

Low-pathogenicity avian influenza (LPAI) viruses circulate in wild birds and can be transmitted to poultry. LPAI viruses can mutate to become highly pathogenic avian influenza (HPAI) viruses causing severe disease and death in poultry. Little is known about this switch to high pathogenicity. We isolated an LPAI H7N3 virus from an infected turkey farm and showed that this contains small amounts of HPAI virus. The HPAI virus rapidly outcompeted the LPAI virus in chickens that were experimentally infected with this mixture of viruses. We analyzed the genome sequences of the LPAI and HPAI viruses and identified several changes that may be important for a virus to become highly pathogenic. This knowledge may be used for timely identification of LPAI viruses that pose a risk of becoming highly pathogenic in the field.
KEYWORDS: avian influenza virus, adaptive mutations, pathotypes, virulence determinants
ABSTRACT
Low-pathogenicity avian influenza (LPAI) viruses of subtypes H5 and H7 have the ability to spontaneously mutate to highly pathogenic (HPAI) virus variants, causing high mortality in poultry. The highly pathogenic phenotype is caused by mutation of the hemagglutinin (HA) cleavage site, but additional mutations may play a role. Evidence from the field for the switch to high pathogenicity remains scarce. This study provides direct evidence for LPAI-to-HPAI virus mutation during H7N3 infection of a turkey farm in the Netherlands. No severe clinical symptoms were reported at the farm, but deep sequencing of isolates from the infected turkeys revealed a minority of HPAI virus sequences (0.06%) in the virus population. The HPAI virus contained a 12-nucleotide insertion in the HA cleavage site that was likely introduced by a single event as no intermediates with shorter inserts were identified. This suggests nonhomologous recombination as the mechanism of insertion. Analysis of different organs of the infected turkeys showed the largest amount of HPAI virus in the lung (4.4%). The HPAI virus was rapidly selected in experimentally infected chickens after both intravenous and intranasal/intratracheal inoculation with a mixed virus preparation. Full-genome sequencing revealed that both pathotypes contained a deletion in the stalk region of the neuraminidase protein. We identified additional mutations in HA and polymerase basic protein 1 (PB1) in the HPAI virus, which were already present as minority variants in the LPAI virus population. Our findings provide more insight into the molecular changes and mechanisms involved in the emergence and selection of HPAI viruses.
IMPORTANCE Low-pathogenicity avian influenza (LPAI) viruses circulate in wild birds and can be transmitted to poultry. LPAI viruses can mutate to become highly pathogenic avian influenza (HPAI) viruses causing severe disease and death in poultry. Little is known about this switch to high pathogenicity. We isolated an LPAI H7N3 virus from an infected turkey farm and showed that this contains small amounts of HPAI virus. The HPAI virus rapidly outcompeted the LPAI virus in chickens that were experimentally infected with this mixture of viruses. We analyzed the genome sequences of the LPAI and HPAI viruses and identified several changes that may be important for a virus to become highly pathogenic. This knowledge may be used for timely identification of LPAI viruses that pose a risk of becoming highly pathogenic in the field.
INTRODUCTION
Aquatic wild birds are the natural host reservoir for avian influenza A (AI) viruses (1). Based on antigenic properties of their hemagglutinin (HA) and neuraminidase (NA) surface glycoproteins, AI viruses are classified into subtypes. To date, 16 different HA and 9 different NA subtypes, which can be found in various combinations, have been described in wild birds (2). A further classification into low-pathogenicity and highly pathogenic (LP/HP) viruses refers to their virulence in chickens (3). LPAI viruses are circulating in their natural hosts without causing severe clinical symptoms. Introduction of LPAI viruses into poultry can cause mild disease. Some LPAI viruses of the subtypes H5 and H7 have the ability, under natural conditions, to spontaneously mutate to the HP phenotype. These HPAI viruses cause severe disease in poultry, with up to 100% mortality (4). As only H5 and H7 subtypes have been observed to acquire HP virus mutations, infections of poultry with these viruses are therefore considered to be notifiable regardless of their pathogenicity. Control of notifiable subtypes H5 and H7 in poultry aims to prevent the mutation to HPAI virus and the spread of these viruses that are associated with large economic losses in poultry production.
The major determinant of virulence is the HA gene (3, 5) although other genes may contribute to increased pathogenicity (6). In the subtypes H5 and H7, the HP phenotype correlates with the presence of multiple basic amino acids at the endoproteolytic cleavage site (CS) of the HA protein. The HA precursor protein is cleaved by trypsin-like proteases which recognize the monobasic cleavage motif. Replication of LPAI viruses is therefore restricted to the respiratory and gastrointestinal tract where these enzymes are expressed. Polybasic cleavage motifs are recognized by ubiquitous proteases, such as furin. These enzymes are expressed in most avian tissues, which enables HPAI viruses to replicate systemically in their hosts (7). There is insufficient knowledge on the mechanisms involved in the emergence and selection of HPAI viruses, but replication of the virus in gallinaceous birds is considered a critical part of the process.
For H5 subtypes, the mutation from an LPAI to HPAI virus can occur by replacement of nonbasic with basic amino acids, but most commonly one or more additional basic amino acids are inserted at the cleavage site. These insertions are likely caused by duplication events during RNA replication (8). All H7 HPAI viruses have insertions of 2 to 10 additional basic amino acids at the cleavage site. These could be introduced by duplication events, but for several viruses, insertions by nonhomologous recombination appear more likely (9–11). These recombination events may occur with either viral or cellular RNA sequences. Almost all H7 HPAI outbreak viruses appear to have become highly pathogenic by unique events at the cleavage site, which makes prediction of pathogenicity by sequence alone more difficult for H7 subtypes. It is currently unknown why only H5 and H7 subtypes mutate and how the HPAI virus is selected in poultry.
In the Netherlands, LPAI H7N3 virus was detected by isolation and sequencing on a turkey farm after it scored positive in a serological monitoring program performed in 2003 (12). Two neighboring free-ranging chicken farms also tested positive for antibodies against H7. No clinical signs were reported at the chicken farms, and only mild clinical symptoms were seen at the turkey farm. Surprisingly, when the virus isolated from the infected turkeys was inoculated into chickens, it rapidly induced disease and death, resulting in an intravenous-pathogenicity index (IVPI) of 2.48. Sequencing of the virus from the dead chickens with a high IVPI revealed an HP cleavage site (12). Simultaneous detection of both the HPAI virus and its LPAI precursor virus on the same infected poultry holding is very rare. Therefore, this outbreak presents a unique opportunity to study the emergence and selection of the HPAI virus. We performed deep sequencing to study the emergence and selection of the HPAI virus variant in different organs of the infected turkeys retrieved from the farm and in experimentally infected chickens. We identified several mutations in the HPAI viral genome, in addition to the mutation of the HA cleavage site. Determining the drivers of emergence of HPAI viruses is crucial for a better understanding of why and when certain LPAI viruses pose a risk of becoming highly pathogenic.
RESULTS
Pathogenicity of the H7N3 virus.
The H7N3 virus was isolated from pooled tracheal tissue of five turkeys from the outbreak by inoculation into embryonated chicken eggs (i.e., isolation method 1). The virus was subtyped as LP H7N3 by Sanger sequencing, with the cleavage site sequence PEIPKG(R/G)LF. In this study, we infected 10 chickens by intravenous injection with the virus, after which clinical symptoms were monitored for 10 days to determine the intravenous-pathogenicity index (IVPI). This resulted in an IVPI score of 2.68, which is very similar to the score previously determined (IVPI of 2.48) using virus preparation 1 (12), and classified this virus as highly pathogenic. Sequencing of the virus retrieved from lung tissue of dead chickens revealed an HP virus cleavage site, with the sequence PEIPKGSRVR(R/G)LF (boldface indicates a 4-amino-acid [aa] insertion). These results suggest that either an HPAI virus was generated in the chickens during these IVPI experiments or, alternatively, that small amounts of HPAI virus may already have been present in the virus stock derived from the infected turkeys, which was then selected and amplified in the inoculated chickens.
To investigate this, a second virus stock was prepared by 1,000-fold dilution of the tracheal tissue sample before inoculation into embryonated eggs (i.e., isolation method 2). Ten chickens were infected with this second virus preparation by intravenous injection. No severe symptoms or deaths were observed during 10 days of observation, resulting in an IVPI score of 0.07, which is consistent with an LPAI virus. Antibodies against H7 were detected in the chickens using a hemagglutination inhibition (HI) assay, which demonstrated that the virus did replicate in these animals. A second group of six chickens was inoculated, and two animals were sacrificed at 24, 48, and 96 h after infection. Sanger sequencing of the HA cleavage site was performed for lung samples of the infected chickens. At every time point after infection, sequencing revealed an LP virus cleavage site. The HPAI virus thus did not emerge in the chickens during this experiment. These results suggest that the initial virus preparation derived from the infected turkeys contained a mixture of viruses. The small amount of HPAI virus was probably diluted out in this second virus preparation, resulting in a pure LPAI virus population.
Detection of a minority population HPAI virus in the infected turkeys.
The two virus isolations used for the IVPI experiments were analyzed by deep sequencing to detect potential HPAI virus minority sequences in the virus population. Part of the HA gene was amplified (nucleotides [nt] 802 to 1075) and sequenced using an Illumina platform. Virus recovered from tracheal tissue using isolation method 1 was found to contain 0.06% of HPAI virus sequences (32/53,568 reads), whereas virus prepared by method 2 did not contain HPAI virus sequences (0/54,450 reads).
Subsequently, the presence of HPAI minority species was analyzed in pooled organ samples of five infected turkeys using deep sequencing of the HA gene. The results are shown in Fig. 1. Analysis of sequences showed that the cleavage site of the highly pathogenic virus, PEIPKGSRVRRGLF (14 amino acids in length, 4-aa insertion in bold), was present in very small amounts (<0.07%) in the trachea, cloaca, spleen, liver, pancreas, and brains of the infected turkeys. Interestingly, around 4.4% (1,990/45,384 reads) of HP virus cleavage site sequences were detected in lung tissue. This suggests that the HPAI virus may have first emerged, or was selected, in the lung of the LPAI H7N3-infected turkeys.
FIG 1.
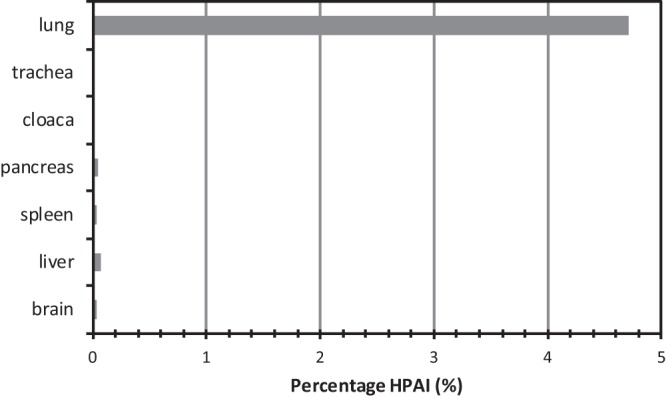
Percentage of HP virus cleavage site sequences in different organs of the H7N3-infected turkeys as determined by deep sequencing of part of the HA gene.
The sequence variation at the cleavage site was determined and is shown for the lung and spleen samples (Table 1). Around 2.5% of the sequences have one (or more) amino acid changes in the 10 amino acids in the cleavage site of the low-pathogenic virus, PEIPKGRGLF. These mutations may have been introduced during RNA genome replication as the viral RNA polymerase is known to be error prone and lacks a proofreading function. However, it cannot be excluded that a proportion of these mutations are artifacts of the PCR or sequencing procedure, and it is unknown if these variants represent infectious viruses. Interestingly, no sequences were detected containing insertions of 1, 2, or 3 amino acids at the cleavage site. Similar results were obtained for trachea, cloaca, liver, pancreas, and brain samples of the infected turkeys. These results suggest that the additional 4 amino acids (12 nucleotides) were introduced by a single event.
TABLE 1.
Sequence variation at the HA cleavage site in spleen and lung samples of H7N3-infected turkeys
| Spleen CS |
Lung CS |
||
|---|---|---|---|
| Sequencea | No. (%) of reads (n = 35,220) | Sequencea | No. (%) of reads (n = 45,384) |
| PEIPKGRGLF | 34,402 (97.68) | PEIPKGRGLF | 42,271 (93.14) |
| PEIPKGRGLL | 91 (0.26) | PEIPKGSRVRRGLFb | 1,990 (4.38) |
| PEIPKGGGLF | 84 (0.24) | PEIPRGRGLF | 116 (0.26) |
| PEIPRGRGLF | 66 (0.19) | PEIPKGGGLF | 112 (0.25) |
| PEISKGRGLF | 55 (0.16) | PEIPKGRGLL | 108 (0.24) |
| PEILKGRGLF | 41 (0.12) | PETPKGRGLF | 59 (0.13) |
| PEIPKGRGPF | 37 (0.11) | PEILKGRGLF | 56 (0.12) |
| PETPKGRGLF | 36 (0.10) | PEIPKGRGPF | 51 (0.11) |
| PEIPEGRGLF | 26 (0.07) | PEVPKGRGLF | 36 (0.08) |
| PEVPKGRGLF | 26 (0.07) | SEIPKGRGLF | 35 (0.08) |
| PEIPKGRGLS | 25 (0.07) | PEIPKGRGLS | 34 (0.07) |
| SEIPKGRGLF | 23 (0.07) | PEIPEGRGLF | 34 (0.07) |
| PEIPKRRGLF | 21 (0.06) | PEIPKGRSLF | 30 (0.07) |
| TEIPKGRGLF | 21 (0.06) | PEIPKGKGLF | 28 (0.06) |
| LEIPKGRGLF | 15 (0.04) | PEIPKGRDLF | 27 (0.06) |
| PGIPKGRGLF | 14 (0.04) | LEIPKGRGLF | 26 (0.06) |
| PKIPKGRGLF | 13 (0.04) | PEISKGRGLF | 25 (0.06) |
| PEIPKGRDLF | 13 (0.04) | PEIPKRRGLF | 24 (0.05) |
| PEIPKGSRVRRGLFb | 12 (0.03) | TEIPKGRGLF | 22 (0.05) |
| PEIPKGRSLF | 11 (0.03) | PKIPKGRGLF | 20 (0.04) |
| Other | 188 (0.53) | Other | 280 (0.62) |
| Total | 35,220 (100.00) | Total | 45,384 (100.00) |
All sequences listed, except as noted, were 10 aa in length.
Highly pathogenic HA cleavage site (14 aa).
Selection of HPAI virus is independent of the route of inoculation.
The minority HPAI virus was selected rapidly in the chickens in the IVPI experiment as they all died on day 2 or 3 after inoculation. This may be caused by the injection of the virus directly into the bloodstream of the chickens. We therefore performed an experiment in which intravenous injection was compared to intranasal/tracheal inoculation of the virus. A group of 16 chickens was inoculated by intravenous injection with virus isolated using method 1, and a second group was subjected to intranasal/tracheal inoculation (Fig. 2). At 24 h postinoculation, four chickens of both groups were euthanized for further analysis. The remaining 12 chickens in both groups rapidly developed disease and died. All of the intravenously injected chickens died (or were euthanized because they reached humane endpoints) within 32 h (Fig. 2A); disease was somewhat delayed for the chickens that received intranasal/tracheal inoculations, but all died within 48 h (Fig. 2B). Sequencing of lung tissue samples from these chickens confirmed the presence of an HP virus HA cleavage site for both groups. These results suggest that the HPAI virus is selected rapidly in chickens, independent of the route of inoculation.
FIG 2.

Clinical symptoms and death of chickens infected by intravenous injection (A) and intranasal/tracheal inoculation (B) with H7N3 virus isolation 1, at several hours after infection. Sixteen chickens were inoculated, of which 4 were euthanized at 24 h postinfection for further analysis. Clinical conditions are indicated according to the color legend as follows: normal, without symptoms; sick, mild clinical symptoms; very sick, severe clinical symptoms.
Rapid selection of HP virus sequences after experimental infection of chickens.
Deep sequencing of the HA gene was performed to analyze the rapid selection of the HPAI virus in the experimentally infected chickens. Two chickens euthanized 24 h after intravenous and intranasal/tracheal inoculation were selected. To study potential differences in the selection of HPAI virus in different organs, we analyzed liver, colon, kidney, spleen, lung, trachea, and brain samples from these chickens. The percentage of HP virus cleavage site sequences was between 89.5% and 99.9% in the different organs of the chickens that were intravenously inoculated (Fig. 3, chickens IV1 and IV2). For the chickens infected by intranasal/tracheal inoculation, a lower percentage (35% to 73%) of HP virus sequences was detected in the lung and trachea (Fig. 3, chickens INT1 and INT2). These results suggest that the HPAI virus is selected and rapidly spreads systemically in the chickens. The higher number of LP virus sequences in the lung and trachea of chickens INT1 and INT2 is likely due to sustained local replication of the LPAI virus after intranasal and intratracheal inoculation.
FIG 3.
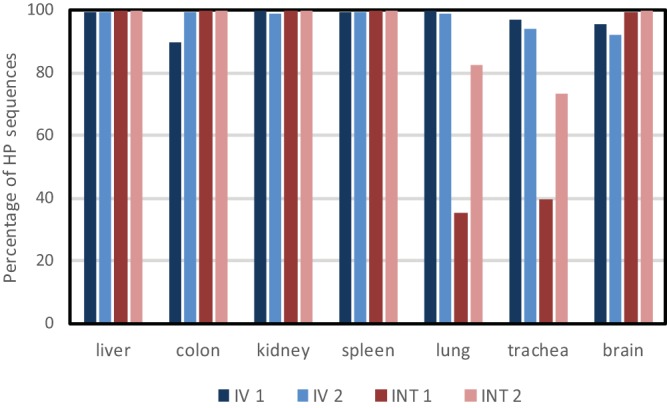
Percentage of HP virus cleavage site sequences in different organs of two chickens infected by intravenous injection (IV1 and IV2) and by intranasal/tracheal inoculation (INT1 and INT2) with H7N3 virus (prepared using isolation method 1), as determined by deep sequencing of part of the HA gene.
Additional adaptations in the H7N3 HPAI virus genome.
Additional changes in the genome of the H7N3 LPAI virus, besides the mutation of the HA cleavage site, may contribute to the pathogenicity of the virus. To analyze this, we performed deep sequencing of the complete virus genomes present in the virus isolations, in organs of the infected turkeys, and in organs of chickens after experimental infection. The analysis showed that both the LPAI and the HPAI viruses contain a deletion at nucleotide positions 130 to 207 in the NA gene, resulting in a 26-amino-acid deletion in the stalk region of the NA protein. The consensus and minority changes compared to the sequence of virus isolation 1 are shown in Table 2. The presence of an HP virus cleavage site sequence appears correlated with mutation A340G in the PB1 gene, which results in a threonine-to-alanine amino acid change at position 110 in the protein. This mutation was already present as a minority variant in the virus inoculum (virus isolation 1) and in most organs of the infected turkeys. In addition, three additional nucleotide changes in HA (A650G, T1191C, and A1243G) were detected, of which two result in amino acid changes (E214G and I412V). These mutations were already found as minority variants in the virus inoculum and were already present as minority or majority sequences in organs of the infected turkeys. The percentage of the nucleotide change A297G in the matrix protein (MP) gene varied but did not appear to correlate with the percentage of HP virus cleavage site sequence detected. None of the identified substitutions has been assigned previously to specific functions related to virus replication or pathogenicity.
TABLE 2.
Additional consensus and minority nucleotide changes in the virus genomes compared to virus isolation 1
| Pathotype and/or sample source | % of reads with the indicated nucleotide (amino acid) change in: |
|||||
|---|---|---|---|---|---|---|
| HA CS (12 nt [4 aa] insertion) | HA |
PB1 (A340G [Thr110Ala]) | MP (A297G [Lys95Arg]) | |||
| A650Ga (Glu214Gly) | T1191Ca (silent) | A1243Ga (Ile412Val) | ||||
| Virus isolation 1b | 0.1 | 15 | 26 | 25 | 7 | 45 |
| Virus isolation 2, dilutedc | 0 | 14 | 14 | 15 | 15 | 32 |
| LP | ||||||
| Infected turkey lung | 2.1 | 85 | 85 | 86 | 6 | 82 |
| Infected turkey trachea | 0 | 30 | 45 | 45 | 11 | 43 |
| Infected turkey brain | 0 | 86 | 53 | 88 | 0 | 94 |
| HP | ||||||
| IVPI experiments | ||||||
| Chicken 1 lung | 99.9 | 100 | 100 | 100 | 83 | 72 |
| Chicken 1 brain | 100 | 100 | 100 | 100 | 87 | 64 |
| Chicken 5 lung | 99.6 | 100 | 100 | 100 | 80 | 73 |
| Chicken 5 braind | 100 | 100 | 100 | 100 | 93 | 25 |
| Chicken IV1 lung | 99.9 | 100 | 100 | 100 | 87 | 74 |
| Chicken IV2 lung | 99.9 | 100 | 100 | 100 | 87 | 77 |
| Chicken INT17 lung | 23.4 | 62 | 63 | 64 | 34 | 60 |
| Chicken INT19 lung | 90.1 | 92 | 93 | 93 | 78 | 67 |
LPAI numbering in the H7 gene.
Virus isolation used as the inoculum in infection experiments and used as a reference sequence for this genetic analysis.
The following additional nucleotide changes were found: PA A1841G and HA G445T.
The following additional nucleotide changes were found: PB1 C310T, HA A485G, and NA T190C.
RNA secondary structure analysis.
The RNA secondary structure of the HA gene is thought to play a role in the emergence of the HP virus mutation. We therefore compared the RNA structure in the HA cleavage site region of the LPAI and HPAI virus. A stretch of RNA nucleotides encoding the HA cleavage site was predicted to fold into a stem-loop structure for both the LP and HP variants of the H7N3 virus (Fig. 4). The insertion leading to the creation of the multibasic cleavage site in the HPAI H7N3 virus occurred in the loop of the hairpin. The 12-nucleotide insertion results in a larger loop region and a slight increase in the thermodynamic stability of the RNA structure. The additional mutations in HA present in the HPAI virus did not change the RNA secondary structure of the cleavage site region or other regions of the HA segment (results not shown). A BLAST search for the 12-nucleotide insertion indicates that the sequence may be derived from the turkey major histocompatibility complex (MHC) B locus mRNA. The location of the insertion site in the hairpin loop is suggestive of nonhomologous recombination as the mechanism for insertion.
FIG 4.
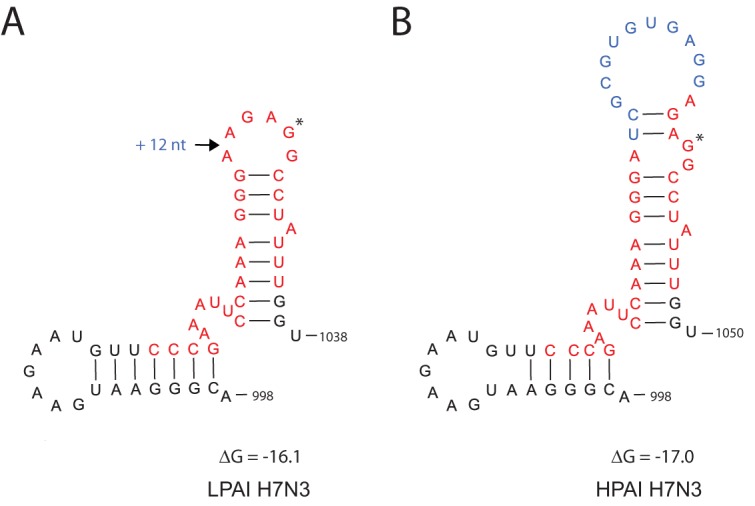
(A and B) RNA secondary structure prediction for the HA cleavage site region of the LPAI and the HPAI H7N3 viruses. The cleavage site is marked in red, and the GGC codon for the first Gly residue of the HA2 protein downstream of the cleavage site is marked by an asterisk. The 12-nt insertion in the cleavage site is marked in blue. Numbering of nucleotides corresponds to the LPAI H7 segment. ΔG, folding free energy for the depicted positive-sense RNA region (kilocalories/mole).
DISCUSSION
In this study, the H7N3 virus that caused an outbreak on a commercial turkey farm in the Netherlands in 2002 was investigated. The isolated virus showed low pathogenicity on the farm, but the subsequent IVPI determination using this virus preparation demonstrated high pathogenicity. Deep sequencing revealed the presence of a minority population (0.06%) of viruses with an HP virus HA cleavage site sequence in the H7N3 virus population. We show that this minority HPAI H7N3 virus is rapidly selected in chickens after intravenous or intratracheal/intranasal inoculation. The experimentally infected chickens died within 2 days after infection. Consistent with this highly pathogenic phenotype, mainly HP virus HA cleavage site sequences were detected in the organs of experimentally infected chickens. Experimental infection of chickens with a pure LPAI H7N3 virus population (in which no HPAI virus sequences were detected) did not result in generation or selection of HPAI virus. These results show that small amounts of HPAI H7N3 virus (0.06%) present in a mixed population rapidly outcompete the LPAI virus in infected chickens. The replication advantage found for the H7N3 virus may not be a general feature of HPAI viruses as a study on H7N7 reported competition between HPAI and LPAI viruses in experimentally coinfected chickens (13). In the 10-day-old embryonated chicken eggs used for H7N3 virus isolation, the HPAI virus apparently did not have a replication advantage in the allantoic fluid; other embryonal tissues were not analyzed. This may be due to the immature immune response in the early chicken embryo (14). A previous study showed that highly pathogenic H5N2 viruses have a replication advantage in 14-day-old embryonated chicken eggs but not in 10-day-old eggs. Our results demonstrate a strong replication advantage for the HPAI H7N3 virus compared to that of the LPAI virus in experimentally infected chickens. This study also underlines the importance of the obligatory IVPI experiment for all H5 and H7 subtype viruses that are detected in poultry as this may assist in the early detection of HPAI viruses. Sanger sequencing does not allow the detection of a minority of HPAI sequences in an LPAI virus population; therefore, deep sequencing of notifiable LPAI virus cases may be a valuable addition to routine diagnostics.
Deep-sequencing analysis was also performed to study the virus population in different organs of the turkeys retrieved from the infected farm. For this analysis, organ tissues derived from five infected turkeys were pooled. We show that only 4.4% of the HA cleavage site sequences contained the HP virus mutation, whereas even smaller amounts were present in the other organs tested (<0.07%). This suggests that the HPAI H7N3 virus may have first emerged, or was selected, in the lung of infected turkeys. As the tissues of five turkeys were pooled for this analysis, it is unknown whether one or more turkeys were infected with LPAI or HPAI virus or a mixture of these viruses. No mortality or clinical signs consistent with HPAI virus infection were reported on the farm with the infected turkeys. In the monitoring program, blood samples collected a few months later tested positive for antibodies against H7 virus, indicating that the flock had overcome the infection. Thus, on the turkey farm the HPAI H7N3 virus did not outcompete the LPAI virus. Possibly, the index turkey(s) in which the HPAI virus emerged was euthanized for this study and did not infect other turkeys in the flock. Or the replication advantage of the HPAI virus is dependent on the host and less prominent in turkeys than in chickens. But most likely, the HPAI virus mutation occurred relatively late in the infection of the turkey flock. The protective immune response elicited by the initial infection with LPAI H7N3 virus may have prevented the selection and subsequent spread of the HPAI virus within the turkey flock. In the serological monitoring program, two neighboring chicken farms also tested positive for antibodies against H7 a few months later. The mechanism and direction of this putative between-farm transmission of the H7N3 virus are unknown. But no clinical signs were reported at these chicken farms, indicating that the HPAI virus did not spread. Virological identification of a matching pair of the LPAI precursor virus and its HPAI virus descendant at the index poultry holding is a rare event. The simultaneous detection of both pathotypes has been observed for only a few H7 outbreaks in the past (6). During the H7N1 outbreak in Italy in 1999, the LPAI virus circulated for several months in poultry before mutating to HPAI virus. The HPAI viruses contained a 12-nt insert in the HA cleavage site. In addition, a 22-amino-acid deletion in the NA stalk region and a truncation of the NS1 gene were identified, and some viruses contained additional glycosylation sites in HA (15). In 2002, an outbreak of H7N3 occurred on a chicken farm in Chile, and the LPAI and HPAI viruses cocirculated for some period in the index premises. The HPAI viruses contained a 30-nt insert in the HA cleavage site that was attributed to nonhomologous recombination with the nucleoprotein (NP) gene of the virus (9). In 2004, an outbreak of H7N3 occurred in Canada, and both LPAI and HPAI viruses were isolated from different infected poultry farms. The HPAI viruses contained a 21-nt insert in the HA cleavage site, which may be attributed to nonhomologous recombination with the matrix gene of the virus. No NA stalk deletions were observed (10). In the United Kingdom in 2008, H7N7 viruses of low and high pathogenicity were detected on a single poultry premises. Sequencing analysis identified LP virus cleavage site sequences in samples taken from two chickens. However, the LPAI virus could not be not isolated. Three different HP virus cleavage site motifs were detected in viruses derived from different chickens, suggestive of ongoing evolution of the cleavage site. No NA stalk deletions were identified, but potential additional glycosylation sites in HA may be present in some HPAI viruses (16). In 2015, two farms in Germany became infected with LPAI H7N7 virus, after which HPAI H7N7 was detected on the second farm. The HPAI virus contained an insertion of 6 nt in the HA cleavage site and 10 amino acid changes compared to the sequence of the LPAI precursor virus in different genome segments (17). Finally, in 2016 an outbreak of HPAI H7N8 was reported on a turkey farm in the United States. The LPAI precursor virus was detected during surveillance at surrounding farms and in wild birds. Deep sequencing did not identify LPAI virus sequences in the HPAI virus-infected farm, and HPAI virus sequences were not identified in the surrounding LPAI virus-infected farms. Possibly, the HPAI virus emerged and rapidly outcompeted the LPAI virus at the index farm. The HPAI virus contained a 9-nt insertion in the HA cleavage site and induced high mortality in both experimentally infected turkeys and chickens (18, 19). This study also identified three mutations in HA associated with the HPAI virus: E95K, F251L, and E269K. In 2017, HPAI H7N9 viruses were detected at two poultry farms in the United States (20). Deep sequencing was performed for the HPAI viruses and for the LPAI H7N9 viruses that were previously detected in poultry. Phylogenetic analysis predicted a theoretical precursor virus, which suggests that the LPAI H7N9 virus circulated undetected in the area before the initial HPAI virus detection. Combined, these studies suggest that NA stalk deletions, HA glycosylation sites, and additional mutations in HA may be involved in adaptation of the virus to poultry and selection of HPAI viruses.
We performed deep-sequencing analysis, which demonstrated that the 12-nt insert in the HA cleavage site of the H7N3 virus was likely introduced by one single event as no intermediates with shorter inserts were identified. This is in contrast to studies on H5 viruses, for which stepwise changes in the HA cleavage site were reported, likely introduced by slippage of the polymerase complex during replication (21–23). Previously, nonhomologous recombination has been proposed as a mechanism for cleavage site mutation for several H7 viruses. Two studies describe nonhomologous recombination as a mechanism for insertion under experimental conditions (24, 25), and three studies describe field observations (9–11). These observations are all linked to American viruses of the H7 subtype. Moreover, for the H7N1 outbreak in Italy, nonhomologous recombination was suggested as the mechanism for generation of the HPAI virus (26). The RNA secondary structure analysis we performed for the H7N3 virus predicts folding of HA cleavage site sequence in a stem-loop structure. Stem-loop structures in the cleavage site region were also predicted for other H7 viruses (27). The insertion site is located within the loop region, which is suggestive for hairpin-induced template switching, as in recombination of retroviral genomes (28) and discontinuous RNA transcription in arteriviruses (29). Homology between the 12-nt insert and the sequence of the turkey major histocompatibility complex (MHC) locus B was found, suggesting that the insert may have been derived from this mRNA. Analysis of insertions in the HA cleavage site of other HPAI H7 viruses identified viral sequences or cellular rRNA or mRNA sequences as putative donors (9–11). Our analysis suggests that the 12-nt insertion in the cleavage site of the H7N3 virus detected on a turkey farm in the Netherlands was introduced by nonhomologous recombination.
Analysis of the full-genome sequences of the LPAI and HPAI viruses was performed to identify potential genomic features that may underlie the switch to the HP phenotype. This analysis identified a 26-amino-acid deletion in the stalk region of the NA protein. The stalk is a structure that separates the enzymatically active head domain from the membrane domain, but little is known about its function. Previous studies have shown that deletions in the NA stalk region influence virus replication and virulence (30–32). Stalk deletions may represent an adaptation to gallinaceous hosts although these deletions are also found in nonpoultry hosts (33). NA stalk deletions are often accompanied by mutations in the HA protein to maintain the functional balance between HA and NA activity, which is required for virus replication (34–37). Additional HA glycosylation sites have been reported for several HPAI viruses containing NA stalk deletions (6) but were not observed for the HPAI H7N3 virus. However, three nucleotide changes in HA were detected, of which two result in amino acid changes (E214G and I412V). In addition, the presence of an HP virus cleavage site sequence appears to correlate with mutation A340G in the PB1 gene, which results in a threonine-to-alanine amino acid change at position 110 in the protein. The HA and PB1 mutations were already present as minority variants in the LPAI virus population. It is currently unknown if these mutations result in functional changes or whether their selection is due to genetic hitchhiking (38). Further research will be required to investigate this. The H7N3 virus also contained mutation 251L in the HA gene that was previously described for the HPAI H7N8 virus detected on a turkey farm in the United States (18, 19) and may be linked to host adaption from wild birds to poultry. This study provides insight into the genetic context of LP H7 viruses converting to the HP phenotype and the mutation mechanism and selection of the HP virus cleavage site sequence. This knowledge may contribute to identification and early detection of LPAI viruses that are likely to become HPAI viruses in the field.
MATERIALS AND METHODS
Virus isolation.
Virus was isolated from pooled tracheal tissues of five H7N3-infected turkeys. Using virus isolation method 1, a 10% tracheal tissue suspension was inoculated into the allantoic cavity of 10-day-old embryonated specific-pathogen-free (SPF) chicken eggs (MSD Animal Health), according to standard protocols (39). Using virus isolation method 2, the eggs were inoculated with a 1,000-fold dilution of 10% tracheal suspension. Eggs were incubated at 37°C and candled daily for embryo vitality. After embryo death, the allantoic fluids were harvested and assayed for hemagglutination activity, according to standard protocols (39). The subtype of the virus was determined by hemagglutination inhibition (HI) assay (39) and confirmed by Sanger sequencing as described previously (40).
Animal experiments.
The intravenous-pathogenicity index (IVPI) for the virus was determined using 10 SPF chickens (MSD Animal Health). The 6-week-old SPF chickens were infected by intravenous injection, according to standard protocols (39). The chickens were inoculated with virus prepared using isolation method 1. The chickens were monitored daily for clinical signs and mortality during 10 days to determine the IVPI score. A similar experiment was performed in which 10 chickens were inoculated with virus prepared using isolation method 2. Six additional chickens were inoculated with this virus (isolation method 2), and two animals each were euthanized at 24, 48, and 96 h after infection. We isolated blood, lung, trachea, colon, and brain samples for further analysis.
In a second experiment, two groups of 16 4-week-old chickens were inoculated by either the intravenous or intranasal/tracheal route with the virus prepared using method 1 with a dose of 106.6 50% egg infective doses (EID50)/chicken. Four chickens from each group were euthanized at 24 h after inoculation. We isolated blood, liver, colon, kidney, spleen, lung, trachea, and brain samples for further analysis. Clinical symptoms were monitored and scored according to the IVPI protocol (39). All experiments were performed in biosecurity level 3 facilities under the approval of the Central Animal Experiments Committee (license numbers 2012139.b, 2016024.b, and AVD401002015317) in the Netherlands.
Deep sequencing of viral RNA.
Viral RNA was extracted from 10% suspensions of organ tissues using a High Pure Viral RNA kit (Roche). Part of the HA gene was amplified by reverse transcription (RT-PCR) (positions 802 to 1075) using the forward primer H7FA (5′‐CGT GCA AGT TTT CTG AGA GG‐3′) and reverse primer B H7R (5′‐GAC CTT CCC ATC CAT TTT CA‐3′), as previously described (41). Full-genome sequences were amplified using universal eight segment-specific primers, as described previously (42). The purified HA amplicons and full genomes were sequenced at high coverage using the Nextera library preparation method and Illumina MiSeq paired-end 150-bp sequencing (average of >40,000 and >5,000 base coverage, respectively, per nucleotide position). Quality control-passed sequence reads were mapped using a ViralProfiler-Workflow, an extension of the CLC Genomics Workbench (Qiagen, Germany) as previously described (40). Minority sequences were also detected using this workflow. A recent study suggested a limit of 0.5% for reliable detection of single-nucleotide polymorphisms in the influenza virus genome, based on the error rate of the next-generation sequencing (NGS) procedure (43). In our study, we used a conservative cutoff of 2.0% occurrence per base position having a minimum coverage of 50 reads to ensure reliable detection of minority variants in the virus genome. No cutoff was used for the detection of the HP virus cleavage site sequence as this is a specific insertion of 12 nucleotides. The detection of this large insertion is unlikely to be affected by erroneous single-nucleotide changes during the PCR and/or sequencing procedure.
RNA secondary structure prediction.
RNA secondary structures were predicted using the Zuker algorithm (44) on the Mfold Web server (45). RNA secondary structure drawings were created using RNAviz software (46).
Data availability.
The majority consensus sequences generated in this study were submitted to the GISAID database (http://gisaid.org) under accession numbers EPI_ISL_391795 to EPI_ISL_391799.
ACKNOWLEDGMENTS
We thank Sylvia Pritz-Verschuren, Diana van Zoelen-Bos, and Cynthia Baars-Lorist for technical assistance. We acknowledge Arjan Stegeman and Francisca Velkers for collection of the turkeys from the infected farm and Ruth Bouwstra for helpful discussions. We thank the animal caretakers of Wageningen Bioveterinary Research for their support during the animal experiment.
This work was funded by the Dutch Ministry of Agriculture, Nature and Food Quality (projects WOT-01-003-012 and WOT-01-003-059).
REFERENCES
- 1.Stallknecht DE, Brown JD. 2008. Ecology of avian influenza in wild birds, p 43–58. In Swayne DE. (ed), Avian influenza. John Wiley & Sons, New York, NY. [Google Scholar]
- 2.Olsen B, Munster VJ, Wallensten A, Waldenstrom J, Osterhaus AD, Fouchier RA. 2006. Global patterns of influenza a virus in wild birds. Science 312:384–388. doi: 10.1126/science.1122438. [DOI] [PubMed] [Google Scholar]
- 3.Webster RG, Rott R. 1987. Influenza virus A pathogenicity: the pivotal role of hemagglutinin. Cell 50:665–666. doi: 10.1016/0092-8674(87)90321-7. [DOI] [PubMed] [Google Scholar]
- 4.Alexander DJ. 2000. A review of avian influenza in different bird species. Vet Microbiol 74:3–13. doi: 10.1016/s0378-1135(00)00160-7. [DOI] [PubMed] [Google Scholar]
- 5.Bottcher-Friebertshauser E, Garten W, Matrosovich M, Klenk HD. 2014. The hemagglutinin: a determinant of pathogenicity. Curr Top Microbiol Immunol 385:3–34. doi: 10.1007/82_2014_384. [DOI] [PubMed] [Google Scholar]
- 6.Abdelwhab el SM, Veits J, Mettenleiter TC. 2013. Genetic changes that accompanied shifts of low pathogenic avian influenza viruses toward higher pathogenicity in poultry. Virulence 4:441–452. doi: 10.4161/viru.25710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garten W, Braden C, Arendt A, Peitsch C, Baron J, Lu Y, Pawletko K, Hardes K, Steinmetzer T, Böttcher-Friebertshäuser E. 2015. Influenza virus activating host proteases: identification, localization and inhibitors as potential therapeutics. Eur J Cell Biol 94:375–383. doi: 10.1016/j.ejcb.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 8.Nao N, Yamagishi J, Miyamoto H, Igarashi M, Manzoor R, Ohnuma A, Tsuda Y, Furuyama W, Shigeno A, Kajihara M, Kishida N, Yoshida R, Takada A. 2017. Genetic predisposition to acquire a polybasic cleavage site for highly pathogenic avian influenza virus hemagglutinin. mBio 8:e02298-16. doi: 10.1128/mBio.02298-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suarez DL, Senne DA, Banks J, Brown IH, Essen SC, Lee CW, Manvell RJ, Mathieu-Benson C, Moreno V, Pedersen JC, Panigrahy B, Rojas H, Spackman E, Alexander DJ. 2004. Recombination resulting in virulence shift in avian influenza outbreak, Chile. Emerg Infect Dis 10:693–699. doi: 10.3201/eid1004.030396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pasick J, Handel K, Robinson J, Copps J, Ridd D, Hills K, Kehler H, Cottam-Birt C, Neufeld J, Berhane Y, Czub S. 2005. Intersegmental recombination between the haemagglutinin and matrix genes was responsible for the emergence of a highly pathogenic H7N3 avian influenza virus in British Columbia. J Gen Virol 86:727–731. doi: 10.1099/vir.0.80478-0. [DOI] [PubMed] [Google Scholar]
- 11.Maurer-Stroh S, Lee RT, Gunalan V, Eisenhaber F. 2013. The highly pathogenic H7N3 avian influenza strain from July 2012 in Mexico acquired an extended cleavage site through recombination with host 28S rRNA. Virol J 10:139. doi: 10.1186/1743-422X-10-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Velkers FC, Bouma A, Matthijs MG, Koch G, Westendorp ST, Stegeman JA. 2006. Outbreak of avian influenza H7N3 on a turkey farm in the Netherlands. Vet Rec 159:403–405. doi: 10.1136/vr.159.13.403. [DOI] [PubMed] [Google Scholar]
- 13.Graaf A, Ulrich R, Maksimov P, Scheibner D, Koethe S, Abdelwhab EM, Mettenleiter TC, Beer M, Harder T. 2018. A viral race for primacy: co-infection of a natural pair of low and highly pathogenic H7N7 avian influenza viruses in chickens and embryonated chicken eggs. Emerg Microbes Infect 7:204. doi: 10.1038/s41426-018-0204-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davison TF. 2003. The immunologists’ debt to the chicken. Br Poult Sci 44:6–21. doi: 10.1080/0007166031000085364. [DOI] [PubMed] [Google Scholar]
- 15.Banks J, Speidel ES, Moore E, Plowright L, Piccirillo A, Capua I, Cordioli P, Fioretti A, Alexander DJ. 2001. Changes in the haemagglutinin and the neuraminidase genes prior to the emergence of highly pathogenic H7N1 avian influenza viruses in Italy. Arch Virol 146:963–973. doi: 10.1007/s007050170128. [DOI] [PubMed] [Google Scholar]
- 16.Seekings AH, Slomka MJ, Russell C, Howard WA, Choudhury B, Nunez A, Londt BZ, Cox W, Ceeraz V, Thoren P, Irvine RM, Manvell RJ, Banks J, Brown IH. 2018. Direct evidence of H7N7 avian influenza virus mutation from low to high virulence on a single poultry premises during an outbreak in free range chickens in the UK, 2008. Infect Genet Evol 64:13–31. doi: 10.1016/j.meegid.2018.06.005. [DOI] [PubMed] [Google Scholar]
- 17.Dietze K, Graaf A, Homeier-Bachmann T, Grund C, Forth L, Pohlmann A, Jeske C, Wintermann M, Beer M, Conraths FJ, Harder T. 2018. From low to high pathogenicity—characterization of H7N7 avian influenza viruses in two epidemiologically linked outbreaks. Transbound Emerg Dis 65:1576–1587. doi: 10.1111/tbed.12906. [DOI] [PubMed] [Google Scholar]
- 18.Killian ML, Kim-Torchetti M, Hines N, Yingst S, DeLiberto T, Lee DH. 2016. Outbreak of H7N8 low-pathogenic avian influenza in commercial turkeys with spontaneous mutation to highly pathogenic avian influenza. Genome Announc 4:00457-16. doi: 10.1128/genomeA.00457-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee DH, Torchetti MK, Killian ML, Swayne DE. 2017. Deep sequencing of H7N8 avian influenza viruses from surveillance zone supports H7N8 high pathogenicity avian influenza was limited to a single outbreak farm in Indiana during 2016. Virology 507:216–219. doi: 10.1016/j.virol.2017.04.025. [DOI] [PubMed] [Google Scholar]
- 20.Lee DH, Torchetti MK, Killian ML, Berhane Y, Swayne DE. 2017. Highly pathogenic avian influenza A(H7N9) virus, Tennessee, USA, March 2017. Emerg Infect Dis 23:1860–1863. doi: 10.3201/eid2311.171013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luczo JM, Tachedjian M, Harper JA, Payne JS, Butler JM, Sapats SI, Lowther SL, Michalski WP, Stambas J, Bingham J. 2018. Evolution of high pathogenicity of H5 avian influenza virus: haemagglutinin cleavage site selection of reverse-genetics mutants during passage in chickens. Sci Rep 8:11518. doi: 10.1038/s41598-018-29944-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abolnik C. 2017. Evolution of H5 highly pathogenic avian influenza: sequence data indicate stepwise changes in the cleavage site. Arch Virol 162:2219–2230. doi: 10.1007/s00705-017-3337-x. [DOI] [PubMed] [Google Scholar]
- 23.Garcia M, Suarez DL, Crawford JM, Latimer JW, Slemons RD, Swayne DE, Perdue ML. 1997. Evolution of H5 subtype avian influenza A viruses in North America. Virus Res 51:115–124. doi: 10.1016/s0168-1702(97)00087-7. [DOI] [PubMed] [Google Scholar]
- 24.Khatchikian D, Orlich M, Rott R. 1989. Increased viral pathogenicity after insertion of a 28S ribosomal RNA sequence into the haemagglutinin gene of an influenza virus. Nature 340:156–157. doi: 10.1038/340156a0. [DOI] [PubMed] [Google Scholar]
- 25.Orlich M, Khatchikian D, Teigler A, Rott R. 1990. Structural variation occurring in the hemagglutinin of influenza virus A/turkey/Oregon/71 during adaptation to different cell types. Virology 176:531–538. doi: 10.1016/0042-6822(90)90023-k. [DOI] [PubMed] [Google Scholar]
- 26.Monne I, Fusaro A, Nelson MI, Bonfanti L, Mulatti P, Hughes J, Murcia PR, Schivo A, Valastro V, Moreno A, Holmes EC, Cattoli G. 2014. Emergence of a highly pathogenic avian influenza virus from a low-pathogenic progenitor. J Virol 88:4375–4388. doi: 10.1128/JVI.03181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gultyaev AP, Spronken MI, Richard M, Schrauwen EJ, Olsthoorn RC, Fouchier RA. 2016. Subtype-specific structural constraints in the evolution of influenza A virus hemagglutinin genes. Sci Rep 6:38892. doi: 10.1038/srep38892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moumen A, Polomack L, Unge T, Veron M, Buc H, Negroni M. 2003. Evidence for a mechanism of recombination during reverse transcription dependent on the structure of the acceptor RNA. J Biol Chem 278:15973–15982. doi: 10.1074/jbc.M212306200. [DOI] [PubMed] [Google Scholar]
- 29.van den Born E, Posthuma CC, Gultyaev AP, Snijder EJ. 2005. Discontinuous subgenomic RNA synthesis in arteriviruses is guided by an RNA hairpin structure located in the genomic leader region. J Virol 79:6312–6324. doi: 10.1128/JVI.79.10.6312-6324.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hossain MJ, Hickman D, Perez DR. 2008. Evidence of expanded host range and mammalian-associated genetic changes in a duck H9N2 influenza virus following adaptation in quail and chickens. PLoS One 3:e3170. doi: 10.1371/journal.pone.0003170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castrucci MR, Kawaoka Y. 1993. Biologic importance of neuraminidase stalk length in influenza A virus. J Virol 67:759–764. doi: 10.1128/JVI.67.2.759-764.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Munier S, Larcher T, Cormier-Aline F, Soubieux D, Su B, Guigand L, Labrosse B, Cherel Y, Quere P, Marc D, Naffakh N. 2010. A genetically engineered waterfowl influenza virus with a deletion in the stalk of the neuraminidase has increased virulence for chickens. J Virol 84:940–952. doi: 10.1128/JVI.01581-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J, Zu Dohna H, Cardona CJ, Miller J, Carpenter TE. 2011. Emergence and genetic variation of neuraminidase stalk deletions in avian influenza viruses. PLoS One 6:e14722. doi: 10.1371/journal.pone.0014722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu C, Eichelberger MC, Compans RW, Air GM. 1995. Influenza type A virus neuraminidase does not play a role in viral entry, replication, assembly, or budding. J Virol 69:1099–1106. doi: 10.1128/JVI.69.2.1099-1106.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu B, Zhou H, Ye D, Kemble G, Jin H. 2005. Improvement of influenza A/Fujian/411/02 (H3N2) virus growth in embryonated chicken eggs by balancing the hemagglutinin and neuraminidase activities, using reverse genetics. J Virol 79:6763–6771. doi: 10.1128/JVI.79.11.6763-6771.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagner R, Wolff T, Herwig A, Pleschka S, Klenk HD. 2000. Interdependence of hemagglutinin glycosylation and neuraminidase as regulators of influenza virus growth: a study by reverse genetics. J Virol 74:6316–6323. doi: 10.1128/jvi.74.14.6316-6323.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo H, Rabouw H, Slomp A, Dai M, van der Vegt F, van Lent JWM, McBride R, Paulson JC, de Groot RJ, van Kuppeveld FJM, de Vries E, de Haan C. 2018. Kinetic analysis of the influenza A virus HA/NA balance reveals contribution of NA to virus-receptor binding and NA-dependent rolling on receptor-containing surfaces. PLoS Pathog 14:e1007233. doi: 10.1371/journal.ppat.1007233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith JM, Haigh J. 2007. The hitch-hiking effect of a favourable gene. Genet Res 89:391–403. doi: 10.1017/S0016672308009579. [DOI] [PubMed] [Google Scholar]
- 39.World Organisation for Animal Health (OIE). 2015. Avian influenza (infection with avian influenza viruses, p 1–24. OIE terrestrial manual 2015. World Organisation for Animal Health (OIE), Paris, France. [Google Scholar]
- 40.Beerens N, Heutink R, Bergervoet SA, Harders F, Bossers A, Koch G. 2017. Multiple Reassorted Viruses as Cause of Highly Pathogenic Avian Influenza A(H5N8) Virus Epidemic, the Netherlands, 2016. Emerg Infect Dis 23:1974–1981. doi: 10.3201/eid2312.171062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Slomka MJ, Pavlidis T, Coward VJ, Voermans J, Koch G, Hanna A, Banks J, Brown IH. 2009. Validated RealTime reverse transcriptase PCR methods for the diagnosis and pathotyping of Eurasian H7 avian influenza viruses. Influenza Other Respir Viruses 3:151–164. doi: 10.1111/j.1750-2659.2009.00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jonges M, Welkers MR, Jeeninga RE, Meijer A, Schneeberger P, Fouchier RA, de Jong MD, Koopmans M. 2014. Emergence of the virulence-associated PB2 E627K substitution in a fatal human case of highly pathogenic avian influenza virus A(H7N7) infection as determined by Illumina ultra-deep sequencing. J Virol 88:1694–1702. doi: 10.1128/JVI.02044-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van den Hoecke S, Verhelst J, Vuylsteke M, Saelens X. 2015. Analysis of the genetic diversity of influenza A viruses using next-generation DNA sequencing. BMC Genomics 16:79. doi: 10.1186/s12864-015-1284-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuker M. 1989. On finding all suboptimal foldings of an RNA molecule. Science 244:48–52. doi: 10.1126/science.2468181. [DOI] [PubMed] [Google Scholar]
- 45.Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Rijk P, Wuyts J, De Wachter R. 2003. RnaViz 2: an improved representation of RNA secondary structure. Bioinformatics 19:299–300. doi: 10.1093/bioinformatics/19.2.299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The majority consensus sequences generated in this study were submitted to the GISAID database (http://gisaid.org) under accession numbers EPI_ISL_391795 to EPI_ISL_391799.