

Abstract
Site-selective nitrene transfer to di- and polyene substrates has been achieved using designed peptide-embedded bioxazoline ligands capable of binding copper. In model 1,3-diene substrates, the olefinic position proximal to a directing group was selectively functionalized. Additional studies indicate that this selectivity stems from non-covalent substrate-catalyst interactions. The peptide-mediated nitrene transfer was also applied to polyene natural product retinol and selective proximal functionalization allowed access to a cis-pyrroline modified retinoid.
Keywords: peptide ligands, site-selectivity, polyene natural products, nitrene transfer, aziridination
Graphical Abstract
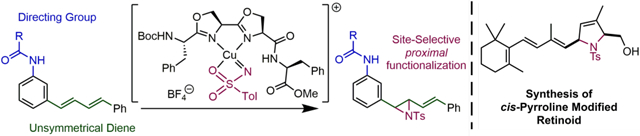
Chemo- and site-selective functionalization of complex molecules via incorporation of nitrogen is a valuable tool for diversifying biologically active natural products.[1] In this context, nitrene transfer to olefins—resulting in aziridination[2]—can potentially be applied to a large variety of natural products since carbon-carbon double bonds are a prevalent structural feature. However, selective olefin modification is challenging as a result of their non-polar properties. On the other hand, selective nitrene transfer to polyene natural products[3] (see Figure 1A for examples) would offer access to derivatives with potentially altered biological activity[4] and additionally the vinylaziridine products would allow for a variety of follow-up chemistry.[5] In most reports on diene aziridination, site-selectivity, if observed at all, has been a result of inherent selectivity.[6] The first studies on catalyst-control in diene functionalization were reported by Pérez and coworkers using silver complexes of homoscorpionate ligands (Tp*,Br) which allowed for directing nitrene transfer to the closer olefinic position, presumably as a result of catalyst interactions with the OH-group (Figure 1B, I).[7] In a related study, Schreiner and coworkers successfully designed a RhII-catalyst for aziridination of the central olefinic position in a farnesol derivative, mediated through hydrogen bonding (Figure 1B, II).[8]
Figure 1.

Polyene natural products and site-selective aziridination strategies. TcesNH2: 2,2,2-Trichloroethoxysulfonamide.
On the basis of precedents for reactions catalyzed by peptidyl-copper complexes,[9,10] the use of amides as directing groups in aziridinations,[11] and for bis(oxazoline) ligands influencing the course of site-selective reactions,[12] we wondered whether short peptide sequences could be designed that would act as ligands for transition metals and thereby allow for achieving site-selective nitrene transfer as a function of non-covalent substrate-catalyst interactions. With the aim of subsequent application in oligo- and polyene natural product functionalization, we were especially interested in conjugated olefin substrates (Figure 1C).
Oxazolines were also chosen as promising metal coordinating residues due to the analogy to bisoxazolines and related L2-type ligands[13] that are highly effective ligands in copper-catalyzed aziridinations. Furthermore, as a common structural feature in peptide-based natural products, oxazolines can be embedded in peptides via cyclodehydration of serine residues—a post-translational modification that results in alternation of hydrogen bonding networks and rigidification of peptide structure.[14] Naturally occurring oxazoline-containing peptides have been found to bind transition metals in peptide siderophores,[15] westiellamides, and patellamides (Figure 2A).[16] However, to the best of our knowledge, no catalytic activity for these naturally occurring complexes has been reported so far. The enzymatic mechanism of cyclodehydration is initiated via nucleophilic attack of the serine OH-group,[16b] while synthetic strategies include the aza-Wittig reaction,[17] and (di-ethylamino)sulfur trifluoride (DAST) mediated dehydration (Figure 2B).[18] Related to the recently reported and elegant approach of Meldal and Diness,[19] we hypothesized that double cyclodehydration of a Ser-Ser sequence would lead to oxazoline peptides that are structurally related to bidentate bioxazoline (BiOx) ligands (Figure 2B). Indeed, treatment of linear peptide 1 with DAST resulted in clean formation of bioxazoline 2 (Figure 2C). Complexation of 2 to Pd(CH3CN)2Cl2—square planar d8 configuration analogous to CuIII nitrene species—was studied in order to probe for transition metal binding, and clean formation of one single species was observed via NMR spectroscopy, presumably the N,N-chelate 3 (Figure 2C, see SI for details). These results allowed for preparation of designed bioxazolines embedded in peptide structures.
Figure 2.

Examples of metal complexes of naturally occurring oxazolines and synthesis of oxazoline peptide ligands. DAST: (diethylamino)sulfur trifluoride.
We began our catalytic studies with unsymmetrical 1,4-diarylbuta-1,3-dienes as model substrates since we expected these compounds to share similarities with polyene natural products in terms of reactivity, while being easier to handle and less prone to cis/trans isomerization. Also, they have previously been investigated in terms of their analogy to retinoids.[20] Non-covalent interaction sites were installed in the aryl meta-position, since para-functionalization was anticipated to be too remote while ortho-modified substrates showed high intrinsic selectivity as a result of electronic perturbance of the diene unit. We focused on studying amide interaction sites, and isovaleryl amide substrate 4, unlike similar compounds, showed excellent solubility properties and was therefore chosen for screening. However, initial studies were hindered by the high reactivity of the vinylaziridine products, which made their isolation challenging. We considered this an opportunity to develop a clean one-pot reaction sequence for the functionalization of dienes. Using parent 1,4-diphenyl-1,3-butadiene we were able to identify and isolate the vinyl aziridine products and found that a palladium–catalyzed hydrogenation with formic acid[21] resulted in highly selective cleavage of the allylic C-N bond while leaving the olefinic double bond intact (Figure 3A, see SI for details). In a control experiment we observed that 2,5-dimethyl-1-tosyl-3-pyrrolidine is not converted to the homoallylic amine under analogous reaction conditions (see SI for details) and, therefore, the involvement of pyrroline species stemming from formal [4+1] addition of the nitrene may be less likely. Interestingly, this one-pot reaction sequence to homoallylic amines complements methods for site-selective hydroamination of dienes.[22]
Figure 3.

Investigation of unsymmetrical 1,4-diarylbuta-1,3-dienes as polyene model substrates in an aziridination-hydrogenation sequence. [a]: Average of two catalysis trials. [b]: Determined by NMR versus trimethyl trimesic acid internal standard. [c]: CH3CN/CH2Cl2 (4:1) instead of CH2Cl2 as solvent. [d]: 10 mol% copper catalyst.
In order to assess intrinsic selectivity with bidentate oxazoline ligands, we initially used bioxazoline 7 as a truncated mimic of our oxazoline peptide ligands.[23] Almost equal amounts of proximal and distal products were formed (Figure 3B, entry 1). A quick screen of commercial ligands revealed that isopropylidene bisoxazolines performed slightly better but still only with low site-selectivity (Figure 3B, entry 3). Our study of peptide ligands began with optimizing the geometry of the chelating binding site through a screen of serine epimers (Figure 3B, entry 4–6) that revealed superior performance of homochiral ligands both in terms of yield and site-selectivity compared to other epimers and also commercial ligands. Investigation of a large variety of C- and N-terminal modified peptide ligands showed optimal results in cases with aromatic amino acid side chains (Figure 3B, entry 7–12), which led to the identification of the Boc-Phe-[Ser]-[Ser]-NH-CHPh2 sequence that resulted in good yield and selectivity greater than 1 : 5, favouring proximal functionalization. Notably, high levels of selectivity were maintained concomitant with only a slight decrease in yield at lowered catalyst loading (Figure 3B, entry 13).[24] Direct comparison of experiments in CH2Cl2 and CH3CN (Figure 3B, entry 1,2 & 7,8) revealed that site-selectivity in the peptide ligand catalyses is fully depleted when using a polar organic solvent capable of interfering with defined non-covalent substrate-catalyst interactions.
In order to gain more detailed insights into the origins of the site-selectivity enhancement, we expanded our study to similar meta-substituted 1,4-diaryl-1,3-butadiene substrates. We were especially curious whether indications for a hydrogen bond between the substrate amide NH and the catalyst could be found. In this context, we studied trifluoroacetamide 10 in direct comparison to the N-methylated analogue 11 (Figure 4, A). Bn-BiOx, again, resulted in an unselective reaction with 10, and screening (see SI for details) of our peptide oxazoline library led to the identification of a sequence—Boc-Phe-[Ser]-[Ser]-3,3-Dpa-OBn—with significantly improved site-selectivity (Figure 4B, Entry 1–2). Intriguingly, none of this selectivity enhancement was observed in the case of N-methylated substrate 11, which even showed a slight inherent selectivity towards the distal product (Figure 4B, Entry 3–4).
Figure 4.

Study of non-covalent substrate-catalyst interactions as origin for the increased site-selectivity. The distal functionalization product of 10 in the solid state[28] is depicted. [a] Results are the average of two catalytic trials. [b] 10 mol% catalyst loading, yields and site-selectivity determined via UPLC-MS versus trimethyl trimesic acid internal standard. [c] 20 mol% catalyst loading, yields and site-selectivity determined via NMR versus trimethyl trimesic acid internal standard. Dpa: 3,3-Diphenylalanine.
This observation supports involvement of the NH group in hydrogen bonding interaction between substrate and catalyst. Additionally, we studied 12 bearing an electron-rich 4-methoxybenzoyl group that could potentially engage in secondary π-stacking interactions. Ligand Boc-Phe-[Ser]-[Ser]-NH-CHPh2 (see SI for all data points) resulted in a significant site-selectivity increase to 1 : 6.4 favouring the proximal product, which corresponds to almost fourfold intrinsic selectivity (Figure 4B, Entries 5–6).
On the basis of these results, we turned our attention towards an application of peptide oxazolines to the site-selective functionalization of the polyene natural product retinol, as postulated in Figure 1A. Interest in derivatives of the latter mainly stems from activity in dermatology and cancer treatment. In this context, synthetic derivatives offer the possibility to increase stability towards cis/trans isomerism as well as selectivity enhancement for certain types of receptors.[25] Also, retinol contains a primary alcohol functionality that we hypothesized could act as non-covalent directing group with the peptide ligands. Following the general reaction scheme (Figure 5A), we aimed for functionalization of the proximal, polar site. Using UPLC-MS analysis, the reaction using Bn-BOX 8 was found to result in multiple species each exhibiting MS characteristics consistent with reaction of retinol with the nitrene source (see SI for details).
Figure 5.

Site-selective functionalization of retinol. (i) 1.0 equiv. retinol, 1.0 equiv. TsNIPh, 24 mol% ligand, 20 mol% [Cu(CH3CN)4]BF4, 10 equiv. Na2SO4, 33 mM in CH2Cl2, 0 °C to r.t., overnight, 50%.
However, using Boc-Phe-[Ser]-[Ser]-Phe-OMe, formation of all but two products was suppressed, which were identified as cis-13 and trans-13 (in almost equal amounts), two diastereomers of proximal functionalization. Most probably this overall [4+1] cycloaddition occurs via ring expansion of an initially formed vinyl aziridine intermediate (Figure 5A, top right).[26,27]
In order to probe for hydrogen bonding interactions, we also studied CH3CN as a polar solvent, which fully inhibited the Bn-BOX-mediated reaction while the peptide variant still showed formation of 13, albeit with significantly reduced yield and selectivity (see SI for details). Intriguingly, the isolated pyrroline retinoid trans-13 underwent reversible ring-opening in CDCl3 solution resulting in complete, clean epimerization to cis-13 over the course of several days (Figure 5B).
In contrast, isolated cis-13 remained unchanged under analogous conditions and the increased thermodynamic stability of cis-13 has been confirmed by DFT studies on a model pyrroline (see SI for details). This product epimerization implies that the peptide-catalyzed version of this reaction overall results in exclusive formation of cis-13 in 50% yield (NMR versus internal standard) without using either retinol or TsNIPh in excess.
In this study, peptide embedded oxazolines were designed and applied as ligands for site-selective nitrene transfer reactions with model dienes and—at the proof-of-concept level—the polyene natural product retinol. Additional studies strongly point towards non-covalent interactions between substrate and catalyst being the origin of highly increased selectivity for reactive sites close to directing groups capable of hydrogen-bonding. The successful selective functionalization of retinol via clean formation of stable pyrroline products opens up a variety of possible applications of this catalytic system, for example nitrene transfer to polyene antibiotics. Furthermore, this new class of bidentate oxazoline ligands could be useful in a variety of different catalytic transformations, with their C1-symmetry allowing for diverse tuning in the context of addressing challenges in stereo- and site-selective transformations.
Experimental Section
The Supporting Information contains experimental details for substrate synthesis, characterization of all catalysts and products, copies of NMR spectra, additional data regarding the analysis of catalytic experiments and assignments, as well as details on DFT investigations.
General procedure for the DAST mediated cyclodeydration
The serine peptide (1.0 equiv.) was dissolved in CH2Cl2 (anhydrous, 0.1 M) and the solution was cooled to −78 °C. DAST (3.0 equiv.) was added and the reaction mixture was stirred for 1 h at −78 °C and then slowly warmed to r.t. in the cooling bath over additional 3 h (excess dry ice was removed). The homogeneous, clear solution was then cooled down to −78 °C again, K2CO3 (anhydrous, 6.0 equiv.) was added, and stirring was continued for 1 h. Subsequently, the reaction mixture was poured into NaHCO3 solution (aqueous, saturated, fivefold amount w.r.t. CH2Cl2) and diluted with CH2Cl2 (fourfold amount w.r.t. reaction solvent). The organic phase was separated and the aqueous phase was extracted with CH2Cl2. The combined organic phases were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified via automated reversed phase chromatography (load in CH3CN; C18 silica; CH3CN/water; 1 CV: 30%, 10 CV: 30–100%, 1 CV: 100%; product containing fractions were combined, extracted with CH2Cl2, dried over Na2SO4, filtered, and concentrated under reduced pressure). The pure product was precipitated from Et2O/npentane, dried in vacuo overnight, and stored in a glove box to prevent decomposition.
General procedure for the aziridination-hydrogenation sequence
A Schlenk flask loaded with Na2SO4 (anhydrous, 0.50 mmol, 10 equiv.) was heated with a heat gun and conditioned with N2 while cooling down to room temperature. Ligand (12 μmol, 24 mol%) and [Cu(CH3CN)4]BF4 (10.0 μmol, 20 mol%) were added, and the flask was evacuated and backfilled with N2. Then, CH2Cl2 (anhydrous, 2.0 mL, 25 mM w.r.t. nitrene source) was added and the catalyst solution was stirred at room temperature for 1 h. Subsequently, diene substrate (75.0 μmol, 1.5 equiv.) was added and the solution was cooled to 0 °C (ice bath) followed by addition of TsNIPh (50.0 μmol, 1.0 equiv.). The reaction was stirred at 0 °C for 1 h. While still in the cooling bath, the reaction mixture was diluted with THF (anhydrous, 2.0 mL), and DIPEA (anhydrous, 1.50 mmol, 30 equiv.), Pd2(dba)3 (10.0 μmol, 20 mol%), and formic acid (1.50 mmol, 30 equiv.) were added. The ice bath was removed and the reaction mixture was stirred at room temperature for 2 h. Then, it was passed through a plug of silica (Pasteur pipette, 5 cm, rinsed with 5 CV hexanes/EtOAc (50:50)) and concentrated under reduced pressure. The crude products were purified via automated normal phase chromatography (silica; CH2Cl2/MeOH; 1 CV: 0%, 12 CV: 0–15%; all fractions containing product—as determined by UPLC–MS analysis—were collected). Then, 350 μL internal standard solution (trimethyl trimesic acid in deacidified CDCl3, 47.7 mM, 16.7 μmol, 0.33 equiv.) was added, the sample was diluted with CDCl3, and analyzed via NMR spectroscopy (10 s relaxation time).
Site-Selective Retinol Functionalization
A Schlenk flask loaded with Na2SO4 (anhydrous, 1.00 mmol, 10 equiv.) was heated with a hot air gun and conditioned with N2 while cooling down to room temperature. Ligand (12 μmol, 12 mol%) and [Cu(CH3CN)4]BF4 (10.0 μmol, 10 mol%) were added, and the flask was evacuated and backfilled with N2. Then, CH2Cl2 (anhydrous, 3.0 mL, 33 mM w.r.t. nitrene source) was added and the catalyst solution was stirred at room temperature for 1 h. Subsequently, diene substrate (100 μmol, 1.0 equiv.) was added and the solution was cooled to 0 °C (ice bath) followed by addition of TsNIPh (100 μmol, 1.0 equiv.). The reaction mixture was let warm up in the cooling bath overnight. Subsequently, it was passed through a plug of silica (Pasteur pipette, 5 cm, rinsed with 5 CV hexanes/EtOAc (50:50)). See Figure SI-21 for UPLC-MS analysis. Then, 700 μL internal standard solution (trimethyl trimesic acid in deacidified CDCl3, 47.7 mM, 33.3 μmol, 0.33 equiv.) were added and the yield was determined via NMR spectroscopy (10 s relaxation time).
Supplementary Material
Acknowledgements
This work is supported by the National Institutes of Health (NIH R35-GM132092). GS is also very grateful to the Deutsche Forschungsgemeinschaft (DFG) for a postdoctoral fellowship (STO 1175/1-1). We also thank Dr. Brandon Q. Mercado for assistance with X-ray crystallography.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201
References
- [1].a) Roizen JL, Harvey ME, Du Bois J, Acc. Chem. Res 2012, 45, 911–922; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Alderson JM, Corbin JR, Schomaker JM, Acc. Chem. Res 2017, 50, 2147–2158; [DOI] [PubMed] [Google Scholar]; c) Clark JR, Feng K, Sookezian A, White MC, Nat. Chem 2018, 10, 583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Minakata S, Acc. Chem. Res 2009, 42, 1172–1182; [DOI] [PubMed] [Google Scholar]; b) Zhu Y, Wang Q, Cornwall RG, Shi Y, Chem. Rev 2014, 114, 8199–8256; [DOI] [PubMed] [Google Scholar]; c) Degennaro L, Trinchera P, Luisi R, Chem. Rev 2014, 114, 7881–7929. [DOI] [PubMed] [Google Scholar]
- [3].a) Rychnovsky SD, Chem. Rev 1995, 95, 2021–2040; [Google Scholar]; b) Thirsk C, A. Whiting, J. Chem. Soc., Perkin Trans 1 2002, 999–1023. [Google Scholar]
- [4].Ismail FMD, Levitsky DO, Dembitsky VM, Eur. J. Med. Chem 2009, 44, 3373–3387. [DOI] [PubMed] [Google Scholar]
- [5].a) Sweeney JB, Chem. Soc. Rev 2002, 31, 247–258; [DOI] [PubMed] [Google Scholar]; b) Watson IDG, Yu L, Yudin AK, Acc. Chem. Res 2006, 39, 194–206; [DOI] [PubMed] [Google Scholar]; c) Ohno H, Chem. Rev 2014, 114, 7784–7814. [DOI] [PubMed] [Google Scholar]
- [6].a) Knight JG, Muldowney MP, Synlett 1995, 949–951; [Google Scholar]; b) Nishimura M, Minakata S, Thongchant S, Ryu I, Komatsu M, Tetrahedron Lett. 2000, 41, 7089–7092; [Google Scholar]; c) Atkinson RS, Meades CK, J. Chem. Soc., Perkin Trans 1 2001, 1518–1527; [Google Scholar]; d) Piangiolino C, Gallo E, Caselli A, Fantauzzi S, Ragaini F, Cenini S, Eur. J. Org. Chem 2007, 743–750; [DOI] [PubMed] [Google Scholar]; e) Mack DJ, Njardarson JT, Chem. Sci 2012, 3, 3321–3325; [Google Scholar]; f) Armstrong A, Pullin RDC, Jenner CR, J. N. Scutt, J. Org. Chem 2010, 75, 3499–3502. [DOI] [PubMed] [Google Scholar]
- [7].a) Llaveria J, Beltrán Á, Díaz-Requejo MM, Matheu MI, Castillón S, Pérez PJ, Angew. Chem. Int. Ed 2010, 49, 7092–7095; [DOI] [PubMed] [Google Scholar]; b) Llaveria J, Beltrán Á, Sameera WMC, Locati A, Díaz-Requejo MM, Matheu MI, Castillón S, Maseras F, Pérez PJ, J. Am. Chem. Soc 2014, 136, 5342–5350. [DOI] [PubMed] [Google Scholar]
- [8].Berndt J-P, Radchenko Y, Becker J, Logemann C, Bhandari DR, Hrdina R, Schreiner PR, Chem. Sci 2019, 10, 3324–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Brown MK, Degrado SJ, Hoveyda AH, Angew. Chem. Int. Ed 2005, 44, 5306–5310. [DOI] [PubMed] [Google Scholar]
- [10].a) Kim B, Chinn AJ, Fandrick DR, Senanayake CH, Singer RA, Miller SJ, J. Am. Chem. Soc 2016, 138, 7939–7945; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chinn AJ, Kim B, Kwon Y, Miller SJ, J. Am. Chem. Soc 2017, 139, 18107–18114; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kwon Y, Chinn AJ, Kim B, Miller SJ, Angew. Chem. Int. Ed 2018, 57, 6251–6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhong F, Bach T, Chem. Eur. J 2014, 20, 13522–13526. [DOI] [PubMed] [Google Scholar]
- [12].Allen CL, Miller SJ, Org. Lett 2013, 15, 6178–6181. [DOI] [PubMed] [Google Scholar]
- [13].a) Li Z, Conser KR, Jacobsen EN, J. Am. Chem. Soc 1993, 115, 5326–5327; [Google Scholar]; b) Evans DA, Faul MM, Bilodeau MT, Anderson BA, Barnes DM, J. Am. Chem. Soc 1993, 115, 5328–5329; [Google Scholar]; c) Desimoni G, Faita G, Jørgensen KA, Chem. Rev 2011, 111, PR284–PR437. [DOI] [PubMed] [Google Scholar]
- [14].a) Abbenante G, Fairlie DP, Gahan LR, Hanson GR, Pierens GK, van den Brenk AL, J. Am. Chem. Soc 1996, 118, 10384–10388; [Google Scholar]; b) Sinha Roy R, Gehring AM, Milne JC, Belshaw PJ, Walsh CT, Nat. Prod. Rep 1999, 16, 249–263; [DOI] [PubMed] [Google Scholar]; c) Nolan EM, Walsh CT, ChemBioChem 2009, 10, 34–53; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Walsh CT, Malcolmson SJ, Young TS, ACS Chem. Biol 2012, 7, 429–442; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Yang X, van der Donk WA, Chem. Eur. J 2013, 19, 7662–7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Snow GA, Bacteriol. Rev 1970, 34, 99–125; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Luo M, Fadeev EA, Groves JT, Nat. Chem. Biol 2005, 1, 149–153. [DOI] [PubMed] [Google Scholar]
- [16].a) van den Brenk AL, Byriel KA, Fairlie DP, Gahan LR, Hanson GR, Hawkins CJ, Jones A, Kennard CHL, Moubaraki B, Murray KS, Inorg. Chem 1994, 33, 3549–3557; [Google Scholar]; b) Wipf P, Venkatraman S, Miller CP, Geib SJ, Angew. Chem. Int. Ed 1994, 33, 1516–1518. [Google Scholar]
- [17].Riedrich M, Harkal S, Arndt H-D, Angew. Chem. Int. Ed 2007, 46, 2701–2703. [DOI] [PubMed] [Google Scholar]
- [18].a) Phillips AJ, Uto Y, Wipf P, Reno MJ, Williams DR, Org. Lett 2000, 2, 1165–1168; [DOI] [PubMed] [Google Scholar]; b) Brandstätter M, Roth F, Luedtke NW, J. Org. Chem 2015, 80, 40–51. [DOI] [PubMed] [Google Scholar]
- [19].Jacobsen CB, Nielsen DS, Meldal M, Diness F, J. Org. Chem 2019, DOI: 10.1021/acs.joc.9b00732. [DOI] [PubMed] [Google Scholar]
- [20].a) M. T. Allen, Whitten DG, Chem. Rev 1989, 89, 1691–1702; [Google Scholar]; b) Yang L.-y., Liu RSH, Boarman KJ, Wendt NL, Liu J, J. Am. Chem. Soc 2005, 127, 2404–2405. [DOI] [PubMed] [Google Scholar]
- [21].Satake A, Shimizu I, Yamamoto A, Synlett 1995, 64–68. [Google Scholar]
- [22].a) Löber O, Kawatsura M, Hartwig JF, J. Am. Chem. Soc 2001, 123, 4366–4367; [DOI] [PubMed] [Google Scholar]; b) Pawlas J, Nakao Y, Kawatsura M, Hartwig JF, J. Am. Chem. Soc 2002, 124, 3669–3679; [DOI] [PubMed] [Google Scholar]; c) Brouwer C, He C, Angew. Chem. Int. Ed 2006, 45, 1744–1747; [DOI] [PubMed] [Google Scholar]; d) Huang L, Arndt M, Gooßen K, Heydt H, Gooßen LJ, Chem. Rev 2015, 115, 2596–2697; [DOI] [PubMed] [Google Scholar]; e) Yang X-H, Dong VM, J. Am. Chem. Soc 2017, 139, 1774–1777; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Yang X-H, Lu A, Dong VM, J. Am. Chem. Soc 2017, 139, 14049–14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Although the focus of this present study was on site-selectivity, enantio-selectivity was also determined. Proximal product 6 was formed racemic applying ligand 7, with 35% ee using (S,S)-tBu-BOX, and with 32% ee in the case of Boc-Phe-[Ser]-[Ser]-Phe-OMe. [Google Scholar]
- [24].Further lowering of the catalyst loading to 5 mol% resulted in no significant TON increase.
- [25].a) Barnard JH, Collings JC, Whiting A, Przyborski SA, Marder TB, Chem. Eur. J 2009, 15, 11430–11442; [DOI] [PubMed] [Google Scholar]; b) Álvarez R, Vaz B, Gronemeyer H, de Lera ÁR, Chem. Rev 2014, 114, 1–125. [DOI] [PubMed] [Google Scholar]
- [26].a) Fugami K, Y. Morizawa, Ishima K, Nozaki H, Tetrahedron Lett. 1985, 26, 857–860; [Google Scholar]; b) Brichacek M, Lee D, Njardarson JT, Org. Lett 2008, 10, 5023–5026; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Brichacek M, Navarro Villalobos M, Plichta A, Njardarson JT, Org. Lett 2011, 13, 1110–1113; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wu Q, Hu J, Ren X, Zhou J, Chem. Eur. J 2011, 17, 11553–11558. [DOI] [PubMed] [Google Scholar]
- [27].It has to be noted that based on the pyrroline product, initial aziridination of the first or second double bond could funnel to the same product species.
- [28].CCDC-1943272 contains the supplementary crystallographic data and it can be obtained free of charge from The Cambridge Crystallographic Data Centre via. www.ccdc.cam.ac.uk/data_request/cif.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.