

Abstract
In response to signals associated with infection or tissue damage, macrophages undergo a series of dynamic phenotypic changes. Here we show that during the response to LPS and interferon-γ stimulation, metabolic reprogramming in macrophages is also highly dynamic. Specifically, the TCA cycle undergoes a two-stage remodeling: the early stage is characterized by a transient accumulation of intermediates including succinate and itaconate, while the late stage is marked by the subsidence of these metabolites. The metabolic transition into the late stage is largely driven by the inhibition of pyruvate dehydrogenase complex (PDHC) and oxoglutarate dehydrogenase complex (OGDC), which is controlled by the dynamic changes in lipoylation state of both PDHC and OGDC E2 subunits and phosphorylation of PDHC E1 subunit. This dynamic metabolic reprogramming results in a transient metabolic state that strongly favors HIF-1α stabilization during the early stage, which subsides by the late stage; consistently, HIF-1α levels follow this trend. This study elucidates a dynamic and mechanistic picture of metabolic reprogramming in LPS and interferon-γ stimulated macrophages, and provides insights into how changing metabolism can regulate the functional transitions in macrophages over a course of immune response.
Introduction
In response to external stimuli, macrophages are capable of rapidly adopting a multitude of cellular functions; this plasticity fashions macrophages as a key part of innate immunity. In response to signals associated with microbial infection or tissue damage, such as lipopolysaccharide (LPS) and interferon-γ (IFN-γ), naïve macrophages rapidly adopt pro-inflammatory phenotypes. This classical (M1) activation is characterized by an increased production of pro-inflammatory cytokines and nitric oxide, which is important for eradicating the invading pathogen.1,2 Prolonged inflammation, however, can lead to cellular damage.3 To prevent this damage and promote tissue repair, when exposed to stimuli repeatedly or for extended periods, macrophages develop more M2-like phenotypes.4 This state is characterized by decreased production of some pro-inflammatory cytokines and increased production of anti-inflammatory cytokines.5–7 Transition into the proper phenotypic state is critical for macrophage function in innate immunity, and dysregulation of this process plays an important role in diseases such as autoimmunity and sepsis.8,9
Increasing evidence suggests that the transition of macrophages into diverse functional states is accompanied by, and reliant on, metabolic reprogramming. This reprogramming can provide energy and precursors required for specific functions as well as modulate the level of signaling metabolites.10,11 The metabolism of classically activated macrophages is most notably characterized by unique changes in arginine metabolism, increased dependence on glycolysis, and altered mitochondrial respiration, as compared to naïve and M2 macrophages.11 Particularly, recent studies have highlighted the importance of TCA cycle remodeling in classical macrophage activation through regulation of isocitrate dehydrogenase (IDH) and succinate dehydrogenase (SDH). This is coupled to rapid accumulation of itaconate and succinate, as well as an altered cellular redox state.10,12 Emerging evidence demonstrates these metabolites have important immunoregulatory roles, and changes in their levels can impact the inflammatory status of macrophages.11–19
Although our knowledge of metabolic features associated with acute classical activation is expanding, little is known about metabolic reprogramming in macrophages upon repeated or prolonged stimulation. Since the functional phenotype changes dynamically as macrophages respond to stimulation, it is likely that major metabolic reprogramming continues to accompany and support the evolving functions. Furthermore, the ability of macrophages to undergo transitions through pro-inflammatory state and anti-inflammatory state is mediated by the remodeling of epigenetic landscape and transcriptional activity.10,20–23 As recent studies point to the ability of key metabolites to modulate transcriptional programs,24–27 it is possible that shifts in metabolic state play an important role in these transitions by influencing transcriptional program.
Using metabolomics and isotopic labeling approaches, we elucidated the metabolic reprogramming in murine bone marrow-derived macrophages (BMDM) and RAW 264.7 cells throughout a time-course of LPS and IFN-γ stimulation, and determined the underlying mechanisms driving the metabolic transitions. We found that after an extended period (48–72h), macrophages transit into a unique metabolic and functional state that is distinct from what observed during the initial response to stimulation (6–24h) or in unstimulated (0h) macrophages. Disruption of flux through two mitochondrial enzyme complexes, pyruvate dehydrogenase complex (PDHC) and oxoglutarate dehydrogenase complex (OGDC), is critical for the adoption of this metabolic state. This flux modulation leads to profound and dynamic shifts in important metabolites, which correlate strongly with the production of several pro-inflammatory cytokines and HIF-1α protein levels. These results elucidate the highly dynamic metabolic reprogramming in macrophages in response to LPS and IFN-γ stimulation, and highlights the important temporal connection between metabolism and inflammatory state.
Results
Macrophages undergo dynamic metabolomic changes in response to LPS and interferon-γ stimulation.
To characterize the metabolic reprogramming in macrophages throughout a course of immune response, we first quantified the intracellular and extracellular metabolomic profile in RAW 264.7 cells and BMDMs continually exposed to LPS and IFN-γ for up to 72h. At the same time, the production of nitric oxide and several pro-inflammatory cytokines was measured as indicators of inflammatory status. The production rates of these cytokines and nitric oxide changed over time, and the dynamic trends were cytokine-specific (Figure 1a): Nitric oxide release increased dramatically 24h after stimulation and remained elevated throughout the rest of the measured time-course. In contrast, production of IL-6 and TNF-α increased soon after stimulation but subsided after 48h, while IL-1β release increased most profoundly at later time points of stimulation. The reduced release of pro-inflammatory cytokines TNF-α and IL-6 at late time points is consistent with previous studies in vitro and in vivo, which showed their production is suppressed upon repeated or prolonged LPS stimulation.6,7,23
Figure 1. Macrophages undergo dynamic metabolomic and functional changes in response to LPS and IFN-γ stimulation.
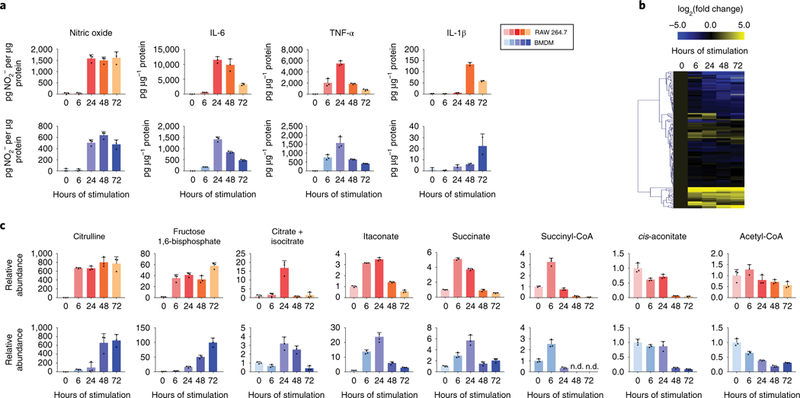
a. Release of cytokines and nitric oxide by RAW 264.7 cells or BMDMs stimulated with LPS and IFN-γ for indicated time. Bar graph with error bar represents mean + SD, dots represent individual values (n=3 independent samples for nitric oxide, IL-6 and TNFα; n=2 for IL-1α).
b. Metabolomic changes in RAW 264.7 cells after exposed to LPS and IFN-γ for 0–72 hours, as indicated. Relative abundance of metabolite is compared to unstimulated cells and displayed on a log2 scale as a heatmap. Each box represents the mean (n=3 independent samples).
c. Relative intracellular abundance of metabolites in RAW 264.7 cells and BMDMs after stimulation with LPS and IFN-γ.Data are normalized to protein content and expressed relative to abundance in unstimulated cells (0h). Bar graph with error bar represents mean + SD (n=3 independent samples), dots represent individual values. (nd indicates non-detectable)
As cytokine production rates change profoundly and dynamically, the levels of a wide variety of intracellular metabolites also changed substantially (Figure 1b). Most of these metabolites showed consistent kinetic trends in the two macrophage cell models investigated, and compounds in the same metabolic pathway often clustered together based on their dynamic behavior. Among the most profound alterations was the increase in citrulline level (Figure 1c). Increased arginine metabolism via inducible nitric oxide synthase (iNOS), which produces nitric oxide and citrulline, is a well-established characteristic of classically activated macrophages.28,29 The rapid increase and continued elevation of citrulline throughout the time-course mirrored the dynamics of nitric oxide production (Figure 1a). Similarly, as early as 6h, intracellular levels of many glycolytic intermediates, such as fructose-1,6-bisphosphate, showed significant increases, which is consistent with preceding work indicating glycolysis is increased with classical activation; 10,26,30 this increase in glycolytic intermediates persisted throughout the stimulation time-course (Figure 1c).
In contrast to these persisting changes, many other metabolites showed drastic differences in early and late phases post stimulation. It is particularly intriguing that several important metabolites in, or immediately derived from, the TCA cycle, including citrate, succinate, succinyl-CoA, and itaconate, showed dynamic patterns similar to the production of the cytokines TNF-α and IL-6 - a profound increase after 6–24h of stimulation followed by a reduction, to levels similar to, or below, that in unstimulated macrophages (Figure 1c). Conversely, total levels of cis-aconitate, and acetyl-CoA showed little change at early time points, but decreased at later time points (Figure 1c). These highly dynamic metabolic changes were not due to changes in general cell health and viability, as the macrophages remained highly viable throughout the time-course (Supplementary Figure 1).
Release of extracellular metabolites was also highly dynamic (Supplementary Figure 2). After stimulation, the release of lactate and citrulline increased and remained high until the end of the measured time-course, consistent with increased glycolysis and iNOS activity. Release of itaconate transiently increased, mirroring the trend of intracellular itaconate levels, and pyruvate release decreased with increasing stimulation time. We did not observe any detectable extracellular ATP or adenosine, two potential signaling compounds that can regulate immune response.31 However, we found that another purine metabolism product, xanthine, is released at a low rate, which increased at later time points.
Taken together, these data illustrate that upon stimulation with LPS and IFN-γ, the metabolic landscape in macrophages, similar to cytokine production, is highly dynamic, and that these dynamic trends are diverse and pathway-specific.
Metabolic flux through TCA cycle is dynamically reprogrammed.
Among the diverse dynamic behaviors in different metabolites, we were particularly interested in the transient increase in TCA cycle intermediates, especially given several recent studies highlighting the importance of these intermediates, particularly itaconate and succinate, in regulating the inflammatory status of macrophages.12–16,19 To test if the transient increase in succinate and itaconate level is robust, we performed the experiment in RAW 264.7 cells in two other ways. In studies described above, fresh media containing LPS and IFN-γ were refreshed regularly throughout the time course, allowing for continual exposure to stimuli. In the alternative protocols we did the following: (i) cells were stimulated with a single dose of LPS and IFN-γ for 2 hours, after which the stimuli were completely removed and cells were cultured in fresh media without LPS and IFN-γ (acute stimulation, Supplementary Figure 3a); and (ii) cells were stimulated with a single dose of LPS and IFN-γ, then metabolism was monitored for 48h without any media changes (no media change, Supplementary Figure 3b). We found in both cases succinate and itaconate showed a similar transient increase, suggesting this dynamic trend is not dependent on the replenishment of media containing LPS and IFN-γ, and is not largely affected by the possible autocrine effects of cytokines.
To elucidate the mechanisms driving these robust changes in metabolite levels, we utilized a U-13C-glucose tracer to probe flux changes through the TCA cycle over the time-course (Figure 2a). As early as 6h of stimulation, the fraction of labeled α-ketoglutarate was dramatically reduced as compared to unstimulated macrophages (Figure 2b). The labeling of TCA cycle metabolites downstream of α-ketoglutarate, such as malate, also reflected this early loss of labeling, while total labeling of TCA cycle metabolites upstream of α-ketoglutarate, such as cis-aconitate, remained high at this point (Figure 2b). This reduction in α-ketoglutarate labeling suggested reduced relative flux into α-ketoglutarate from citrate. Previous work has reported that after 24h of LPS and IFN-γ stimulation, macrophages experience a break in the TCA cycle at isocitrate dehydrogenase (IDH), due to reduced Idh expression.10,16 We confirmed that Idhl and Idh2 expression reduced rapidly after stimulation, and found this persisted throughout the time-course (Supplementary Figure 4). The early reduction in IDH flux drove the drastic accumulation in citrate/isocitrate level after 6–24 h of stimulation (Figure 1c). The block of IDH flux, along with increased IRG1 expression (Supplementary Figure 5),10 also diverted TCA cycle flux from glucose into production of itaconate, and resulted in the increased itaconate levels observed at this time point (Figure 1c).
Figure 2. Glucose labeling reveals dynamic reprogramming of TCA cycle flux in LPS and IFN-γ stimulated macrophages.
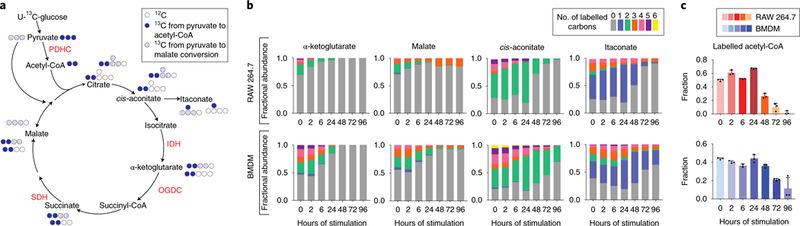
a. Schematic showing the labeling incorporation from U-13C-glucose into the TCA cycle. Possible isotopomers labeled within one TCA cycle turn are shown next to the metabolite. PDHC (pyruvate dehydrogenase complex), IDH (isocitrate dehydrogenase), OGDC (oxoglutarate dehydrogenase complex), SDH (succinate dehydrogenase).
b. Labeling pattern of intracellular metabolites in RAW 264.7 cells and BMDMs after stimulation with LPS and IFN-γ. Bar graph represents the mean (n=3 independent samples).
c. The fraction of acetyl-CoA containing 2-labeled acetyl moiety. The labeled fraction was determined based on measured labeling pattern of acetyl-CoA and coenzyme A, as specified in Methods. Bar graph with error bar represents mean + SD (n=3 independent samples), dots represent individual values.
After 48h of stimulation, the labeling of cis-aconitate and itaconate, especially labeled forms primarily derived from labeled acetyl-group (cis-aconitate M+2, M+5; itaconate M+1, M+4), decreased significantly (Figure 2b). In addition, during this late stage, the labeling incorporation from glucose into the acetyl moiety in acetyl-CoA was greatly reduced (Figure 2c), although lower glycolysis remained fully labeled. These results, together with the reduction in total acetyl-CoA level (Figure 1c), suggest a block in glucose oxidation flux through pyruvate dehydrogenase complex (PDHC) in macrophages during the late stage.
Reduction in PDHC flux would cause a reduction of glucose-derived citrate production. Subsequently, because itaconate is produced downstream of citrate by aconitate decarboxylation by IRG1,32,33 limited influx to citrate can also limit the downstream production of itaconate. Consistently, reduced levels of citrate, cis-aconitate, and itaconate were observed at later time points (Figure 1c), despite sustained IRG1 levels (Supplementary Figure 5). Recent studies have illustrated that itaconate is a competitive inhibitor of succinate dehydrogenase (SDH), and in classically activated macrophages, high levels of itaconate causes accumulation of succinate.12,13,34 Consistent with this, we observed an increase in succinate during early time points (6–24h). However, succinate levels subsided at later time points, following the trend of itaconate levels (Figure 1C). This suggests the inhibition of PDHC is also likely to contribute to the fall of succinate levels at this stage, by limiting itaconate production flux from glucose and thus releasing the competitive inhibition of SDH.
To overcome the limitation of very low labeling enrichment in the metabolites downstream of α-ketoglutarate from the U-13C-glucose tracer, we further used a U-13C15N glutamine tracer to examine TCA cycle flux through reactions downstream of IDH (Figure 3a). Mirroring the U-13C-glucose labeling result, when labeled with U-13C15N-glutamine, there is a complete loss of 3-labeled α-ketoglutarate from 4-labeled citrate after 24h of stimulation, reflecting reduced oxidative IDH flux (Figure 3b). Furthermore, the lack of 5-labeling in citrate/isocitrate while 5-labeled α-ketoglutarate is abundant indicates minimal to no reductive IDH activity (Figure 3b). These data are consistent with the persistently low Idh expression throughout the time-course (Supplementary Figure 4).
Figure 3. Glutamine labeling reveals remodeling of TCA cycle flux at the late stage.
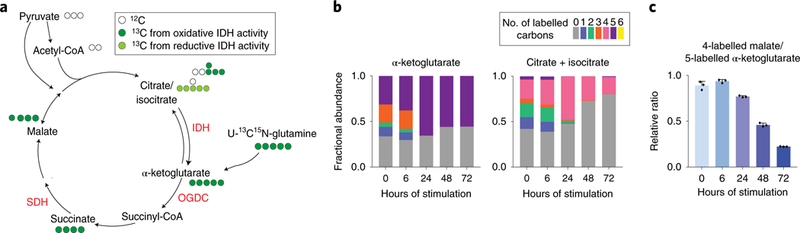
a. Schematic showing the labeling incorporation from U-13C15N-glutamine into TCA cycle. Possible isotopomers labeled within one TCA cycle turn are shown next to the metabolite. IDH (isocitrate dehydrogenase), OGDC (oxoglutarate dehydrogenase complex), SDH (succinate dehydrogenase).
b. Labeling pattern of metabolites in BMDMs after stimulation with LPS and IFN-γ for indicated time. Bar graph represents the mean (n=3 independent samples).
c. Ratio between the fractions of fully labeled malate to fully labeled α-ketoglutarate in BMDMs labeled with U-13C15N-glutamine, after stimulation with LPS and IFN-γ for indicated time. Bar graph with error bar represents mean + SD (n=3 independent samples), dots represent individual values.
Interestingly, we found that profound remodeling of TCA flux downstream of IDH occurred during the late stage of response to stimulation. The ratio of 4-labeled malate to 5-labeled α-ketoglutarate reflects the relative contribution of oxidative TCA cycle flux to the pool of malate. This ratio remained high in unstimulated macrophages through 24h of stimulation, but decreased profoundly after 48 to 72h (Figure 3c). This pointed to a block in the TCA cycle between α-ketoglutarate and malate at the late stage. Furthermore, we observed succinyl-CoA levels depleted at these time points (Figure 1c), suggesting the block likely occurs before succinyl-CoA, at oxoglutarate dehydrogenase complex (OGDC). A block at this step would cut off succinate production flux from a-ketoglutarate, and contribute to the observed drop in succinate level at this stage (Figure 1c).
Activities of PDHC and OGDC are reduced in response to LPS and IFN-γ
As the metabolic labeling data pointed to decreased flux through PDHC and OGDC, we further investigated the activity of these enzymes in lysates of RAW 264.7 cells. Enzymatic activity of both PDHC and OGDC was drastically reduced after 24h stimulation with LPS and IFN-γ (approximately 85% for PDHC and 55% for OGDC) compared to control cells mock treated for 24h. The activities of both enzymes continued to decrease over time, which were nearly undetectable by 48h, and remained so through 72h (Figure 4a,b). These assays were performed in conditions which substrates were not limited and potential small molecule regulators were diluted out from the lysate, suggesting the observed activity reduction was likely controlled by altered enzyme level and/or covalent modification.
Figure 4. Activities of PDHC and OGDC decrease in response to LPS and IFN-γ exposure.
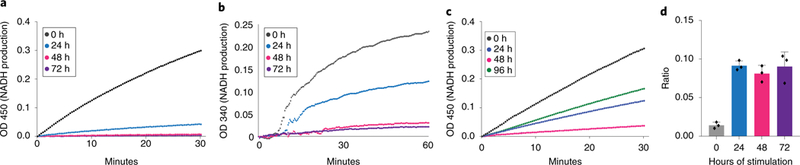
a, b. Activity of PDHC (a) and OGDC (b) in RAW 264.7 cells cultured in presence of LPS and IFN-γ for indicated time. Activity is measured by the rate of pyruvate- or α-ketoglutarate-dependent NADH production, as specified in Methods. Unstimulated cells (0h) were cultured, without stimulation, for the same length of time as cells stimulated for 24h. Experiments were repeated twice independently with similar results.
c. Activity of PDHC in RAW 264.7 cells after acute stimulation. Cells were incubated with LPS and IFN-γ for 2 hours, after which cells were washed and culture media were replaced with fresh media without stimuli. Cells were lysed for activity assay at indicated time after initial stimulation. Experiment was performed once.
d. Ratio of intracellular NADH to NAD+ in RAW 264.7 cells. Bar graph with error bar represents mean + SD (n=3 independent samples), dots represent individual values.
We further tested whether the activity of PDHC also decreased in macrophages stimulated with a single, acute dose of LPS and IFN-γ. Similar to the continual exposure to LPS and IFN-γ, after 2h of acute stimulation, PDHC activity continued to decrease over the next 24–48h. At 48h, activity of PDHC was nearly undetectable, even though LPS and IFN-γ had been completely removed for 46h. This striking result strongly suggested that the inhibition of PDHC is triggered at the initial stimulation and is independent of the continual presence of LPS and IFN-γ. This loss of PDHC activity can drive the decrease of itaconate at later time points in the acute stimulation setting, in a similar manner as observed in the continual stimulation setting. Interestingly, 96h after acute stimulation, PDHC activity recovered to approximately half of the activity in unstimulated macrophages (Figure 4c), indicating the inhibition of PDHC is reversible.
Both PDHC and OGDC reactions are coupled with NAD+ to NADH conversion. We found the NADH/NAD+ ratio showed a sustained increase throughout the time-course in both BMDM and RAW 264.7 cells (Figure 4d, Supplementary Figure 6). This suggests in addition to profoundly reduced PDHC and OGDC maximal enzyme activity, which was measured in lysate in the presence of saturating levels of NAD+, the increased intracellular NADH/NAD+ ratio could further contribute to the decreased flux through PDHC and OGDC in cells in the late stage. Furthermore, the increased NADH/NAD+ ratio may also contribute to the inhibition of IDH flux, in addition to the observed decreases in Idh expression.
Fluctuation in key metabolites is linked to dynamic changes in transcriptional program
Our findings revealed dynamic alterations in TCA cycle flux and metabolite levels in macrophages throughout the response time-course to LPS and IFN-γ stimulation. Soon after stimulation, the reduction in IDH led to the dramatic increase in intracellular citrate, and together with increased expression of IRG1, diverted flux from citrate into itaconate. High itaconate levels in turn caused accumulation of succinate via competitive inhibition of SDH. However, during the late stage, greatly reduced PDHC activity cut off production flux from glucose into citrate and itaconate. The fall of itaconate levels, which released the competition with succinate for SDH, together with the great reduction in OGDC flux at this time, allowed for the normalization of succinate to similar levels as in unstimulated macrophages. While this model explains the dynamic modulation of the TCA cycle, we further sought to understand the functional implications of these dynamics.
The dynamic changes in the relative levels of two TCA cycle metabolites, succinate and α-ketoglutarate, were of particular interest, as recent studies suggested their ratio is important in determining the phenotypic state of macrophages.14–16 One important mechanism is that the ratio of these two metabolites can impact the activities of a family of 2-oxoglutarate-dependent dioxygenases (2-OGDDs).15,35 Included in this enzyme family are the prolyl hydroxylases (PHD1–3), which act to promote degradation of HIF-1α (Figure 5a), a transcription factor that has been shown to promote pro-inflammatory phenotypes. In addition to inhibiting PHD-dependent degradation of HIF-1α, high levels of succinate may also further increase HIF-1α by promoting ROS production via SDH.14 Our data showed the succinate to α-ketoglutarate ratio increased significantly as early as 6h, peaked at 24h, and decreased after 48h of stimulation in both BMDMs and RAW 264.7 cells (Figure 5b, Supplementary Figure 7a). The transient high succinate to α-ketoglutarate ratio can stabilize HIF-1α by inhibiting PHDs.14–16 Indeed, we observed that patterns in HIF-1α protein abundance closely correlated with the ratio of succinate to α-ketoglutarate throughout the time-course (Figure 5c, Supplementary Figure 7b). In the acute (2h) stimulation setting, we also observed a very similar transient increase of HIF-1α that correlated with the metabolic change (Supplementary Figure 7c, Supplementary Figure 3a), indicating this dynamic trend is highly robust.
Figure 5. Changes in key metabolite levels correlate with changes in HIF-lα protein and histone methylation.
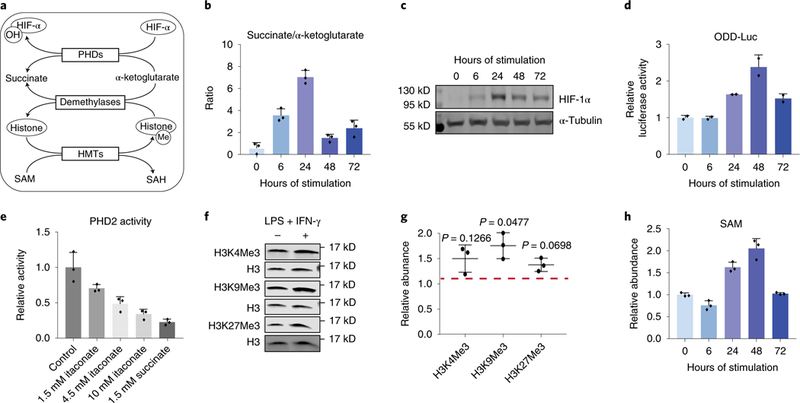
a. The role of key metabolites in regulating HIF-1α and histone methylation. HIF-lα can be hydroxylated by PHDs, using α-ketoglutarate as the substrate and producing succinate. Hydroxylated HIF-lα is then targeted for degradation. The level of histone methylation is controlled by the competing actions of histone methyltransferases (HMTs) and demethylases. HMTs require S-adenosyl-L-methionine (SAM) as the methyl donor. Demethylation by the JmjC family of demethylases uses α-ketoglutarate as the substrate and produces succinate.
b. Ratio of intracellular succinate to α-ketoglutarate in BMDM stimulated with LPS and IFN-γ for indicated time. Bar graph with error bar represents mean + SD (n=3 independent samples), dots represent individual values.
c. HIF-la protein levels in BMDMs stimulated with LPS and IFN-γ for time indicated. Similar results were observed in at least 6 independent experiments.
d. Luciferase activity measured in BMDMs derived from ODD-luc mice after stimulation with LPS and IFN-γ for indicated time. Data were normalized to protein content and expressed relative to the activity in unstimulated (0h) BMDMs. Bar graph with error bar represents mean + SD (n=2 independent samples), dots represent individual values.
e. Activity of recombinant human PHD2 in the presence of itaconate or succinate of indicated concentration. Bar graph with error bar represents mean + SD (n=3 independent samples), dots represent individual values.
f. Representative western blot of histone trimethylation marks in BMDMs stimulated with LPS and IFN-γ for 24h. Experiments were performed independently three times, and quantitation of all these blots is represented in g. Relative abundance of indicated histone trimethylation was first normalized to total H3 level, then expressed as relative change compared to unstimulated BMDMs. Center value with error bar represents mean ± standard deviation (n=3 independent samples), dots represent individual values. p-value was determined via one-sample t-test (H0: μ=1; two tailed).
h. Relative abundance of intracellular SAM in BMDMs after stimulated with LPS and IFN-γ for indicated time. Bar graph with error bar represents mean + SD (n=3 independent samples), dots represent individual values.
To further investigate whether altered PHD activity is responsible for the dynamic change of HIF-1α, we measured PHD activity using BMDMs derived from “ODD-luc” bioluminescent reporter knock-in mice. These cells constitutively express the oxygen-dependent degradation domain (ODD) of HIF-1α, which is the region hydroxylated by PHDs leading to HIF-1α degradation, fused to firefly luciferase (luc).36,37 ODD-luc BMDMs displayed a transient increase in luciferase activity, indicating decreased PHD activity (Figure 5d). This supports the hypothesis that dynamic changes in metabolite levels modulate PHD activity and subsequently HIF-1α protein level.
We noticed that the level of itaconate also showed strong correlation with HIF-1α throughout the time-course. Furthermore, itaconate is structurally similar to succinate, and due to this similarity, can competitively inhibit succinate dehydrogenase.12,13,34 Therefore, we tested the hypothesis that itaconate can similarly inhibit PHD activity and thus contribute to the dynamic regulation of HIF-1α. To determine the ability of itaconate to inhibit PHD in vitro, we measured the rate of HIF-1α peptide hydroxylation catalyzed by recombinant human PHD2, in the presence of varying amounts of itaconate. We found that itaconate inhibited PHD2 activity in vitro in a dose-dependent manner (Figure 5e). The inhibition by itaconate is weaker than the inhibition by succinate (Figure 5e). However, at its peak, intracellular itaconate levels can reach above the highest concentration tested in the in vitro assay (10mM), while peak intracellular succinate levels are similar to or below the level tested in the in vitro assay (1.5mM).13 This suggests the dynamic changes in itaconate level can also directly regulate HIF-1α level by modulating PHD activity, in addition to the indirect effect on HIF-1α by affecting SDH activity and thus succinate level.12 To test the effects of itaconate on HIF-1α stability in cells, we treated BMDMs derived from ODD-luc mice with exogenous itaconate, and found itaconate supplementation increased luciferase activity (i.e. ODD stability) (Supplementary Figure 7d). Itaconate treatment also increased the expression of HIF-1α targets, Hk2 and Vegfa (Supplementary Figure 7e), while the expression of these targets was decreased in Irg1−/− BMDMs (Supplementary Figure 7f).
Beyond stabilizing HIF-1α, the transient increase in succinate to α-ketoglutarate ratio would also favor inhibition of the JmJC-family of histone demethylases, additional members of 2-OGDDs, which could lead to increased methylation of histone lysine residues. Indeed, we observed a general increase in lysine trimethylation on H3 24h after stimulation (Figure 5f,g, Supplementary Figure 7g). However, the magnitude of change is less than that of HIF-1α and the dynamic trend at later time points is not as consistent in the two macrophage cell types investigated. In addition to demethylation, histone methylation state is also controlled by the flux of the methylation reaction, which can be influenced by the abundance of the methyl-donor S-adenosyl-methionine (SAM).24,38 We found that intracellular level of SAM also increased during the early stage and decreased at the late stage (Figure 5h, Supplementary Figure 7h), showing a similar dynamic trend as the succinate to α-ketoglutarate ratio, although this is likely through an independent mechanism. The dynamic trends of SAM and succinate/ α-ketoglutarate together create a metabolic environment that favors histone hypermethylation during the early stage of response to stimulation. It is also worth noting that the level of 2-hydroxyglutarate, another metabolite that can act as a competitive inhibitor of histone demethylases 39–42, also increased significantly in BMDMs in response to LPS and IFN-γ stimulation (Supplementary Figure 7i) which may also contribute to the changes in histone methylation.
Control of PDHC and OGDC flux in LPS and IFN-γ stimulated macrophages
Our studies revealed that the control of PDHC flux was important for mediating the metabolic transition into the late stage and controlling the levels of functionally important metabolites in macrophages. We therefore sought to investigate the molecular mechanism by which PDHC flux was regulated. Activity assays indicated that PDHC activity may be under the control of post-translational modifications. A well-characterized mechanism of PDHC regulation occurs via inhibitory phosphorylation of the E1 subunit (PDH) at one or multiple serine residues. Phosphorylation of PDH was not significantly elevated early (6h) after stimulation, which is consistent with previous reports.43 However, after 24h, phosphorylation at S293 and S300 were elevated in both BMDMs and RAW 264.7 cells, while total PDH levels remained mostly constant, indicating phosphorylation may contribute to the reduction in PDHC flux (Figure 6a). Phosphorylation of PDH is catalyzed by pyruvate dehydrogenase kinases (PDKs), which can be transcriptionally upregulated by HIF-1α44,45 Indeed, we observed a transient increase in transcription of Pdk3 that followed the dynamic trend of HIF-1α level, and the increase in Pdk3 transcription occurred right before the appearance of increased PDH phosphorylation (Figure 6b). At later time points (48–72h), PDH phosphorylation remained high and PDHC activity remained low, while Pdk3 transcript levels already subsided, suggesting that the mechanisms act to normalize the effect of PDK, such as PDH dephosphorylation or turnover, may act more slowly.
Figure 6. Dynamic regulation of PDHC and OGDC in LPS and IFN-γ stimulated macrophages.
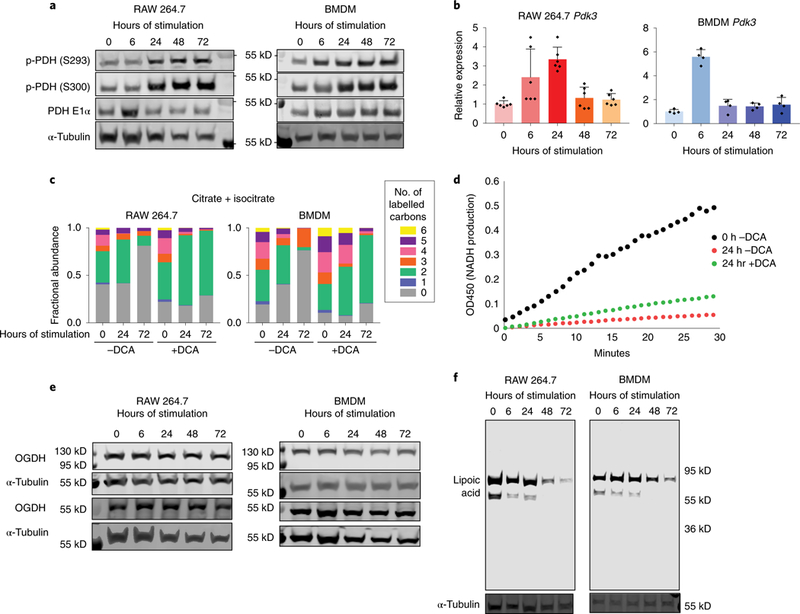
a. PDH phosphorylation levels in RAW 264.7 cells and BMDMs stimulated with LPS and IFN-γ for indicated time. Each experiment was performed independently twice with similar results.
b. Relative mRNA level of Pdk3 in RAW 264.7 cells and BMDMs stimulated with LPS and IFN-γ for indicated time. Data are normalized to expression in unstimulated macrophages (0h). Bar graph with error bar represents mean + SD, dots represent individual values. (n=6 independent samples for RAW 264.7 cells, n=3 independent samples for BMDM).
c. Labeling patterns of intracellular citrate+isocitrate in RAW 264.7 cells and BMDMs incubated with U-13C glucose and stimulated with LPS and IFN-γ, with or without treatment of dichloroacetate (DCA). Data represents the mean (n=3 independent samples).
d. Activity of PDHC in RAW 264.7 cells stimulated with LPS and IFN-γ for 24h or cultured for 24h without stimulation (0h). Stimulated cells were either treated with 10mM DCA from 24h prior to stimulation until the time of collection (+DCA), or treated with vehicle control (-DCA). Experiments were performed twice independently with similar results.
e. Protein levels of the two splice variants of the E1 subunit of OGDC (OGDH) in RAW 264.7 cells and BMDMs stimulated with LPS and IFN-γ for indicated time. Experiment was performed once.
f. Protein lipoylation state in RAW 264.7 cells and BMDMs stimulated with LPS and IFN-γ for indicated time. E2 subunit of PDHC is ~69 kD and E2 subunit of OGDC is ~55 kD. Experiments were performed independently three times in RAW 264.7 cells and twice in BMDMs with similar results.
To test the hypothesis that phosphorylation has a role in regulating PDHC flux, we treated unstimulated macrophages and macrophages stimulated for 24h or 72h with dichloroacetate (DCA), an inhibitor of PDKs. DCA treatment successfully eliminated PDH phosphorylation (Supplementary Figure 8a). The loss of labeling incorporation from U-13C-glucose into citrate and the reduction of citrate, itaconate, and succinate levels during the late stage was restored by DCA treatment (Figure 6c, Supplementary Figure 8b), while DCA treatment also increased baseline citrate labeling. Measurement of PDHC activity in the lysate from macrophages stimulated with LPS and IFN-γ for 24h, showed that DCA treatment causes a recovery of PDHC activity as compared to the untreated control (Figure 6d). However, DCA treatment was not capable of completely restoring activity to that observed in unstimulated macrophages. These data indicate that phosphorylation contributes to the modulation of flux through PDHC in stimulated macrophages, while additional mechanisms also play a significant role.
Interestingly, the inhibition of PDHC, partially mediated by HIF-1α –PDK, leads to diminished levels of itaconate and succinate, the metabolites which stabilize HIF-1α. This ultimately creates a feedback loop. Consistently, we found DCA treatment increases the stability of HIF-1α, as indicated by increased luciferase activity in DCA treated ODD-luc BMDMs (Supplementary Figure 8c). To further test the functional significance of PDHC flux control, particularly via its ability to regulate itaconate level, we measured the expression of Il-1β, a pro-inflammatory cytokine whose production has been shown to be inhibited by itaconate.12 DCA treatment, which increases the intracellular itaconate level, significantly decreased the expression of Il-1β. The effect is similar to the supplementation of exogenous itaconate, and opposite to that in Irg1−/− BMDMs (Supplementary Figure 8d).
As the enzyme activity assay of DCA treated macrophages indicated PDH phosphorylation is only partially responsible for the PDHC activity loss, we sought to identify other mechanisms contributing to the inhibition, as well as the mechanism for inhibition of OGDC. Not only are OGDC and PHDC both profoundly inhibited at the late stage of response to LPS and IFN-γ, they have similar catalytic mechanisms and belong to the same family of dehydrogenase complexes; therefore, we hypothesized a common mechanism contributes to the regulation of both OGDC and PDHC in macrophages. OGDC and PHDC are composed of three subunits: E1 subunits, OGDH and PDH, decarboxylate α-ketoglutarate and pyruvate, respectively; E2 subunits, DLST and DLAT, transfer succinyl- and acetyl-group to the thiol of coenzyme A, with a lipoic acid covalently bound to lysine residue as the required catalytic mediator; and E3 subunits oxidize dihydrolipoate to back to lipoic acid on the E2 subunit and produce NADH. Similar to PDH, we found the level of total OGDH proteins (both splice variants) did not change significantly throughout the time-course (Figure 6e), and mRNA level of different OGDH isoforms did not show any profound change that correlate with the drastic activity change (Supplementary Figure 9a). However, strikingly, we found that the level of catalytically active lipoic acid on the E2 subunit of both OGDC and PDHC decreased drastically at later time points (Figure 6f), while the level of total PDHC E2 protein or OGDC E2 and E3 transcript did not show any significant reduction (Supplementary Figure 9b,c). Furthermore, when cells were stimulated with a single, acute (2h) dose of LPS and IFN-γ, the lipoylation of both PDHC and OGDC E2 subunits first decreased drastically, then recovered 72h after the stimulation was applied and removed (Supplementary Figure 9d), which correlates with the dynamics of PDHC activity loss and recovery in this experimental setting (Figure 4c). These data indicate changes of E2 subunit lipoylation state is a critical mechanism for regulating the flux through OGDC and PDHC in response to LPS and IFN-γ stimulation.
Dynamic changes in substrate contribution to acetyl-CoA production
Studies above revealed that the inhibition of PDHC and OGDC at the late stage profoundly limited the ability of cells to metabolize glucose and glutamine in the TCA cycle to supply important intermediates such as citrate and acetyl-CoA. Consistent with this, we found when cells are simultaneously given both fully labeled glucose and glutamine, a large fraction of citrate remained unlabeled at later time points (Figure 7a). We then sought to investigate the capability of other substrates in supplying two-carbon units.
Figure 7. Alternative substrate utilization in LPS and IFN-γ stimulated macrophages.
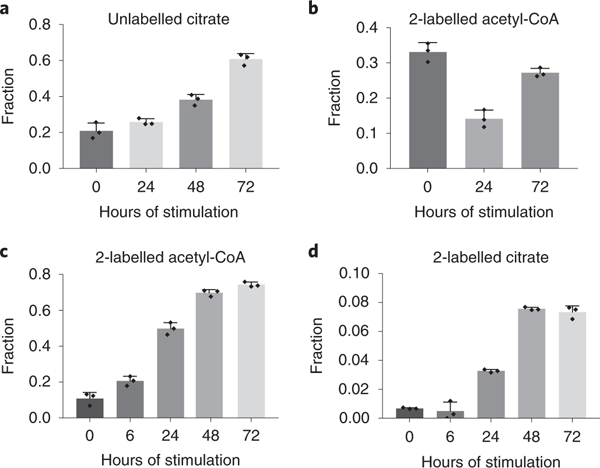
a. Fraction of citrate that remains unlabeled in RAW 264.7 cells stimulated with LPS and IFN-γ for indicated time, after 24h incubation with both U-13C-glucose and U-13C15N-glutamine. Bar graph with error bar represents mean + SD (n=3 independent samples), dots represent individual values.
b. Fraction of 2-labeled acetyl-CoA in RAW 264.7 cells stimulated with LPS and IFN-γ for indicated time, after 24h incubation with 40μM BSA-conjugated U-13C-palmitic acid. Bar graph with error bar represents mean + SD (n=3 independent samples), dots represent individual values.
c,d. Fraction of 2-labeled acetyl-CoA (c) and citrate (d) in RAW 264.7 cells stimulated with LPS and IFN-γ for indicated time, after 24h incubation with 500μM U-13C-acetate. Bar graph with error bar represents mean + SD (n=3 independent samples), dots represent individual values.
Fatty acid oxidation can produce acetyl-CoA in mitochondria and support respiration. To measure the contribution of fatty acid oxidation to acetyl-CoA production, we supplemented cells with BSA conjugated U-13C-palmitate. Throughout the time-course, labeling of palmitoyl-carnitine remains constant (Supplementary Figure 10a), while the labeling incorporation from palmitate to acetyl-CoA decreases significantly at 24h, and recovers 72h after stimulation (Figure 7b). To investigate the possible changes in fatty acid oxidation upon LPS and IFN-γ stimulation, we also measured oxygen consumption rate (OCR) throughout the time-course in absence or presence of a fatty acid oxidation inhibitor, etomoxir (ETO). Overall oxygen consumption rate decreased as soon as 24h after stimulation, and continued to decrease at later time points to nearly undetectable levels (Supplementary Figure 10b). This is consistent with the disrupted TCA cycle in both early and late stages of response. The decrease of oxygen consumption rates caused by ETO treatment indicates fatty acid dependent respiration. This rate is a minor fraction of overall respiration rate in unstimulated macrophages (Supplementary Figure 10b), and it decreases quickly after upon LPS and IFN-γ stimulation. After 24h, ETO treatment resulted in minimal reduction of OCR. (Supplementary Figure 10b). These results suggest fatty acid oxidation rate, similar to overall respiration rate, decreases quickly after stimulation, resulting in a lower relative contribution of fatty acid oxidation to acetyl-CoA production at the early stage. At the late stage, the recovery of acetyl-CoA labeling from U-13C-palmitate is likely due to largely reduced relative contribution from glucose at this time (Figure 1c, Figure 2c).
As fatty acid oxidation cannot explain the fraction of acetyl-CoA that is not produced from glucose or glutamine at the late stage, we investigated the contribution of other potential substrates. Acetate has been shown to contribute significantly to acetyl-CoA production in many biological situations, especially when acetyl-CoA production from glucose is limited.46,47 Unlike glucose and most amino acids, acetate can supply acetyl-CoA production without generating NADH, which may be favored when the NADH/NAD+ ratio raises significantly after LPS and IFN-γ stimulation (Figure 4d). When cells were incubated with U-13C-acetate, acetyl-CoA labeling increased greatly throughout the stimulation time-course (Figure 7c). Similarly, the fraction of citrate that is 2-labeled increases over time (Figure 7d). However, the labeled fraction in citrate is less than that observed in acetyl-CoA, suggesting the labeled acetyl-CoA formed from acetate is likely mainly cytosolic. These data indicate that during the later stage of response, acetate can become an important substrate for acetyl-CoA supply, when the contribution from glucose decreases drastically.
Discussion
Here we have demonstrated that in response to LPS and IFN-γ stimulation, metabolic reprogramming in macrophages is highly dynamic. In particular, we revealed a two-stage remodeling of the TCA cycle. The early stage (6–24h) is concurrent with increased TNFα and IL6 production. This metabolic state, consistent with previous reports,10,12,13,48 is characterized by a flux breakpoint at IDH and accumulation of citrate, itaconate, and succinate. The late stage (48–72h) occurs at the same time as diminished production of TNFα and IL6, and is largely driven by decreased flux through PDHC and OGDC. This reduced PDHC and OGDC flux leads to normalization of citrate, itaconate, and succinate levels, and decreased acetyl-CoA and succinyl-CoA. Aconitase activity is also likely to be somewhat reduced at this point, due to continuously high NO production which can destabilize the required Fe-S cluster.49,50 This would also contribute to the depletion of aconitate level at the late stage, although our results indicated the flux is mainly controlled upstream of aconitase. Interestingly, we observed that after a prolonged time of stimulation, many key TCA cycle metabolites return to similar levels as those observed in unstimulated macrophages. However, this metabolic state is driven by different underlying metabolic fluxes—macrophages at the late stage, similar to acutely activated macrophages, and unlike naïve and M2 macrophages,10 display a disrupted TCA cycle (Figure 8).
Figure 8. Two-stage remodeling of TCA cycle in LPS and IFN-γ stimulated macrophages.
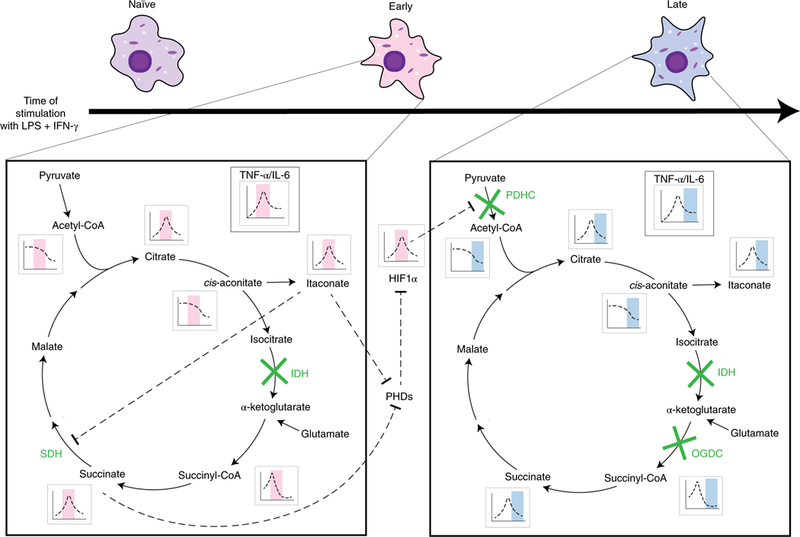
During the early stage of the response to LPS and IFN-γ stimulation, isocitrate dehydrogenase (IDH) is blocked, upstream metabolite citrate/isocitrate accumulates, and TCA flux is diverted to the production of itaconate, which competitively inhibits succinate dehydrogenase (SDH), leading to the buildup of succinate and succinyl-CoA. The changes in metabolite levels, especially succinate and itaconate accumulation, leads to increased level of HIF-1α via the inhibition of prolyl hydroxylases (PHDs). HIF-1α stabilization results in increased Pdk3 transcription, which partially mediates reduced flux through pyruvate dehydrogenase complex (PDHC). In the late stage, substantial inhibition of PDHC contributes to the reduction in glucose- driven acetyl-CoA, citrate, cis-aconitate and itaconate production, and inhibition of OGDC contributes to the reduction in succinyl-CoA and succinate production. The levels of these metabolites reduce drastically, and HIF-1α level normalizes.
Many of the key metabolites, whose levels are dynamically regulated throughout the time-course, have immunoregulatory roles through a diversity of mechanisms. The relative level of succinate and α-ketoglutarate can regulate 2-oxoglutarate-dependent dioxygenases and therefore modulate HIF-1α and histone methylation.15,16 Itaconate has immunoregulatory effects through its ability to inhibit SDH 12,13, alkylate proteins, induce electrophilic stress 51,52, and as we show here, act as modulators of PHDs. Acetyl-CoA, succinyl-CoA, and SAM are the substrates and key regulators for protein acetylation, succinylation, and methylation respectively.15,21,24,38,53–55 Additionally, TCA cycle remodeling also results in elevated level of ROS (Supplementary Figure 11); another group of important signaling molecules.14,56,57 Therefore, the overall effects of metabolic rewiring on macrophage functions is a composite result of various mechanisms. Here we focused on how the dynamic changes of metabolic state regulate HIF-1α in a time dependent manner. The important functional implications of dynamic metabolic reprogramming through other mechanisms, such as regulating protein acetylation and succinylation, requires further in-depth studies.
Our study also revealed the mechanism for the inhibition of PDHC and OGDC, which drives the metabolic transition into the late stage. Both these enzymes are regulated by the dynamic change of lipoylation status. Several molecular mechanisms may contribute to this change. Lipoic acid synthesis relies on the Fe-S cluster containing enzyme lipoic acid synthase, and proper Fe-S cluster insertion can be inhibited by nitric oxide or by decreased expression of Fe-S cluster biogenesis components.58 Additionally, lipoylation on the E2 subunit may be removed,59 or the lipoyl group may be reversibly or irreversibly modified and inactivated by reactive species including 4-hydroxynonenal, glutathione and nitric oxide.60–62 The changes in metabolic landscape in macrophages upon stimulation, including increased ROS and NOS production, likely supports multiple mechanisms that contribute to decreased catalytically active lipoylation. Additionally, PHDC activity is also regulated by E1 subunit phosphorylation, which demonstrates metabolic feedback in this sequence of metabolic transitions.
Overall, our study elucidates a dynamically remodeled metabolic program in macrophages over the course of an immune response. It expands our current knowledge in macrophage metabolism beyond the association with different activation signals, and reveals a temporal connection between metabolic state and regulators of immune function. This provides important insights into how dynamic metabolic reprogramming in macrophages orchestrates the transitions through different phenotypical phases.
Methods
Cell Culture:
RAW 264.7 cells (ATCC) were cultured in RPMI 1640, 25mM Hepes, 1% penicillin/streptomycin and 10% dialyzed FBS at 37°C and 5% CO2. To stimulate the cells, cells were incubated with 50ng/mL LPS (E. coli O111 :B4, Sigma) and 10ng/mL IFN-γ (R&D Systems).
Wild-type mouse bone marrow-derived macrophages (BMDMs) isolated from C57Bl/6J mice were used for the majority of experiments. Additional experiments utilized heterozygous FVB.129S6-Gt(ROSA)26Sortm2(HIF1A/luc)Kael/J (ODD-Luc) mice36,37, or homozygous C57BL/6NJ-Acod1em1(IMPC)J/J (Irg1−/−) mice, as indicated. For Irg1−/− experiments, wildtype C57Bl/6NJ mice were used as controls. Mice were bred and maintained according to protocols approved by the University of Wisconsin-Madison Institutional Animal Care and Use Committee. Bone marrow cells were harvested from femurs and tibia of 6–10 week old mice (both male and females used). Cells were differentiated in RPMI 1640 containing 25mM Hepes, 1% penicillin/ streptomycin, 10% FBS and 20% L929 spent media. Three days post isolation, media were changed every day subsequently until 7 days post-isolation. Cells were then plated for experiments at 5*105 cells/well in 6-well plates in RPMI 1640 containing 25mM Hepes, 1% penicillin/streptomycin, 10% dFBS and 20ng/mL M-CSF (R&D Systems). To stimulate BMDM, cells were incubated with 50ng/mL LPS (E. coli O111:B4, Sigma) and 30ng/mL IFN-γ (R&D Systems).
To avoid nutrient depletion and keep continual LPS and IFN-γ stimulation, media for both RAW 264.7 cells and BMDMs were refreshed every day until collection; with LPS and IFN-γ maintained in the media throughout. For metabolomics experiments, media were also refreshed 2h before extraction to further minimize the possible inconsistences due to variations in chemical compositions in spent media. Unstimulated controls (0h) were collected 2 days after plating. Parallel control experiments showed that without stimulation, there was no significant changes in the levels of discussed metabolites across 3 days in culture.
In addition to this stimulation protocol, which is used for most of the experiments in this report unless otherwise indicated, two additional protocols for stimulation were tested. First, acute stimulation was performed by incubating cells with LPS and IFN-γ for 2h only. Then the media containing LPS and IFN-γ were removed, cells were washed, and cell culture was continued in fresh media without LPS and IFN-γ. Here, after the acute stimulation, media without LPS and IFN-γ were refreshed every 24h to avoid nutrient depletion. Second, cells were stimulated with LPS and IFN-γ at time 0, and cells were cultured for 48h, without any media change during this period. In this protocol, cells were cultured with 3× the typical volume of media to avoid nutrient depletion.
Viability Assays:
Macrophage viability following LPS and IFN-γ stimulation was determined using two independent methods: Trypan blue staining and an LDH cytotoxicity assay. To assay viability using Trypan blue staining, cells were detached by 15min incubation with Accutase cell detachment solution (Stemcell Technologies), then the cell suspension was stained with Trypan blue and live/dead cells were counted using a Countess II automated cell counter (Invitrogen). To measure cytotoxicity based on LDH release into media, which is a result of cell death, we used a Pierce Lactate Dehydrogenase (LDH) Cytotoxicity Assay Kit following the manufacturer’s instructions. LDH activity was measured in media incubated with the cells for 8h and expressed relative to total LDH activity, which was obtained via measurement of LDH activity after complete lysis of cells, using provided lysis reagent. The kit couples LDH activity to the formation of a red formazan product which was measured by absorbance at 490nm using a BioTek® Epoch2 microplate reader.
Cytokine and NO production assays:
To measure the release of TNF-α, Il-6, IL-1β and nitric oxide in media, cells were incubated with culture media for 22h prior to indicated collection time. The spent media were spun at 1000×g for 5 min, and supernatant was transferred and frozen at −80°C until analysis. TNF-α and IL6 secretion was determined using DuoSet ELISA kits (R&D Systems) and levels of IL-1β were determined using a Novex ELISA Kit (Invitrogen) as per manufacturer’s instructions. Nitrite was measured by Greiss Assay (Promega). A BioTek® Epoch2 microplate reader was used to for these measurements.
Metabolite analysis:
To analyze intracellular metabolites, cells were washed three times with dPBS and metabolites were extracted with cold LCMS grade 80:20 methanol:H2O (v:v). Samples were dried under nitrogen flow and subsequently dissolved in LCMS grade water or LCMS grade 80:20 ACN: H2O (v:v) for different LCMS analysis methods. To analyze extracellular metabolites, spent media samples were extracted with 4X volume of LCMS grade methanol, and protein pellets were removed by centrifugation. Supernatant containing small molecules was further diluted with either LCMS grade H2O or ACN:H2O (90:10) for different LCMS analysis methods.
Samples were analyzed using a Thermo Q-Exactive mass spectrometer coupled to a Vanquish Horizon UHPLC, using three methods for optimal quantification. Metabolites were separated on either a C18 or HILIC column (details below) at a 0.2 ml/min flow rate and 30°C column temperature. Data was collected on full scan mode at resolution of 70K.
In the first method samples were loaded in water and separated on a 100 × 2.1 mm, 1.7μM Acquity UPLC BEH C18 Column (Waters) with a gradient of solvent A (97:3 H2O:methanol, 10mM TBA, 9mM acetate, pH 8.2) and solvent B (100% methanol). The gradient is: 0 min, 5% B; 2.5 min, 5% B; 17 min, 95% B; 21 min, 95% B; 21.5 min, 5% B. Data was collected on a full scan negative mode. Setting for the ion source were; 10 aux gas flow rate, 35 sheath gas flow rate, 2 sweep gas flow rate, 3.2kV spray voltage, 320°C capillary temperature and 300°C heater temperature.
In the second and third methods, samples were dissolved in 80:20 ACN:H2O and separated on a 150 × 2.1 mm SeQuant PEEK HPLC Column with ZIC-pHILIC (5μm) polymeric beads (Millipore Sigma). In the second method, metabolites were separated with a gradient of solvent A (95% ACN, 5% water) and solvent B (10mM NH4Ac, pH 5.5), and data were collected on a full scan positive mode. Setting for the ion source were; 12 aux gas flow rate, 40 sheath gas flow rate, 1 sweep gas flow rate, 3.5 kV spray voltage, 340°C capillary temperature and 250°C heater temperature. The third method utilized a gradient of solvent A (95% ACN, 5% H2O) and solvent B (10mM NH4Ac, pH 6.8) and collected data on a full scan negative mode. Setting for the ion source were; 10 aux gas flow rate, 35 sheath gas flow rate, 2 sweep gas flow rate, 3.2 kV spray voltage, 320°C capillary temperature and 300°C heater temperature. The gradient for both the second and third methods is: 0 min, 5% B; 2 min, 5% B; 18 min, 60% B; 19 min, 90% B; 24 min, 90% B; 25.5 min, 5% B.
Metabolites reported here were identified based on exact M/z and retention times determined with chemical standards. Data were analyzed with Maven.63,64 Relative metabolite levels were normalized to protein content and expressed relative to levels measured in unstimulated (0h) macrophages. To quantify excretion rate of extracellular metabolites, absolute concentrations were quantified using calibration curves generated with external standards of the corresponding metabolites. These data were from media incubated with cells for 22h prior to collection.
Stable Isotope Labeling:
U-13C-glucose, U-13C15N-glutamine, U-13C-palmitic acid and U-13C-sodium acetate tracers were from Cambridge Isotopes. Cells were incubated with media containing indicated tracers for 24h prior to collection. For glucose or glutamine labeling experiments, the labeled tracer of same concentration replaces the corresponding unlabeled glucose or glutamine in regular media. Media were otherwise chemically identical to regular culture media. Palmitic acid was conjugated to BSA prior to incubation with cells. For labeling experiments with labeled palmitate (40μM) or acetate (500μM), the tracer was supplemented in addition to the regular media. Data from labeling experiments was adjusted for natural abundance of 13C.
Calculation of acetyl moiety labeling in acetyl-CoA:
The isotopic distribution of the acetyl moiety in acetyl-CoA, {MAcetyl}, was calculated based on measured isotopic distribution of acetyl-CoA, {MAcetyl-CoA}, and coenzyme A, {MCoA}, using the following equations:
Where MAcetyl-CoA(i) and MCoA(i) are the measured fraction of acetyl-CoA and coenzyme A, respectively, with i labeled carbons, after incubation with U13C glucose tracer. Labeling of acetyl moiety in acetyl-CoA was calculated by fitting MAcetyl(0), MAcetyl(1), and MAcetyl(2) based on these equations.
Luciferase Assay:
Analysis of luciferase activity in BMDM from ODD-luc mice was performed using a Promega Luciferase Assay system, as per manufacturer’s instructions. Cells were lysed with Cell Culture Lysis Reagent (Promega, 200μL per 60mm plate). After centrifugation (12,000×g, 15sec, room temperature), lysate was combined with Luciferase Assay Reagent (Promega) and luminescence was immediately measured using a BioTek® Cytation3 Cell Imaging Multi-Mode Reader.
Enzyme Activity Assays:
Pyruvate dehydrogenase complex activity was analyzed using PDH enzyme activity microplate assay kit (Abcam), as per manufacturer’s instructions. This kit measures PDHC activity by monitoring pyruvate dependent NADH production. The NADH level was measured by absorbance of NADH coupled dye (450nm) using a BioTek® Epoch2 plate reader.
To evaluate OGDC activity, mitochondria were isolated and the α-ketoglutarate dependent NADH production rate in mitochondria lysate was measured by monitoring NADH absorbance at 340nm, according to a previously established method.65 Cells were lysed in hypotonic lysis buffer (20mM Tris buffer pH=7.4, 1mM EDTA with phosphatase and protease inhibitors (Pierce™ Protease and Phosphatase Inhibitor Tablets)) and incubated on ice for 30 min. Lysate was then homogenized in a dounce homogenizer and then separated via centrifugation at 700 × g, 4°C, for 10 min, in a mannitol sucrose solution (440mM mannitol/140mM sucrose, 20mM Tris, 1mM EDTA, pH 7.4). The mitochondrial fraction was resuspended in RIPA buffer with protease and phosphatase inhibitors (ThermoFisher). Mitochondrial lysate (~30μg protein) was combined with 200μL of assay buffer containing 1mM MgCl, 400μM thiamine diphosphate, 500μM CoA, 2mM NAD+, 1mM DTT, 50mM HEPES pH 8.0, 10% glycerol, 0.05% BSA and 6mM α-ketoglutarate; absorbance was measured at 340nM using a BioTek® Epoch2 microplate reader. As a blank control, absorbance was measured from parallel wells in which α-ketoglutarate was excluded from the assay mix. The blank absorbance values were subtracted in data analysis.
To measure the activity of recombinant human PHD2 (Active Motif, 81065) in the presence of varying amount of itaconate or succinate66, 100μM HIF-1α peptide (DLDLEALAPYIPADDDFQL, GeneScript) was incubated with 300 nM PHD2 protein in 30 μl reaction mix containing 50 mM ammonium acetate buffer at pH 7.5, 50 μM α-ketoglutarate, 100 μM ascorbate, 1mg/mL bovine serum albumin, 50 μM Fe(II) acetate, and varying amounts of itaconate (0, 1.5, 4.5 or 10 mM) or succinate (0 or 1.5mM) as indicated. Activity was determined by measuring level of the hydroxylated HIF-1α peptide using LC-MS. Small aliquots of reaction mix were quenched every 15 min after initiation of reaction (through 90 min), using 90:10 LCMS grade ACN: water. Samples were spun at max speed for 5min to remove protein, and supernatant were analyzed using LC-MS method #3 as indicated above. Reaction rates were calculated by fitting the level of hydroxylated HIF-1α peptide, which was determined by its LCMS signal (M/z=1134), as a function of time using linear regression.
PDK inhibition experiment
To inhibit PDK, cells were pretreated with 10mM sodium dichloroacetate (Sigma) 48h prior to LPS and IFN-γ stimulation, and the sodium dichloroacetate was kept in media throughout the stimulation time-course.
Quantitative real-time PCR:
RNA was extracted using RNAStat60 (Tel-Test) and treated with RQ1 DNAse (Promega). cDNA was synthesized from DNAse treated RNA samples (1–2μg) using SuperScript III (ThermoFisher) following manufacturer’s instructions. Quantitative PCR was performed using KAPA SYBR FAST qPCR MasterMix on a Roche® LightCycler 480.
Relative gene expression level was normalized to the housekeeping gene CycloB. The following
primers (IDT) were used: Idh1 Fwd 5’gtggagatgcaaggagatgaa’3; Idh1 Rev
5’atgcagatccagttccacatag’3; Idh2 Fwd 5,gatgacatctgtgctggtctg’3; Idh2 Rev
5’ccttctggtgttctcggtaatg’3; Pdk3 Fwd 5’gcccaaggcgtgattgagta’3; Pdk3 Rev
5’gggtaagtgtcaccaccaaac’3; Dlst Fwd 5’ctcggcacaaggatgctttc’3; Dlst Rev
5,gggccttctctcctagttcatta’3; Dld Fwd 5’gagactgagtcgtctctacc’3; Dld Rev
5’cctatcactgtcacgtcagcc’3; Hk2 Fwd 5’tgatcgcctgcttattacgg’3; Hk2 Rev
5’aaccgcctagaaatctccaga’3; Vefga Fwd 5’gcacatagagagaatgagcttcc3’; Vegfa Rev
5’ctccgctctgacaaggct3’; Il-1β-Fwd 5’aagtcatatatctccatgagctttct’3: Il-1β Rev
5’ttcttctttgggtattgcttgg’3; Ogdh 1/¾ Fwd 5’agggcatatcagatacgaggg’3; Ogdh 1/¾ Rev
5’ctgtggatgagataatgtcagcg’3; Ogdh 5 Fwd 5’gtttcttcaaacgtggggtcct’3; Ogdh 5 Rev
5’gcatgattccaggggtctcaaa’3; Cyclob Fwd 5’tggaagagcaccaagacagaca’3; Cyclob Rev
5’tgccggagtcgacaatgat’3.
Western Blot:
All Western blots reported here, except the histones, were performed using whole cell lysate. Histone analysis was performed using isolated histones. Whole cell lysate samples were prepared by lysing cells in RIPA buffer with phosphatase and protease inhibitors (Pierce™ Protease and Phosphatase Inhibitor Tablets) after washing with dPBS three times. Isolated histones were extracted as previously described:67 Cells were washed with dPBS three times, scraped from plate, resuspended in hypotonic lysis buffer (10mM Tris HCl, 10mM KCL, 3mM MgCl2, protease and phosphatase inhibitors, pH 7.4), and incubated on ice for 20 min. The cell suspension was then passed through a 27G needle (5×), and resulting lysate was spun down (10000 × g, 4°C, 10min) to pellet nuclei. The pellet was washed with ice cold PBS supplemented with protease and phosphatase inhibitors, and spun down again (10000×g, 4°C, 10min). The pellet was then resuspended in 5 volumes of 0.4 H2SO4 and incubated for 4hours at 4C, with constant gentle shaking. After incubation, the suspension was spun down (3400 ×g, 4C, 5min) and supernatant was transferred to a new tube, to which 1/2 volume of 50% trichloroacetic acid was added. This solution was incubated overnight at 4C to precipitate histones, after which the mixture was spun down (3400×g, 4C, 5min), the pellet was washed twice with acetone, air-dried and then resuspended in water.
Protein content of total cell lysate and histone extract was quantified using BCA assay kit (ThermoFisher). Whole cell lysates were separated on 4–12% Bolt Gel (ThermoFisher), and histone samples were separated on 16% Bis-Tris Gel (ThermoFisher) after heat denaturation in sample buffer (ThermoFisher). Proteins were subsequently transferred to 0.2μM nitrocellulose membrane. Membranes were blocked in 5% nonfat milk in TBS-T buffer and probed with the following primary antibodies (see Reporting Summary for details): anti-HIF-1α (CST 14179), anti-H3 (CST 4499), anti-Tri-methyl H3K27 (CST 9733), anti-Tri-methyl H3K4 (CST 9751), anti-Tri-methyl H3K9 (CST 13969) anti-PDH E1 (CST 2784S), anti-p-PDH Ser 293 (Novus NB100–93479), anti-p-PDH Ser 300 (Calbiochem AP1064), anti-α-tubulin (Abcam ab7291), anti-IRG1 (Abcam 222411), anti PDH E2 (Abcam ab66511), anti lipoic acid (Calbiochem, 467695), anti-OGDH (116kD isoform) (proteintech 15212–1-AP), anti OGDH (48kD isoform) (abcam, ab87057), anti P-actin (CAT 4967). Primary antibody incubation was followed by incubation with Li-Cor® secondary antibodies (goat anti-rabbit 800CW, goat anti-mouse 680RD). Membranes were imaged with a Li-Cor® Odyssey ClX and quantified using ImageStudio™ Lite software. Uncropped images are reported in Supplementary Figure 12.
Oxygen Consumption Rate Analysis:
Oxygen consumption rates (OCR) were measured using an Agilent XF Seahorse Extracellular Flux Analyzer. OCR was measured in cells cultured in RPMI 1640, 25mM Hepes, 1% penicillin/streptomycin and 10% dialyzed FBS, with or without treatment with 40μM etomoxir 30min prior to measurements. Seeding density is 1×106 cells per well. Data was blanked from wells containing media but no cells.
Intracellular ROS Assay:
To measure the level of intracellular ROS, cells were incubated with 500nM CellRox Green in media for 1hour. Following incubation cells were washed twice with PBS and then detached from plates by Accutase (StemCell Technologies) treatment for 15 minutes. Cells were then spun down (1000x g, 3 min) and resuspended in 2% FBS in PBS and stained with 5nM SyTOX Red (ThermoFisher) to measure cell viability. The samples were analyzed on a MACs Quant 10 Analyzer (Miltenyi Biotec) at the University of Wisconsin Carbone Cancer Center Flow Cytometry Laboratory. Resulting data files were analyzed using FlowJo10.4 (Tree Star). Median CellRox Green signal of those cells that were SyTOX Red negative (live cells) was calculated. See example of gating strategy in Supplementary Figure 11b.
Supplementary Material
Acknowledgements:
This work is primarily supported by the Morgridge Institute for Research (start-up fund for Dr. Fan). Additionally, Dr. Eisenstein is supported by NIH R01DK66600, Dr. Pagliarini is supported by NIH R35GM130294. The authors would like to thank the University of Wisconsin Carbone Cancer Center (UWCCC). This work is supported in part by NIH/NCI P30CA014520-UW Cancer Center Support Grant. The authors would also like to thank Dr. Yatrik Shah at the University of Michigan for sharing the ODD-luc mice.
Footnotes
Competing Interests: The authors declare no competing interests.
Data Availability:The data that support the findings of this study are available from the corresponding author upon request.
Reporting Summary:Further information on research design is available in the Reporting Summary linked to this article.
References
- 1.Janeway CA & Medzhitov R Innate immune recognition. Annu. Rev. Immunol. 20, 197–216 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Mosser DM & Edwards JP Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liew FY, Xu D, Brint EK & O’Neill LAJ Negative regulation of toll-like receptor-mediated immune responses. Nat. Rev. Immunol. 5, 446–458 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Ivashkiv LB Inflammatory signaling in macrophages: Transitions from acute to tolerant and alternative activation states. Eur. J. Immunol. 41, 2477–2481 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foster SL, Hargreaves DC & Medzhitov R Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447, 972–978 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Medvedev AE, Kopydlowski KM & Vogel SN Inhibition of Lipopolysaccharide-Induced Signal Transduction in Endotoxin-Tolerized Mouse Macrophages: Dysregulation of Cytokine, Chemokine, and Toll-Like Receptor 2 and 4 Gene Expression. J. Immunol. 164, 5564–5574 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Ziegler-Heitbrock H Molecular mechanism in tolerance to lipopolysaccharide. J. Inflamm. 45, 13–26 (1994). [PubMed] [Google Scholar]
- 8.Kumar V Targeting macrophage immunometabolism: Dawn in the darkness of sepsis. Int. Immunopharmacol. 58, 173–185 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Navegantes KC et al. Immune modulation of some autoimmune diseases: the critical role of macrophages and neutrophils in the innate and adaptive immunity. J. Transl. Med. 15, 36 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jha AK et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Kelly B & O’Neill LA Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 25, 771–784 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lampropoulou V et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 24, 158–166 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cordes T et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J. Biol. Chem. 291, 14274–84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mills EL et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 167, 457–470.e13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tannahill GM et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–42 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu PS et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 18, 985–994 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Janeway CA & Medzhitov R Innate immune recognition. Annu. Rev. Immunol. 20, 197–216 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Williams NC & O’Neill LAJ A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol. 9, 141 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dominguez-Andres J et al. The Itaconate Pathway Is a Central Regulatory Node Linking Innate Immune Tolerance and Trained Immunity. Cell Metab. 29, 211–220.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Medzhitov R & Horng T Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 9, 692–703 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Ivashkiv LB Epigenetic regulation of macrophage polarization and function. Trends Immunol. 34, 216–223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Novakovic B et al. β-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 167, 1354–1368.e14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foster SL, Hargreaves DC & Medzhitov R Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447, 972–978 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Mentch SJ et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 22, 861–873 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Etchegaray J-P & Mostoslavsky R Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 62, 695–711 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newsholme P, Curi R, Gordon S & Newsholme EA Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem. J. 239, 121–5 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fan J, Krautkramer KA, Feldman JL & Denu JM Metabolic Regulation of Histone Post-Translational Modifications. ACS Chem. Biol. 10, 95–108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Munder M, Eichmann K & Modolell M Alternative Metabolic States in Murine Macrophages Reflected by the Nitric Oxide Synthase/Arginase Balance: Competitive Regulation by CD4+ T Cells Correlates with Th1/Th2 Phenotype. J. Immunol. 160, 5347–5354 (1998). [PubMed] [Google Scholar]
- 29.Corraliza IM, Soler G, Eichmann K & Modolell M Arginase induction by suppressors of nitric oxide synthesis (IL-4, IL-10 and PGE2) in murine bone-marrow- derived macrophages. Biochem. Biophys. Res. Commun. 206, 667–673 (1995). [DOI] [PubMed] [Google Scholar]
- 30.Hard GC Some biochemical aspects of the immune macrophage. Br. J. Exp. Pathol. 51, 97–105 (1970). [PMC free article] [PubMed] [Google Scholar]
- 31.Cekic C & Linden J Purinergic regulation of the immune system. Nat. Publ. Gr. 16, (2016). [DOI] [PubMed] [Google Scholar]
- 32.Strelko CL et al. Itaconic Acid Is a Mammalian Metabolite Induced during Macrophage Activation. J. Am. Chem. Soc. 133, 16386–16389 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michelucci A et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. 110, 7820–7825 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ackermann WW & Potter VR Enzyme Inhibition in Relation to Chemotherapy. Exp. Biol. Med. 72, 1–9 (1949). [DOI] [PubMed] [Google Scholar]
- 35.Xiao M et al. Inhibition of a-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 26, 1326–1338 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Safran M et al. Mouse model for noninvasive imaging of HIF prolyl hydroxylase activity: Assessment of an oral agent that stimulates erythropoietin production. Proc. Natl. Acad. Sci. 103, 105–110 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramakrishnan SK et al. Loss of von Hippel-Lindau Protein (VHL) Increases Systemic Cholesterol Levels through Targeting Hypoxia-Inducible Factor 2 and Regulation of Bile Acid Homeostasis. Mol. Cell. Biol. 34, 1208–1220 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mentch SJ & Locasale JW One-carbon metabolism and epigenetics: Understanding the specificity. Ann. N. Y. Acad. Sci. 1363, 91–98 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chowdhury R et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. Nat. Publ. Gr. 12, 463–469 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu W et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Figueroa ME et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–67 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koivunen P et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483, 484–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meiser J et al. Pro-inflammatory macrophages sustain pyruvate oxidation through pyruvate dehydrogenase for the synthesis of itaconate and to enable cytokine expression. J. Biol. Chem. 291, 3932–3946 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim JW, Tchernyshyov I, Semenza GL & Dang CV HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. CellMetab. 3, 177–185 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Lu CW, Lin SC, Chen KF, Lai YY & Tsai SJ Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J. Biol. Chem. 283, 28106–28114 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Comerford SA et al. Acetate dependence of tumors. Cell 159, 1591–1602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamphorst JJ, Chung MK, Fan J & Rabinowitz JD Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab. 2, 23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mills EL & O’Neill LA Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur. J. Immunol. 46, 13–21 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Drapier JC & Hibbs JB Murine cytotoxic activated macrophages inhibit aconitase in tumor cells. Inhibition involves the iron-sulfur prosthetic group and is reversible. J. Clin. Invest. 78, 790–797 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kennedy MC, Antholine WE & Beinert H An EPR investigation of the products of the reaction of cytosolic and mitochondrial aconitases with nitric oxide. J. Biol. Chem. 272, 20340–30347 (1997). [DOI] [PubMed] [Google Scholar]
- 51.Mills EL et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556, 113–117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bambouskova M et al. Electrophilic properties of itaconate and derivatives regulate the IκBζ-ATF3 inflammatory axis. Nature 556, 501–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi L & Tu BP Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr. Opin. Cell Biol. 33, 125–131 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Z et al. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 7, 58–63 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cameron AM, Lawless SJ & Pearce EJ Metabolism and acetylation in innate immune cell function and fate. Seminars in Immunology 28, 408–416 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou R, Yazdi AS, Menu P & Tschopp J A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–226 (2011). [DOI] [PubMed] [Google Scholar]
- 57.Schieber M & Chandel NS ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 24, R453–R462 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tong WH et al. TLR-activated repression of Fe-S cluster biogenesis drives a metabolic shift and alters histone and tubulin acetylation. Blood Adv. 2, 1146–1156 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mathias RA et al. Sirtuin 4 Is a Lipoamidase Regulating Pyruvate Dehydrogenase Complex Activity. Cell 159, 1615–1625 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Humphries KM, Yoo Y & Szweda LI Inhibition of NADH-linked mitochondrial respiration by 4-hydroxy-2- nonenal. Biochemistry 37, 552–557 (1998). [DOI] [PubMed] [Google Scholar]
- 61.O’Brien M, Chalker J, Slade L, Gardiner D & Mailloux RJ Protein S- glutathionylation alters superoxide/hydrogen peroxide emission from pyruvate dehydrogenase complex. Free Radic. Biol. Med. 106, 302–314 (2017). [DOI] [PubMed] [Google Scholar]
- 62.McLain AL, Cormier PJ, Kinter M & Szweda LI Glutathionylation of α-ketoglutarate dehydrogenase: The chemical nature and relative susceptibility of the cofactor lipoic acid to modification. Free Radic. Biol. Med. 61, 161–169 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Clasquin MF, Melamud E & Rabinowitz JD LC-MS data processing with MAVEN: A metabolomic analysis and visualization engine. Curr. Protoc. Bioinforma. 37, 14.11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Melamud E, Vastag L & Rabinowitz JD Metabolomic Analysis and Visualization Engine for LC–MS Data. Anal. Chem. 82, 9818–9826 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Allen EL et al. Differential Aspartate Usage Identifies a Subset of Cancer Cells Particularly Dependent on OGDH. Cell Rep. 17, 876–890 (2016). [DOI] [PubMed] [Google Scholar]
- 66.Hewitson KS, Schofield CJ & Ratcliffe PJ Hypoxia-Inducible Factor Prolyl- Hydroxylase: Purification and Assays of PHD2. Methods Enzymol. 435, 25–42 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Fan J, Baeza J & Denu JM Investigating Histone Acetylation Stoichiometry and Turnover Rate. Methods Enzymol. 574, 125–148 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.