

Abstract
The incidence of human papillomavirus–positive (HPV+) head and neck squamous cell carcinoma (HNSCC) has surpassed that of cervical cancer and is projected to increase rapidly until 2060. The coevolution of HPV with transforming epithelial cells leads to the shutdown of host immune detection. Targeting proximal viral nucleic acid–sensing machinery is an evolutionarily conserved strategy among viruses to enable immune evasion. However, E7 from the dominant HPV subtype 16 in HNSCC shares low homology with HPV18 E7, which was shown to inhibit the STING DNA-sensing pathway. The mechanisms by which HPV16 suppresses STING remain unknown. Recently, we characterized the role of the STING/type I interferon (IFN-I) pathway in maintaining immunogenicity of HNSCC in mouse models. Here we extended those findings into the clinical domain using tissue microarrays and machine learning–enhanced profiling of STING signatures with immune subsets. We additionally showed that HPV16 E7 uses mechanisms distinct from those used by HPV18 E7 to antagonize the STING pathway. We identified NLRX1 as a critical intermediary partner to facilitate HPV16 E7–potentiated STING turnover. The depletion of NLRX1 resulted in significantly improved IFN-I–dependent T cell infiltration profiles and tumor control. Overall, we discovered a unique HPV16 viral strategy to thwart host innate immune detection that can be further exploited to restore cancer immunogenicity.
Keywords: Immunology, Oncology
Keywords: Autophagy, Head and neck cancer, Innate immunity

Introduction
The coevolution of oncogenic viruses with transforming epithelial cells encourages pathogens to develop unique mechanisms that enable immune evasion. The type I interferon (IFN-I) system is an ancient and powerful host first-line antiviral defense strategy, which is conserved from human to bony fish Osteichthyes (1). Thus, viruses have to encode proteins that can block the activation of IFN-I for their replication, intercellular transmission, and/or genome integration. Conceptually, these strategies may directly target the enhanceosome components of IFN-I, such as p65 and IRF3, to directly inhibit IFN-I gene transcription (2, 3). Recently, another major class of viral proteins has surfaced, which alternatively target the more proximal nucleic acid–sensing protein complexes, including the stimulator of interferon genes (STING) complex (4). This strategy adds another layer of viral control of the host innate immune signaling.
Human papillomavirus–associated (HPV-associated) cancers have become an endemic worldwide. HPV is associated with several cancer types, including cervical, anal, penile, and head and neck squamous cell carcinomas. Strikingly, the incidence rate of HPV-associated head and neck squamous cell carcinoma (HNSCC) has surpassed that of cervical cancer and will continue to increase until at least 2060, even after adjustment for the use of HPV vaccines among women (5). Despite the presence of viral epitopes in HPV+ HNSCCs, these tumors exhibit less T cell clonal expansion than HPV– tumors (6). A 2-year long-term follow-up of a randomized phase III trial (CheckMate 141) showed that HNSCC patients can benefit from anti–PD-1 therapy regardless of the HPV status. Notably, the hazard ratios (HRs) were almost identical between the HPV– and HPV+ groups, despite the generally more favorable response profiles to chemotherapy among patients with HPV+ tumors (7, 8). Similarly, pembrolizumab resulted in little survival improvement over standard of care in patients with p16+ tumors enrolled in another randomized phase III trial, KEYNOTE-040 (HR = 0.97; 95% confidence interval, 0.63–1.49) (9). Thus, immunotherapy-mediated survival improvement appears to be lower than expected for HPV+ tumors (10), despite their dense immune infiltrates and association with viral epitopes. Not surprisingly, HPV could encode IFN-I–inhibiting proteins to dampen innate immune sensing. For example, the HPV subtype 18 (HPV18) E7 oncoprotein was shown to bind with STING and inhibit its function (11). However, HPV18 is rare in HNSCCs and accounts for fewer than 3% of HPV+ cases, whereas over 90% of HPV+ HNSCC cancers are instead linked to HPV16 (12). More importantly, the E7 proteins encoded by HPV16 and HPV18 share a low degree of homology, suggesting potential functional divergence. How HPV16 inhibits the IFN-I systems remains unknown.
This presents a significant knowledge gap, as the manipulation of the IFN-I system can yield potentially transformative immune-priming outcomes. The current conceptual framework surrounding antitumor immunity centers on the relationship between cancer mutational load and the number of tumor neoantigen–targeted cytotoxic T lymphocytes (CTLs); it remains unexplained why most HNSCCs are hypoimmunogenic despite the presence of high mutation burdens and the presentation of HPV-associated neoantigens (13). To explain this dichotomy, we recently demonstrated that HNSCC cells evade immune surveillance through suppression of the STING/IFN-I pathway, a key adaptive resistance mechanism that limits effector T cell expansion (14). IFN-I is produced by both cancer cells and antigen-presenting cells (APCs). IFN-I and its downstream chemokines create a strongly Th1/cytotoxic T cell type 1 (Tc1)-skewed milieu to promote APC tumor trafficking and cross-presentation to CD8+ CTLs (15–17). A second messenger and the physiological agonist of STING, cGAMP, has been exploited in different tumor models to improve CTL expansion (18–23). To prevent the expanded CTLs from rapidly entering into an exhaustion state, at least 3 ongoing clinical trials are assessing a combination strategy of STING agonist plus immune checkpoint blockade (NCT02675439, NCT03172936, and NCT03010176, ClinicalTrials.gov). Because of the highly polar molecular properties of cGAMP, which may prevent it from entering the cytoplasm effectively to maximally activate STING, we engineered unique nanoparticles and slow-release delivery systems to improve the pharmacodynamics of STING agonists. These efforts improved the delivery of cGAMP in vivo and sensitized hypoimmunogenic HNSCCs to immune checkpoint blockade (14, 24).
STING is ubiquitously expressed by epithelial cells and immune cells. Thus, an in-depth examination of the mechanisms underpinning HPV16-mediated cancer-specific suppression of STING could reveal novel cancer immune escape strategies and identify potential intervention points to prime cold epithelial malignancies for checkpoint inhibitors. In this study, we sought to comprehensively annotate the relationship between STING/IFN-I signatures and tumor-infiltrating lymphocyte (TIL) subsets in clinical HNSCC specimens and reveal the mechanisms by which HPV16 evades STING-induced IFN-I activation.
Results
Deconvolution of the immune landscape of human HNSCCs.
To better quantify the TIL subsets present within the tumor microenvironment (TME), we engineered a novel and robust machine learning tool, Fast and Robust Deconvolution of Tumor Infiltrating Lymphocyte from Expression Profiles (FARDEEP), which exhibits less susceptibility to data outliers that are universally present in whole-tissue RNA-Seq data sets (25). We quantitated TILs in 520 HNSCC specimens from The Cancer Genome Atlas (TCGA) database, including 97 HPV+ and 420 HPV– tumors that involved oral cavity, oropharynx, and larynx. We found an extensive infiltration of regulatory T cells, resting dendritic cells, and M2-like macrophages in the TME. The infiltration of CD8+ T cells, γδ T cells, activated memory T cells (Tmem), M1-like macrophages, and activated natural killer (NK) cells, which are essential to launching a tumor-specific immunologic attack, was highly variable across the specimens (Figure 1A). To thoroughly characterize the relationship between STING signatures and TIL distribution, we performed a marginal correlation analysis. We noted a robust positive correlation of STING signatures including STING, MX1, CXCL9, CXCL10, ISG15, and ISG54 with intratumoral infiltration of M1-like macrophages, γδ T cells, Tmem, CD8+ T cells, and NK cells. STING signaling was inversely correlated with the presence of naive CD4+ T cells, follicular helper T cells, plasma cells, and neutrophils (Figure 1B), the last of which were found to be a significant prognosticator for a poor outcome in a pan-cancer study (26).
Figure 1. STING correlates with enhanced infiltration of Th1/Tc1-skewed immune subsets in HNSCC and improved patient survival.

(A) Using a machine learning pipeline, we deconvolved tumor-infiltrating lymphocyte (TIL) compositions of 520 human HNSCC specimens in the TCGA database. Each color represents an immune cell subset, and each vertical line represents 1 specimen. (B) The relationship between expression levels of IFN-I signatures and the percentages of TIL subsets was analyzed by Spearman’s rank-order correlation. Positive values indicate positive associations, and negative values indicate inverse associations. (C and D) Kaplan-Meier overall survival analysis was performed based on STING expression in TCGA, presented stratified by age or across all age groups. (E) A tissue microarray (TMA) consisting of 297 HNSCCs with 3 cores for each specimen was stained with STING. Tumor parenchyma and tumor microenvironment (TME) were defined and scored independently using Aperio ImageScope. STING staining scores were available for 264 HNSCC patients. Kaplan-Meier survival curves were compared using a log-rank test. *P < 0.05; **P < 0.01. (F) Representative IHC staining for STING is shown (scale bar: 200 μm).
STING is a favorable prognosticator of HNSCCs.
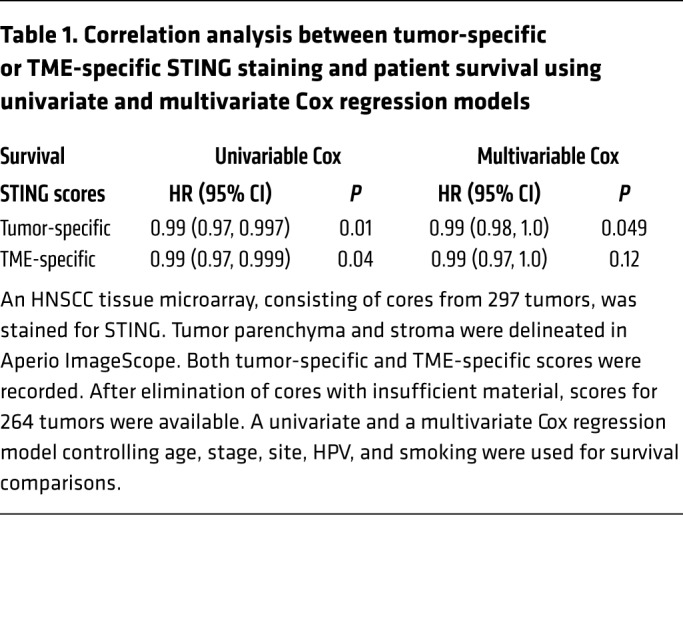
Next, we sought to establish the clinicopathologic correlation of STING expression in HNSCCs. We examined the survival data available from the TCGA, and found that high STING expression levels were significantly associated with improved prognosis in younger patients (P = 0.005) (Figure 1C). When the Kaplan-Meier analysis was extended to all patients (n = 520), STING remained a trending positive prognosticator (P = 0.06) (Figure 1D). STING is broadly expressed in tumor cells and a variety of cell types in the TME. RNA-Seq data cannot distinguish the contribution of STING levels from different sources and may not be entirely concordant with protein expression. Thus, we constructed a tissue microarray using 297 previously nontreated HNSCC specimens, 3 tumor cores for each specimen with a total of 891 cores, representing tumors of the larynx, hypopharynx, oral cavity, oropharynx, and other sites. The median follow-up at the time of analysis was 60.1 months. STING staining in HNSCC parenchyma and TME was independently defined and quantitated using Aperio ImageScope as we described previously (14, 27). Upon removal of cores with insufficient tumor tissue, STING staining scores were available from 264 HNSCC specimens. This cohort contained 32% HPV+ and 60% HPV– HNSCCs, and the demographic details are summarized in Supplemental Table 1 (supplemental material available online with this article; https://doi.org/10.1172/JCI129497DS1). In agreement with the in silico analysis of TCGA patients, we found that higher STING IHC staining scores were significantly correlated with improved patient survival (Figure 1, E and F; P = 0.027). Univariable Cox regression analysis found that higher STING scores in HNSCC parenchyma (P = 0.01) and in TME (P = 0.04) were both positively correlated with improved patient survival. Then, we built a multivariate Cox regression model controlling age, stage, site, HPV, and smoking. We found that STING expression in the tumor parenchyma remained a favorable prognosticator (P = 0.049), while the STING staining scores of the TME were no longer significant (P = 0.12), suggesting the critical importance of STING protein levels in HNSCC cancer cells in overall patient outcome (Table 1).
Table 1. Correlation analysis between tumor-specific or TME-specific STING staining and patient survival using univariate and multivariate Cox regression models.
As the average age of patients with HPV+ tumors is younger, our observation that the expression levels of STING were inversely correlated with patient age (Supplemental Figure 1A) prompted us to investigate whether this was due to an HPV-STING interaction term. Indeed, we identified a significant interaction term between HPV and the protein expression levels of STING (P = 0.046). We also assessed the HPV mRNA levels of STING interaction term using the TCGA database. Interestingly, an interaction was not identified (the corresponding P values for the population with an age less than 60 and all patients were 0.81 and 0.46, respectively), suggesting that HPV is more relevant to the posttranslational regulation of STING expression. After stratification of the tumors by HPV status, STING was strongly associated with patient survival in the HPV+ group but not in the HPV– group using multivariate Cox models (Supplemental Figure 1B and Supplemental Table 2).
HPV16 E7 suppresses STING signaling in HNSCC cells.
The clinical findings prompted us to further investigate the interaction between HPV and STING signaling. HPV likely evolves anti–IFN-I strategies to make the host cells more permissive to viral replication and integration. Indeed, HPV18 E7 was previously shown to interact with and inhibit STING (11). However, only 3% of HPV+ HNSCCs are positive for HPV18, while more than 90% of HPV+ HNSCC is positive for HPV16 (5, 28). More importantly, upon a sequence homology analysis between E7 proteins encoded by HPV16 and HPV18, we found only 40% homology between the two E7 proteins (Supplemental Figure 2), suggestive of molecular and functional divergence. Notably, when we transfected the physiological STING agonist cGAMP into HPV16+ 93VU147T and UMSCC47 cells, no IFN-I signaling induction was observed, in contrast to cGAMP-induced IFN-I signaling in HPV– UMSCC49 cells (Supplemental Figure 3, A–C). To determine the specific role of HPV16 E7 in the regulation of the STING/IFN-I pathway, we first examined whether enhanced expression of HPV16 E7 protein modulates IFN-I signaling in 93VU147T and UMSCC47 cells, which contain low to intermediate genomic copy numbers of HPV16 (29). Upon analysis of the expression of IFNB1 and 2 essential downstream IFN-I target genes, CXCL10 and ISG54, we found that HPV16 E7 potently suppressed STING-induced immune activation (Figure 2, A–F). In agreement, HPV16 E7 similarly inhibited cytoplasmic poly(dA:dT)-induced IFN-I signaling (Supplemental Figure 4, A and B). To minimize the potential contribution of endogenous HPV16 E7 in these cell lines, we repeated these experiments in an HPV– HNSCC cell line, FaDu. Consistent with our previous results, HPV16 E7 suppressed STING-mediated immune activation (Figure 2, G–I). To confirm the findings with IFNB1 transcripts, we performed ELISA to quantitate the protein levels of IFN-β in the supernatant. STING induced the production of IFN-β by 93VU147T, UMSCC47, and FaDu cells; HPV16 E7 largely abolished STING-mediated immune induction (Figure 2, J–L).
Figure 2. HPV16 E7 inhibits STING-induced transcription of IFN-I target genes in HNSCC cells.

(A–I) HPV+ 93VU147T (A–C), HPV+ UMSCC47 (D–F), and HPV– FaDu (G–I) cells were transfected with 1.5 μg/mL STING expression plasmid for 24 hours with or without transfection of 1.5 μg/mL HPV16 E7 plasmid. The mRNA levels of IFNB1, CXCL10, and ISG54 were determined by qPCR. Values displayed indicate the mean ± SEM of 3 biological replicates. The comparisons were made by 2-way ANOVA with Šidák’s multiple-comparisons test (**P < 0.01, ***P < 0.001, ****P < 0.0001). Experiments were performed twice. NT, no treatment. (J–L) 93VU147T (J), UMSCC47 (K), and FaDu (L) cells were transfected with 1.5 μg/mL STING expression plasmid for 24 hours in the absence or presence of 1.5 μg/mL HPV16 E7 plasmid in 3 biological replicate wells. The levels of secreted IFN-β were quantified using ELISA. The comparisons were made by 2-way ANOVA followed by Šidák’s post-test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). Experiments were performed twice.
HPV16 E7 suppresses STING through an interaction-independent mechanism.
Given the low sequence homology between the HPV16 E7 and HPV18 E7 oncoproteins, we speculated that HPV16 E7 could suppress STING signaling through a distinct mechanism from HPV18 E7. To determine whether HPV16 E7 can directly associate with STING, like HPV18 E7, we performed a semi-endogenous coimmunoprecipitation (co-IP) in 93VU147T cells expressing HA-tagged STING. We were unable to detect an HPV16 E7–STING association even in the presence of STING overexpression. The expression of HPV16 E7 was confirmed in the input samples (Figure 3A). A mitochondrial member of the NOD-like receptor (NLR) family, NLRX1, recently shown to directly bind STING (30), was used as a positive control for the quality of co-IP, with NLRX1-deficient cells serving as an additional control.
Figure 3. HPV16 E7 attenuates STING-induced innate immune signaling.

(A) 93VU147T cells were transduced with empty vector (EV) control or shNLRX1-expressing lentiviruses to produce stable control and NLRX1-deficient cell lines, which were then transfected with 1.0 μg/mL HA-tagged STING plasmid and incubated for 16 hours. STING protein complexes were immunoprecipitated using anti-HA affinity matrix followed by immunoblotting for the indicated potential binding partners. Experiments were performed 3 times, and representative blots are shown. (B) The protein lysates of HPV+ 93VU147T, UDSCC2, UMSCC47, and SCC90 as well as HPV– FaDu and PCI-13 cells were harvested on ice and separated by SDS-PAGE. Endogenous expression levels of HPV16 E7 and STING were then detected with respective antibodies. (C–E) 93VU147T, UMSCC47, and FaDu cells were transfected with 1.0 μg/mL STING plasmid and incubated for 24 hours with or without introduction of 1.5 μg/mL HPV16 E7 plasmid. Cell lysates were immunoblotted with HPV16 E7, STING, and markers for IFN-I activation. Densitometry analysis was performed using ImageJ and is shown in the lower panels. Comparisons between 2 groups were made by 2-tailed unpaired t test, while comparisons between multiple groups were conducted by 1-way ANOVA test followed by Tukey’s multiple-comparisons test. Results displayed represent the mean ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). Each immunoblot represents 3 biological repeats, and representative blotting results are shown.
Since HPV16 E7 does not associate with STING, we hypothesized that HPV16 E7 may target STING by interfering with its turnover. Thus, we next investigated whether the levels of HPV16 E7 expression are inversely correlated with those of STING in HNSCC cells. To this end, we screened a panel of HNSCC cells that included 4 HPV+ and 2 HPV– cell lines. We found UMSCC47 to have a low level of expression of HPV16 E7, 93VU147T cells to have an intermediate level of HPV16 E7, and UDSCC2 and SCC90 to have the highest levels of endogenous HPV16 E7 (Figure 3B). These findings are concordant with previous reports of HPV16 genome copy numbers in these cell lines (29). Interestingly, we observed an inverse correlation between HPV16 E7 and STING protein levels, wherein high levels of HPV16 E7 were associated with decreased levels of STING protein (Figure 3B). To validate whether HPV16 E7 inhibits downstream STING signaling, we analyzed whether STING-dependent phosphorylation of TANK-binding kinase 1 (TBK1) was altered by varying expression levels of HPV16 E7. We observed a significant decrease in STING levels in 93VU147T, UMSCC47, and FaDu cells (Figure 3, C–E) following exogenous expression of HPV16 E7. Additionally, we observed a decrease in phosphorylation of TBK1 (phospho-S172, normalized to total TBK1) with increased levels of HPV16 E7 (Figure 3, C–E). These results substantiate HPV16 E7 as an important suppressor of STING and STING-dependent IFN-I responses.
HPV16 E7 promotes autophagy-dependent degradation of STING.
To understand how HPV16 promotes the turnover of STING protein, we examined existing literature for STING regulation. Autophagy, as a central process maintaining cellular homeostasis, is also frequently employed to control overzealous inflammation (31). In fact, innate immune signaling complexes, including STING, are found to be cargos for autophagosomes (14, 32–36). Thus, we posited that HPV16 E7 induces STING degradation via an autophagy-dependent mechanism. To this end, we analyzed LC3B-II, a marker of autophagy induction, in 93VU147T, UMSCC47, and FaDu cells. Consistently, we observed that the expression of HPV16 E7 led to decreased levels of STING protein and a marked increase in LC3B-II (Figure 4, A–C). Further, we repeated these experiments in the presence of bafilomycin A1 (BafA1), a pharmacologic inhibitor of autophagy. We observed that BafA1 could partially reverse the HPV16 E7–induced loss of STING protein in 93VU147T cells (Figure 4D), and that inhibition of autophagy completely abolished HPV16 E7–mediated degradation of STING in UMSCC47 (Figure 4E) and FaDu (Figure 4F) cells. These results suggest that HPV16 E7 accelerates STING turnover through an autophagy-dependent mechanism. To confirm that HPV16 E7–mediated regulation of STING was a posttranslational event, we further assessed whether HPV16 E7 decreased the mRNA levels of STING. We found that HPV16 E7 had only a modest effect on the mRNA transcription of STING in the 3 cell lines (Supplemental Figure 5A).
Figure 4. HPV16 E7 promotes autophagy-dependent degradation of STING.

(A–C) 93VU147T, UMSCC47, and FaDu cells were transfected with 1.5 μg/mL HPV16 E7 for 24 hours, 93VU147T alone was simultaneously transfected with STING, and immunoblotting was performed against HPV16 E7, STING, and LC3B. Each immunoblot represents 3 biological repeats, and representative blotting results are displayed. (D–F) 93VU147T, UMSCC47, and FaDu cells were transfected with 1.5 μg/mL HPV16 E7 and incubated for 24 hours. 93VU147T cells were simultaneously transfected with STING. Half of the groups were then treated with 200 nM bafilomycin A1 (BafA1) and incubated for 8 hours. Cell lysates were immunoblotted for HPV16 E7, STING, and LC3B. Representative blots are shown and represent 3 independent repeats. Densitometric quantitation of STING/β-actin was performed using ImageJ and is shown in the lower panels. Comparisons between multiple groups were determined by 1-way ANOVA test followed by Tukey’s multiple-comparisons test. Results represent mean ± SEM (****P < 0.0001).
Genetic deletion of HPV16 E7 restores STING-mediated IFN-I induction.
To substantiate these findings, we next generated HPV16 E7–/– 93VU147T and UMSCC47 cell lines using CRISPR/Cas9 lentiviruses containing an sgRNA targeting HPV16 E7. Immunoblotting confirmed the loss of E7 protein in both knockout cell lines (Figure 5, A and B). Notably, loss of HPV16 E7 led to reduced autophagy, a striking restoration of STING protein levels, and elevated levels of phospho-TBK1 in 93VU147T and UMSCC47 cells under basal and induced conditions (Figure 5, A and B). We next performed quantitative PCR (qPCR) to examine the expression of IFN-I signature genes in these cells. We observed a significant increase in both basal and cGAMP-stimulated upregulation of IFNB1, MX1, ISG15, and ISG54 in HPV16 E7–/– HNSCC cells compared with E7+/+ controls (Figure 5, C and D). To validate the transcription profile findings at protein levels, we performed ELISA to detect secreted IFN-β in the supernatant. Transfection of cGAMP could not induce the production of IFN-β in wild-type 93VU147T and UMSCC47 cells; and the production of IFN-β was restored upon deletion of E7 (Figure 5E). Last but not least, to confirm whether autophagy was reduced by removal of HPV16 E7, as a mechanism of stabilizing STING protein levels, control or HPV16 E7–/– UMSCC47 cells were transfected with pEGFP-LC3B and imaged by confocal microscopy 48 hours later. In agreement with LC3B immunoblotting, we observed a significant loss of LC3B-GFP+ puncta (Figure 5F) upon deletion of E7, supporting the notion that loss of HPV16 E7 leads to a corresponding decrease in autophagy.
Figure 5. Deletion of HPV16 E7 restores IFN-I signaling along with reduced autophagic activity.

(A and B) 93VU147T and UMSCC47 cells were transduced with lentivirus of CRISPR/Cas9 targeting E7, and the EV was considered as control. The established cell lines were transfected with STING agonist (cGAMP) or mock for 16 hours, and cell lysates were subjected to immunoblotting for HPV16 E7, STING, LC3B, phospho-TBK1, and TBK1. Representative blots of 2 repeats are presented. (C and D) 93VU147T and UMSCC47 cells with or without the expression of E7 were transfected with cGAMP for 16 hours, and total RNA was isolated. qPCR was then performed to quantitate the mRNA levels of indicated IFN-I signature genes. Values represent mean ± SEM of 3 biological replicates. Comparisons were made by 2-way ANOVA followed by Šidák’s post-test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). Experiments were performed 3 times. (E) 93VU147T and UMSCC47 cells with or without the expression of HPV16 E7 were transfected with cGAMP for 16 hours, and the protein levels of IFN-β from supernatant were determined by ELISA. Comparisons were made by 2-way ANOVA with Šidák’s post-test (***P < 0.001, ****P < 0.0001). Experiments were performed twice. (F) Left panel: Laser confocal analysis was conducted in EV or HPV16 E7–/– UMSCC47 cells, which were transfected with pEGFP-LC3B for 48 hours before the images were captured. Scale bars: 10 μm. Right panel: Quantitation of EGFP-LC3 puncta in each cell section of both groups was conducted. Comparisons between the 2 sets were completed using an unpaired 2-tailed t test. Values represent mean ± SEM (****P < 0.0001). n = 20 cell sections from 2 repeats.
HPV16 E7 specifically interacts with NLRX1.
Our previous studies discovered an NLRX1-centered molecular complex that potentiates autophagosome formation (27, 33). To determine whether HPV16 E7 intersects with this pathway as a mechanism promoting STING turnover, we first performed endogenous co-IP experiments (Figure 6, A and B). After pulling down endogenous HPV16 E7–interacting protein complexes from 93VU147T and SCC90 cell lysates, we identified a specific interaction with endogenous NLRX1. Three types of controls were included: (a) isotype control antibody was used for the mock pull-down; (b) starting lysates from the isotype control group and HPV16 E7 antibody group were identical; (c) protein specificity controls were also included. HPV16 E7 does not interact with 3 abundant proteins that are localized in mitochondria (VDAC), cytoplasm (p63), or endoplasmic reticulum (STING). As a previous study suggests that HPV18 E7 interacts with STING as a mechanism of inhibiting STING signaling (11), we sought to determine whether HPV16 E7 engagement with NLRX1-mediated autophagic machinery is specific to HPV16. We transduced HPV– UMSCC49 cells with HPV18 E7, and indeed identified an interaction between HPV18 E7 and STING. However, HPV18 E7 did not interact with NLRX1 and other aforementioned control proteins under our stringent buffer conditions (Figure 6C). Thus, here we show that HPV16 and HPV18 E7 proteins use distinct mechanisms to inhibit STING-mediated immune activation. Next, we performed a confocal imaging colocalization study to further confirm the interaction between HPV16 E7 and NLRX1. We stained 93VU147T cells with MitoTracker, NLRX1, and HPV16 E7 (Figure 6D). As validation of the staining quality, we show that NLRX1 colocalized with mitochondria, as we previously reported (37). Interestingly, HPV16 E7 colocalized with NLRX1 and mitochondria, in agreement with our findings in Figure 6, A and B.
Figure 6. HPV16 E7 specifically interacts with NLRX1.

(A and B) 93VU147T and SCC90 cells were lysed, precleared, and incubated with an isotype control antibody and anti-HPV16 E7. Immunoprecipitation was performed using Protein A/G UltraLink Resin, and immunoprecipitated protein complexes were washed before SDS-PAGE. Immunoblotting of NLRX1 and specificity control proteins was carried out. (C) The whole-cell lysates of HPV18 E7–expressing UMSCC49 cells were precleared and incubated with IgG2a isotype control or anti–HPV18 E7, followed by incubation with Protein A/G UltraLink Resin for 2 hours at room temperature. Immunoprecipitated protein complexes were washed and subjected to SDS-PAGE. Immunoblotting of STING and specificity control proteins was performed. Experiments were performed 3 times, and representative results are shown. (D) 93VU147T cells were stained with MitoTracker, followed by fixation, permeabilization, and staining with NLRX1 and HPV16 E7. Nuclei were counterstained with Hoechst. Representative images and colocalization overlay are shown (scale bars: 10 μm). Experiments were performed twice.
NLRX1 inhibits STING/IFN-I signaling in HPV16+ HNSCC cells.
Despite the well-characterized core proteins involved in autophagosome membrane initiation and elongation, the regulatory mechanisms are complex and cell type–dependent. Recently, we identified NLRX1 as an important contributor to autophagy in HNSCC (27), functioning as a protein complex scaffold recruiting autophagy-promoting molecules including ATG12–ATG5 and BECN1 (27, 33). As HPV16 E7 specifically interacts with NLRX1, we sought to determine whether HPV16 E7–induced STING turnover in cancer cells uses an NLRX1-dependent mechanism. We first examined the expression of NLRX1 in 4 HPV+ HNSCC cell lines and found that 3 of 4 (93VU147T, UMSCC47, and SCC90) had high endogenous levels of NLRX1 protein (Figure 7A). To determine whether NLRX1 contributes to STING degradation in these HPV+ cell lines, we generated stable NLRX1-deficient 93VU147T, UMSCC47, and SCC90 cell lines through shRNA transduction (shNLRX1; or empty vector [EV] as a control). As a result of low endogenous levels of STING in 93VU147T and SCC90 cells (Figure 3A), STING was expressed in EV and shNLRX1 cells 24 hours before harvest. Compared with EV controls, shNLRX1 cells showed reduced autophagy and substantially increased protein levels of STING (Figure 7, B–D). Consistently, a defect in NLRX1 resulted in a significant increase in the expression of the IFN-I signature genes IFNB1, ISG15, CXCL9, and CXCL10 (Figure 7, E and F), an effect that was consistent across the HNSCC cell lines analyzed. As a control, we found that NLRX1 did not affect the mRNA levels of STING (Supplemental Figure 5B), suggesting that NLRX1-mediated inhibition of STING signaling occurs at post-transcriptional levels. We also analyzed the phosphorylation of TBK1 and p65 to determine whether downstream STING signaling was restored in shNLRX1 93VU147T and SCC90 cells. In agreement, we observed increased ratios of phospho-TBK1 to TBK1 and phospho-p65 to p65 in shNLRX1 93VU147T and SCC90 cells compared with EV controls (Figure 7G).
Figure 7. NLRX1 potentiates autophagy-mediated inhibition of STING/IFN-I signaling in HPV16+ HNSCC cells.

(A) Cell lysates of 4 HPV+ HNSCC cell lines were immunoblotted for NLRX1, HPV16 E7, STING, and β-actin. (B–D) UMSCC47, 93VU147T, and SCC90 cells were transduced with lentiviruses carrying an empty vector (EV) control construct or a construct expressing NLRX1-targeted shRNA. Stable cell lines were generated through puromycin selection. 1.0 μg/mL EV or 1.0 μg/mL STING plasmid was then introduced into 93VU147T and SCC90 cells and incubated for 24 hours. Cell lysates were separated by SDS-PAGE and immunoblotted for the indicated proteins. Immunoblots were performed twice, and representative blots are shown. (E and F) EV control or NLRX1-deficient 93VU147T and SCC90 cells were stimulated by 1.0 μg/mL STING for 16 hours, and qPCR was performed to determine the mRNA levels of IFNB1, ISG15, CXCL9, and CXCL10. Values represent mean ± SEM of 3 biological replicates. The comparisons were made by 2-way ANOVA with Šidák’s multiple-comparisons test (**P < 0.01, ***P < 0.001, ****P < 0.0001). (G) Control and shNLRX1 93VU147T and SCC90 cells were transfected with 1.0 μg/mL STING plasmid and incubated for 16 hours. Cell lysates were then subjected to immunoblotting for markers of IFN-I activation. Immunoblots represent 2 independent repeats.
NLRX1 suppresses STING-mediated immune detection of E7-expressing tumors in vivo.
Recently, we characterized a new HPV16 E6/E7–expressing HNSCC mouse model, MOC2-E6/E7, which is syngeneic to C57BL/6 (14, 24, 38). These tumors grow aggressively and are completely resistant to immune checkpoint blockade therapy (14, 39). To determine whether NLRX1 inhibits cancer immunogenicity by targeting the STING/IFN-I pathway, we generated stable NLRX1-deficient MOC2-E6/E7 cells using lentiviruses targeting NLRX1 (or empty vector lentivirus as a control, EV). Wild-type and NLRX1-deficient tumor cells were transfected with a STING agonist, poly(dA:dT). Then, we analyzed the expression levels of IFN-I signatures Ifnb1, Mx1, Isg15, and Isg54 16 hours after induction (Figure 8A). In agreement with the results obtained in the human cell lines, we observed that NLRX1-deficient MOC2-E6/E7 cells exhibited significantly enhanced STING activation. To further support these results, we expressed murine STING in control and NLRX1-deficient MOC2-E6/E7 cells and confirmed that NLRX1 deficiency resulted in enhanced STING signaling, evidenced by increased phospho-TBK1 (Figure 8B).
Figure 8. NLRX1 in cancer cells inhibits STING signaling in vivo and excludes functional effectors from TME.

(A) Control and shNLRX1 MOC2-E6/E7 cells were stimulated by 1.0 μg/mL poly(dA:dT) for 16 hours, and qPCR was performed to quantitate the mRNA levels of indicated IFN-I signature genes. Experiments were performed 3 times. Comparisons between 2 groups were made using a 2-tailed unpaired t test (**P < 0.01, ****P < 0.0001). (B) Control and NLRX1-deficient MOC2-E6/E7 cells were transfected with 1.0 μg/mL expression plasmid encoding murine STING and incubated for 16 hours. Cell lysates were immunoblotted against the indicated markers. Immunoblotting results represent 2 independent repeats. (C) The proliferation of EV control and shNLRX1 MOC2-E6/E7 cells was measured by an alamarBlue assay. Each group included 5 replicate wells. Experiments were performed twice. (D) One million EV control or NLRX1-deficient MOC2-E6/E7 cells were implanted subcutaneously in the right flank of C57BL/6 hosts. Tumor measurements were performed every 2–3 days. Tumor burden was compared using the generalized estimating equations model (n = 8 in each group; *P < 0.05). In vivo experiments were performed 3 times with n = 19 total in each group. A representative set is shown. (E) Total RNA was isolated from 1 representative set of tumors and subjected to qPCR. (F) After harvesting of tumors, TILs of 1 representative set were isolated and analyzed by flow cytometry (n = 8 in control group, n = 4 in shNLRX1 group due to tumor rejection). (G) Lymphocytes were isolated from draining lymph nodes of 1 representative set and assessed by flow cytometry (n = 5 in each group). Comparisons between 2 groups from E–G were made using a 2-tailed unpaired t test (*P < 0.05, **P < 0.01). Quantifications indicate the mean ± SEM. Results represent 3 independent experiments.
Depletion of NLRX1 had minimal effect on the rates of cell proliferation (Figure 8C). However, NLRX1-deficient tumors exhibited a significant reduction in tumor burden in wild-type C57BL/6 hosts (Figure 8D; P = 0.02), with 14 of 19 NLRX1-deficient tumors completely rejected. Whole tumors were homogenized and qPCR analysis performed. We found that NLRX1-deficient tumors showed significantly elevated expression levels of Tnf, Il6, and Ifnb1, key markers for STING-mediated downstream effector activation (Figure 8E and ref. 40). IL-10 was identified as a prominent immunosuppressive factor in MOC2-derived tumors (41); thus, we additionally assessed the transcription levels of IL-10. We found that IL-10 was trending decreased in the NLRX1-deficient tumors (P = 0.08) (Supplemental Figure 6). Consistently, we examined the histology of control and NLRX1-deficient tumors and found that more inflammatory infiltrates were present in the tumor-stroma interface in the NLRX1-deficient tumors than in control specimens (Supplemental Figure 7A). We stained the specimens with anti-CD8 and noted an increase in CD8+ immune cells in the TME of NLRX1-deficient tumors (Supplemental Figure 7B). To better characterize the immune infiltrates, TILs and immune cells from draining lymph nodes were purified from mice with EV– or shNLRX1–MOC2-E6/E7 tumors via a Ficoll-Paque gradient and analyzed by flow cytometry. We found that NLRX1-deficient tumors harbored significantly reduced CD3+CD4+Tim3+ and CD3+CD8+Tim3+ T cells (Figure 8F), which are functionally exhausted in HNSCC specimens (42). Mice bearing NLRX1-deficient tumors also showed better CTL expansion in the draining lymph nodes (Figure 8G).
NLRX1-mediated inhibition of antitumor immunity is IFN-I–dependent.
As autophagy has pleiotropic effects on a variety of pathways, we next sought to determine whether the shNLRX1-mediated tumor rejection we observed in vivo was indeed T cell– and IFN-I–dependent. To this end, we first depleted CD8+ T cells using a monoclonal antibody; and depletion was confirmed using flow cytometry (Supplemental Figure 8). We found that depletion of CD8+ T cells largely rescued NLRX1 deficiency–mediated tumor rejection, although NLRX1-deficient tumors were slightly smaller, suggesting the involvement of other TIL subsets in addition to CTL (Figure 9A). No difference was observed for a panel of effector cell markers (Figure 9B). To more thoroughly test the phenotype dependence on adaptive immune response, we established EV– or shNLRX1–MOC2-E6/E7 tumors in B cell– and T cell–deficient Rag1–/– hosts. In contrast to the results in wild-type C57BL/6 hosts (Figure 8D), we observed no difference in tumor growth between control and NLRX1-deficient tumors (Figure 9C), as well as comparable levels of transcription of STING signature genes from whole-tumor homogenates (Figure 9D). Then, we repeated these experiments in Ifnar1–/– hosts, which contain a deletion of the IFN-I receptor Ifnar1 and show abolished IFN-I signaling. Similarly, we did not observe any differences in tumor volumes between EV– and shNLRX1–MOC2-E6/E7 tumors (Figure 9E), or in the transcription of STING signature genes within the TME (Figure 9F).
Figure 9. NLRX1-potentiated tumor immune escape is IFN-I–dependent.

(A) C57BL/6 hosts were given 0.5 mg of anti-CD8 or PBS intraperitoneally daily for 3 days before the tumor implantation and then twice per week for 2 weeks. The overall tumor burden was compared using the generalized estimating equations model (n = 7 in each group; *P < 0.05). (B) Tumors were harvested and total RNA isolated for qPCR detection of the indicated STING signature genes. Values represent mean ± SEM. Comparisons between groups were assessed using an unpaired t test. (C) One million EV control or shNLRX1 MOC2-E6/E7 cells were inoculated subcutaneously in the right flank of Rag1–/– mice. Tumors were monitored and compared as described above (n = 6 in each group). (D) After euthanasia, tumors were harvested and total RNA was isolated. qPCR was conducted to quantify the mRNA levels of indicated genes. Values represent mean ± SEM. Comparisons between groups were assessed using an unpaired t test. (E) One million EV control or shNLRX1 MOC2-E6/E7 cells were inoculated subcutaneously in the right flank of Ifnar1–/– mice (n = 5 in control group, n = 6 in shNLRX1 group). Tumor growth was monitored and compared as described above. Experiments were performed twice, and 1 representative set is shown. (F) After euthanasia, all tumors were harvested and total RNA isolated for qPCR analysis. Values represent mean ± SEM. Comparisons between groups were assessed using an unpaired t test.
In order to further characterize the relationship between NLRX1 and TIL distribution in HNSCCs, we performed a correlation analysis of NLRX1 expression and the quantity of different TIL subsets in 78 HPV16+ HNSCC specimens in the TCGA database. We found that the expression levels of NLRX1 were significantly inversely correlated with the infiltration of CD8+ T cells (Spearman ρ = –0.42, P = 0.00018), CD4+ activated memory T cells (Spearman ρ = –0.40, P = 0.00039), and activated NK cells (Spearman ρ = –0.27, P = 0.022). A negative correlation with M1-like macrophages was also observed with a marginal P value (Spearman ρ = –0.19, P = 0.099) (Figure 10A). As proof of high-fidelity rendering of the immune infiltrate, we also performed correlation analysis between the expression levels of STING and different TIL subsets. We show that STING is significantly positively correlated with CD8+ T cells (Spearman ρ = 0.24, P = 0.040), CD4+ memory T cells (Spearman ρ = 0.36, P = 0.0016), γδ T cells (Spearman ρ=0.41, P = 0.00031), and M1-like macrophages (Spearman ρ = 0.33, P = 0.0039) (Figure 10B). Interestingly, these correlations are more prominent in HPV+ HNSCC specimens compared with HPV– HNSCC specimens (Supplemental Figure 9), possibly owing to the role of NLRX1 in potentiating HPV16 E7–mediated STING suppression.
Figure 10. NLRX1 is negatively correlated with antitumor immune subsets in HPV+ HNSCC specimens.

Spearman’s correlation analysis was performed to assess the relationship between the expression levels of NLRX1 (A) or STING (B) among 78 HPV16+ HNSCC specimens in the TCGA database and the frequencies of major TIL subsets; Spearman’s correlation coefficients and P values are indicated in each panel. Each dot represents 1 HPV16+ HNSCC specimen. This figure relates to Supplemental Figure 9.
Discussion
Throughout the course of coevolution, the host has developed a battery of germline-encoded pattern recognition receptors (PRRs) to detect double-stranded DNA (dsDNA) viruses (43). Based on sequence homology and subcellular localization, the nucleic acid–sensing PRRs are classified into 4 major families: Toll-like receptors (TLRs), NLRs (also known as nucleotide-binding domain and leucine-rich repeats containing proteins), cGAS-STING cytoplasmic sensors, and AIM2 inflammasome. An array of DNA viral proteins targets PRR members to establish persistent infections. Conceptually, 3 viral strategies are well established to disrupt host DNA-sensing pathways. (a) The DNA viruses may encode proteins to occupy host DNA sensor binding pockets to sequester viral genome from PRRs (44). (b) Viral proteins may directly interact with innate immune signaling complexes, such as the STING-TBK1 complex, to disrupt downstream signaling. HPV18 E7 uses this strategy to control the IFN-I system (11). (c) DNA viral proteins may directly target distal IFN-I enhanceosome components to interfere with target gene transcription (2, 3).
This study reveals a novel dsDNA viral strategy to control the STING/IFN-I axis — protein destabilization. HPV16 and HPV18 are among the most common high-risk HPV subtypes that drive carcinogenesis. A plethora of studies on the biology of their oncoproteins leads to the revelation of the pathologically critical pRb interactome (45). Emerging evidence shows that HPV oncoproteins also exhibit pleiotropic effects on the host innate immune system. The nonkeratinizing oropharyngeal squamous cell carcinomas are frequently HPV16-related and arise in the tonsillar crypts of palatine and lingual tonsils. This unique anatomic location is characterized by dense immune cell infiltration, which should constitute a potent immune defense. Thus, in order to establish an invasive front, the transforming basal layer cells need to develop a strategy to mitigate innate immune detection. Indeed, the normal basal layer of squamous epithelium expresses STING (46), and the HPV16-induced turnover of STING likely dampens the “visibility” of the invasive fronts to the immune system. HPV16 E7 has only a low degree of homology with its HPV18 counterpart and does not interact with the STING complex as HPV18 E7 does. Instead, HPV16 E7 hijacks a PRR member, NLRX1, to destabilize STING. The unique presence of viral proteins in HPV+ tumors triggered enthusiasm for the development of therapeutic vaccines that enhance HPV protein-specific antitumor immunity (47). The viral epitopes were considered to be highly immunogenic. However, the clinical response of HPV+ HNSCC to checkpoint immunotherapy is surprisingly low (8). In fact, HPV+ HNSCC contains less CTL clonal expansion and exhibits lower levels of antigen-presenting machinery than HPV– tumors (6). Hence, the findings from this study pinpoint a previously unknown strategy that HPV16 uses to inhibit the host processing of viral neoepitopes.
Harnessing the checkpoints for IFN-I induction could yield significant immune-priming benefits. Our recent unbiased screen using HNSCC cell–effector immune cell coculture identified the IFN-I pathway as a central signal that maintains HNSCC sensitivity to immune killing (14). IFN-I downstream cytokines and chemokines improve APC and effector trafficking to the tumor mass and maintain a TME that favors APC maturation and cross-priming of CD8+ CTLs (16, 19, 22, 41, 46–49), thereby driving antitumor immunity. A number of strategies are being assessed to activate STING-mediated innate immune sensing and enhance immune priming. For example, ionizing radiation triggers DNA damage, which in turn activates the STING pathway. Preclinical models show that radiotherapy can prime the TME for a better response to checkpoint immunotherapy (19, 50). This combination has also entered into a clinical trial for HPV– HNSCC (NCT03635164). Another emerging approach is to inhibit the DNA damage repair pathway. Inhibition of either ataxia telangiectasia–mutated protein (ATM) or ataxia telangiectasia– and Rad3-related protein (ATR), 2 essential signaling components of the DNA damage response pathway, can induce IFN-I signaling and prime tumors for checkpoint blockade (51, 52). Cotargeting immunosuppressive pathways, such as IL-10 signaling, can also improve IFN-I–mediated antitumor activity (41). However, as we show in this study, HPV16 E7 functions as a rheostat for IFN-I signaling, which likely limits the cancer cell–specific response to the aforementioned neoadjuvant therapies. Thus, identification of novel checkpoints of the STING/IFN-I axis informs rational design of immune-priming combinations.
Here we show that NLRX1 is a pivotal intermediary partner that links HPV16 E7 to the suppression of IFN-I. Although the NLR family was initially thought to represent a cytoplasmic counterpart of the TLR family detecting intracellular microbes, accumulating evidence suggests that NLRs fundamentally control the homeostasis of inflammatory signaling and metabolism in addition to sensing microbial threats (53). We first characterized the role of NLRX1 in promoting autophagy in response to RNA virus infection (33). This autophagy-promoting function was later found to be also a central mechanism regulating bacteria- and fungi-host interactions (54, 55). Now we show that the NLRX1-centered autophagy-promoting molecular complex keeps dsDNA virus–induced immune activation in check by increasing the turnover of autophagosome cargos that include STING. The production of IFN-I and its downstream target chemokines de novo is a metabolically demanding process. Thus, autophagy recycles excessive proteins and damaged organelles to maintain nutrient supply, and, at the same time, increases the turnover of innate immune signaling complexes to keep the cytokine production under control. Together with our previous findings, these results suggest that NLRX1 broadly controls IFN-I production mediated by cytoplasmic PRRs such as STING and MAVS (30, 33, 56). This signaling pathway is hijacked by HPV16 to suppress TIL infiltration. Indeed, NLRX1 deficiency results in many spontaneous rejections of tumors even using the highly aggressive MOC2-derived tumor models. Depletion of NLRX1 in the tumor cells significantly reduces the exhausted T cell populations in an IFN-I–dependent fashion. The levels of NLRX1 also correlate with T cell exclusion among clinical HPV16+ HNSCC specimens (Figure 10).
Overall, this study delineates a novel dsDNA virus strategy by which HPV16 E7 functions as a powerful “degrader” of the central innate immune sensing signaling adaptor STING. Our study identifies the NLRX1-mediated autophagic machinery as a potential intervention point to restore the immunogenicity of HPV16+ HNSCCs. These findings represent a conceptual advance by complementing the current neoantigen-centered framework of cancer immunogenicity.
Methods
Clinical samples and tissue microarray.
The University of Michigan Head and Neck Cancer Specialized Program of Research Excellence (SPORE) recruited patients with previously untreated HNSCC from 2008 to 2012 for a longitudinal study. The patient demographic information is summarized in Supplemental Table 1. The tumors from 297 patients, with a median follow-up of 60.1 months, were incorporated into a tissue microarray (TMA). For each tumor, 3 representative 5-μm cores, identified by head and neck pathologist Jonathan McHugh (Department of Pathology, University of Michigan, Ann Arbor, MI, USA), were included and stained with anti-STING antibody (LS-B9374, LifeSpan BioSciences). The secondary antibody was biotinylated goat anti-rabbit antibody (Vectastain ABC HRP Kit, PK-4001, Vector Laboratories). Immunohistochemical (IHC) staining density was quantitated using Aperio ImageScope and averaged from the 3 cores. Missing cores and those with insufficient tumor parenchyma were excluded from analysis, and STING IHC scores were available for 264 patients.
Animals.
Eight-week-old C57BL/6 (strain 000664), Ifnar1–/– (strain 32045-JAX), and Rag1–/– (strain 002216) mice were purchased from The Jackson Laboratory and housed under specific pathogen–free conditions in a temperature- and light-controlled environment. As sex is not a known prognosticator for HNSCC, both sexes were used. Syngeneic HNSCC models were established by inoculation of 1 million empty vector control or shNLRX1 MOC2-E6/E7 cells subcutaneously in the right flank. Beginning on day 7 after tumor implantation, tumors were measured using a digital Vernier caliper every 2–3 days, and tumor volume was calculated according to the formula 1/2 (length × width2). All mice were euthanized at the indicated time points. After euthanasia, the tumors, TILs, lymph nodes, and spleens of the mice were harvested for subsequent analysis.
Cell culture.
PCI-13 and SCC90 HNSCC lines were acquired from the University of Pittsburgh. UMSCC47 and UMSCC49 were obtained from the University of Michigan. 93VU147T was provided by Renske Steenbergen at VU University Medical Center Amsterdam (Amsterdam, Netherlands) (57). FaDu was purchased from ATCC (HTB-43). All cell lines were authenticated and maintained in DMEM (10-013-CV, Corning) supplemented with 10% FBS (Gibco, Life Technologies), 100 U/mL penicillin (Gibco), and 100 mg/mL streptomycin (Gibco). The MOC2-E6/E7 cell line was cultured in 60% IMDM (SH30228.01, HyClone) with 30% F12 nutrient mix (11764-054, Gibco), 5% FBS, 4 μg/mL puromycin, 5 μg/mL insulin, 40 ng/mL hydrocortisone, 5 ng/mL EGF, 100 U/mL penicillin, and 100 mg/mL streptomycin. All cells were maintained in a humidified incubator at 37°C with 5% CO2.
Plasmids, antibodies, inhibitors, and antibiotics.
The pcDNA3.1–human STING–HA and pcDNA3.1–murine STING–HA expression plasmids were provided by Glen N. Barber at the University of Miami (Miami, Florida, USA). The CMV–HPV16 E7, pEGFP-LC3B, and lentiCRISPRv2 plasmids were acquired from Addgene (catalog 13686, 24920, and 98290, respectively). The single-guide RNA (sgRNA) targeting HPV16 E7 sequence was 5′-CACCGGCAAGTGTGACTCTACGCTT-3′. The pcDNA3.1 empty vector (EV) was described previously (33). Poly(dA:dT) (tlrl-patn-1) and cGAMP (tlrl-nacga23-1) were purchased from InvivoGen. Human pGIPZ-shNLRX1-puro and pGIPZ–empty vector–puro lentiviral construct glycerol stocks were obtained from Thermo Fisher Scientific (NC0744603 and NC9619315, respectively). Mouse pLKO.1-shNLRX1-puro and pLKO.1–empty vector–puro lentiviral constructs were obtained from Sigma-Aldrich (catalog SHCLND-NM_178420 and SHC001V). MSCV-C HPV18 E7 retroviral construct (catalog 37886) and its corresponding control empty vector (catalog 24828) were obtained from Addgene. Lentiviral and retroviral packaging vectors including psPAX2, pMLV gag-pol, and VSV-G were provided by Jenny P.Y. Ting at the University of North Carolina at Chapel Hill (Chapel Hill, North Carolina, USA).
Primary antibodies used for immunoblotting, immunoprecipitation, and immunofluorescence were as follows: β-actin (ab49900, Abcam), HPV16 E7 (sc-65711, Santa Cruz Biotechnology), HPV18 E7 (sc-365035, Santa Cruz Biotechnology), normal mouse IgG2a (sc-3878, Santa Cruz Biotechnology), NLRX1 for immunoblotting (04-146, MilliporeSigma), NLRX1 for immunofluorescence (ab105412, Abcam), phospho-p65 (Ser536) (3033S, Cell Signaling Technology), p65 (8242S, Cell Signaling Technology), phospho-TBK1 (Ser172) (5483S, Cell Signaling Technology), TBK1 (3504S, Cell Signaling Technology), STING (13647S, Cell Signaling Technology), LC3B (2775S, Cell Signaling Technology), HA-HRP (2999S, Cell Signaling Technology), VDAC (4661S, Cell Signaling Technology), and p63 (39692S, Cell Signaling Technology). The secondary antibodies used for immunoblotting were goat anti-rabbit IgG–HRP (ab97051, Abcam) and goat anti-mouse IgG–HRP (ab97023, Abcam). The secondary antibodies for immunofluorescence were donkey anti-mouse IgG antibody (Alexa Fluor 488, 715-545-150, Jackson ImmunoResearch) and donkey anti-rabbit IgG antibody (Alexa Fluor 647, 711-605-152, Jackson ImmunoResearch). Bafilomycin A1 was purchased from Sigma-Aldrich (B1793, Ronkonkoma). Puromycin was purchased from InvivoGen (ant-pr-1).
H&E staining, immunohistochemistry, and immunofluorescence.
After the harvest of 5 EV and 5 shNLRX1 tumors from C57BL/6 mice, tumors were fixed with 4% paraformaldehyde (15710, Electron Microscopy Sciences) for 24 hours before dehydration and paraffin embedding. The 5-μm tumor sections were deparaffinized at 58°C for 20 minutes and then rehydrated. H&E and IHC staining were performed as we described. For immunofluorescence staining, cells seeded in chamber slides were incubated with 200 nM MitoTracker (M36008, Life Technologies) for 45 minutes before fixation. After permeabilization with 0.1% Triton X-100 for 10 minutes, the cells were washed and incubated with primary antibodies against HPV16 E7 and NLRX1 at 1:50 dilution at 4°C overnight. Samples were then incubated with secondary antibodies and counterstained with Hoechst 33342 (catalog H3570, Life Technologies) at room temperature. Images of cells were visualized and captured by a Nikon A1si confocal microscope.
Lentiviral transduction and stable cell line generation.
Lentivirus packaging and retrovirus packaging were performed as we have previously described (33). Stable human or mouse NLRX1-knockdown HNSCC cells (including 93VU147T, UMSCC47, SCC90, and MOC2-E6/E7 cell lines) were generated by lentiviral transduction followed by puromycin selection. The cells transduced with empty vector constructs were used as negative controls. The selecting concentration of puromycin was determined first by establishment of a kill curve by addition of various doses of puromycin into the wild-type HNSCC cells and determination of the lowest concentration that could kill all live cells. The concentrations used were 15 μg/mL puromycin for 93VU147T, UMSCC47, and SCC90 cell lines, and 120 μg/mL puromycin for MOC2-E6/E7 cells. Immunoblotting was performed to validate the knockdown efficiency. In addition, HPV16 E7–knockout cells were established by transduction of HNSCC cells (93VU147T, UMSCC47) with CRISPR/Cas9 lentivirus, and the cells transduced with empty vector virus were considered as a negative control. The knockout of HPV16 E7 was validated by immunoblotting. HPV18 E7–expressing UMSCC49 cells were established by an HPV18 E7 expression retrovirus, with empty vector retrovirus–transduced tumor cells as a negative control.
Gene expression qPCR and ELISA.
Total RNA was extracted using QIAshredder and the RNeasy Plus Mini Kit (catalog 79654 and 74134, respectively; Qiagen). RNA concentration was measured using a Nanodrop Spectrophotometer (Thermo Fisher Scientific). RNA was reverse-transcribed into cDNA using High-Capacity cDNA Reverse Transcription Kit and RNAse inhibitor (4368814 and N8080119, Applied Biosystems). The primers were synthesized by Integrated DNA Technologies, and most of the sequences have been previously described (14). Other primers were as follows: Isg15 forward 5′-TGACTGTGAGAGCAAGCAGC-3′, reverse 5′-CCCCAGCATCTTCACCTTTA-3′; Isg54 forward 5′-TCTGGTCACCTGGGGAAACTATG-3′, reverse 5′-TTCTCAATCCTGTAGGGGCTGG-3′; Tnf forward 5′-ATGAGAAGTTCCCAAATGGC-3′, reverse 5′-CTCCACTTGGTGGTTTGCTA-3′; Il6 forward 5′-CTCTGGGAAATCGTGGAAAT-3′, reverse 5′-CCAGTTTGGTAGCATCCATC-3′; Il10 forward 5′-TAACTGCACCCACTTCCCAG-3′, reverse 5′-AGGCTTGGCAACCCAAGTAA-3′; MX1 forward 5′-CAATCAGCCTGCTGACATTG-3′, reverse 5′-TGTCTCCTGCCTCTGGATG-3′; STING forward 5′-AGCATTACAACAACCTGCTACG-3′, reverse 5′-GTTGGGGTCAGCCATACTCAG-3′. IFN-β in the supernatant was quantified using a high-sensitivity ELISA kit (41415, PBL Assay Science).
Immunoblotting and immunoprecipitation assays.
Whole-cell lysates in each well were harvested on ice in RIPA buffer (50 mM Tris-HCl pH 8.0, 1% Triton X-100, 0.05% SDS, 0.25% deoxycholate, 150 mM NaCl, and 50 mM NaF) supplemented with a protease inhibitor cocktail (11836170001, Roche) and Halt Phosphatase Inhibitor Cocktail (78420, Thermo Fisher Scientific). The dilutions for the primary antibodies were as follows: β-actin, 1:100,000; HPV16 E7, 1:200; HPV18 E7, 1:200; and 1:1000 for other antibodies. All antibodies were diluted in 5% skim milk. To assess co-IP between HPV16 E7 and STING, control and HA-STING expression plasmids were transfected into empty vector control and NLRX1-deficient 93VU147T cells. After lysates were harvested on ice in IP buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, and 0.1% NP-40) supplemented with a protease inhibitor cocktail (11836170001, Roche), the samples were rotated at 4°C for 30 minutes and centrifuged at 13,000 g for 15 minutes at 4°C. One-third of the lysates were retained as input controls, while two-thirds were used for IP. Supernatants were incubated with anti-HA beads (26181, Thermo Fisher Scientific) at 4°C overnight, gently washed 3 times in IP buffer, and subjected to a short spin to obtain immunoprecipitated protein complexes. For endogenous co-IP experiments, protein lysates were incubated with HPV16/HPV18 E7–targeted antibodies or IgG2a isotype control, followed by Protein A/G UltraLink Resin (53132, Thermo Fisher Scientific) pull-down. The protein complexes were resuspended in 1× LDS sample buffer, boiled at 95°C for 5 minutes, and then subjected to immunoblotting. Densitometric analysis was conducted relative to the indicated band (specified in the figure legends) using ImageJ (NIH).
AlamarBlue assay.
Tumor cells were seeded at a density of 500 cells per well in 96-well microplates with a flat black bottom (3904, Corning). Every 24 hours from day 1 to day 6, corresponding wells were supplemented with 10% alamarBlue (DAL1025, Invitrogen), and the plate was subsequently incubated at 37°C for 4 hours. The fluorescence intensity (excitation 560, emission 590 nm) of these wells was measured using a Biotek plate reader and Gen5 program (version 2.09), and 5 replicates per group were examined simultaneously.
Flow cytometry.
Immune cells from tumors, lymph nodes, spleens, and peripheral blood were purified as we have previously described (14), followed by staining for multi-fluorophore flow cytometric analysis with the following antibodies: anti-CD45 (clone 30-F11, BioLegend), anti-CD3 (clone 17A2, BD Biosciences), anti-CD4 (clone RM4-5, BioLegend), anti-CD8 (clone 53-6.7, BioLegend), CD16/32 (93, eBioscience), and CD366 (RMT3-23, BioLegend). Cells were also stained for viability using Fixable Viability Dye (FVD) eFluor 780 (65-0865-14, Thermo Fisher Scientific) or Zombie Aqua (423101, BioLegend) diluted 1:1000 in PBS at 4°C for 30 minutes. Acquisition and compensation were conducted on a Beckman Coulter CyAn ADP flow cytometer. FlowJo version 10 software was used for data analysis.
Data availability.
HNSCC TCGA data are available through http://firebrowse.org/ TCGA raw data are stored in the database of Genotypes and Phenotypes (dbGaP) with the accession number phs000178.
Statistics.
Statistical analysis between 2 independent groups was made using unpaired, 2-tailed Student’s t tests. Comparisons between more than 2 groups were made using 2-way ANOVA with Šidák’s multiple-comparisons post hoc analysis. Spearman’s rank-order correlation was performed to identify correlations between immune cells and IFN-I signature genes. Univariate Cox linear regression modeling was used to identify the association between STING expression in TMAs and patient age. The association between STING expression scores and patient survival was conducted using multivariate Cox regression models. Statistical significance in survival probability between STING-low and STING-high TMAs was evaluated by Kaplan-Meier survival curves and a log-rank test. Tumor burden between groups was compared using the generalized estimating equation model as we have previously described (14). Statistical significance is indicated in all figures according to the following scale: *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001. All graphs are presented as the mean ± SEM.
Study approval.
All animal procedures were performed in accordance with the protocol approved by the Institutional Animal Care and Use Committee at the University of Michigan (PRO00008517). The clinical protocol to obtain HNSCC specimens was approved by the University of Michigan Institutional Review Board (HUM00042189 and HUM00113038), with informed consent obtained from all patients.
Author contributions
X. Luo conducted experiments, acquired and analyzed data, and wrote the manuscript. CRD acquired and analyzed data and wrote the manuscript. TM, BRH, LAD, YH, YST, WG, X. Lin, and YX conducted experiments, acquired and analyzed data, and reviewed the manuscript. EB and BAK analyzed data and reviewed the manuscript. MAM, RLF, JCB, TEC, LC, MEP, SY, HW, PJP, GTW, and QC provided critical reagents and contributed to the experimental design. YLL reviewed the data, wrote the manuscript, and was responsible for the overall direction of the project.
Supplementary Material
Acknowledgments
This work was supported by NIH grants R01 DE026728, R00 DE024173, R03 DE027399, U01 DE029255, F31 DE028740, and T32 AI007413; a Rogel Cancer Center research committee grant; National Natural Science Foundation of China grants 81520108009, 81621062, and 81730030; and the 111 MOE project of China. We thank the many investigators in the University of Michigan Head and Neck SPORE for their contributions to patient enrollment, tissue procurement, and TMA generation. We also thank the patients and their families who tirelessly participated in the study.
Version 1. 12/24/2019
In-Press Preview
Version 2. 02/24/2020
Electronic publication
Version 3. 04/01/2020
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(4):1635–1652.https://doi.org/10.1172/JCI129497.
Contributor Information
Xiaobo Luo, Email: xiaobol@umich.edu.
Christopher R. Donnelly, Email: chdonnel@umich.edu.
Wang Gong, Email: wagong@umich.edu.
Blake R. Heath, Email: heathbr@umich.edu.
Yuning Hao, Email: yuningh0411@gmail.com.
Lorenza A. Donnelly, Email: loriad@umich.edu.
Toktam Moghbeli, Email: toktam_moghbeli@yahoo.com.
Yee Sun Tan, Email: yeestan@umich.edu.
Xin Lin, Email: nilnix@umich.edu.
Emily Bellile, Email: lighte@med.umich.edu.
Benjamin A. Kansy, Email: benjamin.kansy@uk-essen.de.
Thomas E. Carey, Email: careyte@med.umich.edu.
J. Chad Brenner, Email: chadbren@med.umich.edu.
Lei Cheng, Email: chengleidentist@163.com.
Peter J. Polverini, Email: neovas@umich.edu.
Meredith A. Morgan, Email: mmccrack@umich.edu.
Haitao Wen, Email: Haitao.Wen@osumc.edu.
Mark E. Prince, Email: mepp@umich.edu.
Robert L. Ferris, Email: ferrisrl@upmc.edu.
Yuying Xie, Email: xyy@egr.msu.edu.
Simon Young, Email: simon.young@uth.tmc.edu.
Gregory T. Wolf, Email: gregwolf@umich.edu.
Qianming Chen, Email: qmchen@scu.edu.cn.
Yu L. Lei, Email: leiyuleo@umich.edu.
References
- 1.Secombes CJ, Zou J. Evolution of interferons and interferon receptors. Front Immunol. 2017;8:209. doi: 10.3389/fimmu.2017.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doehle BP, Chang K, Rustagi A, McNevin J, McElrath MJ, Gale M. Vpu mediates depletion of interferon regulatory factor 3 during HIV infection by a lysosome-dependent mechanism. J Virol. 2012;86(16):8367–8374. doi: 10.1128/JVI.00423-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Unterholzner L, et al. Vaccinia virus protein C6 is a virulence factor that binds TBK-1 adaptor proteins and inhibits activation of IRF3 and IRF7. PLoS Pathog. 2011;7(9):e1002247. doi: 10.1371/journal.ppat.1002247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma Z, Ni G, Damania B. Innate sensing of DNA virus genomes. Annu Rev Virol. 2018;5(1):341–362. doi: 10.1146/annurev-virology-092917-043244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gillison ML, Chaturvedi AK, Anderson WF, Fakhry C. Epidemiology of human papillomavirus-positive head and neck squamous cell carcinoma. J Clin Oncol. 2015;33(29):3235–3242. doi: 10.1200/JCO.2015.61.6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saloura V, et al. Characterization of the T-cell receptor repertoire and immune microenvironment in patients with locoregionally advanced squamous cell carcinoma of the head and neck. Clin Cancer Res. 2017;23(16):4897–4907. doi: 10.1158/1078-0432.CCR-17-0103. [DOI] [PubMed] [Google Scholar]
- 7.Ferris RL, et al. Nivolumab vs investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck: 2-year long-term survival update of CheckMate 141 with analyses by tumor PD-L1 expression. Oral Oncol. 2018;81:45–51. doi: 10.1016/j.oraloncology.2018.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferris RL, et al. Nivolumab in patients with recurrent or metastatic squamous cell carcinoma of the head and neck: efficacy and safety in CheckMate 141 by prior cetuximab use. Clin Cancer Res. 2019;25(17):5221–5230. doi: 10.1158/1078-0432.CCR-18-3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen EEW, et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): a randomised, open-label, phase 3 study. Lancet. 2019;393(10167):156–167. doi: 10.1016/S0140-6736(18)31999-8. [DOI] [PubMed] [Google Scholar]
- 10.Bauman JE, et al. Immunotherapy of head and neck cancer: emerging clinical trials from a National Cancer Institute Head and Neck Cancer Steering Committee Planning Meeting. Cancer. 2017;123(7):1259–1271. doi: 10.1002/cncr.30449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lau L, Gray EE, Brunette RL, Stetson DB. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science. 2015;350(6260):568–571. doi: 10.1126/science.aab3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dufour X, Beby-Defaux A, Agius G, Lacau St Guily J. HPV and head and neck cancer. Eur Ann Otorhinolaryngol Head Neck Dis. 2012;129(1):26–31. doi: 10.1016/j.anorl.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 13.Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan YS, et al. Mitigating SOX2-potentiated immune escape of head and neck squamous cell carcinoma with a STING-inducing nanosatellite vaccine. Clin Cancer Res. 2018;24(17):4242–4255. doi: 10.1158/1078-0432.CCR-17-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15(7):405–414. doi: 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- 16.Lei Y, et al. Telltale tumor infiltrating lymphocytes (TIL) in oral, head & neck cancer. Oral Oncol. 2016;61:159–165. doi: 10.1016/j.oraloncology.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol. 2015;33:445–474. doi: 10.1146/annurev-immunol-032414-112043. [DOI] [PubMed] [Google Scholar]
- 18.Fu J, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7(283):283ra52. doi: 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng L, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41(5):843–852. doi: 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng L, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. 2014;124(2):687–695. doi: 10.1172/JCI67313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corrales L, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015;11(7):1018–1030. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baird JR, et al. Radiotherapy combined with novel STING-targeting oligonucleotides results in regression of established tumors. Cancer Res. 2016;76(1):50–61. doi: 10.1158/0008-5472.CAN-14-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore E, et al. Established T cell-inflamed tumors rejected after adaptive resistance was reversed by combination STING activation and PD-1 pathway blockade. Cancer Immunol Res. 2016;4(12):1061–1071. doi: 10.1158/2326-6066.CIR-16-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leach DG, et al. STINGel: controlled release of a cyclic dinucleotide for enhanced cancer immunotherapy. Biomaterials. 2018;163:67–75. doi: 10.1016/j.biomaterials.2018.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hao Y, Yan M, Heath BR, Lei YL, Xie Y. Fast and robust deconvolution of tumor infiltrating lymphocyte from expression profiles using least trimmed squares. PLoS Comput Biol. 2019;15(5):e1006976. doi: 10.1371/journal.pcbi.1006976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gentles AJ, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938–945. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lei Y, et al. EGFR-targeted mAb therapy modulates autophagy in head and neck squamous cell carcinoma through NLRX1-TUFM protein complex. Oncogene. 2016;35(36):4698–4707. doi: 10.1038/onc.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gillison ML. Human papillomavirus-related diseases: oropharynx cancers and potential implications for adolescent HPV vaccination. J Adolesc Health. 2008;43(4 suppl):S52–S60. doi: 10.1016/j.jadohealth.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olthof NC, et al. Viral load, gene expression and mapping of viral integration sites in HPV16-associated HNSCC cell lines. Int J Cancer. 2015;136(5):E207–E218. doi: 10.1002/ijc.29112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo H, et al. NLRX1 sequesters STING to negatively regulate the interferon response, thereby facilitating the replication of HIV-1 and DNA viruses. Cell Host Microbe. 2016;19(4):515–528. doi: 10.1016/j.chom.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Degenhardt K, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kimura T, et al. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J Cell Biol. 2015;210(6):973–989. doi: 10.1083/jcb.201503023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lei Y, et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity. 2012;36(6):933–946. doi: 10.1016/j.immuni.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jounai N, et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci U S A. 2007;104(35):14050–14055. doi: 10.1073/pnas.0704014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu D, et al. STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 2019;26(9):1735–1749. doi: 10.1038/s41418-018-0251-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang Q, et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe. 2014;15(2):228–238. doi: 10.1016/j.chom.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore CB, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451(7178):573–577. doi: 10.1038/nature06501. [DOI] [PubMed] [Google Scholar]
- 38.Dharmaraj N, et al. Anti-tumor immunity induced by ectopic expression of viral antigens is transient and limited by immune escape. Oncoimmunology. 2019;8(4):e1568809. doi: 10.1080/2162402X.2019.1568809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shah S, Caruso A, Cash H, Waes CV, Allen CT. Pools of programmed death-ligand within the oral cavity tumor microenvironment: variable alteration by targeted therapies. Head Neck. 2016;38(8):1176–1186. doi: 10.1002/hed.24269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahn J, Ruiz P, Barber GN. Intrinsic self-DNA triggers inflammatory disease dependent on STING. J Immunol. 2014;193(9):4634–4642. doi: 10.4049/jimmunol.1401337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baird JR, et al. Evaluation of explant responses to STING ligands: personalized immunosurgical therapy for head and neck squamous cell carcinoma. Cancer Res. 2018;78(21):6308–6319. doi: 10.1158/0008-5472.CAN-18-1652. [DOI] [PubMed] [Google Scholar]
- 42.Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology. 2017;6(1):e1261779. doi: 10.1080/2162402X.2016.1261779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5(10):987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 44.Peters NE, et al. A mechanism for the inhibition of DNA-PK-mediated DNA sensing by a virus. PLoS Pathog. 2013;9(10):e1003649. doi: 10.1371/journal.ppat.1003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wiest T, Schwarz E, Enders C, Flechtenmacher C, Bosch FX. Involvement of intact HPV16 E6/E7 gene expression in head and neck cancers with unaltered p53 status and perturbed pRb cell cycle control. Oncogene. 2002;21(10):1510–1517. doi: 10.1038/sj.onc.1205214. [DOI] [PubMed] [Google Scholar]
- 46.Baird JR, et al. STING expression and response to treatment with STING ligands in premalignant and malignant disease. PLoS One. 2017;12(11):e0187532. doi: 10.1371/journal.pone.0187532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tan YS, Sansanaphongpricha K, Prince MEP, Sun D, Wolf GT, Lei YL. Engineering vaccines to reprogram immunity against head and neck cancer. J Dent Res. 2018;97(6):627–634. doi: 10.1177/0022034518764416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woo SR, Corrales L, Gajewski TF. The STING pathway and the T cell-inflamed tumor microenvironment. Trends Immunol. 2015;36(4):250–256. doi: 10.1016/j.it.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu MM, et al. Dendritic cells but not macrophages sense tumor mitochondrial DNA for cross-priming through signal regulatory protein α signaling. Immunity. 2017;47(2):363–373.e5. doi: 10.1016/j.immuni.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Woo SR, et al. STING-dependent cytosolic DNA mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41(5):830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Q, et al. Inhibition of ATM increases interferon signaling and sensitizes pancreatic cancer to immune checkpoint blockade therapy. Cancer Res. 2019;79(15):3940–3951. doi: 10.1158/0008-5472.CAN-19-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vendetti FP, et al. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell-dependent antitumor activity following radiation. J Clin Invest. 2018;128(9):3926–3940. doi: 10.1172/JCI96519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lupfer C, Kanneganti TD. The expanding role of NLRs in antiviral immunity. Immunol Rev. 2013;255(1):13–24. doi: 10.1111/imr.12089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat Immunol. 2019;20(4):433–446. doi: 10.1038/s41590-019-0324-2. [DOI] [PubMed] [Google Scholar]
- 55.Huang JH, et al. NLRX1 facilitates Histoplasma capsulatum-induced LC3-associated phagocytosis for cytokine production in macrophages. Front Immunol. 2018;9:2761. doi: 10.3389/fimmu.2018.02761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lei Y, Wen H, Ting JP. The NLR protein, NLRX1, and its partner, TUFM, reduce type I interferon, and enhance autophagy. Autophagy. 2013;9(3):432–433. doi: 10.4161/auto.23026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Steenbergen RD, et al. Integrated human papillomavirus type 16 and loss of heterozygosity at 11q22 and 18q21 in an oral carcinoma and its derivative cell line. Cancer Res. 1995;55(22):5465–5471. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
HNSCC TCGA data are available through http://firebrowse.org/ TCGA raw data are stored in the database of Genotypes and Phenotypes (dbGaP) with the accession number phs000178.