical PCR (18 cycles) of single clones carrying WT and/or BRCA1 E638K mutant alleles derived from the BRCA1 E638K mutant cell population shown in (C). WT (4, not edited), heterozygous (1), and homozygous (2) BRCA1 mutant clones identified by DTECT are indicated.
(H) Sanger sequencing of WT and heterozygous and homozygous mutant amplicons shown in (G). The targeted dinucleotide is indicated in green, and part of the sequence of the AcuI-tagging primer is indicated in purple.
(I) Genotyping by DTECT-based analytical PCR of Bard1 S563F (left) and Brca1 S1598F (right) knockin mutant mice (Bard1, 18 PCR cycles; Brca1, 20 PCR cycles). gDNA for DTECT analysis was obtained from mouse tail samples. WT (Bard1 8 and Brca1 5) mice and heterozygous (Bard1 2 and Brca1 2) and homozygous (Bard1 3) mutant mice identified by DTECT are indicated. No homozygous Brca1 S1598F mutant mice were identified in the analyzed mouse litters due to sub-Mendelian birth ratios (Billing et al., 2018).
(J) Sanger sequencing of WT and heterozygous and homozygous mutant amplicons shown in (I).
See also Figures S6, S7, and S9.
Given the ability of DTECT to correctly determine the genotype of cellular clones, we then tested whether DTECT could be applied to mouse genotyping. To this end, we obtained tail DNA samples from genetically engineered mice carrying knockin mutations in Brca1 (S1598F) and its partner protein Bard1 (S563F) (Billing et al., 2018). As shown in Figures 5I, 5J, and S7B, DTECT accurately determined the genotype of 24 Bard1 S563F mutant mice and 16 Brca1 S1598F mutant mice. These findings indicate that DTECT can be employed to rapidly determine the genotype of genetically engineered mice.
DTECT Identifies the Presence of Oncogenic Mutations in Cancer Mouse Models and Human Cancer Patient Samples
Precise and rapid detection of pathogenic variants in patients is critical for accurate diagnosis and personalized therapy. Given the ability of DTECT to identify genetic variants rapidly and accurately, we tested whether DTECT could be used to expedite the identification of pathogenic variants in pre-clinical and clinical settings. In particular, we examined whether DTECT could identify the presence of oncogenic variants in various biological systems. In our studies, we focused our attention on the JAK2 V617F variant, which is present in most patients with myeloproliferative neoplasm (MPN) (Levine et al., 2005). Mice transplanted with Jak2 V617F mutant bone marrow cells develop MPN and recapitulate the human disease (Mullally et al., 2010). Therefore, we analyzed the Jak2 V617F variant in the peripheral blood of mice transplanted with a mixture of bone marrow cells that do or do not carry an inducible Jak2 V617F variant (Bhagwat et al., 2014) (Figure S8A). As shown in Figures S8B and S8C, DTECT readily distinguished WT from V617F mutant Jak2 in the examined mouse blood samples, as detected using any of the four distinct AcuI-tagging primers specific for the targeted bases. These experiments show that DTECT can identify oncogenic signatures of interest in mouse tissues in a marker-free manner, enabling the tracking of genetic variants in mouse models without requiring complex selection markers.
We next examined whether DTECT can identify the presence of specific oncogenic mutations in human samples from patients diagnosed with acute lymphoblastic leukemia (ALL), the most common form of childhood cancer (Inaba et al., 2013). Although most ALL patients respond to chemotherapy, ~20% suffer a relapse as a result of resistance to chemotherapy (Bhojwani and Pui, 2013). Moreover, secondary genetic alterations that promote chemoresistance, including mutations in the NT5C2 gene (Tzoneva et al., 2018; Tzoneva et al., 2013), are found in a large fraction of ALL relapse cases (Dieck and Ferrando, 2019; Oshima et al., 2016). To test whether DTECT can identify these relapse-specific oncogenic signatures, we obtained matched DNA samples from the bone marrow of ALL patients at diagnosis and relapse and analyzed them for the presence of three common NT5C2 mutations (R238W, K359Q, and R367Q) (Figures 6A and 6B). Remarkably, DTECT unambiguously identified the presence of oncogenic NT5C2 variants in all five patient samples (patient 1, R238W; patients 2, 4, and 5, R367Q; and patient 3, K359Q) and accurately quantified their frequency in a manner comparable to NGS (Figures 6B, 6C, and S8D). Moreover, DTECT identified the preceding NT5C2 variants in the PDX models generated from these relapsed ALL patients (Figures 6A and 6D). These studies demonstrate that DTECT can identify oncogenic mutations of interest in PDX models and cancer patient samples.
Figure 6. Detection of Oncogenic Signatures in Human Clinical Samples Using DTECT.
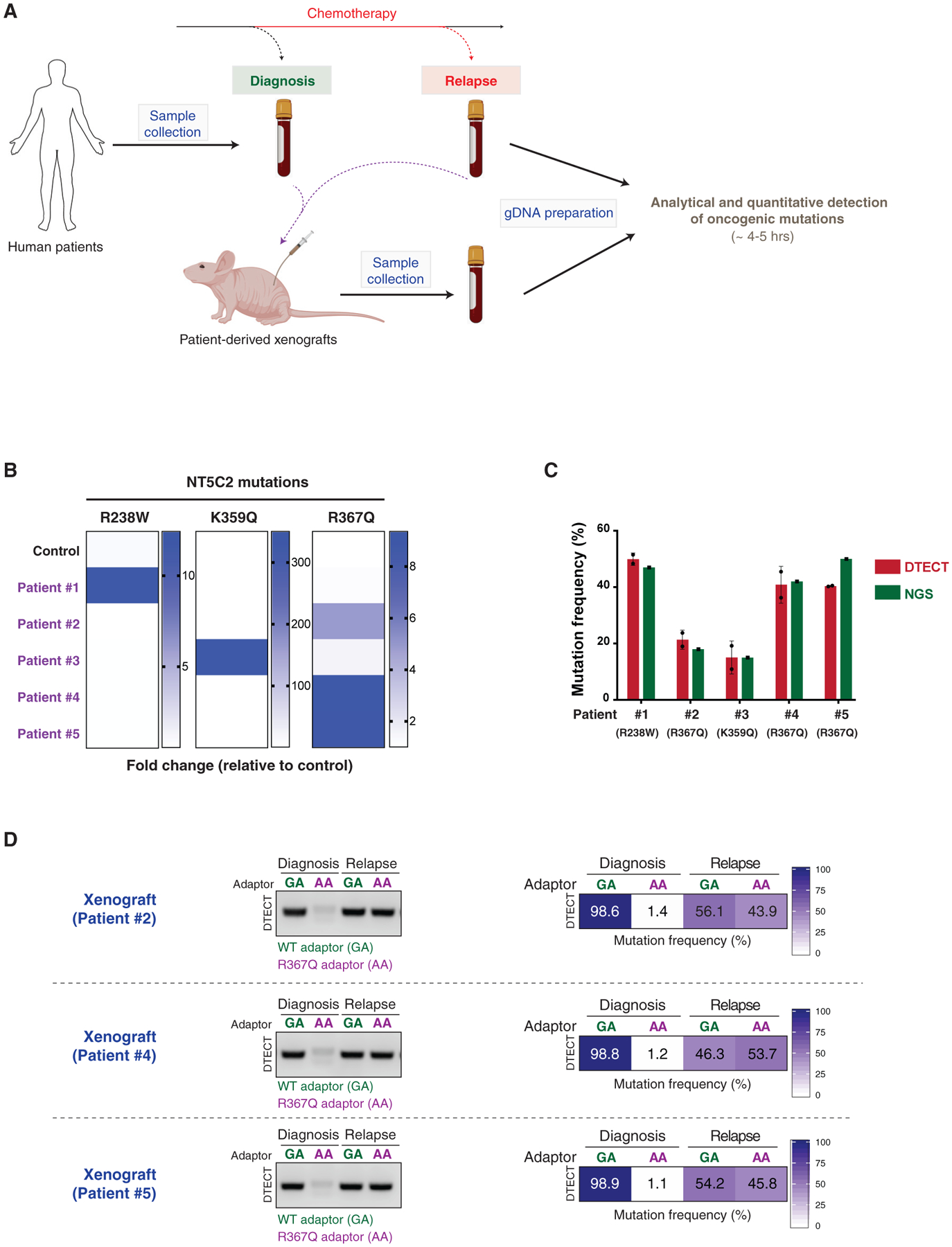
(A) Schematic representation of the experiments conducted on ALL patient-derived samples. Bone marrow samples from ALL patients were collected at diagnosis and after chemotherapy. PDXs were generated from the patient samples. gDNA was recovered from patient samples and PDX mouse models and subjected to analytical and quantitative detection of NT5C2 oncogenic mutations using DTECT.
(B) Heatmap showing the detection of NT5C2 oncogenic mutations in patient samples and a control sample using DTECT. Bone marrow samples from 5 patients were collected, and gDNA was prepared and tested for the presence of 3 frequent NT5C2 mutations responsible for relapse to chemotherapy. A non-patient-derived gDNA sample was used as a control to estimate the levels of non-specific background in the DTECT assay. Data are shown as fold change in the frequency of mutant signatures in the patient samples relative to the control sample.
(C) Graphical representation of the frequency of NT5C2 mutations determined by DTECT (red) and NGS (green) in the 5 human patient samples analyzed in (B). Error bars indicate the SD of 2 independent DTECT replicates.
(D) Analytical and quantitative detection of the NT5C2 R367Q mutation in PDX models generated from ALL tumors of patients 2, 4, and 5 at diagnosis and after chemotherapy relapse. WT and mutant variants were captured using adaptors specific for the WT (GA, green) or mutant (AA, purple) allele and subjected to analytical PCR (left, 18 cycles) and qPCR (right).
See also Figure S8.
DISCUSSION
In this study, we establish DTECT as a sensitive method for the identification of gDNA signatures. In particular, we show that DTECT readily identifies precision genome editing events induced by CRISPR-dependent HDR, base editing, and prime editing, including low-abundance and complex genomic changes. In addition, we show that DTECT can be employed to identify pathogenic lesions of interest, such as oncogenic mutations, in cancer mouse models, PDXs, and cancer patient specimens. DTECT is a rapid (~4–5 h) and easy-to-perform detection method that relies on standard molecular biology techniques (PCR, DNA digestion, and ligation) and common laboratory reagents. This methodology is not labor intensive, given that it entails short periods (5–10 min) of sample processing followed by hands-free incubations. DTECT assays use a unique and common set of adaptors that includes positive and negative controls to ensure specificity and accuracy.
Although highly robust, DTECT has three potential limitations. First, AcuI-induced dinucleotide byproducts can be generated if a genomic AcuI restriction site located close to the targeted dinucleotide is incorporated into the amplicon of the targeted locus. However, an analysis of the ClinVar database revealed that genomic AcuI sites occur relatively infrequently and 95% of clinically relevant variants (404,393 variants) are compatible with DTECT (Figures S9A and S9B). Second, dinucleotide byproducts may occur because of AcuI slippage activity, resulting in the cleavage of DNA molecules 13 (−1) or 15 (+1), instead of 14, bases from the AcuI recognition site. Nonetheless, we found that DTECT is able to identify AcuI slippage events, which occur mostly at position +1 relative to the standard AcuI cleavage site (Lundin et al., 2015; Figure S10A). It is reasonable to anticipate that future optimization of AcuI architecture and improvements in the AcuI digestion protocol will limit its slippage activity. In addition, AcuI byproducts resulting from either genomic AcuI motifs or AcuI slippage activity are easily predictable based on the sequence of the nucleotides flanking the targeted dinucleotide, and they can be avoided by optimal design of the AcuI-tagging primer and appropriate adaptor selection, as shown in Figures S9C and S10B. Third, indel mutations formed at DSB sites generated by Cas nucleases in CRISPR-mediated HDR experiments can result in defective PCR amplification of indel-containing loci that have not undergone HDR and therefore cause an overestimation of the frequency of HDR events by DTECT (Figures S11A and S11B). However, given that the mutagenic spectrum of indel mutations induced by any sgRNA is predictable (Allen et al., 2019; Leenay et al., 2019; Shen et al., 2018; van Overbeek et al., 2016; inDelphi, https://indelphi.giffordlab.mit.edu/), the negative impact of indel mutations on DTECT-based quantification of CRISPR-mediated HDR events can be avoided by introducing the desired genomic changes in indel-free regions adjacent to CRISPR-induced cut sites (Figures S11C and S11D). This limitation does not affect the detection of CRISPR-mediated base editing and prime editing events or naturally occurring genetic variants, which are accompanied by either very low frequency (Anzalone et al., 2019; Gaudelli et al., 2017; Komor et al., 2017; Yeh et al., 2018) or absence of DSB-induced indel formation, respectively.
In addition to its ease of use, speed, and cost efficiency, DTECT has several advantages compared with other detection methods. A major benefit of DTECT is its versatility, which allows the detection and quantification of nucleotide substitutions, precise base indels using the same small set of 16 predefined adaptors (Figures 1B and 7). Each editing event can be identified using 4 distinct signatures resulting from AcuI-mediated digestion of gDNA amplicons, indicating that the design of DTECT studies is flexible (Figures 2E, 2F, S8B, and S8C). These features distinguish DTECT from strategies that employ allele-specific DNA oligonucleotides or probes to identify SNVs, which work with variable efficiency because of the competition between WT and mutant alleles and the number of variant DNA bases, thus requiring a unique experimental design for the detection of each genetic variant. Given that both WT and mutant DNA signatures are captured from the same AcuI-digested PCR amplicon and that a common set of PCR primers is used for both analytical and quantitative detection of all variants (Figure 1A, step VI), DTECT exhibits limited technical variability across distinct experimental conditions. This aspect differentiates DTECT from Sanger-sequencing-based detection methods, such as ICE and EditR, in which efficiency depends on the quality of the sequencing reads, which can vary greatly among sequencing platforms, samples, and reactions (Figure S5B). In addition, DTECT displays greater sensitivity and flexibility compared with RFLP-based assays (Figure S4) and exhibits similar precision to NGS (Figure 3C) at a lower cost and with a faster turnaround time (hours versus days/weeks). Finally, DTECT directly identifies genetic variants independently of genomic markers, enabling the analysis of scarless and marker-free cellular and animal models generated by precision genome editing. Given its ability to identify multiple independent genetic variants simultaneously (Figure 4), DTECT could also expedite the generation of complex genomic changes, especially for genetic interaction studies, synthetic biology applications, and molecular recording (Farzadfard and Lu, 2018).
Figure 7. DTECT Applications for the Detection of Precision Genome Editing and Genetic Variation.
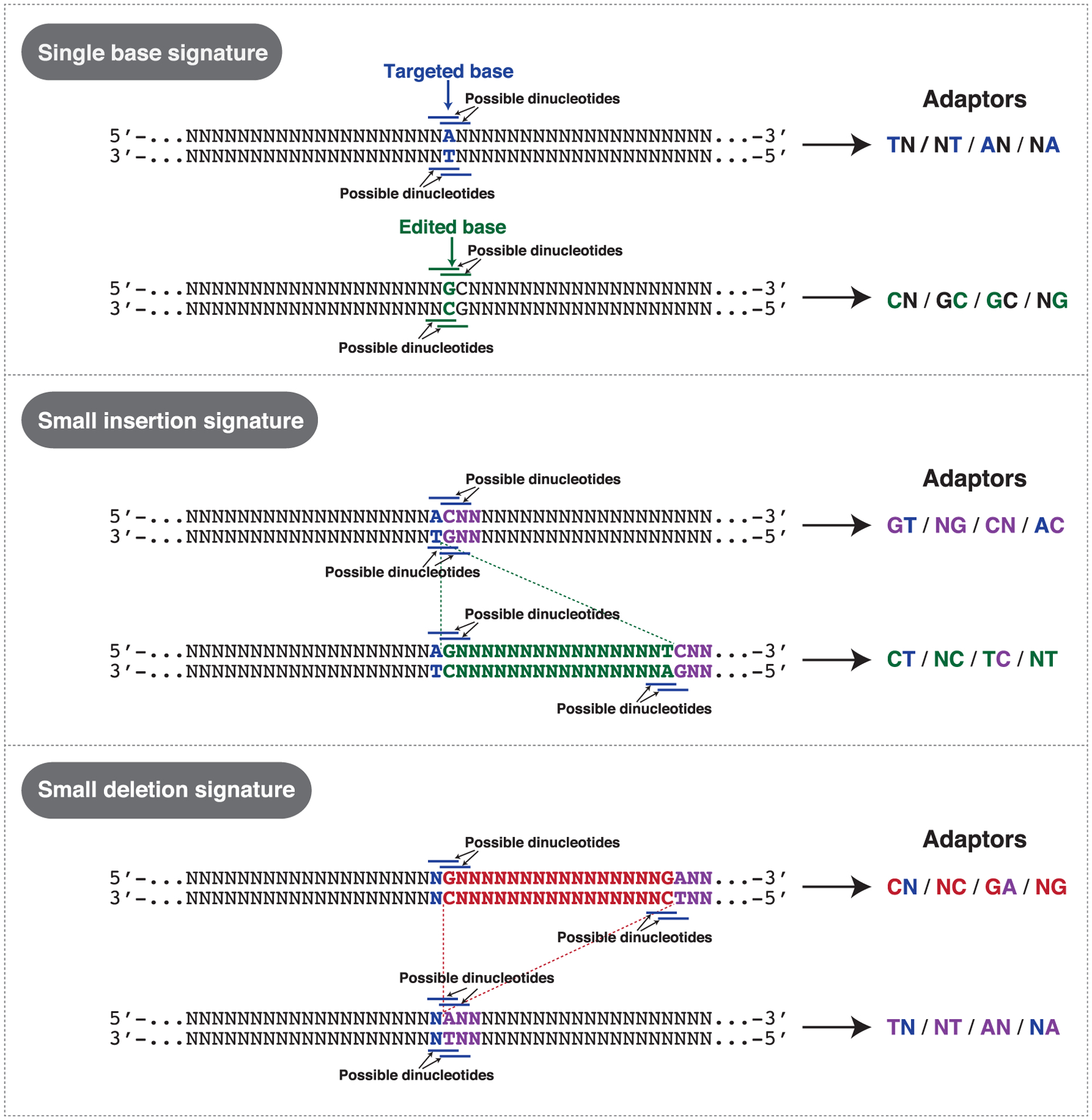
Schematic representation of examples of targeted dinucleotide signatures generated by single base edits and small indels that can be detected using DTECT. Examples of adaptors that can be used to detect the indicated genome editing events are shown on the right.
The ability to model clinically relevant mutations in a marker-free manner is critical for assessing their potential pathogenicity, especially in the case of genes, such as BRCA1 and BRCA2, that have thousands of clinically associated SNVs. Recent studies have led to the development of high-throughput saturation genome editing (SGE) to examine en masse the pathogenicity of BRCA1 variants (Findlay et al., 2018). Although highly useful for classifying BRCA1 SNVs, SGE requires the use of haploid cells and is therefore not compatible with the study of the functional impact of BRCA1 mutations in heterozygosity, as observed in BRCA1 mutation carriers (Apostolou and Fostira, 2013). BRCA1/2 heterozygous mutations have been shown to cause genome instability induced by DNA replication stress (Billing et al., 2018; Pathania et al., 2014; Tan et al., 2017). By facilitating the derivation of both heterozygous and homozygous BRCA1/2 mutant cells and animal models (Figure 5), DTECT could help elucidate the underlying mechanisms by which genome instability causes breast and ovarian cancer development in BRCA1/2 mutation carriers.
In addition to facilitating precision genome editing, we show that DTECT can be used to detect pathogenic variants in pre-clinical and clinical settings. In particular, DTECT can rapidly identify the presence of oncogenic variants in cancer mouse models (Figure S8), facilitating the study of cancer pathogenesis and the development of novel cancer therapies. Furthermore, DTECT can identify oncogenic mutations in samples from cancer patients and PDX mouse models (Figure 6). The speed by which DTECT accurately and unambiguously identifies pathogenic variants could accelerate cancer diagnosis and expedite the testing of cancer therapies in PDX models, leading to more effective cancer treatments. We envision that future developments and implementations of the DTECT protocol may further simplify the detection of desired genomic signatures and increase the sensitivity of DTECT, expanding the number of possible DTECT applications and enabling early diagnosis of cancer and hereditary disorders through the detection of pathogenic variants in circulating cell-free tumor and fetal DNA (Zhang et al., 2019).
Collectively, our work establishes DTECT as a facile, rapid, and cost-effective method for identifying genomic variants in various biological systems, such as mammalian cell lines, organoids, mouse tissues, PDX models, and human patient samples. Given the growing number of genetic variants identified in the human population (Lek et al., 2016) and in human genetic disorders (McClellan and King, 2010), this versatile method for the detection of genomic signatures should facilitate the study of human genetic variation and expedite the diagnosis and treatment of human disease.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Plasmids for DTECT quantification and expression of base editing sgRNAs targeting BRCA1, BRCA2 and FANCD2 and have been deposited to Addgene (#139321–139333, and 139511). Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alberto Ciccia (ac3685@cumc.columbia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell line generation and single clone isolation
HEK293T and DLD1 cell lines were obtained from ATCC. Cells were cultured in DMEM (ThermoFisher Scientific) supplemented with 10% Fetalgro bovine growth serum (BGS, RMBIO) and 1% penicillin-streptomycin (ThermoFisher Scientific). Cells were grown at 37°C with 5% CO2 and tested regularly for mycoplasma. NIH/3T3 were maintained in DMEM supplemented with 10% bovine calf serum. Organoids were isolated and cultured as previously described (Zafra et al., 2018). To generate cells constitutively expressing FNLS-BE3-P2A-BlastR, HEK293T cells were infected with a lentivirus expressing the above construct. Viruses were produced in HEK293T in 6-well plates by transfecting 2 μg of FNLS-BE3-P2A-BlastR, 0.2 μg of Tat, 0.2 μg of Gag/Pol, 0.2 μg of Rev, 0.4 μg of VSV-G expressing plasmids in 250 μl of DMEM without serum. 9 μl of TransIT-293 (Mirus) were added to the DNA, mixed and incubated for 15 min at room temperature. The DNA transfection reagent mix was added dropwise to the cells and incubated at 37°C with 5% CO2. The next day the cell medium was replaced and cells were incubated for 48 hours. The medium containing lentiviruses was then collected and utilized to infect new HEK293T cells. 48 hours after infection, blasticidin was added to the medium until the uninfected control cells were killed. FNLS-BE3 expression was determined by western blot and the base editing activity of the construct was tested using previously validated sgRNAs. Single HEK293T clones were selected for high base editing efficiency. Clones were isolated by trypsinization of the initial cell population into individual cells. Cell density was evaluated by counting the cells with a hemocytometer and cells were diluted to approximately 0.13 cells/μl, equivalent to 20 cells per 150 μl. Serial dilutions were prepared and 150 μl of the diluted cell mixture were seeded into 96-well plates. Single clones were expanded and further examined for FNLS-BE3 expression and activity.
Editing of cell lines, organoids and mice
To induce CRISPR-mediated HDR editing, HEK293T cells were seeded at 50%–70% confluency into 24-well plates and reverse transfected with 0.25 μg of sgRNA and 0.25 μg of Cas9 expressing plasmid (Addgene #42230) with or without 0.5 μl of ssODN (40 μM) into 100 μl of DMEM without Fetalgro BGS and antibiotics. 3 μl of TransIT-293 (Mirus) were added to the DNA, mixed and incubated for 15 min at room temperature. Experiments involving i53 were done by adding 0.25 μg of i53 (Addgene #77939) to the transfection mixture. The gDNAs of cell populations and individual clones were recovered by resuspending the cell pellets in the Quick Extract DNA Extraction Solution (Epicenter), followed by incubation at 65°C for 10 min and 95°C for 5 min. The isolated gDNAs were diluted in H2O, quantified using Nanodrop and stored at −20°C or directly used in PCR reactions. In base editing experiments, we used cells constitutively expressing FNLS-BE3 or transfected with pCMV-BE3 (Addgene #73021) and sgRNAs, as described above. Empty plasmids (Addgene #100708) with no sgRNAs were used as controls. To determine the accuracy of the quantification of variant frequency by DTECT (Figure 2G), STOP codons were introduced into SPRTN, SMARCAL1 and PIK3R1 genes using iSTOP, as previously described (Billon et al., 2017). To isolate the WT alleles, the locus was amplified by PCR and cloned into the pCR-Blunt II-TOPO vector (ThermoFisher Scientific). The STOP alleles were isolated by PCR amplification using gDNA that was partially edited as template. The PCR product was subsequently digested using restriction enzymes that specifically cleave the WT PCR alleles (i.e., PvuII for SPRTN, SfaNI for SMARCAL1 and TaqaI for PIK3R1). The digestion reaction was loaded on a 2% agarose gel and the undigested PCR products were column purified (Zymoclean #D4008). The purified products were subsequently cloned into the pCR-Blunt II-TOPO vector (ThermoFisher Scientific). Cloned WT and STOP PCR fragments were confirmed by Sanger sequencing and are shown in Figure S3B. RFLP assays were conducted by digesting PCR amplicons of the edited genomic loci with enzymes that recognize restriction sites created or disrupted by editing of the targeted loci. Restriction digest products were run on 6% TBE polyacrylamide gels. Gels were run at 160 V in 1X TBE and stained for 5 min using SybrGold diluted in 1X TBE buffer. In prime editing experiments, 1 μg of pCMV-PE2 (Addgene #132775) was transfected into HEK293T cells along with 500 ng of control pegRNA (Addgene #132777) or pegRNA HEK3 insCTT (Addgene #132778). Three days after transfection, genomic DNA was recovered as above and the edited signature was identified with DTECT. Edited DLD1 (FANCF locus) and NIH/3T3 (Pik3ca and Apc loci) cell populations and mouse intestinal organoids (Pik3ca and Apc loci) were previously described (Zafra et al., 2018). Genomic DNA from the edited cell populations was used to quantify the editing efficiency by DTECT (Figure S5A).
In order to introduce multiple variants into the BRCA1 and BRCA2 genes, HEK293T cells expressing FNLS-BE3 were seeded at 50%–70% confluency into 24-well plates and reverse transfected with 1 μg of sgRNA into 100 μl of DMEM without Fetalgro BGS and antibiotics. 3 μl of TransIT-293 (Mirus) were added to the DNA, mixed and incubated for 15 min at room temperature. The DNA transfection mix was added dropwise to the cells and incubated at 37°C with 5% CO2 for 4 days. Single clones were generated and the gDNAs of cell populations and individual clones were recovered as describe above. Genomic loci were Sanger sequenced by Eton Bioscience or Genewiz. Sanger sequencing data were analyzed using Serial Cloner and viewed by Snapgene Viewer. The sequencing profiles shown in this manuscript were generated by SnapGene Viewer. Quantitative detection of the editing level using the AcuI-tagged amplicon was done blindly.
In vivo mouse editing was performed as previously described (Zafra et al., 2018). Briefly, eight week-old C57BL/6N female mice (Charles River) were injected with 0.9% sterile sodium chloride solution containing 20 μg of pLenti-FNLS-P2A-Puro and 10 μg of sgRNA vector. The total injection volume corresponded to 20% of the individual mouse body weight and was injected into the lateral tail vein in 5–7 s. All animal experiments were authorized by the regional board of Karlsruhe, Germany.
Mouse genotyping and bone marrow transplantation
The generation of genetically engineered mice harboring the Brca1 S1598F and Bard1 S563F alleles was previously described (Billing et al., 2018; Shakya et al., 2011). Mouse genotyping was performed using DTECT on genomic DNA extracted from the tails of both male and female mice. AcuI-tagging of the targeted loci was performed using 50 ng of gDNA (see DTECT protocol). Genotyping experiments were conducted blindly.
Competitive transplantation experiments were performed to assess chimerism of Jak2 V617F mutant cells in relation to wild-type support. Specifically, Mx1-Cre+;CD45.2 Jak2V617F/+ and Mx1Cre+;CD45.1 wild-type female mice were dosed with polyinosine-polycytosine (PIPC) 8 weeks prior to sacrifice to induce MPN in mutant mice. On day of sacrifice, dissected femurs and tibias were isolated and bone marrow flushed with a syringe into PBS. Red blood cells (RBCs) were lysed in ammonium chloride-potassium bicarbonate lysis buffer for 10 min on ice. 1.5 × 106 filtered whole donor Mx1-Cre+;Jak2V617F/+ bone marrow cells (CD45.2) were then mixed with wild-type 1.5 × 106 competitor bone marrow cells (CD45.1) and transplanted via tail vein injection into lethally irradiated (2 × 550 Rad) CD45.1 host female mice. Mice were then monitored serially for the development of MPN based on blood counts and donor chimerism by retroorbital bleed draws using heparinized microhematocrit capillary tubes (ThermoFisher Scientific). After 3 consecutive hematocrits of > 65%, mice were then sacrificed for peripheral blood fluorescence-activated cell sorting (FACS) analysis and DNA extraction. All animal procedures were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees at Memorial Sloan Kettering Cancer Center. The conditional Mx1-Cre+;Jak2V617F/+ mice are all C57BL/6 background and have been previously described (Mullally et al., 2010). Automated peripheral blood counts were obtained using a ProCyte Dx (IDEXX Laboratories) according to the manufacturer’s protocol. For surface flow cytometry of mouse peripheral blood, bone marrow, and spleen, RBCs were lysed and stained with monoclonal antibodies in PBS plus 1% BSA for 1 hour on ice. For flow cytometry of erythroid lineage, bone marrow or splenic cells were stained without RBC lysis. DAPI was used for live/dead cell analysis. Cell populations were analyzed using an LSR Fortessa (Becton Dickinson), and data were analyzed with FlowJo software (Tree Star). DNA extraction was performed using the QIAamp DNA Micro Kit (QIAGEN) per manufacturer’s protocol.
Analysis of ALL patient samples and PDXs
DNA samples from leukemic ALL blasts obtained at diagnosis and after relapse were provided by multiple institutions, as previously described (Oshima et al., 2016). Informed consent was obtained at study entry and samples were collected under the supervision of local Institutional Review Boards for participating institutions and analyzed under the supervision of the Columbia University Irving Medical Center Institutional Review Board. Research was conducted in compliance with ethical regulations. Patient samples are anonymous and sex information is not available. ALL patients received standard combination chemotherapy at diagnosis. Diagnosis and relapse samples were harvested from bone marrow. High molecular weight genomic DNA from matched diagnosis and relapse samples of ALL patients was extracted from patient leukemic blasts or from xenografts using the DNeasy Blood & Tissue Kit (QIAGEN) or the AllPrep DNA/RNA Mini Kit (QIAGEN). Primary human xenograft ALL cells were passaged and harvested from the spleens of NRG (NOD.Cg-ag1tm1MomIl2rgtm1Wjl/SzJ, The Jackson Laboratory) female mice. Whole exome sequencing was performed and analyzed as previously described (Oshima et al., 2016).
METHOD DETAILS
Vector construction and cloning
sgRNAs were synthesized as complementary oligonucleotides (IDT) compatible with BbsI restriction sites located into the B52 plasmid (Addgene #100708). Oligonucleotides were designed as previously described (Billon et al., 2017). Cloned sgRNAs were verified by Sanger sequencing. Sequences of the sgRNAs are available in Table S1. ssODNs used in HDR experiments were synthesized as ultramer oligos (IDT) and their sequences are available in Table S1. To generate the FNLS-BE3-P2A-BlastR plasmid, the pLenti-FNLS-P2A-Puro plasmid (Addgene #110841) (Zafra et al., 2018) was modified by replacing the puromycin resistance gene with the blasticidin resistance gene. Briefly, the blasticidin resistance gene coding sequence was amplified by PCR and recombined using Gibson assembly into FNLS-BE3-P2A. The FNLS-BE3-P2A-BlastR sequence was verified by Sanger sequencing.
AcuI-tagging primer design
The AcuI-tagging oligonucleotide enables the insertion of an AcuI motif (5′-CTGAAG-3′) 14 bp away from a targeted dinucleotide. This motif is inserted as a hairpin in the middle of a sequence complementary to the targeted genomic locus. The AcuI-tagging oligonucleotide is 60 bp-long and contains a non-complementary handle sequence of 20–25 bp. Common handle sequences used are PB547 (5′-GATCCTCTAGAGTCGACCTG-3′) or PB1072 (5′-GCAATTCCTCACGAGACCCGTCCTG-3′) (Table S1). The oligonucleotide sequence complementary to the targeted genomic locus plus the AcuI motif has the following sequence: 5′-N(20) CTGAAGN(14)-3′ or 5′-N(15)CTGAAGN(14)-3′, with “N” corresponding to A, T, G or C bases complementary to the targeted locus. Reverse primers used in AcuI-tagging reactions were designed by Primer 3 (http://bioinfo.ut.ee/primer3-0.4.0/) using the default parameters with the following changes: Mispriming library = “HUMAN” for amplifying from human genomic DNA or Mispriming library = “RODENT” for amplifying from mouse genomic DNA, Primer size “min = 25, Opt = 27, Max = 30, ” Primer Tm “Min = 57.0°C, Opt = 60.0°C, Max = 63.0°C. ” Reverse primers are located > 100 bp away from the targeted dinucleotides. All sequences of the primers used in this study are available in Table S1.
Adaptor library generation and characterization
A set of 17 individual oligonucleotides constitutes the full adaptor library. This library contains: a) One constant oligonucleotide with the following sequence: 5′-CTGGGGCACGGGTAAGAAGCATTCTGTCTCTcttctaagaattcgagctcggtacccg-3′. The lowercase nucleotide sequence located at the 3′ end of the constant oligonucleotide (5′-cttctaagaattcgagctcggtacccg-3′) corresponds to the handle sequence used to detect the ligated products with either PB548 (5′-cgggtaccgagctcgaattc-3′) or PB1073 (5′-cgggtaccgagct cgaattcttagaag-3′); b) 16 variable oligonucleotides that contain a sequence complementary to the constant oligonucleotide plus one of 16 different dinucleotides at their 3′ end. The variable oligonucleotides have the following sequence: 5′-cgggtaccgagct cgaattcttagaagAGAGACAGAATGCTTCTTACCCGTGCCCCAGNN-3′. NN, with N = A, C, G or T, corresponds to the dinucleotide that is different for each of the 16 oligos. The adaptor sequences are available in Table S1. The constant oligonucleotide and each variable oligonucleotide were resuspended at a concentration of 100 μM in H2O. 2.5 μl of constant oligonucleotide and 2.5 μl of each variable oligonucleotide were mixed with 1X ligase buffer (ThermoFisher Scientific) and water in a 20 μl reaction. The reactions were placed in a thermocycler and oligonucleotides were annealed by incubating them for 5 min at 95°C, followed by a gradual temperature decrease from 95°C to 15°C. After annealing was completed, 100 μl of water were added to dilute the adaptors in a 120 μl final volume. Adaptors were frozen and stored at −20°C.
The adaptor library was tested at two independent loci, as shown in Figure S2C. In this assay, AcuI-tagging oligonucleotides targeting the ampicillin resistance gene were designed following the rules detailed above (Table S1). First, we linearized the pUC19 plasmid as follows: 1.5 μg of pUC19, 1X CutSmart Buffer (NEB) and 0.75 μl of BamHI-HF were mixed in a 30 μl reaction and incubated for 2 hours at 37°C. The digested plasmid was subsequently purified on column (Zymoclean #D4008) and used as a template in PCR reactions with each AcuI-tagging primer and a constant reverse primer (5′-CCAATGCTTAATCAGTGAGG-3′) located at the 3′-side of the ampicillin resistance gene. The PCRs were performed in a 25 μl reaction containing: 1 μM forward and reverse primers, 0.1 μM dNTP (NEB #N0447L), 1X Q5 buffer (NEB), 20 ng of digested pUC19, 1 unit of Q5 polymerase (NEB) and water. The PCR program used was the following: 95°C for 1 min, 40 cycles of 95°C for 10 s, 58°C for 10 s, 72°C for 45 s and a final amplification step of 1 min at 72°C. PCR reactions were loaded on a 2% agarose gel, extracted from gel and purified on column (Zymoclean #D4008). Finally, the DTECT protocol was applied as described below. Briefly, 0.5 pmol of AcuI-tagging PCR products were digested by AcuI for 30 min at 37°C. 10 μl of the digested products were purified with 18 μl of solid phase reversible immobilization magnetic beads (Beckman Coulter #A63881). 20 μl of supernatant (unbound fraction) were recovered and 0.5 μl of this supernatant were ligated using complementary and negative control adaptors for 1 hour at 25°C, followed by T4 ligase inactivation for 10 min at 65°C. The complementary and negative control adaptors used in Figure S2C are the following: AA #1 (Specific adaptor: TT, Non-specific adaptor: CC), AA #2 (TT, CC), AC #1 (GT, AC), AC #2 (GT, AA), AG #1 (CT, GA), AG #2 (CT, GA), AT #1 (AT, GG), AT #2 (AT, GG), CA #1 (TG, CA), CA #2 (TG, CA), CC #1 (GG, CC), CC #2 (GG, CC), CG #1 (CG, AA), CG #2 (CG, AA), CT #1 (AG, TT), CT #2 (AG, TT), GA #1 (TC, GA), GA #2 (TC, GA), GC #1 (GC, TT), GC #2 (GC, TT), GG #1 (CC, TT), GG #2 (CC, TT), GT #1 (AC, TG), GT #2 (AC, TG), TA #1 (TA, GG), TA #2 (TA, GG), TC #1 (GA, CT), TC #2 (GA, CT), TG #1 (CA, TG), TG #2 (CA, TG), TT #1 (AA, GG) and TT #2 (AA, GG). The ligated products were subsequently detected by PCR amplification using the primers PB547 (5′-gatcctctagagtcgacctg-3′) and PB1073 (5′-cgggtaccgagct cgaattcttagaag-3′). All primer sequences are listed in Table S1.
The measurement of the dinucleotide capture efficiency of each adaptor (Figures 2J and 2K) was determined by ligating the 16 different adaptors to annealed oligonucleotides containing complementary dinucleotides. To mimic the 5′ phosphorylation induced by AcuI in DTECT experiments, the reverse oligonucleotide (PB1449: 5′-gtagttcgccagttCTTCAGaatagtttgcgcaCAGGACGGGTCTCGTGAGGAATTGC-3′) was phosphorylated with PNK (NEB). The phosphorylation reaction was conducted as follows: 5 μl of PB1449 (100 μM), 4 ml of 5X ligase buffer, 0.5 μl of PNK in a 20 μl reaction. Phosphorylation was obtained upon incubation for 1 hour at 37°C, followed by heat inactivation of PNK for 20 min at 65°C. After incubation, the phosphorylated oligonucleotide PB1449 was annealed to 16 complementary oligonucleotides with the following sequence: 5′-GCAATTCCTCACGAGACCCGTCCTGTGCGCAAACTATTCTGAAGAACTGGCGAACTACNN-3′. The two Ns indicate the dinucleotide that is different for each of the 16 oligos, with N = A, C, G or T. In the annealing reaction, 40 μl of 5X ligase buffer and 130 μl of H2O were added to the phosphorylation reaction. 9.5 μl of this mix were used for annealing with 0.5 μl of each of the above 16 oligos (50 μM). Annealing, which was performed as described above for the library of adaptors, resulted in a 5′-phosphorylated double-stranded DNA with an overhang of 2 nucleotides, mimicking the product of AcuI digestion. The ligation between the adaptors and the phosphorylated products was performed as follows: 1 μl of annealed oligonucleotides, 2 μl of T4 ligase buffer, 0.5 μl of T4 ligase and 0.5 μl of adaptors in a 10 μl reaction. The ligation reaction was incubated for 1 hour at 25°C and 10 min at 65°C. Detection was performed using qPCR as described below in the DTECT protocol.
The assay performed to measure the efficiency of DNA ligation (Figure S3F) was conducted in a master mix reaction equivalent to 5 μl per time point as follows: 0.5 μl of AcuI digested products, 1 ml of T4 ligase buffer and 0.5 μl of adaptors with or without 0.5 μl of T4 ligase. The reactions were incubated at 25°C. After 5 min, 5 μl were taken from the reaction and the T4 ligase was added for 10 min at 65°C. 1 hour after the start of the ligation reaction, 5 μl were additionally taken from the reaction and heat inactivated. The rest of the reaction was incubated overnight for 16 hours and heat inactivated. The amount of products captured was determined by qPCR as described below.
To calculate the frequency of non-specific dinucleotide capture shown in Figure S3E, AcuI-generated fragments of WT SMARCAL1, SPRTN and PIK3R1 amplicons (obtained as described below) were ligated to each of the 16 library adaptors under the adaptor ligation conditions described above. The frequency of non-specific dinucleotide capture for all the adaptors non-complementary to the SMARCAL1, SPRTN and PIK3R1 dinucleotide signatures was calculated by qPCR analysis, as described below. Adaptors complementary to +1 and −1 AcuI-dependent slippage events were excluded from the analysis.
DTECT protocol
The DTECT protocol consists of 6 steps (I-VI, Figure 1A). I) Design of the AcuI-tagging primer, as described above. II) Amplification of the genomic locus of interest using the AcuI-tagging primer. The genomic DNA (gDNA) is prepared using the Quick Extract Solution (Epicenter) by incubating the cells at 65°C for 10 min and 95°C for 5 min. The genomic DNA is quantified by Nanodrop, diluted to 200 ng/μl in H2O and stored at −20°C or immediately used in PCR reactions. PCRs were performed in a 25 μl or 50 μl solution containing: 1 μM forward and reverse primers, 0.1 mM dNTP (NEB #N0447L), 1X Q5 buffer (NEB), 10–200 ng of gDNA, 1 unit of Q5 polymerase (NEB) and water. PCR reactions were conducted as follows: 95°C for 30 s; 40 cycles of 95°C for 10 s, 58°C for 10 s, 72°C for 45 s; and final amplification at 72°C for 1 min. When the AcuI-tagging PCR did not work on gDNA (< 5% of the cases), a PCR using standard locus-specific primers was performed to amplify the targeted locus and the AcuI-tagging PCR was conducted using this amplicon as template DNA. PCR products were loaded on a 2% agarose gel and run in TAE buffer. PCR products were extracted from gel and column purified (Zymo Research #D4008) and the purified products were subsequently quantified using Nanodrop. III) Digestion of the AcuI-tagged genomic amplicon with AcuI. The purified PCR products were digested by 0.25 μl AcuI (NEB #0641L) in a 20 μl reaction containing 1X CutSmart Buffer (NEB) supplemented with 40 μM S-adenosylmethionine (SAM) and 100 ng of purified PCR product. The reaction was incubated for 1 hour at 37°C with heat inactivation at 65°C for 20 min. IV) Isolation of the AcuI-digested genomic amplicon by solid phase reversible immobilization (SPRI). 10 μl of the digestion reaction were subsequently mixed with 18 μl of Agencourt AMPure XP magnetic beads (Beckman Coulter #A63881) by pipetting up and down the beads 10 times (volume ratio of DNA:beads = 1:1.8) and then incubated at room temperature for 5 min. This procedure resulted in the binding of the larger digestion fragment (> 100 bp) to the beads, while the smaller digested fragment (60 bp) remained in the supernatant. After incubation, the supernatant was isolated using a magnetic rack. 20 μl of the supernatant were recovered, diluted in 40 ml of H2O and stored at −20°C or immediately used for capture with DNA adaptors. V) Capture of the digested 60 bp-long products using DNA adaptors. The purified 60 bp-long DNA fragments were ligated to DNA adaptors generated as described above. The adaptors and the purified products were ligated in the following reaction: 6.5 μl of water, 2 μl of 5X ligase buffer (ThermoFisher Scientific), 0.5 μl of T4 ligase (ThermoFisher Scientific), 0.5 μl of adaptors and 0.5 μl of purified DNA product. The ligation reaction was performed for 1 hour at 25°C in a thermocycler, followed by inactivation of the T4 ligase for 10 min at 65°C. The ligated products were stored at −20°C or used directly for detection of the captured material. VI) Analytical or quantitative detection of the captured DNA products by PCR amplification. For analytical detection, the amplification of the captured material was performed by PCR in a 12.5 or 25 μl reaction volume containing 0.5 μM forward and reverse primers, 0.05 μM dNTP (NEB #N0447L), 1X Q5 buffer (NEB), 0.5–1 μl of ligated product, 0.1–0.2 μl of Q5 polymerase (NEB), 0.5–1 μl ligation reaction and water. PCR primers (PB1072 and PB1073) contained sequences complementary to the adaptor and handle (see above). The PCR program used was the following: 95°C for 1 min, and different number of cycles (indicated in each figure legend) of 95°C for 10 s, 65°C for 5 s, 72°C for 7 s. Detection of low abundant genomic variants (%1% frequency) was generally obtained with 23–25 PCR cycles, while detection of greater amounts of edited products was achieved with 17–22 PCR cycles. 5 μl of the PCR reactions were incubated with SYBR Gold (Thermofisher Scientific #S-11494), loaded on a 2% agarose gel and run in 1X TAE buffer until the DNA was separated. Gels were developed using LI-COR Odyssey. qPCR was performed using QuantStudio 3 (Applied Biosystems). qPCR reactions were performed as follows: 5 μl of 2X SYBR Gold master mix (ThermoFisher Scientific #4367659), 0.1 μl of forward and reverse primers (PB1072 and PB1073, 100 μM) and 1 μl of ligated products (diluted 1:100 in H2O) in a 10 μl reaction. The PCR program used in the qPCR reaction was the following: 95°C for 10 s and 40 cycles of 60°C 30 s, 95°C 15 s. Quantification of the frequency of genomic variants was conducted as described below (Quantification and Statistical Analysis section).
Next-generation sequencing
Samples for NGS were prepared by amplifying the edited regions of interest by PCR. Samples were sequenced by the Genome Sciences Facility at The Pennsylvania State College of Medicine or by Genewiz and the results were analyzed by Genewiz, or by using an R-based script of the Ciccia laboratory or CRISPResso2 (Clement et al., 2019). To ensure that no biases were introduced during DTECT assays, the AcuI-tagging amplicons for the BRCA1 and BRCA2 mutant samples were sequenced by NGS and analyzed using an R-based script. In this analysis, 7 sequences with > 6000 reads were filtered out from the analysis due to incorrect sequence. The editing frequency from the NGS results were determined using the formula: [(Number of reads for the edited dinucleotide) / (total number of reads)] × 100. Oligonucleotides used for PCR amplifications, Illumina sequencing adaptors and indexes are listed in Table S1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Technical duplicates of each sample were performed in each qPCR reaction. A standard curve to determine the concentration of the captured material was generated using predefined concentrations of a DTECT ligation product (Figure 1, step V) cloned into the pCR-Blunt II-TOPO vector (ThermoFisher Scientific; B650 plasmid, Addgene #139333) and oligos PB1072 and PB1073 (Table S1). The calculated standard curve corresponds to a linear curve with the following parameters: y = −3.3245x + 7.5504 and R2 = 0.99819. Quantification of the frequency of genomic variants was determined by calculating the mean Ct score (Mean Ct) of the two technical duplicates for each sample. The concentration of the captured material for each sample was determined using the following formula: Concentration = 10^[(Mean Ct −7.5504)/−3.3245]. The relative abundance between WT and mutant signatures was determined as follows: FrequencyMutant = [ConcentrationMutant / (ConcentrationMutant + ConcentrationWT)] × 100 and FrequencyWT = [ConcentrationWT / (ConcentrationMutant + ConcentrationWT)] × 100.
DATA AND CODE AVAILABILITY
R-based scripts of the Ciccia laboratory for analysis of NGS reads and ClinVar datasets are available upon request. The accession number for the raw NGS reads of edited DLD1 and NIH/3T3 cells, organoids and liver samples reported in this paper is SRA: SRP151111. The accession number for the raw NGS reads of edited HEK293T cells reported in this paper is BioProject: PRJNA603357. All uncropped gels, raw qPCR data and Sanger sequencing reads are available in Mendeley Data (https://doi.org/10.17632/gtkk6sthtw.1).
Supplementary Material
Highlights.
DTECT captures dinucleotide signatures revealed by the restriction enzyme AcuI
DTECT uses common adaptors to identify precise base changes, deletions, and insertions
DTECT quantifies the frequency of DNA variants generated by precision genome editing
DTECT identifies oncogenic mutations in mouse models and patient samples
ACKNOWLEDGMENTS
We thank Samuel H. Sternberg and Alejandro Chavez (Columbia University Irving Medical Center) for critical reading of the manuscript. We thank Eric E. Bryant (Columbia University) for providing help with the analysis of the ClinVar database, Andrew Palacios and Anuj Gupta for technical support, and Foon Wu-Baer and Madeline Wong for providing mouse tail DNA. pCMV-BE3, pCMV-PE2, pU6-pegRNA-GG acceptor, and pU6-Sp-pegRNA-HEK3_CTT_ins were gifts from David Liu (Addgene 73021, 132775, 132777, and 132778); pX330-U6-Chimeric_BB-CBhhSpCas9 was a gift from Feng Zhang (Addgene 42230); and pCDNA3-Flag::UbvG08 I44A, deltaGG was a gift from Daniel Durocher (Addgene 74939). Graphical abstract and figures were created using BioRender (https://biorender.com/). This work was supported by NIH grants R01 GM117064 and R01 CA197774 to A.C. and MSKCC Support Grant/Core Grant P30 CA008748. A.F. was supported by NIH grants R35 CA210065, R01 CA216981, and P30 CA013696 (Flow Cytometry Shared Resource and Genomics Shared Resource, Herbert Irving Comprehensive Cancer Center); Leukemia and Lymphoma Society grants LLS 6531-18 and 120749-14; and the Chemotherapy Foundation. R.B. was supported by NIH grants R01 CA172272 and P01 CA174653. K.O. was supported by a postdoctoral research fellowship from the Rally Foundation.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.068.
DECLARATION OF INTERESTS
P.B. and A.C. have filed a provisional patent for the invention described in this paper. R.L.L. is on the supervisory board of QIAGEN and is a scientific advisor to Loxo, Imago, C4 Therapeutics, and Isoplexis, each of which includes an equity interest. R.L.L. receives research support from and has consulted for Cel-gene and Roche; he has received research support from Prelude Therapeutics, and he has consulted for Incyte, Novartis, Astellas, Morphosys, and Janssen. He also has received honoraria from Lilly and Amgen for invited lectures and from Gilead for grant reviews.
REFERENCES
- Allen F, Crepaldi L, Alsinet C, Strong AJ, Kleshchevnikov V, De Angeli P, Palenikova P, Khodak A, Kiselev V, Kosicki M, et al. (2019). Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol 37, 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, and Liu DR (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolou P, and Fostira F (2013). Hereditary breast cancer: the era of new susceptibility genes. BioMed Res. Int 2013, 747318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri EM, Muir P, Akhuetie-Oni BO, Yellman CM, and Isaacs FJ (2017). Precise Editing at DNA Replication Forks Enables Multiplex Genome Engineering in Eukaryotes. Cell 171, 1453–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bath AJ, Milsom SE, Gormley NA, and Halford SE (2002). Many type IIs restriction endonucleases interact with two recognition sites before cleaving DNA. J. Biol. Chem 277, 4024–4033. [DOI] [PubMed] [Google Scholar]
- Bhagwat N, Koppikar P, Keller M, Marubayashi S, Shank K, Rampal R, Qi J, Kleppe M, Patel HJ, Shah SK, et al. (2014). Improved targeting of JAK2 leads to increased therapeutic efficacy in myeloproliferative neoplasms. Blood 123, 2075–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhojwani D, and Pui CH (2013). Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 14, e205–e217. [DOI] [PubMed] [Google Scholar]
- Billing D, Horiguchi M, Wu-Baer F, Taglialatela A, Leuzzi G, Nanez SA, Jiang W, Zha S, Szabolcs M, Lin CS, et al. (2018). The BRCT Domains of the BRCA1 and BARD1 Tumor Suppressors Differentially Regulate Homology-Directed Repair and Stalled Fork Protection. Mol. Cell 72, 127c–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billon P, Bryant EE, Joseph SA, Nambiar TS, Hayward SB, Rothstein R, and Ciccia A (2017). CRISPR-Mediated Base Editing Enables Efficient Disruption of Eukaryotic Genes through Induction of STOP Codons. Mol. Cell 67, 1068–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Kousholt AN, Harmsen T, Leemans C, Chen T, Jonkers J, and van Steensel B (2018). Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Res. 46, e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canny MD, Moatti N, Wan LCK, Fradet-Turcotte A, Krasner D, Mateos-Gomez PA, Zimmermann M, Orthwein A, Juang YC, Zhang W, et al. (2018). Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR-Cas9 genome-editing efficiency. Nat. Biotechnol 36, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Sarangi P, and D’Andrea AD (2016). The Fanconi anaemia pathway: new players and new functions. Nat. Rev. Mol. Cell Biol 17, 337–349. [DOI] [PubMed] [Google Scholar]
- Chadwick AC, Wang X, and Musunuru K (2017). In Vivo Base Editing of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) as a Therapeutic Alternative to Genome Editing. Arterioscler. Thromb. Vasc. Biol 37, 1741–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement K, Rees H, Canver MC, Gehrke JM, Farouni R, Hsu JY, Cole MA, Liu DR, Joung JK, Bauer DE, and Pinello L (2019). CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol 37, 224–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, and Zhang F (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieck CL, and Ferrando A (2019). Genetics and mechanisms of NT5C2-driven chemotherapy resistance in relapsed ALL. Blood 133, 2263–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow LE (2015). Modeling Disease In Vivo With CRISPR/Cas9. Trends Mol. Med 21, 609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzadfard F, and Lu TK (2018). Emerging applications for DNA writers and molecular recorders. Science 361, 870–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, Janizek JD, Huang X, Starita LM, and Shendure J (2018). Accurate classification of BRCA1 variants with saturation genome editing. Nature 562, 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Tao Y, Lamas V, Huang M, Yeh WH, Pan B, Hu YJ, Hu JH, Thompson DB, Shu Y, et al. (2018). Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 553, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, and Liu DR (2017). Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germini D, Tsfasman T, Zakharova VV, Sjakste N, Lipinski M, and Vassetzky Y (2018). A Comparison of Techniques to Evaluate the Effectiveness of Genome Editing. Trends Biotechnol 36, 147–159. [DOI] [PubMed] [Google Scholar]
- Guo X, Chavez A, Tung A, Chan Y, Kaas C, Yin Y, Cecchi R, Garnier SL, Kelsic ED, Schubert M, et al. (2018). High-throughput creation and functional profiling of DNA sequence variant libraries using CRISPR-Cas9 in yeast. Nat. Biotechnol 36, 540–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba H, Greaves M, and Mullighan CG (2013). Acute lymphoblastic leukaemia. Lancet 381, 1943–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M, and Haber JE (2016). The democratization of gene editing: Insights from site-specific cleavage and double-strand break repair. DNA Repair (Amst.) 44, 6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluesner MG, Nedveck DA, Lahr WS, Garbe JR, Abrahante JE, Webber BR, and Moriarity BS (2018). EditR: A Method to Quantify Base Editing from Sanger Sequencing. CRISPR J 1, 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor AC, Kim YB, Packer MS, Zuris JA, and Liu DR (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor AC, Zhao KT, Packer MS, Gaudelli NM, Waterbury AL, Koblan LW, Kim YB, Badran AH, and Liu DR (2017). Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv 3, eaao4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenay RT, Aghazadeh A, Hiatt J, Tse D, Roth TL, Apathy R, Shifrut E, Hultquist JF, Krogan N, Wu Z, et al. (2019). Large dataset enables prediction of repair after CRISPR-Cas9 editing in primary T cells. Nat. Biotechnol 37, 1034–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. ; Exome Aggregation Consortium (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, et al. (2005). Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 7, 387–397. [DOI] [PubMed] [Google Scholar]
- Levy JM, Yeh WH, Pendse N, Davis JR, Hennessey E, Butcher R, Koblan LW, Comander J, Liu Q, and Liu DR (2020). Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng 4, 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay H, Burger A, Biyong B, Felker A, Hess C, Zaugg J, Chiavacci E, Anders C, Jinek M, Mosimann C, and Robinson MD (2016). Crisp-RVariants charts the mutation spectrum of genome engineering experiments. Nat. Biotechnol 34, 701–702. [DOI] [PubMed] [Google Scholar]
- Liu Z, Lu Z, Yang G, Huang S, Li G, Feng S, Liu Y, Li J, Yu W, Zhang Y, et al. (2018). Efficient generation of mouse models of human diseases via ABE- and BE-mediated base editing. Nat. Commun 9, 2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin S, Jemt A, Terje-Hegge F, Foam N, Pettersson E, Käller M, Wirta V, Lexow P, and Lundeberg J (2015). Endonuclease specificity and sequence dependence of type IIS restriction enzymes. PLoS ONE 10, e0117059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashal RD, Koontz J, and Sklar J (1995). Detection of mutations by cleavage of DNA heteroduplexes with bacteriophage resolvases. Nat. Genet 9, 177–183. [DOI] [PubMed] [Google Scholar]
- McClellan J, and King MC (2010). Genetic heterogeneity in human disease. Cell 141, 210–217. [DOI] [PubMed] [Google Scholar]
- Mullally A, Lane SW, Ball B, Megerdichian C, Okabe R, Al-Shahrour F, Paktinat M, Haydu JE, Housman E, Lord AM, et al. (2010). Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell 17, 584–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima K, Khiabanian H, da Silva-Almeida AC, Tzoneva G, Abate F, Ambesi-Impiombato A, Sanchez-Martin M, Carpenter Z, Penson A, Perez-Garcia A, et al. (2016). Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 113, 11306–11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania S, Bade S, Le Guillou M, Burke K, Reed R, Bowman-Colin C, Su Y, Ting DT, Polyak K, Richardson AL, et al. (2014). BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nat. Commun 5, 5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen BS, Mandal PK, Frock RL, Boyraz B, Yadav R, Upadhyayula S, Gutierrez-Martinez P, Ebina W, Fasth A, Kirchhausen T, et al. (2017). Ectopic expression of RAD52 and dn53BP1 improves homology-directed repair during CRISPR-Cas9 genome editing. Nat. Biomed. Eng 1, 878–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinello L, Canver MC, Hoban MD, Orkin SH, Kohn DB, Bauer DE, and Yuan GC (2016). Analyzing CRISPR genome-editing experiments with CRISPResso. Nat. Biotechnol 34, 695–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P, Shandilya H, D’Alessio JM, O’Connor K, Durocher J, and Gerard GF (2004). Mutation detection using Surveyor nuclease. Biotechniques 36, 702–707. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees HA, and Liu DR (2018). Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet 19, 770–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy KR, Smith JD, Vonesch SC, Lin G, Tu CS, Lederer AR, Chu A, Suresh S, Nguyen M, Horecka J, et al. (2018). Multiplexed precision genome editing with trackable genomic barcodes in yeast. Nat. Biotechnol 36, 512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu SM, Koo T, Kim K, Lim K, Baek G, Kim ST, Kim HS, Kim DE, Lee H, Chung E, and Kim JS (2018). Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol 36, 536–539. [DOI] [PubMed] [Google Scholar]
- Shakya R, Reid LJ, Reczek CR, Cole F, Egli D, Lin CS, deRooij DG, Hirsch S, Ravi K, Hicks JB, et al. (2011). BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 334, 525–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen MW, Arbab M, Hsu JY, Worstell D, Culbertson SJ, Krabbe O, Cassa CA, Liu DR, Gifford DK, and Sherwood RI (2018). Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563, 646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CQ, Jiang T, Richter M, Rhym LH, Koblan LW, Zafra MP, Schatoff EM, Doman JL, Cao Y, Dow LE, et al. (2020). Adenine base editing in an adult mouse model of tyrosinaemia. Nat. Biomed. Eng 4, 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SLW, Chadha S, Liu Y, Gabasova E, Perera D, Ahmed K, Constantinou S, Renaudin X, Lee M, Aebersold R, et al. (2017). A Class of Environmental and Endogenous Toxins Induces BRCA2 Haploinsufficiency and Genome Instability. Cell 169, 1105–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschaharganeh DF, Xue W, Calvisi DF, Evert M, Michurina TV, Dow LE, Banito A, Katz SF, Kastenhuber ER, Weissmueller S, et al. (2014). p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 158, 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzoneva G, Perez-Garcia A, Carpenter Z, Khiabanian H, Tosello V, Allegretta M, Paietta E, Racevskis J, Rowe JM, Tallman MS, et al. (2013). Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat. Med 19, 368–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzoneva G, Dieck CL, Oshima K, Ambesi-Impiombato A, Sánchez-Martín M, Madubata CJ, Khiabanian H, Yu J, Waanders E, Iacobucci I, et al. (2018). Clonal evolution mechanisms in NT5C2 mutant-relapsed acute lymphoblastic leukaemia. Nature 553, 511–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Overbeek M, Capurso D, Carter MM, Thompson MS, Frias E, Russ C, Reece-Hoyes JS, Nye C, Gradia S, Vidal B, et al. (2016). DNA Repair Profiling Reveals Nonrandom Outcomes at Cas9-Mediated Breaks. Mol. Cell 63, 633–646. [DOI] [PubMed] [Google Scholar]
- Villiger L, Grisch-Chan HM, Lindsay H, Ringnalda F, Pogliano CB, Allegri G, Fingerhut R, Häberle J, Matos J, Robinson MD, et al. (2018). Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med 24, 1519–1525. [DOI] [PubMed] [Google Scholar]
- Wang L, Xue W, Yan L, Li X, Wei J, Chen M, Wu J, Yang B, Yang L, and Chen J (2017). Enhanced base editing by co-expression of free uracil DNA glycosylase inhibitor. Cell Res. 27, 1289–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh WH, Chiang H, Rees HA, Edge ASB, and Liu DR (2018). In vivo base editing of post-mitotic sensory cells. Nat. Commun 9, 2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M, Koteliansky V, Sharp PA, Jacks T, and Anderson DG (2014). Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol 32, 551–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Song CQ, Dorkin JR, Zhu LJ, Li Y, Wu Q, Park A, Yang J, Suresh S, Bizhanova A, et al. (2016). Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol 34, 328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafra MP, Schatoff EM, Katti A, Foronda M, Breinig M, Schweitzer AY, Simon A, Han T, Goswami S, Montgomery E, et al. (2018). Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol 36, 888–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Li J, Saucier JB, Feng Y, Jiang Y, Sinson J, McCombs AK, Schmitt ES, Peacock S, Chen S, et al. (2019). Non-invasive prenatal sequencing for multiple Mendelian monogenic disorders using circulating cell-free fetal DNA. Nat. Med 25, 439–447. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
R-based scripts of the Ciccia laboratory for analysis of NGS reads and ClinVar datasets are available upon request. The accession number for the raw NGS reads of edited DLD1 and NIH/3T3 cells, organoids and liver samples reported in this paper is SRA: SRP151111. The accession number for the raw NGS reads of edited HEK293T cells reported in this paper is BioProject: PRJNA603357. All uncropped gels, raw qPCR data and Sanger sequencing reads are available in Mendeley Data (https://doi.org/10.17632/gtkk6sthtw.1).