

Abstract
Visceral adipose tissue plays a critical role in numerous diseases. Although imaging studies often show adipose involvement in abdominal diseases, their outcomes may vary from being a mild self-limited illness to one with systemic inflammation and organ failure. We therefore compared the pattern of visceral adipose injury during acute pancreatitis and acute diverticulitis to determine its role in organ failure. Acute pancreatitis–associated adipose tissue had ongoing lipolysis in the absence of adipocyte triglyceride lipase (ATGL). Pancreatic lipase injected into mouse visceral adipose tissue hydrolyzed adipose triglyceride and generated excess nonesterified fatty acids (NEFAs), which caused organ failure in the absence of acute pancreatitis. Pancreatic triglyceride lipase (PNLIP) increased in adipose tissue during pancreatitis and entered adipocytes by multiple mechanisms, hydrolyzing adipose triglyceride and generating excess NEFAs. During pancreatitis, obese PNLIP-knockout mice, unlike obese adipocyte-specific ATGL knockouts, had lower visceral adipose tissue lipolysis, milder inflammation, less severe organ failure, and improved survival. PNLIP-knockout mice, unlike ATGL knockouts, were protected from adipocyte-induced pancreatic acinar injury without affecting NEFA signaling or acute pancreatitis induction. Therefore, during pancreatitis, unlike diverticulitis, PNLIP leaking into visceral adipose tissue can cause excessive visceral adipose tissue lipolysis independently of adipocyte-autonomous ATGL, and thereby worsen organ failure.
Keywords: Gastroenterology, Inflammation
Keywords: Adipose tissue, Fatty acid oxidation, Mouse models

Introduction
In humans, visceral adipose tissue is commonly involved in acute diseases like acute diverticulitis (1, 2), appendicitis (3, 4), and acute pancreatitis (5–7), but the extent of visceral adipose involvement only parallels acute pancreatitis severity (1–7). This involvement is commonly imaged as stranding around the inflamed organs. While both acute diverticulitis and acute pancreatitis have a sudden onset, involve visceral fat, and occur in a similar demographic, diverticulitis is often treated in the outpatient setting and rarely causes organ failure (8), whereas acute pancreatitis requires hospitalization and has a higher risk of progression to organ failure (9). The mechanisms underlying these differences are unknown.
Visceral fat necrosis worsens acute pancreatitis outcomes in obesity (10–14). However, models mimicking acute diverticulitis (e.g., cecal ligation, puncture) have variable outcomes in obese mice (15, 16), with some reporting that visceral adipose tissue plays a protective role (17). A clue to this discrepancy may lay in acute nonesterified fatty acid (NEFA) elevations in acute pancreatitis (18, 19). High NEFAs can inhibit mitochondrial complexes I and V, cause necrosis of the pancreas, renal tubular injury, acute respiratory distress syndrome (ARDS), and result in multisystem organ failure (13, 14, 20). NEFA infusion can induce renal injury (21, 22) and ARDS (23, 24). The lipase mediating this rapid and excessive NEFA generation is therefore of interest.
Lipolysis within adipocytes is normally a highly regulated process involving adipocyte triglyceride lipase (ATGL) and hormone-sensitive lipase (25). The pancreas itself has 3 lipases: pancreatic triglyceride lipase (PNLIP), the major contributor to the pancreas’s lipolytic activity (26, 27); pancreatic lipase-related protein 2 (PNLIPRP2); and carboxyl ester lipase (CEL) (28). These normally digest dietary fat in the intestine. Although pharmacologic lipase inhibition improves pancreatitis outcomes (13, 14), whether adipocyte or pancreatic lipases mediate this NEFA generation is unknown. ATGL is normally the initiating and rate-limiting lipolytic enzyme in adipocytes (29). Its adipocyte-specific deletion decreased adipocyte lipolysis and serum lipids in mice (30). More recently, pharmacologic inhibition of ATGL has been shown to slow progression of heart failure in mice (31). Because cardiovascular failure, shock (32, 33), and lipotoxicity from NEFAs (14, 34) can contribute to severe acute pancreatitis, we therefore also considered whether ATGL can mediate severe acute pancreatitis.
We therefore started by comparing the pathophysiology of NEFA generation in human acute diverticulitis to acute pancreatitis, and roles of pancreatic lipases versus ATGL in the progression to organ failure. For this we used in vitro and in vivo models of acute pancreatitis, and tested the efficacy of the specific ATGL inhibitor Atglistatin (35), an adipocyte-specific ATGL deletion (36), or genetic deletion of PNLIP (37) in preventing the cascade of lipolysis resulting in organ failure. Because CEL cannot hydrolyze long-chain triglycerides, such as those present in visceral fat (38–40), it was not studied here.
Genetically obese mice, homozygous for mutant leptin (B6.Cg-Lepob/J; ob/ob), have increased visceral fat (41) and unlike lean mice develop pancreatitis-associated multisystem organ failure similar to human severe acute pancreatitis (10, 13, 42). Therefore, we mated ATGL or PNLIP knockouts with the leptin mutants to generate dual ATGL-floxed Cre-positive (ATGL-KO) ob/ob mice or PNLIP-knockout (PNLIP-KO) ob/ob mice. These KO ob/ob mice matched the control ob/ob littermates in physical parameters and body composition. This allowed us to study the lipase(s) causing acute lipolysis of visceral adipose tissue and consequent local and systemic lipotoxic injury during acute pancreatitis. Interestingly, we note that acute visceral adipose tissue lipolysis changes pancreatitis outcomes without affecting its initiation independently of the mechanisms regulating lipolysis within adipocytes.
Results
During human acute pancreatitis, unlike diverticulitis, PNLIP accumulates in visceral adipose tissue and may hydrolyze it.
Because both acute pancreatitis and acute diverticulitis have a sudden onset, involve visceral fat, and occur in a similar demographic but have very different outcomes, we first aimed to understand why visceral adipose tissue involvement during acute pancreatitis can worsen it, unlike diverticulitis. For this we compared the fat necrosis of acute pancreatitis to that of diverticulitis. An example of the CT appearance of visceral adipose tissue involvement at the time of diagnosing acute diverticulitis or acute pancreatitis is shown in Figure 1, A–D. The day from disease onset is mentioned below the images. Although both diseases had visceral adipose tissue involvement around the colon and pancreas, respectively (yellow outline Figure 1, A–D), this increased in size over time during acute pancreatitis (Figure 1, B–D). Patients with acute pancreatitis had a higher prevalence of organ failure than those with acute diverticulitis (5 of 8 vs. 0 of 8, P = 0.03) despite being younger (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI132767DS1). The interval from the onset of disease to the time of surgery averaged 2 months in both cases (Supplemental Figure 1). We then analyzed the visceral fat samples removed at the time of surgery for these diseases. Uninvolved fat was not tested.
Figure 1. Comparison of biochemical parameters, histology of acute pancreatitis and diverticulitis in humans.

Cross-sectional CT scan images on the first day of acute diverticulitis (A) or acute pancreatitis (B–D) with fat stranding of visceral adipose tissue (yellow outline) around the colon (C), or the pancreas (P). Images in B–D show progression of visceral fat involvement (yellow line) on the different days after onset of pancreatitis mentioned at the bottom. (E) A representative example of a serial section of human tissue stained for myeloperoxidase during acute diverticulitis showing the colon on the right and lower edges of section. (E–K) Acute diverticulitis (E–H) and acute pancreatitis (I–K) stained with H&E (F and I), calcium stained with von Kossa (G and J), and IHC for PNLIP (H and K) are also shown. Higher-magnification views of the insets are shown below. Note von Kossa– and PNLIP-positive areas overlap (arrows) in areas of fat necrosis (FN), but not normal fat (green outline). Also note positive von Kossa staining in necrosed pancreas (Panc), which has loss of cell outlines (above the dotted red line). This localizes the saponified NEFAs in the necrosed pancreas. Scale bars: 500 μm (E–H) and 200 μm (I–K). (L) Thin layer chromatography comparing the relative amount of NEFAs and TG in samples from patients with acute pancreatitis or diverticulitis. Amylase (M), lipase (N), and trypsin (O) activities measured in these samples. Error bars represent SEM. (P) Western blot images comparing detectable ATGL and perilipin 1 (Peri-1), and adiponectin (Adipo) bands in these samples.
Histologically, acute diverticulitis showed intense adipose tissue acute inflammation with myeloperoxidase-positive infiltrate (Figure 1E). However, the adipocytes appeared normal (Figure 1F) and stained negative for calcium (no black staining in von Kossa; Figure 1G) and PNLIP (Figure 1H). On a 0–3 scale, a blinded pathologist scored these 0 and 0-1, respectively, in diverticulitis (Table 1). Fat necrosis during acute pancreatitis bordered pancreatic necrosis and appeared chalky pink on hematoxylin and eosin (H&E) staining (Figure 1I), as shown previously (14, 43). The necrosed adipocytes, unlike normal ones (green outline), stained positive for calcium (black stain, black arrows in Figure 1J, scored as 3 out of 3), consistent with saponified NEFAs in the necrotic fat (14, 43). Serial sections scored 3 out of 3 for PNLIP (brown arrows in brown areas (Figure 1K) in von Kossa–positive areas, unlike the fat in diverticulitis. Thus, there was histologic evidence of PNLIP leakage from the inflamed pancreas into necrotic fat (Figure 1K) unlike during diverticulitis (Figure 1, F–H). This has been noted previously in human (44–46) and experimental pancreatitis (47–49).
Table 1. von Kossa and PNLIP positivity scores.
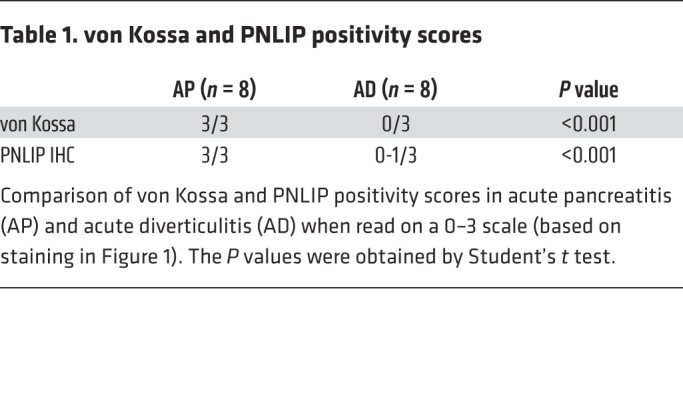
On thin layer chromatography, the involved fat in acute diverticulitis remained predominantly triglyceride (Figure 1L), but was hydrolyzed to NEFAs in acute pancreatitis, with NEFA concentrations of 3.5 ± 2.9 mM. Similar to previous reports (14), oleic acid (OA, C18:1) was the principal NEFA, comprising 38% ± 10%. The fat in acute pancreatitis contained higher activity of pancreatic lipase, amylase, and trypsin compared with diverticulitis (Figure 1, M–O). Interestingly, on Western blotting, there was no detectable ATGL or perilipin 1 protein in the necrotic fat, while these proteins were present in diverticulitis (Figure 1P). Adiponectin was used as a marker for adipocytes and was present in both pancreatitis and diverticulitis. This result suggested that acute pancreatitis caused leakage of pancreatic hydrolases into the visceral adipose tissue, which degraded proteins and triglycerides normally present in visceral adipose tissue. Such leakage has previously been shown in human pancreatitis (44–46) and experimental pancreatitis (47–49). Overall, these findings support the need to verify that visceral adipose tissue could undergo lipolysis independently of ATGL, and to understand the mechanisms underlying this lipolysis that results in organ failure.
Pancreatic lipase hydrolyzes visceral adipose tissue causing fat necrosis, organ failure, and worsening inflammation.
We went on to investigate whether a nonadipocyte lipase can cause visceral adipose tissue necrosis and organ failure. For this we first injected pancreatic lipase extracted from pigs (50) into the fat pads of ob/ob mice alone or along with the lipase inhibitor orlistat. Lipase injection dramatically reduced survival over 24 hours (Figure 2A). Grossly, fat necrosis was seen as round white deposits all over the visceral fat of the lipase-treated mice, which was prevented by orlistat (Figure 2B). The lipase injection also increased fat pad pancreatic lipase activity (Figure 2C), and hydrolyzed adipocyte triglycerides into to NEFAs (Figure 2D). This lipolysis resulted in a 66 ± 44-fold increase in various long-chain NEFAs in visceral fat, with OA (C18:1) being the main one (84 ± 44-fold, Figure 2E). All these changes were prevented by orlistat. This lipolysis also resulted in a large increase in serum cytokines IL-6, MCP-1, and TNF-α (Figure 2, F–H) and blood urea nitrogen (BUN) elevation signifying renal injury (Figure 2I), which were measured at the time of euthanasia. These increases were prevented or reduced by orlistat (Figure 2, F–I). Additionally, lipolysis caused a large drop in rectal temperature and carotid pulse distension, implying hypothermia and shock (Figure 2, J and K), which are a part of the severe systemic inflammatory response syndrome. These parameters, which were measured 12–16 hours after injection and before euthanasia was initiated, were also normalized by lipase inhibition.
Figure 2. Effect of visceral fat lipolysis by pancreatic lipase injection in ob/ob mice without pancreatitis.

(A) Survival curves of the mice given lipase alone (red) or lipase along with the lipase inhibitor orlistat (Lipase+orli; black line). (B) Gross appearance of the pancreas (yellow oval) and visceral fat (yellow rectangle) in situ at the time of necropsy. (C) Pancreatic lipase activity in the gonadal fat pads of these mice at the time of necropsy. (D) Thin layer chromatography comparing the relative amount of NEFAs and triglyceride in these fat pads. STD., standards; TG, glyceryl trioleate (triglyceride standard); NEFA; oleic acid, a NEFA standard. (E) Individual NEFA concentrations in the fat pads of the mice as measured by gas chromatography. (F–H) Serum cytokines at the time of necropsy in controls (Con) and other groups mentioned below the respective bars. (I) Serum BUN measured at the time of necropsy, and rectal temperature (J) and carotid pulse distention (pulse dist.) (K) measured in the lipase-treated group at the last monitoring time before necropsy (12–16 hours after first injection) or in the orlistat group after 24 hours, and just before elective euthanasia. #P < 0.05, indicates significant increase over untreated controls; *P < 0.05, indicates a significant difference between the lipase and Lipase+orli. groups. Significance determined by Mann-Whitney test. Error bars represent SD. There were 7 mice per group.
Because NEFA generation immediately follows lipolysis, we went on to test whether NEFA alone could result in an inflammatory response and organ failure. Noting that OA (C18:1) comprises a high proportion of NEFA in both human pancreatic necrosis collections and fat necrosis in mice, we studied the effect of administering OA to lean (CD-1) mice. OA (0.3% body weight), when given intraperitoneally to simulate visceral fat necrosis, increased serum OA concentrations (311 ± 103 μM vs. 132 ± 53 μM in controls, P < 0.003; Figure 3A). OA-treated mice became progressively less active, requiring euthanasia at 48 ± 13 hours. At this time there was a large increase noted in serum cytokines IL-6, MCP-1, and TNF-α (Figure 3, B–D) and BUN (Figure 3E) in the OA-treated group versus controls. Prior to euthanasia the OA-treated mice were in shock and hypothermic (Figure 3, F and G). OA treatment increased TUNEL-positive cells in the lung from 0.2% ± 0.1% to 2.7% ± 1.4% (P < 0.001, Figure 3H), similar to acute respiratory distress syndrome in rodents (24) and humans (51).
Figure 3. Relevance of oleic acid to systemic injury in mice and during human severe pancreatitis.

(A–E) Blood levels of serum oleic acid (OA) (A), IL-6 (B), MCP-1 (C), TNF-α (D), and BUN (E) in the control (Con) and OA-treated groups of mice at the time of euthanasia. (F) Carotid pulse distention and (G) rectal temperatures prior to euthanasia of these mice. (H) Representative images from lung histologic sections stained by TUNEL (green) with nuclei stained red. Arrows point to the positive-staining nuclei. Scale bars: 250 μm. There were 8 mice in each group. (I) Serum OA levels in the control patient group (Con Pts.) and severe acute pancreatitis (SAP) patients from the PROOF cohort. *P < 0.05 by Mann-Whitney test. Error bars represent SD.
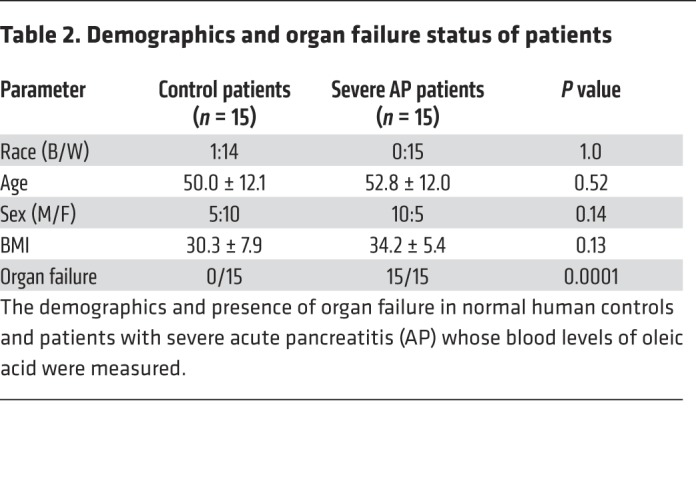
To test the clinical relevance of our findings with OA in a blinded fashion, OA concentrations were measured in a separate cohort of patients at the University of Pittsburgh (detailed in Table 2). Some of these had severe acute pancreatitis, which is characterized by the presence of organ failure for more than 48 hours (52). OA levels in severe acute pancreatitis patients (n = 15) were significantly higher versus normal controls (n = 15) who were demographically similar (Figure 3I). OA levels in patients were similar to the levels noted in OA-treated and control CD-1 mice (Figure 3A).
Table 2. Demographics and organ failure status of patients.
Cumulatively, these results show that during acute pancreatitis, unlike diverticulitis, pancreatic lipase can hydrolyze visceral adipose tissue and generate NEFAs, which cause multisystem organ failure independently of intrapancreatic phenomena. We further validated these findings using the standard caerulein acute pancreatitis model in lean and obese (ob/ob) mice. In ob/ob mice, fat necrosis was first noted at 12 hours near the pancreas (Supplemental Figure 2A, red rectangles). This increased in amount and distance from the pancreas over time and appeared similar to what was noted after lipase injection (Figure 2B). Increased PNLIP and PNLIPRP2 were noted in the fat by 6 hours (Supplemental Figure 2B), and reduced amounts of ATGL and perilipin 1 (Supplemental Figure 2K) were noted after 12 hours, which correlated with an increase in pancreatic enzymes like trypsin and amylase in the necrotic fat (Supplemental Figure 2L). Trypsin has been previously shown to hydrolyze ATGL (53). Circulating amylase and lipase activity, which are markers of ongoing acute pancreatitis, were similarly elevated during acute pancreatitis (Supplemental Figure 2, C and D) in ob/ob and lean C57BL/6J mice. However, unlike lean mice (body fat 2.8 ± 0.2 g vs. 17.1 ± 1.8 g) that developed little or no fat necrosis and organ failure (Supplemental Figure 3), serum NEFAs increased in ob/ob mice (Supplemental Figure 2E) in parallel with serum IL-6, MCP-1, TNF-α, BUN, and reduced survival (Supplemental Figure 2, F–J).
To understand how pancreatic lipases enter adipocytes, we exposed 3T3-LI cells to PNLIP-mCherry in the presence of phospholipase A2, several isoforms of which are involved in acute pancreatitis (54–56). This allowed the entry of PNLIP into the adipocytes (Supplemental Figure 4A). Similarly, PNLIPRP2, which has a phospholipase-like activity (27), allowed the entry of a live/dead marker into these cells (Supplemental Figure 4B). The injurious role of PNLIPRP2 (unlike its inactive S148G mutant or PNLIP) was supported by its expression in HEK293 cells causing a significant increase in LDH leakage versus PNLIP (Supplemental Figure 4C). Lastly, exposure of 3T3-L1 adipocytes to linoleic acid (LA), previously shown to be present at approximately 2 mM in acute pancreatitis collections, also injured adipocytes, leading to entry of PNLIP into the cells (Supplemental Figure 4D). Thus, pancreatic enzymes including lipases, which may leak from a disrupted duct (57, 58) or basolaterally during human (44–46) and experimental pancreatitis (47–49), can enter the surrounding visceral adipocytes in multiple ways and cause organ failure. Therefore, we went on to identify the relative contribution of pancreatic lipases versus ATGL to acute pancreatitis–associated fat necrosis, and consequent pancreatic acinar necrosis, the source of pancreatic lipase leakage.
Pharmacological inhibition or genetic deletion of ATGL is not sufficient to prevent adipocyte-induced lipotoxic acinar injury.
As previously shown, NEFAs injure pancreatic acini (14, 59) and other cell types such as the kidney cell line HEK293 (40). To initially identify the lipase(s) mediating the cell injury resulting from fat necrosis, we used a previously described method (14, 43). Using this method, we cocultured wild-type (WT) adipocytes and pancreatic acini alone or in the presence of Atglistatin (50 μM), the ATGL-specific inhibitor (35) that does not inhibit pancreatic lipases, or the generic lipase inhibitor orlistat (50 μM) (Figure 4, A–D). As previously shown (14, 43), during coculture these were separated by a 3-μm pore size mesh (Figure 4A). This system, therefore, allows the macromolecules released from one cell type to interact with the other cell type, thus simulating the leakage of pancreatic lipases into adipose tissue noted previously during human (44–46) and mouse acute pancreatitis (14, 43). This model is relevant to the current study (Figure 1, Figure 2, and Supplemental Figures 2–4) because it allows the leaked pancreatic enzymes access to adipocytes and their triglycerides in the lower chamber (Figure 4A), and conversely allows glycerol and NEFAs from adipocytes access to acini in the upper chamber, where these molecules can be measured. Injury to acini (or its prevention thereby) can thus be measured by staining the acini in the upper chamber with a live/dead marker such as trypan blue or propidium iodide, as shown previously (14, 43).
Figure 4. Comparison of the effects of pharmacologic or genetic inhibition of ATGL or PNLIP on adipocyte-induced acinar injury.

(A) Schematic showing the setup of the pancreatic acini/adipocyte coculture experiment using a Transwell system with a 3-μm mesh separating the acini from the adipocytes. (B–E) Effect of the generic lipase inhibitor orlistat (50 μM) and the ATGL-specific inhibitor Atglistatin (50 μM) on the pancreatic lipase activity in the medium (B), glycerol generation in the medium (C), and propidium iodide (PI) uptake (D) and trypan blue staining (E) of pancreatic acini at the end of 6 hours of coculture. (F) Activity of pancreatic lipase and amylase in the media from acini of WT or PNLIP-KO mice. Effect of coculturing acini from WT and PNLIP-KO mice with WT adipocytes on glycerol generation (G), propidium iodide uptake (H), and trypan blue staining (I) of acini at the end of 6 hours of coculture. Effect of culturing WT acini alone, with WT adipocytes or ATGL-KO adipocytes (with and without 50 μM orlistat [orli]) on the measurable pancreatic lipase activity in the medium (J), glycerol concentration in the medium (K), propidium iodide uptake (L), and trypan blue staining (M) by the acini. Original magnification, ×10 (E, I, and M). Each experiment was performed 3–7 times separately. Each symbol represents a different experiment. *P < 0.05 by ANOVA, indicating a significant change compared with other conditions in the group. †P < 0.05 by Student’s t test, indicating a significant and selective reduction with orlistat (but not Atglistatin or deletion of ATGL as may be relevant to the experimental design). Error bars represent SEM.
We initially noted that the generic lipase inhibitor orlistat inhibited pancreatic lipase activity, and reduced the lipolytic generation of glycerol and consequent lipotoxic necrosis of acini (14), as measured by their uptake of propidium iodide and trypan blue (Figure 4, B–E). Atglistatin, however, did not inhibit these phenomena despite potent inhibition of isoproterenol induced lipolysis (Supplemental Figure 5). In contrast, the medium in which PNLIP-KO acini were suspended had reduced pancreatic lipase compared with medium from WT acini, whereas amylase activity in the 2 media was similar (Figure 4F). Coculture of PNLIP-KO acini with WT adipocytes reduced glycerol generation, propidium iodide uptake, and trypan blue staining (Figure 4, G–I). Although adipocytes from ATGL-KO acini did have reduced lipolytic response to isoproterenol (Supplemental Figure 5), as previously shown (30), coculture of adipocytes from ATGL-KO mice with WT acini caused no reduction in pancreatic lipase activity, glycerol generation, or protection from acinar necrosis (Figure 4, J–M). These data suggest that PNLIP, rather than ATGL, mediates the unregulated lipolysis of adipocyte triglycerides, the subsequent NEFA release, and lipotoxic cell injury (14, 43). These findings, along with (a) in vivo evidence that visceral fat necrosis is mediated by pancreatic lipases (Figure 2) and contains PNLIP, PNLIPRP2, but less ATGL (Figure 1, I, J, N, and P, and Supplemental Figure 2B); and results showing that (b) visceral fat necrosis generates NEFAs (Figure 1L and Figure 2, D and E); (c) NEFAs cause organ failure (Figure 3, A–H); and (d) NEFAs are increased in the sera (Figure 3I) and pancreatic collections of patients with severe acute pancreatitis (14, 20, 60), but not diverticulitis (Figure 1L) in whom organ failure is rare, led us to compare the role of pancreatic lipases and ATGL in mediating the organ failure of severe acute pancreatitis in vivo.
PNLIP (but not PNLIPRP2) mediates lipotoxic NEFA generation without affecting lipotoxic NEFA signaling.
We first chose to identify the pancreatic lipase to be targeted. Pancreatic necrosis collections are enriched in NEFAs with 16 or more carbons such as OA and LA (14, 34, 60). Triglycerides esterified to these long-chain fatty acids serve as poor substrates for the bile salt–dependent CEL (38–40). Thus, we focused our attention on PNLIP (black/white/gray) and PNLIPRP2 (burgundy/brownish green) by comparing their relative contributions in lipolytic release of lipotoxic mediators relevant to acute pancreatitis (Figure 5). At equimolar amounts, both enzymes from mice, similar to the human forms (40), were effective in hydrolyzing the triglyceride of LAs, i.e., glyceryl trilinoleate (GTL) (Figure 5, A and B). The lipase activity in pancreatic homogenates of PNLIP-KO mice, however, was only 10% of the WT mice (Figure 5C), in contrast to the activity in homogenates of PNLIPRP2-WT and -KO mice, which were similar. Consistent with this finding, PNLIPRP2-WT and -KO acini had similar glycerol generations and injury (LDH leakage) after 2 hours of incubation with 300 μM GTL (Figure 5, D and E). PNLIP-KO acini, however, generated less glycerol and caused less injury than WT acini (Figure 5, F and G) despite being equally susceptible to the direct lipotoxic effect of LA (600 μM). Real-time analysis of mitochondrial depolarization and cytosolic calcium increase (Figure 5, H and I) revealed PNLIP-KO acini to have a blunted response to GTL, while responding similarly to LA. These protective effects were not due to a generic decrease in exocrine enzyme release by the cells because only the lipase activity, but not amylase activity, was reduced in the medium of the PNLIP-KO acini, resulting in a reduced NEFA generation (Figure 5, J–L). Thus, the protection in PNLIP-KO acini is conferred by reducing triglyceride lipolysis and not by interference with the lipotoxic mechanisms of NEFAs. We next sought to compare the role of PNLIP versus ATGL in severe acute pancreatitis–associated fat necrosis using obese ATGL-KO or obese PNLIP-KO mice.
Figure 5. Comparison of lipolysis by PNLIP and PNLIPRP2, and their roles in acinar injury due to triglyceride lipolysis or direct fatty acid lipotoxicity.

NEFA and glycerol generation from hydrolysis of glyceryl trilinoleate (GTL, 300 μM) by recombinant murine PNLIP (A) and PNLIPRP2 (B), each at 1 μg/mL. The y axis is labeled 100% lipolysis, signifying complete lipolysis of the added GTL (300 μM) generating 300 μM glycerol or 900 μM fatty acid. (C) Comparison of pancreatic lipase activity/tissue weight in homogenates of PNLIP-KO (gray) or PNLIPRP2-KO (brownish green) mice versus their WT litter mates (white and burgundy, respectively). Bar graphs showing the effect of incubating acini from WT mice or PNLIPRP2-KO mice on glycerol concentrations (D and F) and LDH leakage (E and G) in the medium. CON indicates under control state without added GTL, and GTL indicates after incubating with 300 μM GTL for 2 hours. LA indicates 600 μM linoleic acid. (H–L) Representative graphs comparing mitochondrial depolarization using JC-1 (H) and cytosolic calcium increase using Fura-2 AM (I) in WT and PNLIP-KO acini, and the lipase release (J), amylase release (K), and NEFAs present (L) at the end of the 15-minute incubation in the medium of the respective cuvettes. *P < 0.05 by Student’s t test. Each experiment was performed 3–5 times separately. Error bars represent SEM.
Deficiency of PNLIP, but not ATGL, prevents visceral adipose tissue lipolysis during severe acute pancreatitis and improves outcomes.
To confirm homogeneity between different animal groups, we first compared the body weight, adipose weight, and fatty acid composition of the gonadal fat pads (that become necrotic) of ob/ob ATGL-KO and ob/ob PNLIP-KO mice and their ob/ob littermates (Figure 6, A and B). These parameters were similar in all 3 groups. Similarly, the increase in pancreatic edema (measured as percentage water weight/tissue wet weight) and circulating amylase 24 hours after induction of pancreatitis were similar in all 3 groups (Figure 6, C and D). These results indicate that all groups had a similar susceptibility to acute pancreatitis. The PNLIP-KO group, however, had lower circulating pancreatic lipase levels at baseline and throughout the course of acute pancreatitis (Figure 6E). During acute pancreatitis, ob/ob mice and ob/ob ATGL-KO mice developed comparable fat necrosis involving about one-third of the fat in and around the pancreas (Figure 6, F and G, and Supplemental Figures 6 and 7). The pancreas adjacent to the fat necrosis was also similarly necrosed (referred to as perifat acinar necrosis [PFAN]; yellow rectangles in Figure 6F), contributing to half the acinar necrosis. These findings are morphologically similar to human acute pancreatitis (Figure 1, I–K) and supported by previous studies in humans (14, 43). Obese PNLIP-KO mice, however, had dramatically reduced fat necrosis, PFAN, and total necrosis (Figure 6, F and G, and Supplemental Figures 6 and 7). These results are supported by findings of the triglyceride in fat pads of the PNLIP-KO mice being unhydrolyzed (Supplemental Figure 8A). Instead, by the fifth day of pancreatitis, when these mice were electively sacrificed, they had developed acinar-ductal metaplasia involving about half the pancreas and extensive fibrosis (Supplemental Figures 8B and 9), consistent with sustained caerulein pancreatitis, which can progress to chronic pancreatitis (61, 62). These findings suggested that genetic deletion of PNLIP does not affect the induction of acute pancreatitis by caerulein but rather influences the progression via protection from fat necrosis.
Figure 6. Parameters of obesity and caerulein-induced acute pancreatitis in genetically obese littermate (ob/ob; black) mice, ob/ob mice with genetically deleted ATGL (ATGL-KO; blue), or ob/ob mice with genetically deleted PNILP (PNLIP-KO; gray).

(A) Body weight and total fat content of the mice at baseline. (B) NEFAs composing visceral triglyceride (TG). (C) Pancreatic edema (percentage water content in wet weight of the pancreas), in control mice (CON) and those with acute pancreatitis (AP). Serum amylase (D) and lipase (E) levels at baseline, 24 hours after pancreatitis and after euthanasia in the 3 strains of mice. (F) H&E staining of pancreas and surrounding fat. Note the presence of fat necrosis (FN) in the ob/ob and ob/ob ATGL-KO mice with acute pancreatitis. This is adjacent to the pancreas, which is severely damaged (peri-fat acinar necrosis; PFAN, shown in dashed yellow rectangles). The non-necrosed fat is listed as “fat.” Dashed yellow oval in the ob/ob PNLIP-KO mouse panel highlights the dilated lumens of the acini (acinar-ductal metaplasia), consistent with the chronic caerulein hyperstimulation over the 5 days. Scale bars: 75 μm. (G) Box plots comparing the areas of pancreatic necrosis, fat necrosis, PFAN, and acinar ductal metaplasia in the 3 groups with pancreatitis. Box plots depict mean (dashed line), median (solid line), 25th and 75th percentiles (2 boxes), 10th and 90th percentiles (whiskers), and outliers (dots). (J) Kidney TUNEL staining (brown nuclei, arrows highlight some) of the various groups. *P < 0.05, indicates a significant increase over controls or baseline from the same genetic group without pancreatitis by ANOVA. #P < 0.05, indicates a significant difference in the PNLIP-KO mice versus other genetic background littermate mice in the same group of pancreatitis by ANOVA. Each group had 8–10 mice.
In contrast to PNLIP, the lack of ATGL’s role in lipolytic fat necrosis during acute pancreatitis was further supported by the ob/ob ATGL-KO mice having grossly visible visceral fat necrosis similar to the ob/ob mice (Figure 7A) with pancreatitis. ATGL-KO mice had a similar increase in pancreatic lipase activity (Figure 7B) and PNLIP (Figure 7C) in the visceral fat by Western blotting, along with an increase in serum C18:1 (OA) levels (Figure 7D), unlike ob/ob PNLIP-KO mice with acute pancreatitis. Interestingly, the serum C18:1 (OA) levels in ob/ob mice with pancreatitis (276 ± 60 μM) and ATGL-KO mice with pancreatitis (296 ± 92 μM) were similar to those noted in humans with severe acute pancreatitis (Figure 3I) and CD-1 mice administered OA (Figure 3A). ATGL-KO mouse pancreatitis also had a pattern of proinflammatory cytokine mRNA expression in visceral adipose tissue and serum similar to the ob/ob mice with pancreatitis (Figure 7, E–H). Similarly, systemic injury reflected by elevated BUN, kidney, and lung TUNEL positivity, shock (noted as a drop in carotid artery pulse distension), and/or hypothermia were equally severe in the ob/ob and ob/ob ATGL-KO mice with acute pancreatitis (Figure 7, I–L, and Supplemental Figure 10). This was unlike in the ob/ob PNLIP-KO mice, whose parameters for organ failure, even at the end of the study, were no different from control mice without acute pancreatitis and which had a 100% survival rate (Figure 7M).
Figure 7. Inflammation and severity in ob/ob PNLIP-KO (gray), ob/ob littermates (black), and ob/ob ATGL-KO (blue) mice with caerulein-induced acute pancreatitis (CER AP).

(A) Peritoneal cavity of controls and during pancreatitis. Black rectangle shows the pancreas, and adjacent kidney (K), perinephric fat. Note pancreas (P) (magnified view of inset in lower panel) is edematous, lobulated in pancreatitis. Arrows highlight fat necrosis around the kidneys, which is lacking in PNLIP-KO mice. (B) Pancreatic lipase activity in the gonadal fat pads. (C) Western blot of visceral adipose for ATGL with GAPDH as a loading control (same gel, noncontiguous) and a contemporaneously run gel for PNLIP with a vinculin loading control (same gel, noncontiguous). Dotted lines depict splicing junctions. (D) Serum NEFAs in pancreatitis and controls (white bar). (E) mRNA levels of cytokines in the visceral adipose tissue from mice with pancreatitis showing differences in the levels versus control mice without pancreatitis. Serum (Sr.) IL-6 (F), TNF-α (G), MCP-1 (H), and BUN (I) in the various groups. *P < 0.05 versus controls without pancreatitis by ANOVA. Box plots depict mean (dashed line), median (solid line), 25th and 75th percentiles (2 boxes), 10th and 90th percentiles (whiskers), and outliers (dots). (J) Kidney TUNEL staining (brown nuclei, arrows highlight some) of the various groups. Note the increase in the ob/ob and ob/ob ATGL-KO mice with pancreatitis. Scale bars: 100 μm. Trends of the carotid pulse distension (K) to measure shock, rectal temperature (L) to measure organ failure (shown as mean ± SEM), and survival (M) in the various groups. The dashed lines are PNLIP-KO mice with pancreatitis. *P < 0.05 versus other groups by ANOVA. †P < 0.05, indicates a significant reduction in the ob/ob PNLIP-KO group with acute pancreatitis versus the ob/ob group by ANOVA. Each group had 8–10 mice.
We went on to test whether the role of PNLIP was relevant to a mechanistically different acute pancreatitis model using IL-12 and -18 (Supplemental Figure 11), which are cytokines increased in human severe acute pancreatitis (63, 64). All ob/ob mice with acute pancreatitis were moribund by day 4 after induction (Supplemental Figure 11A), as shown previously. The ob/ob PNLIP-KO mice, however, were fully protected over the 5-day course of the study and had nearly normal circulating amylase and lipase (Supplemental Figure 11, B and C) at the time of elective euthanasia. On necropsy, these mice had grossly reduced visceral fat necrosis and no increase in measurable lipase activity in the fat pads (Supplemental Figure 11, D and G). This was verified microscopically and was associated with reduced pancreatic necrosis (Supplemental Figure 11, E and F), even though acinar-ductal metaplasia was increased. The severe visceral adipose tissue necrosis was associated with a large increase in circulating NEFAs, including C18:1, and a severe inflammatory response, both of which were reduced in the PNLIP-KO mice (Supplemental Figure 11, H–K). Additionally, ob/ob PNLIP-KO mice with IL-12– and IL-18–induced acute pancreatitis were protected from renal failure, shock, and hypothermia (Supplemental Figure 11, L–N), all of which occurred in the ob/ob mice prior to them becoming moribund. Therefore, PNLIP lipolytically worsens fat necrosis and acute pancreatitis outcomes in 2 mechanistically distinct acute pancreatitis models.
Deficiency of PNLIP activity does not affect the signaling that leads to the initiation of acute pancreatitis.
Because acute pancreatitis is initiated in pancreatic acinar cells (65), we went on to check whether PNLIP affected the induction of acute pancreatitis by comparing the signaling induced by caerulein in the acini of WT and PNLIP-KO mice (gray, Figure 8, A–C). Caerulein acts via the cholecystokinin receptor and at high doses (>10–9 M) relevant to acute pancreatitis, it causes a sustained increase in cytosolic calcium (66, 67). The 2 types of acini showed no difference in the caerulein-induced increase in cytosolic calcium, pattern of exocrine enzyme amylase secretion, or the generation of trypsin (Figure 8, A–C). Thus, the protection seen in the ob/ob PNLIP-KO mice during caerulein-induced acute pancreatitis is not due to interference with the caerulein signaling leading to acute pancreatitis or the function of cells in which acute pancreatitis is initiated. We further went on to study the validity of this in vivo by comparing lean WT and lean PNLIP-KO mice during a short 10-hour course of caerulein-induced acute pancreatitis. Here, the PNLIP-KO mice were not protected from the increase in circulating amylase (Figure 8D), pancreatic edema (measured as percentage water weight/wet weight; Figure 8E), and acinar necrosis (arrows showing diffuse pink cytoplasm and loss of cell outlines, Figure 8F), despite preventing the increase in circulating lipase compared with WT mice (Figure 8G). Consistent with the short course of acute pancreatitis and relative paucity of visceral adipose tissue in lean mice, neither strain had an increase in serum NEFAs, BUN, or loss of thermoregulation with acute pancreatitis (Supplemental Figure 12, A–C). Similarly, during IL-12– and IL-18–induced pancreatitis, PNLIP-KO mice had a similar increase in serum amylase at 24 hours and pancreatic edema versus the WT mice (Figure 8, H and I). However, the increase in circulating lipase was completely prevented in the PNLIP-KO mice (Figure 8J). Again, serum BUN and rectal temperature were similar (Supplemental Figure 12, D and E) between WT and PNLIP-KO mice. Based on these findings, we can conclude that the protection noted in the PNLIP-KO mice is not due to interference with the initiation of acute pancreatitis. Rather, these mice are protected from the lipotoxicity that ensues from excessive visceral adipose tissue lipolysis mediated by the leakage of PNLIP into the visceral adipose tissue (Figure 9), which causes systemic inflammation and organ failure.
Figure 8. Comparison of acinar cell signaling and early events during pancreatitis in PNLIP-KO (gray) versus WT mice (black).

(A) Cytosolic calcium increase in response to 100 nM caerulein (arrow) as measured in Fura-2 AM–loaded acini (mean ± SEM of 4 different experiments). (B and C) Comparison of physiologic amylase release into the medium (B) and pathologic conversion of trypsinogen to active trypsin (C) in response to physiologic (1 × 10–10 M) and supraphysiologic (pathologic, 1 × 10–7 M) doses of caerulein. Each symbol represents a separate experiment. (D and E) Circulating (plasma, Pl.) amylase (D) and pancreatic edema expressed as the ratio of the water weight to pancreas wet weight (%) (E). (F) Pancreatic histology showing necrosis (arrows). Scale bars: 50 μm. (G) Circulating pancreatic lipase levels measured at the end of 10 hours of caerulein-induced acute pancreatitis versus controls. Note similar parameters in the 2 groups, except lipase. (H–J) Circulating amylase (H), pancreatic edema in control mice and at the time of necropsy after IL-12– and IL-18–induced acute pancreatitis (I), and circulating pancreatic lipase (J) levels measured 24 hours after the first IL-12/IL-18 injection. Note similar parameters in the 2 groups, except lipase. Box plots depict mean (dashed line), median (solid line), 25th and 75th percentiles (2 boxes), 10th and 90th percentiles (whiskers), and outliers (dots). *P < 0.05 by Student’s t test versus controls or basal levels. Each group had 8–10 mice.
Figure 9. Diagrammatic representation of the difference in pathophysiology of acute diverticulitis and acute pancreatitis.

Shown here are the pathophysiology explaining the histological, biochemical, and clinical findings noted in diverticulitis (left) and pancreatitis (right). In the upper part of the left image is shown a perforated diverticulum in the colon with spillage of contents into the visceral adipose tissue. In the lower panel is the zoomed-in view showing the consequent intense local inflammation in the pericolonic visceral adipose tissue. However, the triglycerides (red E) within adipocytes are not degraded and the ATGL (green dots) remains intact. This is associated with a low prevalence of organ failure during diverticulitis. On the right is shown the involvement of visceral adipose tissue in pancreatitis, with the zoomed-in view at the bottom showing the leakage of pancreatic enzymes (dark blue), including PNLIP (light blue), into the visceral adipose tissue. The resulting adipocyte injury results in entry of PNLIP into the injured adipocytes, resulting in hydrolysis of adipocyte triglycerides into NEFAs (red wavy lines), and degradation of ATGL seen as loss of green dots. The entry of NEFAs into the systemic circulation causes elevated cytokines, organ failure including acute respiratory distress syndrome (ARDS), acute renal failure (ARF), and cardiovascular failure (CVF).
Discussion
In this study we note that acute, excessive visceral adipose tissue lipolysis by pancreatic lipase can cause systemic injury, and then identify PNLIP as the principal lipase mediating this during acute pancreatitis. This explains the higher prevalence of organ failure in acute pancreatitis compared with diverticulitis, despite both diseases being acute, occurring in a similar demographic, and having visceral fat involvement. Diverticulitis, however, does not have ongoing visceral fat lipolysis despite the radiographically apparent visceral fat involvement and intense inflammation seen histologically. As summarized in Figure 9, these studies show that during acute pancreatitis, PNLIP leaks from the injured pancreas into the surrounding adipocytes, with its entry facilitated by multiple mechanisms (Supplemental Figure 4). PNLIP-mediated lipolysis results in the release of large quantities of NEFA-like OA, which cause inflammation and elevated cytokines, thus worsening lung injury, kidney injury, and shock, culminating in multisystem organ failure and reduced survival. We additionally note that PNLIP has no role in the mechanisms leading to the initiation of acute pancreatitis or the signaling mediated by the lipotoxic NEFAs such as LA generated by lipolysis. Remarkably, this cascade of events leading to organ failure is independent of the principal adipocyte lipase, ATGL, which is present in diverticulitis but not pancreatitis.
Previous studies have shown that lipotoxic NEFAs enriched in necrotic pancreatic fluid collections can cause mitochondrial damage, noted as swelling and disorganization of their ultrastructure, along with inhibiting complexes I and V (14). The systemic injury we note as TUNEL-positive cells in the lungs and kidneys has previously been shown using OA (21, 23, 24) and LA (40, 68), and during human ARDS (51). Here we identify PNLIP as the principal lipase mediating this uncontrolled generation of NEFAs from visceral fat lipolysis.
Although visceral adipose stranding is radiologically seen around the affected tissue during acute pancreatitis, appendicitis, acute diverticulitis (69), epiploic appendicitis, and omental infarction (1, 2), its role in predicting severity has only been noted in acute pancreatitis (5–7). This study shows that PNLIP mediates this severity by hydrolyzing visceral fat during acute pancreatitis but not acute diverticulitis, despite the latter having intense inflammation (Figure 1E). This pathophysiology is evidenced by (a) triglycerides being the predominant lipid class in the fat of acute diverticulitis, but NEFAs being the one in acute pancreatitis (Figure 1L); (b) histological evidence of PNLIP in fat necrosis (Figure 1K); (c) biochemical evidence of PNLIP in fat necrosis (Figure 7, B and C, and Supplemental Figure 2B); (d) PNLIP-positive histologic areas staining positive for calcium on von Kossa (Figure 1J and Supplemental Figure 6), signifying saponified NEFAs; (e) pancreatic lipase injection into visceral fat causing fat necrosis (Figure 2B) and generating large amounts of NEFA (Figure 2, D and E), resulting in systemic inflammation and organ failure (Figure 2, F–K); (f) OA that is generated in this fat necrosis, alone being able to induce systemic inflammation and organ failure (Figure 3, A–H); (g) the levels of OA noted in the serum of mice with organ failure being the same as patients with pancreatitis who have organ failure (Figure 3, A and I); and lastly (h) the in vitro cellular models (Figures 4 and 5) and mechanistically distinct mouse pancreatitis models (Figure 6, Figure 7, and Supplemental Figure 11) showing that genetic deletion of PNLIP protects from lipotoxic local and systemic injury.
The mechanism underlying the reduction in adipocyte proteins including ATGL and perilipin 1 in acute pancreatitis–associated fat necrosis (Figure 1P and Supplemental Figure 2, B and K) remains unclear. Trypsin is produced in the pancreas, and we note it to be present in necrotic fat (Figure 1O and Supplemental Figure 2L) along with PNLIP and amylase. Trypsin can degrade ATGL (53), which may explain ATGL reduction in the fat during severe necrotizing pancreatitis (Figure 1P, Figure 7C, and Supplemental Figure 2B). This finding, along with acute pancreatitis–associated fat necrosis in ATGL-KO mice having increased PNLIP resulting in high cytokines, serum NEFA increase, and organ failure (Figure 7), supports these outcomes to be independent of ATGL. This is further supported by the coculture system in which we note adipocyte-induced acinar injury to be dependent on lipolysis (since it is inhibited by orlistat, and in PNLIP-KO mouse acini), which is unaffected by using Atglistatin or ATGL-KO adipocytes (Figure 4). Therefore, although ATGL does play a major role in regulated lipolysis of adipocyte triglycerides such as those whose release is mediated by isoproterenol (Supplemental Figure 5), its role in acute pancreatitis–associated visceral adipose tissue necrosis and organ failure is unlikely.
Among the pancreatic lipases, we note that PNLIP, which contributes to 80%–90% of the pancreas’s lipolytic activity, has a bigger role than PNLIPRP2 (Figure 5, A–E) in lipotoxic cell injury due to the hydrolysis of triglycerides. This relative abundance of PNLIP can explain the protective phenotype we observe in vitro (Figure 5, F–I) and in vivo (Figure 6, Figure 7, and Supplemental Figure 11). The role of another lipase is possible, noting the minimal fat necrosis in the PNLIP-KO mice during IL-12– and IL-18–induced acute pancreatitis (Supplemental Figure 11D), and partial protection from GTL-induced injury in acini from the PNLIP-KO mice (Figure 5, F and G). However, PNLIPRP2’s clinical relevance is likely to be low because about 50% of Europeans are heterozygous and 20% homozygous for a common PNLIPRP2 truncation variant that results in nonsense-mediated decay, thus inhibiting expression of PNLIPRP2 at the mRNA level (70). Because CEL cannot effectively hydrolyze triglycerides with fatty acids of greater than 16 carbon chains (38, 39) and requires bile acid concentrations much higher than in pancreatic necrosis collections (40), it is unlikely to play a major role in fat necrosis.
The protection afforded in the PNLIP-KO acini and in mice is unique because PNLIP does not interfere with mechanisms initiating pancreatitis such as caerulein-induced increases in cytosolic calcium or trypsinogen activation (Figure 8, A and C). This supports the hypothesis that inhibiting or neutralizing a disease modifier (in this case PNLIP) can change disease outcomes even after disease initiation. This finding also allows PNLIP neutralization or inhibition to be a therapeutic strategy, because fat necrosis becomes visible after 12 hours of pancreatitis (Supplemental Figure 2A) and precedes organ failure (Supplemental Figure 2I).
The data from the randomly accrued series of human samples, while blinded, are limited by the study design being observational, small in size, and us not controlling for the use of intravenous lipids. Despite these limitations, the differences between diverticulitis and pancreatitis outcomes are significant. Future studies to validate these may be helpful. In the mouse studies, the small group size (n = 3–7) for each sex, may have limited our ability to detect such differences. We also do not focus on the other factors that may influence PNLIP action. For example, we do not detail amount or type of phospholipase(s) or the dose response and time course of PNLIPRP2 that allow PNLIP entry into adipocytes (Supplemental Figure 4). Similarly, we did not study the role of co-lipase, which is PNLIP’s cofactor that increases its activity 3- to 5-fold (26). Additionally, while adiponectin was detectable, ATGL or perilipin 1 underwent proteolysis. This is potentially explained by progressive accumulation of trypsin during fat necrosis in murine acute pancreatitis (Supplemental Figure 2L), and the detectable trypsin activity (Figure 1O) in human acute pancreatitis–associated visceral adipose tissue necrosis. Overall, the genetic and pharmacologic evidence provided make PNLIP and not ATGL the likely mediator of the acute visceral adipose tissue lipolysis that worsens acute pancreatitis.
In summary, we note PNLIP, but not ATGL, to be the principal mediator of excessive and unregulated visceral adipose tissue lipolysis during acute pancreatitis. This phenomenon does not take place in diverticulitis and explains why the visceral adipose tissue involvement in acute pancreatitis parallels disease severity. This also explains the prognostic role of visceral adipose tissue involvement in acute pancreatitis but not diverticulitis and perhaps other acute abdominal diseases. Visceral adipose tissue lipolysis by PNLIP generates a large amount of NEFAs, which mediate severity of acute pancreatitis independently of its initiation, potentially making PNLIP a pharmacologic target to improve outcomes after disease onset.
Methods
Human samples
We compared acute diverticulitis to acute pancreatitis for the following reasons: (a) both are acute, (b) both occur in the same demographic, (c) both have radiographic (e.g., CT scans) and pathologic involvement of visceral fat, yet (d) their outcomes are very different, with a high prevalence of organ failure in pancreatitis versus diverticulitis. We wanted to understand the mechanism of higher organ failure in pancreatitis. All biochemical studies (October 2015 through May 2017) were done as part of a prospectively done observational study using consecutively procured patient waste residual samples remaining after drainage or after testing and processing had been completed on the tissue (in the pathology or microbiology departments) for medically indicated procedures independent of this research, and the residual was ready to be discarded. For example, the samples used for the current studies were residual material from pancreatic necrosis drained as per the revised Atlanta criteria (52) via surgical, endoscopic, or percutaneous debridement. The debridement procedures were clinically indicated and we had no control over what surgery the patient had, or which sample was designated as waste. These specimens were deidentified by an honest broker and the analysts were blinded to the patients’ clinical data until all biochemical analyses and histological analyses were complete. Histology was done on formalin-fixed, paraffin-embedded samples of surgically resected tissue (diverticulitis) of the same patients whose samples were analyzed biochemically (n = 8), or acute pancreatitis sections (n = 8) retrieved from an autopsy database search described in a previous study (14) while excluding autolysis and postmortem changes, as described therein (14). Transportation from the clinical source to the research lab was within 30 minutes of being released. These were kept on ice, aliquoted, and then frozen at –80°C or fixed in formalin. Once 8 samples from each group had been procured and analyzed, the clinical data were mined. Organ failure was defined by a modified Marshall score of 2 or higher for the respiratory, renal, or cardiovascular system, which is used to stratify patients in the revised Atlanta criteria (52).
University of Pittsburgh severe acute pancreatitis patient cohort.
This cohort was used to compare the serum levels of OA in humans with severe acute pancreatitis to the mice with organ failure in a blinded and independent fashion. Samples were from the Pancreatitis-associated Risk of Organ Failure (PROOF) study. PROOF is an observational study at the University of Pittsburgh Medical Center of subjects with acute pancreatitis early in their hospitalization, which recently completed enrollment. The PROOF protocol has been approved by the Institutional Review Board (IRB) of the University of Pittsburgh (Pro00000496) and submitted to the US National Library of Medicine (ClinicalTrials.gov Identifier: NCT03075605). The clinical protocol and cohort characteristics have been previously described (71). For this analysis, serum samples of 15 randomly chosen patients with severe acute pancreatitis and demographically matched controls without pancreatic diseases enrolled in PROOF between 2012 and 2015 were collected at enrollment and subsequent days when available. OA was extracted and quantified using gas chromatographic separation with flame ionization detection, as previously described (72). Data on demographics, etiology, and clinical course were prospectively collected. Severity was defined by the revised Atlanta classification (52).
Mouse studies
Genetic background.
C57BL/6J WT mice, mice with a targeted null allele of Pnlip (PNLIP-KO mice; B6.129(Cg)-Pnliptm1Dyh/J, stock 008884) congenic on C57BL/6J (originally generated by Huggins et al.; ref. 37), and mice with the null Lepob allele of leptin (wt/ob or ob/ob mice; B6.Cg-Lepob/J, stock 00632) congenic on C57BL/6J were obtained from the Jackson Laboratory and mated as described below. Mice with a LoxP-modified Atgl/Pnpla2 allele (Atglfl mice; B6N.129S-Pnpla2tm1Eek/J, stock 24278, originally generated by Sitnick et al.; ref. 36) and mice expressing Cre recombinase under the control of the adipocyte-specific Adipoq promoter (Adipoq-cre mice; ref. 73) (B6;FVB-Tg(Adipoq-cre)1Evdr/J, stock 010803) were likewise obtained from the Jackson Laboratory. Atglfl mice and Adipoq-cre mice were mated to generate adipocyte-specific ATGL-KO (30) and subsequently made congenic on C57BL/6Ntac. To generate PNLIP- and ATGL-KO mice with genetic obesity due to leptin mutation, the breeding schemes in Supplemental Figures 13 and 14, respectively, were used. PNLIP-KO and ATGL-KO mice were serially mated to leptin heterozygous (wt/ob) mice for at least 2 generations with selection of the desired alleles to generate experimental mice in the following groups: (a) ob/ob PNLIP-KO, which were homozygous for both leptin (ob/ob) and PNLIP-KO alleles congenic on C57BL/6J, and (b) ob/ob ATGL-KO, which were homozygous for leptin and ATGLfl alleles and heterozygous for the Cre allele. Although the latter were mixed C57BL/6J (>75%) and C57BL/6Ntac (<25%), confirmatory experiments revealed that these mice had loss of ATGL and its lipolytic action (Figure 7C and Supplemental Figure 5) and were phenotypically similar to the other strains in the study (Figure 6, A and B). Obese littermates homozygous for leptin (ob/ob) and WT for PNLIP or ATGL were used as controls. Mice with a genetic deletion of PNLIPRP2 generated as described previously (74) were used to study its role in comparison to PNLIP. The genotype of all mice was confirmed after agarose gel electrophoresis of polymerase chain reaction amplification of tail DNA following Jackson Laboratory genotyping protocols and using an AccuStart II PCR Genotyping Kit (Quantabio).
PNLIP-KO mice were identified using the following primers: WT forward, CAAACAGCTAATTACTTCAGATGC; KO forward, GCTATCAGGACATAGCGTTGG; and common reverse, GGACAGTGTCTTGCTGGTCTC, yielding a 380-bp band for PNLIP-KO and a 185-bp band for WT. Leptin-deficient ob/ob mice were identified using a 3-primer mix protocol (75) with subsequent confirmation by phenotypic weight gain. ATGLfl mice were identified using the following primers: forward, ATCAGGCAGCCACTCCAAC and reverse, GAGTGCAGTGTCCTTCACCA, yielding a 390-bp band for ATGLfl and a 235-bp band for WT. Adipoq-cre were identified using the following primers: forward, ACGGACAGAAGCATTTTCCA and reverse, GGATGTGCCATGTGAGTCTG, yielding a 200-bp band for Cre.
Mice were housed in a temperature- and humidity-controlled room with 12-hour light/dark cycle, with ad libitum access to standard chow (Purina 5053 diet, LabDiet) and water. Males and females were used in this study.
Body weight and body composition assessment.
Mouse weight, body fat, and lean mass were analyzed by quantitative nuclear magnetic resonance (NMR) as previously described (13) just before the experiments. The remaining methods are described in the supplemental material.
Statistics
Independent variables for in vivo and in vitro studies are shown as bar graphs reported as mean ± SEM. Line graphs were used for continuous variables. Significance levels were evaluated at P < 0.05. Data for multiple groups were compared by 1-way ANOVA versus controls and values significantly different from controls are indicated with asterisks unless otherwise mentioned specifically in the legend (e.g., Figure 2). When comparing 2 groups, a t test or Mann-Whitney test was used, depending on the normality of distribution. Graphing was done using SigmaPlot 12.5 (Systat Software).
Study approvals
All animal protocols used in this study were approved by the Institutional Animal Care and Use Committee at the Mayo Clinic. All human studies were approved by the IRB of the Mayo Clinic (16-000973), University of Pittsburgh (Pro00000496), or Committee for Oversight of Research Involving the Dead, University of Pittsburgh. Written informed consent from participants or their guardians was as per IRB protocol. The rest of the methods are described in the supplemental material.
Author contributions
VPS designed, supervised, and conceptualized the study. Acquisition and analysis of data was facilitated and carried out by CDO, BK, PN, SK, AB, AEP, BB, KSP, AAG, MNM, AM, DMLH, SN, ST, DOF, NF, RP, EEK, MEL, and VPS. DMLH, SN, DOF, GIP, RP, EEK, MEL, and VPS critically evaluated the manuscript. Interpretation of data was done by CDO, BK, PN, SK, AB, AEP, BB, DMLH, GIP, SN, MEL, and VPS. The manuscript was drafted by CDO, BK, AB, SK, and VPS. Statistical analysis was done by CDO, BK, GIP, SK, BB, and VPS. Funding was obtained by VPS. Order of first authorship was based on duration on the project.
Supplementary Material
Acknowledgments
We would like to thank Michael King for the design of Figure 8. The help of the Mayo Clinic Scottsdale, Biospecimens Accessioning and Processing Core, especially Ashley Droddy, Joshua Sandolo, and Darin Posey is greatly appreciated in the efficient procurement and cataloging of the patient samples used in this study. This project was supported by the following grants: R01DK092460 and R01DK119646 from the NIDDK and PR151612 from the DOD (to VPS); and R01DK090166 (to EEK).
Version 1. 01/09/2020
In-Press Preview
Version 2. 03/09/2020
Electronic publication
Version 3. 04/01/2020
Print issue publication
Funding Statement
VPS
VPS
EEK
VPS
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(4):1931–1947.https://doi.org/10.1172/JCI132767.
Contributor Information
Cristiane de Oliveira, Email: crioliva@uol.com.br.
Biswajit Khatua, Email: Khatua.Biswajit@mayo.edu.
Pawan Noel, Email: pnoel@tgen.org.
Sergiy Kostenko, Email: Kostenko.Sergiy@mayo.edu.
Arup Bag, Email: arup.bag@gmail.com.
Bijinu Balakrishnan, Email: bijinu.b@gmail.com.
Krutika S. Patel, Email: krutika2410@gmail.com.
Andre A. Guerra, Email: Guerra.andre@mayo.edu.
Melissa N. Martinez, Email: Martinez.melissa@mayo.edu.
Shubham Trivedi, Email: Trivedi.Shubham@mayo.edu.
Dora M. Lam-Himlin, Email: LamHimlin.dora@mayo.edu.
Sarah Navina, Email: sarahnavina@gmail.com.
Douglas O. Faigel, Email: faigel.douglas@mayo.edu.
Norio Fukami, Email: fukami.norio@mayo.edu.
Rahul Pannala, Email: Pannala.rahul@mayo.edu.
Anna Evans Phillips, Email: evansac3@upmc.edu.
Georgios I. Papachristou, Email: Georgios.Papachristou@osumc.edu.
Erin E. Kershaw, Email: kershawe@pitt.edu.
Mark E. Lowe, Email: lowe@wustl.edu.
Vijay P. Singh, Email: singh.vijay@mayo.edu.
References
- 1.Pereira JM, Sirlin CB, Pinto PS, Jeffrey RB, Stella DL, Casola G. Disproportionate fat stranding: a helpful CT sign in patients with acute abdominal pain. Radiographics. 2004;24(3):703–715. doi: 10.1148/rg.243035084. [DOI] [PubMed] [Google Scholar]
- 2.Tonerini M, Calcagni F, Lorenzi S, Scalise P, Grigolini A, Bemi P. Omental infarction and its mimics imaging features of acute abdominal conditions presenting with fat stranding greater than the degree of bowel wall thickening. Emerg Radiol. 2015;22(4):431–436. doi: 10.1007/s10140-015-1302-0. [DOI] [PubMed] [Google Scholar]
- 3.Avanesov M, et al. Diagnostic prediction of complicated appendicitis by combined clinical and radiological appendicitis severity index (APSI) Eur Radiol. 2018;28(9):3601–3610. doi: 10.1007/s00330-018-5339-9. [DOI] [PubMed] [Google Scholar]
- 4.Kim HY, Park JH, Lee YJ, Lee SS, Jeon JJ, Lee KH. Systematic review and meta-analysis of CT features for differentiating complicated and uncomplicated appendicitis. Radiology. 2018;287(1):104–115. doi: 10.1148/radiol.2017171260. [DOI] [PubMed] [Google Scholar]
- 5.Balthazar EJ, Robinson DL, Megibow AJ, Ranson JH. Acute pancreatitis: value of CT in establishing prognosis. Radiology. 1990;174(2):331–336. doi: 10.1148/radiology.174.2.2296641. [DOI] [PubMed] [Google Scholar]
- 6.Meyrignac O, et al. Acute pancreatitis: extrapancreatic necrosis volume as early predictor of severity. Radiology. 2015;276(1):119–128. doi: 10.1148/radiol.15141494. [DOI] [PubMed] [Google Scholar]
- 7.Bollen TL, et al. A comparative evaluation of radiologic and clinical scoring systems in the early prediction of severity in acute pancreatitis. Am J Gastroenterol. 2012;107(4):612–619. doi: 10.1038/ajg.2011.438. [DOI] [PubMed] [Google Scholar]
- 8.Cirocchi R, et al. Is the outpatient management of acute diverticulitis safe and effective? A systematic review and meta-analysis. Tech Coloproctol. 2019;23(2):87–100. doi: 10.1007/s10151-018-1919-6. [DOI] [PubMed] [Google Scholar]
- 9.Garg PK, Singh VP. Organ failure due to systemic injury in acute pancreatitis. Gastroenterology. 2019;156(7):2008–2023. doi: 10.1053/j.gastro.2018.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sennello JA, et al. Interleukin-18, together with interleukin-12, induces severe acute pancreatitis in obese but not in nonobese leptin-deficient mice. Proc Natl Acad Sci U S A. 2008;105(23):8085–8090. doi: 10.1073/pnas.0804091105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zyromski NJ, et al. A murine model of obesity implicates the adipokine milieu in the pathogenesis of severe acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2008;295(3):G552–G558. doi: 10.1152/ajpgi.90278.2008. [DOI] [PubMed] [Google Scholar]
- 12.Misumi I, Starmer J, Uchimura T, Beck MA, Magnuson T, Whitmire JK. Obesity expands a distinct population of T cells in adipose tissue and increases vulnerability to infection. Cell Rep. 2019;27(2):514–524.e5. doi: 10.1016/j.celrep.2019.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel K, et al. Lipolysis of visceral adipocyte triglyceride by pancreatic lipases converts mild acute pancreatitis to severe pancreatitis independent of necrosis and inflammation. Am J Pathol. 2015;185(3):808–819. doi: 10.1016/j.ajpath.2014.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Navina S, et al. Lipotoxicity causes multisystem organ failure and exacerbates acute pancreatitis in obesity. Sci Transl Med. 2011;3(107):107ra110. doi: 10.1126/scitranslmed.3002573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tschöp J, et al. CNS leptin action modulates immune response and survival in sepsis. J Neurosci. 2010;30(17):6036–6047. doi: 10.1523/JNEUROSCI.4875-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shapiro NI, et al. Leptin exacerbates sepsis-mediated morbidity and mortality. J Immunol. 2010;185(1):517–524. doi: 10.4049/jimmunol.0903975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niiyama S, Takasu O, Sakamoto T, Ushijima K. Intraperitoneal adipose tissue is strongly related to survival rate in a mouse cecal ligation and puncture model. Clin Transl Immunology. 2016;5(2):e64. doi: 10.1038/cti.2016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Domschke S, Malfertheiner P, Uhl W, Büchler M, Domschke W. Free fatty acids in serum of patients with acute necrotizing or edematous pancreatitis. Int J Pancreatol. 1993;13(2):105–110. doi: 10.1007/BF02786078. [DOI] [PubMed] [Google Scholar]
- 19.Sztefko K, Panek J. Serum free fatty acid concentration in patients with acute pancreatitis. Pancreatology. 2001;1(3):230–236. doi: 10.1159/000055816. [DOI] [PubMed] [Google Scholar]
- 20.Durgampudi C, et al. Acute lipotoxicity regulates severity of biliary acute pancreatitis without affecting its initiation. Am J Pathol. 2014;184(6):1773–1784. doi: 10.1016/j.ajpath.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu RP, et al. Protective effect of low potassium dextran solution on acute kidney injury following acute lung injury induced by oleic acid in piglets. Chin Med J. 2012;125(17):3093–3097. [PubMed] [Google Scholar]
- 22.Dettelbach MA, Deftos LJ, Stewart AF. Intraperitoneal free fatty acids induce severe hypocalcemia in rats: a model for the hypocalcemia of pancreatitis. J Bone Miner Res. 1990;5(12):1249–1255. doi: 10.1002/jbmr.5650051210. [DOI] [PubMed] [Google Scholar]
- 23.Lai JP, Bao S, Davis IC, Knoell DL. Inhibition of the phosphatase PTEN protects mice against oleic acid-induced acute lung injury. Br J Pharmacol. 2009;156(1):189–200. doi: 10.1111/j.1476-5381.2008.00020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hussain N, Wu F, Zhu L, Thrall RS, Kresch MJ. Neutrophil apoptosis during the development and resolution of oleic acid-induced acute lung injury in the rat. Am J Respir Cell Mol Biol. 1998;19(6):867–874. doi: 10.1165/ajrcmb.19.6.3118. [DOI] [PubMed] [Google Scholar]
- 25.Wang S, et al. Lipolysis and the integrated physiology of lipid energy metabolism. Mol Genet Metab. 2008;95(3):117–126. doi: 10.1016/j.ymgme.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 26.Giller T, Buchwald P, Blum-Kaelin D, Hunziker W. Two novel human pancreatic lipase related proteins, hPLRP1 and hPLRP2. Differences in colipase dependence and in lipase activity. J Biol Chem. 1992;267(23):16509–16516. [PubMed] [Google Scholar]
- 27.De Caro J, et al. Characterization of pancreatic lipase-related protein 2 isolated from human pancreatic juice. Biochim Biophys Acta. 2004;1701(1-2):89–99. doi: 10.1016/j.bbapap.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 28.Lowe ME. The triglyceride lipases of the pancreas. J Lipid Res. 2002;43(12):2007–2016. doi: 10.1194/jlr.R200012-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Schreiber R, Xie H, Schweiger M. Of mice and men: The physiological role of adipose triglyceride lipase (ATGL) Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864(6):880–899. doi: 10.1016/j.bbalip.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schoiswohl G, et al. Impact of reduced ATGL-mediated adipocyte lipolysis on obesity-associated insulin resistance and inflammation in male mice. Endocrinology. 2015;156(10):3610–3624. doi: 10.1210/en.2015-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parajuli N, et al. Atglistatin ameliorates functional decline in heart failure via adipocyte-specific inhibition of adipose triglyceride lipase. Am J Physiol Heart Circ Physiol. 2018;315(4):H879–H884. doi: 10.1152/ajpheart.00308.2018. [DOI] [PubMed] [Google Scholar]
- 32.Prasada R, Dhaka N, Bahl A, Yadav TD, Kochhar R. Prevalence of cardiovascular dysfunction and its association with outcome in patients with acute pancreatitis. Indian J Gastroenterol. 2018;37(2):113–119. doi: 10.1007/s12664-018-0826-0. [DOI] [PubMed] [Google Scholar]
- 33.Halonen KI, Pettilä V, Leppäniemi AK, Kemppainen EA, Puolakkainen PA, Haapiainen RK. Multiple organ dysfunction associated with severe acute pancreatitis. Crit Care Med. 2002;30(6):1274–1279. doi: 10.1097/00003246-200206000-00019. [DOI] [PubMed] [Google Scholar]
- 34.Noel P, et al. Peripancreatic fat necrosis worsens acute pancreatitis independent of pancreatic necrosis via unsaturated fatty acids increased in human pancreatic necrosis collections. Gut. 2016;65(1):100–111. doi: 10.1136/gutjnl-2014-308043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mayer N, et al. Development of small-molecule inhibitors targeting adipose triglyceride lipase. Nat Chem Biol. 2013;9(12):785–787. doi: 10.1038/nchembio.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sitnick MT, et al. Skeletal muscle triacylglycerol hydrolysis does not influence metabolic complications of obesity. Diabetes. 2013;62(10):3350–3361. doi: 10.2337/db13-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huggins KW, Camarota LM, Howles PN, Hui DY. Pancreatic triglyceride lipase deficiency minimally affects dietary fat absorption but dramatically decreases dietary cholesterol absorption in mice. J Biol Chem. 2003;278(44):42899–42905. doi: 10.1074/jbc.M303422200. [DOI] [PubMed] [Google Scholar]
- 38.Lombardo D, Fauvel J, Guy O. Studies on the substrate specificity of a carboxyl ester hydrolase from human pancreatic juice. I. Action on carboxyl esters, glycerides and phospholipids. Biochim Biophys Acta. 1980;611(1):136–146. doi: 10.1016/0005-2744(80)90049-2. [DOI] [PubMed] [Google Scholar]
- 39.Fontbonne H, Brisson L, Vérine A, Puigserver A, Lombardo D, Ajandouz el H. Human bile salt-dependent lipase efficiency on medium-chain acyl-containing substrates: control by sodium taurocholate. J Biochem. 2011;149(2):145–151. doi: 10.1093/jb/mvq132. [DOI] [PubMed] [Google Scholar]
- 40.Khatua B, et al. Carboxyl ester lipase may not mediate lipotoxic injury during severe acute pancreatitis. Am J Pathol. 2019;189(6):1226–1240. doi: 10.1016/j.ajpath.2019.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 42.Pini M, Sennello JA, Cabay RJ, Fantuzzi G. Effect of diet-induced obesity on acute pancreatitis induced by administration of interleukin-12 plus interleukin-18 in mice. Obesity (Silver Spring) 2010;18(3):476–481. doi: 10.1038/oby.2009.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Acharya C, et al. Fibrosis reduces severity of acute-on-chronic pancreatitis in humans. Gastroenterology. 2013;145(2):466–475. doi: 10.1053/j.gastro.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aho HJ, Sternby B, Nevalainen TJ. Fat necrosis in human acute pancreatitis. An immunohistological study. Acta Pathol Microbiol Immunol Scand A. 1986;94(2):101–105. doi: 10.1111/j.1699-0463.1986.tb02970.x. [DOI] [PubMed] [Google Scholar]
- 45.Aho HJ, Sternby B, Kallajoki M, Nevalainen TJ. Carboxyl ester lipase in human tissues and in acute pancreatitis. Int J Pancreatol. 1989;5(2):123–134. doi: 10.1007/BF02924413. [DOI] [PubMed] [Google Scholar]
- 46.Klöppel G, Dreyer T, Willemer S, Kern HF, Adler G. Human acute pancreatitis: its pathogenesis in the light of immunocytochemical and ultrastructural findings in acinar cells. Virchows Arch A Pathol Anat Histopathol. 1986;409(6):791–803. doi: 10.1007/BF00710764. [DOI] [PubMed] [Google Scholar]
- 47.Watanabe O, Baccino FM, Steer ML, Meldolesi J. Supramaximal caerulein stimulation and ultrastructure of rat pancreatic acinar cell: early morphological changes during development of experimental pancreatitis. Am J Physiol. 1984;246(4 pt 1):G457–G467. doi: 10.1152/ajpgi.1984.246.4.G457. [DOI] [PubMed] [Google Scholar]
- 48.Gaisano HY, et al. Supramaximal cholecystokinin displaces Munc18c from the pancreatic acinar basal surface, redirecting apical exocytosis to the basal membrane. J Clin Invest. 2001;108(11):1597–1611. doi: 10.1172/JCI9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cosen-Binker LI, Lam PP, Binker MG, Reeve J, Pandol S, Gaisano HY. Alcohol/cholecystokinin-evoked pancreatic acinar basolateral exocytosis is mediated by protein kinase C alpha phosphorylation of Munc18c. J Biol Chem. 2007;282(17):13047–13058. doi: 10.1074/jbc.M611132200. [DOI] [PubMed] [Google Scholar]
- 50.Liu X, Wang Y, Duclos RI, O’Doherty GA. Stereochemical structure activity relationship studies (S-SAR) of tetrahydrolipstatin. ACS Med Chem Lett. 2018;9(3):274–278. doi: 10.1021/acsmedchemlett.8b00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol. 2011;6:147–163. doi: 10.1146/annurev-pathol-011110-130158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Banks PA, et al. Classification of acute pancreatitis--2012: revision of the Atlanta classification and definitions by international consensus. Gut. 2013;62(1):102–111. doi: 10.1136/gutjnl-2012-302779. [DOI] [PubMed] [Google Scholar]
- 53.Xie X, et al. Identification of a novel phosphorylation site in adipose triglyceride lipase as a regulator of lipid droplet localization. Am J Physiol Endocrinol Metab. 2014;306(12):E1449–E1459. doi: 10.1152/ajpendo.00663.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nevalainen TJ, Eerola LI, Rintala E, Laine VJ, Lambeau G, Gelb MH. Time-resolved fluoroimmunoassays of the complete set of secreted phospholipases A2 in human serum. Biochim Biophys Acta. 2005;1733(2–3):210–223. doi: 10.1016/j.bbalip.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 55.Nevalainen TJ, Hietaranta AJ, Gronroos JM. Phospholipase A2 in acute pancreatitis: new biochemical and pathological aspects. Hepatogastroenterology. 1999;46(29):2731–2735. [PubMed] [Google Scholar]
- 56.Friess H, et al. Phospholipase A2 isoforms in acute pancreatitis. Ann Surg. 2001;233(2):204–212. doi: 10.1097/00000658-200102000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maléth J, et al. Alcohol disrupts levels and function of the cystic fibrosis transmembrane conductance regulator to promote development of pancreatitis. Gastroenterology. 2015;148(2):427–39.e16. doi: 10.1053/j.gastro.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Dijk SM, et al. Treatment of disrupted and disconnected pancreatic duct in necrotizing pancreatitis: A systematic review and meta-analysis. Pancreatology. 2019;19(7):905–915. doi: 10.1016/j.pan.2019.08.006. [DOI] [PubMed] [Google Scholar]
- 59.Patel K, Durgampudi C, Noel P, Trivedi RN, de Oliveira C, Singh VP. Fatty acid ethyl esters are less toxic than their parent fatty acids generated during acute pancreatitis. Am J Pathol. 2016;186(4):874–884. doi: 10.1016/j.ajpath.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Panek J, Sztefko K, Drozdz W. Composition of free fatty acid and triglyceride fractions in human necrotic pancreatic tissue. Med Sci Monit. 2001;7(5):894–898. [PubMed] [Google Scholar]
- 61.Ding L, Liou GY, Schmitt DM, Storz P, Zhang JS, Billadeau DD. Glycogen synthase kinase-3β ablation limits pancreatitis-induced acinar-to-ductal metaplasia. J Pathol. 2017;243(1):65–77. doi: 10.1002/path.4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bhanot UK, Möller P. Mechanisms of parenchymal injury and signaling pathways in ectatic ducts of chronic pancreatitis: implications for pancreatic carcinogenesis. Lab Invest. 2009;89(5):489–497. doi: 10.1038/labinvest.2009.19. [DOI] [PubMed] [Google Scholar]
- 63.Janiak A, Leśniowski B, Jasińska A, Pietruczuk M, Małecka-Panas E. Interleukin 18 as an early marker or prognostic factor in acute pancreatitis. Prz Gastroenterol. 2015;10(4):203–207. doi: 10.5114/pg.2015.50993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wereszczynska-Siemiatkowska U, Mroczko B, Siemiatkowski A. Serum profiles of interleukin-18 in different severity forms of human acute pancreatitis. Scand J Gastroenterol. 2002;37(9):1097–1102. doi: 10.1080/003655202320378310. [DOI] [PubMed] [Google Scholar]
- 65.Pandol SJ, Saluja AK, Imrie CW, Banks PA. Acute pancreatitis: bench to the bedside. Gastroenterology. 2007;132(3):1127–1151. doi: 10.1053/j.gastro.2007.01.055. [DOI] [PubMed] [Google Scholar]
- 66.Williams JA, Sans MD, Tashiro M, Schäfer C, Bragado MJ, Dabrowski A. Cholecystokinin activates a variety of intracellular signal transduction mechanisms in rodent pancreatic acinar cells. Pharmacol Toxicol. 2002;91(6):297–303. doi: 10.1034/j.1600-0773.2002.910606.x. [DOI] [PubMed] [Google Scholar]
- 67.Williams JA. Intracellular signaling mechanisms activated by cholecystokinin-regulating synthesis and secretion of digestive enzymes in pancreatic acinar cells. Annu Rev Physiol. 2001;63:77–97. doi: 10.1146/annurev.physiol.63.1.77. [DOI] [PubMed] [Google Scholar]
- 68.de Oliveira C, et al. Multimodal transgastric local pancreatic hypothermia reduces severity of acute pancreatitis in rats and increases survival. Gastroenterology. 2019;156(3):735–747.e10. doi: 10.1053/j.gastro.2018.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rao PM, Rhea JT. Colonic diverticulitis: evaluation of the arrowhead sign and the inflamed diverticulum for CT diagnosis. Radiology. 1998;209(3):775–779. doi: 10.1148/radiology.209.3.9844673. [DOI] [PubMed] [Google Scholar]
- 70.Németh BC, et al. The common truncation variant in pancreatic lipase related protein 2 (PNLIPRP2) is expressed poorly and does not alter risk for chronic pancreatitis. PLoS One. 2018;13(11):e0206869. doi: 10.1371/journal.pone.0206869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mounzer R, et al. Comparison of existing clinical scoring systems to predict persistent organ failure in patients with acute pancreatitis. Gastroenterology. 2012;142(7):1476–82; quiz e15. doi: 10.1053/j.gastro.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 72.Kangani CO, Kelley DE, Delany JP. New method for GC/FID and GC-C-IRMS analysis of plasma free fatty acid concentration and isotopic enrichment. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;873(1):95–101. doi: 10.1016/j.jchromb.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eguchi J, et al. Transcriptional control of adipose lipid handling by IRF4. Cell Metab. 2011;13(3):249–259. doi: 10.1016/j.cmet.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lowe ME, Kaplan MH, Jackson-Grusby L, D’Agostino D, Grusby MJ. Decreased neonatal dietary fat absorption and T cell cytotoxicity in pancreatic lipase-related protein 2-deficient mice. J Biol Chem. 1998;273(47):31215–31221. doi: 10.1074/jbc.273.47.31215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ellett JD, Evans ZP, Zhang G, Chavin KD, Spyropoulos DD. A rapid PCR-based method for the identification of ob mutant mice. Obesity (Silver Spring) 2009;17(2):402–404. doi: 10.1038/oby.2008.443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.