

Abstract
Red skin is an important quality trait for pear fruits and is determined by the concentration and composition of anthocyanins. The regulatory mechanism underlying anthocyanin accumulation is a popular topic in fruit research. Red mutants are ideal materials for studying the molecular mechanism of color diversity in pear. Although several red pear mutants have been cultivated and are in production, no exact locus containing the responsible genetic mutation has been identified. In this study, by combining the bulked segregant analysis with whole-genome sequencing, we identified a 14 nucleotide deletion mutation in the coding region of the PpBBX24 gene from the red pear mutant “Zaosu Red”. We further verified that the deletion was present only in the red mutant of “Zaosu” and in its red offspring, which was different from that which occurred in other red pear fruits. This deletion results in a coding frame shift such that there is an early termination of the PpBBX24 gene and loss of key NLS and VP domains from PpBBX24. The lost domains may reduce or alter the normal function of PpBBX24. In addition, we found that the transcript levels of the PpMYB10 and PpHY5 genes in red samples were significantly higher than those in green samples, whereas the results for the normal-type PpBBX24 gene were the opposite. We ultimately revealed that the 14 nucleotide deletion mutation in the coding region of the PpBBX24 gene is associated with the red skin of the “Zaosu Red” pear. This finding of somatic mutational events will be helpful for breeding new red pear cultivars and for understanding the regulatory mechanisms involved in pear skin pigmentation.
Subject terms: Plant molecular biology, Natural variation in plants, Plant hybridization
Introduction
Pear (Pyrus spp.) fruits are very popular and are widely grown in temperate regions all worldwide1. The red skin of pear is one of the most preferred preferences to consumers because of its attractive appearance and nutritional value2, and a study in Australia and New Zealand showed that a large percentage of consumers were willing to buy a novel red-skinned pear despite its poor flavor3. In pear production, red pear cultivars are common for occidental pear (Pyrus communis L.). For oriental pear, especially Chinese sand pear (P. pyrifolia Nakai) and Chinese white pear (P. pyrifolia White Pear Group), red peel color is hard to obtain and is unstable in different regions and under different cultivation conditions4–6; fully red-skinned cultivars are rare. Previous studies have shown that the developmental patterns of red coloration are different among plants and pear cultivation groups1,7,8, and it has been shown that the red color is a dominant characteristic and that the ratio of red-skinned fruits in progeny is 1:1 for the crosses between a yellow pear parent and heterozygous red pear parent9,10.
The concentration and composition of anthocyanins are the main determinants of red coloration of pear fruits; these determinants are regulated by many genes and are affected by various environmental conditions, including light and temperature2,5,11–14. Previous studies have shown that the components of anthocyanins in red-skinned pears mainly include cyanidin-3-galactoside (C-Ga), peonidin-3-galactoside (P-Ga), cyanidin-3-glucoside, cyanidin-3-arabinofuranoside, and peonidin-3-glucoside15. The anthocyanin biosynthetic pathway and its regulatory mechanisms have been well elucidated at the transcriptional level in many plant species. Phenylalanine ammonia-lyase (PAL), chalcone synthase (CHS), chalcone isomerase (CHI), flavanone-3-hydroxylase (F3H), dihydroflavonol 4-reductase, anthocyanidin synthase (ANS), and UDP-glucose: flavonoid 3-glucosyltransferase (UFGT) are the key enzymes involved in anthocyanin biosynthesis in pear and other plant species16–18.
Furthermore, there are numerous reports of transcription factor proteins in the anthocyanin biosynthesis pathway. Among these proteins, R2R3-type MYBs are the most important for anthocyanin accumulation19,20, as they regulate the expression of anthocyanin biosynthesis genes by forming MYB–bHLH–WD40 (MBW) protein complexes combined with basic helix–loop–helix (bHLH) and W40 repeat proteins6,21. In pear, PcMYB10 and PyMYB114 have been verified to have such functions22,23.
In addition to their involvement in MBW complexes, other transcription factors also have important effects on anthocyanin biosynthesis by interacting with the complex. Recently, Tao et al.24 revealed that PpHY5 participated in anthocyanin biosynthesis induced by blue light in red pear. Bai et al.6,25 found that PpBBX16 cooperated with PpHY5 and positively regulated light-induced anthocyanin accumulation by activating MYB10 in red pear; PpBBX18 and PpBBX21 antagonistically regulate anthocyanin biosynthesis via competitive association with PpHY5 in the peel of pear fruits. Ni et al.14 reported that the ethylene response factors Pp4ERF24 and Pp12ERF96 regulate blue light-induced anthocyanin biosynthesis in red pear fruits by interacting with MYB114, and Wu et al.26 indicated that PyMADS18 is likely to be involved in anthocyanin accumulation and the regulation of anthocyanin synthesis in the early development of pear fruits. Thus, the regulatory networks of anthocyanin biosynthesis are intricate and complex.
“Zaosu” is a pear cultivar widely cultivated in China and has crisp, green-skinned fruit (Fig. 1e); this cultivar is a hybrid of “Pingguoli” (female parent, P. pyrifolia White Pear Group) and “Shenbuzhi” (male parent, P. communis). “Zaosu Red,” whose phenotype, including that of its fruits, flowers, stems, and young leaves, is red, originated from the discovery of a red mutant of “Zaosu” (Fig. 1a–f, h, i). The mutant inherits the excellent fruit quality and cultivation traits of “Zaosu” and is an exceptional parent for breeding new varieties with crisp, red-skinned fruits. Because its genetic background is nearly identical to that of its original type, “Zaosu Red” is also an ideal material to study the molecular mechanism of color diversity in pear. Although recent studies have demonstrated that the genetic locus underlying red foliage and fruit skin traits had been mapped to LG4 in “Red Zaosu”27, the exact locus of the genetic mutation remains elusive.
Fig. 1. Flowers, fruits, new shoots, and seedlings of “Zaosu Red”, “Zaosu”, and “Kuala Pear”.

a, c, e, and h respectively show flowers, young fruits, mature fruits, and new shoots of “Zaosu”; b, d, f, and i respectively show flowers, young fruits, mature fruits, and new shoots of “Zaosu Red”; g and j respectively show fruits and new shoots of “Kuala Pear”; k shows seedlings of “Kuala Pear” × “Zaosu Red”
In this study, by combining bulked segregant analysis (BSA) with whole-genome sequencing, we identified a 14 nucleotide deletion that is located in the coding region of the PpBBX24 gene and that is associated with the red color of “Zaosu Red”. This finding of somatic mutational events will be helpful for breeding new red pear cultivars and understanding the regulatory mechanisms involved in pear skin pigmentation.
Results
Anthocyanin content in the leaves and fruit peels of pear
As shown in Fig. 2, four known anthocyanins were detected in the leaves and peels of young fruits of “Zaosu Red”, and the contents in “Zaosu Red” were significantly higher than those in “Zaosu”. No anthocyanins were detected in the peels of young fruits of “Zaosu”, and only a few C-Ga and P-Ga were detected in the leaves of “Zaosu”. Among the four anthocyanins, C-Ga constituted the majority (77.7–95.8%), P-Ga was the next most abundant (4.2–21.2%), and the other two were present at very low levels (<2%). Of the major two kinds of anthocyanins in “Zaosu Red”, the proportions between the leaves and fruits were different. The proportion of C-Ga in the leaves was 91.0%, which was higher than that in fruits (77.7%), while the proportion of P-Ga in the leaves was 7.3%, which was lower than that in fruits (21.2%).
Fig. 2. Anthocyanin concentrations in different samples, as detected by UPLC-PDA-MS/MS.
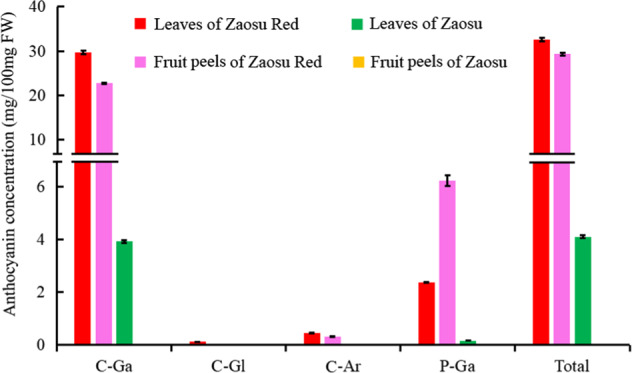
C-Ga, C-Gl, C-Ar, and P-Ga represent cyanidin-3-galactoside, cyanidin-3-O-glucoside, cyanidin-3-O-arabinoside, and peonidin-3-O-galactoside, respectively. Total represents the sum of all the above anthocyanins
Whole-genome resequencing analysis
Here, five leaf samples from two parents (“Zaosu Red” and “Kuala Pear”), two pools (red and green), and “Zaosu” were sequenced. A total of 96.91 Gb of clean bases were obtained, with an average coverage depth of 24.64× the reference genome among two parents and “Zaosu” and 57.80× coverage between the two pools (the reference genome was estimated to be ~511.33 Mb). Approximately 80% of these reads were properly mapped to the reference genome, and they were used for further analysis (Supplementary Table S1). The clean read data have been deposited in the NCBI Sequence Read Archive (SRA) database under accession number SRP225824.
SNP/InDel variant detection and annotation
To obtain accurate variants, the single-nucleotide polymorphism (SNP)/InDel variants were filtered through a series of steps after detection by GATK. In total, 4.86 (“Kuala Pear”) to 6.90 (red pool) million SNPs and 0.92 (“Kuala Pear”) to 1.27 (red pool) million InDels were detected against the reference genome from the test samples; among these data, the frequency of the SNPs was more than five times that of the InDels, the frequency of the transition-type SNPs was approximately two times that of the transversion types, the frequency of insertions was slightly less than that of deletions, and the heterozygosity ratios of SNPs and the homozygosity ratios of InDels were more than 60% in all the test samples (Supplementary Table S2). According to the annotation information, ~30% of the SNPs/InDels were located in the intergenic region, ~40% were in upstream and downstream of genes, ~10% were in introns, and only ~6% of SNPs and 1.5% of InDels were located in the coding sequence (CDS) region in every test sample. Approximately 53 and 45% of the SNPs in the CDS region caused synonymous coding and nonsynonymous coding variants, respectively. Approximately 54% of the InDels in the CDS region caused frame shift variants (Supplementary Tables S3–S6).
Association analysis
To obtain high-quality variants, before association analysis, the SNP/InDel variants that had multiple genotypes, which were supported by less than four reads, whose genotypes are the same in both pools and which are not from the recessive parent in the recessive pool, were filtered, and a total of 731,881 high-quality SNPs and 470,493 high-quality InDels were ultimately obtained (Supplementary Table S7). The euclidean distance (ED) and SNP index values of all the high-quality SNPs/InDels were then calculated and fitted using a sliding window approach with 2 Mb windows sliding in 10 kb steps.
For the ED method, the median values +3 SDs (standard deviations) were used as the associated thresholds, which were 0.21 and 0.14 for the SNP and InDel variants, respectively. Only the regions whose fitted SNP and InDel ED values were higher than the corresponding associated thresholds were defined as candidate-associated regions. By the use of this method, a region of Chr 4 from 7.41 to 22.59 Mb was screened, and the highest associated peak appeared around the region of 17.3 Mb. The results of the SNPs and InDels were consistent (Table 1 and Fig. 3).
Table 1.
Summary of the candidate-associated regions
| Method | Variant type | Chromosome ID | Start (bp) | End (bp) | Size (Mb) | Gene number |
|---|---|---|---|---|---|---|
| ED | SNP | Chr 4 | 7,410,000 | 7,490,000 | 0.08 | 5 |
| Chr 4 | 8,080,000 | 8,780,000 | 0.7 | 57 | ||
| Chr 4 | 8,840,000 | 22,590,000 | 13.75 | 1097 | ||
| InDel | Chr 4 | 7,450,000 | 22,580,000 | 15.13 | 1211 | |
| SNP index | SNP | Chr 4 | 60,000 | 90,000 | 0.03 | 6 |
| Chr 4 | 6,480,000 | 6,520,000 | 0.04 | 4 | ||
| Chr 4 | 6,720,000 | 6,730,000 | 0.01 | 1 | ||
| Chr 4 | 6,850,000 | 6,910,000 | 0.06 | 4 | ||
| Chr 4 | 6,930,000 | 11,000,000 | 4.07 | 310 | ||
| InDel | Chr 4 | 1 | 170,000 | 0.17 | 14 | |
| Chr 4 | 10,070,000 | 10,100,000 | 0.03 | 1 | ||
| Chr 4 | 10,310,000 | 10,380,000 | 0.07 | 5 | ||
| Chr 4 | 10,470,000 | 10,470,000 | 0 | 1 | ||
| Chr 4 | 10,490,000 | 10,990,000 | 0.5 | 30 | ||
| Chr 4 | 7,060,000 | 7,140,000 | 0.08 | 6 | ||
| Chr 4 | 7,240,000 | 7,250,000 | 0.01 | 2 | ||
| Chr 4 | 7,300,000 | 9,390,000 | 2.09 | 163 | ||
| Chr 4 | 9,430,000 | 9,440,000 | 0.01 | 1 | ||
| Chr 4 | 9,490,000 | 9,500,000 | 0.01 | 1 | ||
| Chr 4 | 9,560,000 | 10,000,000 | 0.44 | 29 | ||
| Chr 11 | 1,240,000 | 1,260,000 | 0.02 | 4 | ||
| Chr 11 | 520,000 | 1,220,000 | 0.7 | 102 |
Fig. 3. ED value distribution of InDels and SNPs in chromosomes.

The abscissa represents the chromosome name, the colored dots represent ED values, the black lines represent the fitted ED values (with 2 Mb windows sliding in 10 kb steps), and the red dotted lines represent associated thresholds
For the SNP index method, regardless of the SNPs and InDels, no region was associated when the confidence level was set to 0.90, so we used the 99th percentiles of the fitted ΔSNP index values as the associated threshold, which were 0.14 and 0.06 for SNP and InDel variants, respectively. By the use of this method, six regions of Chr 4 from 1 bp to 11 Mb and two regions of Chr 11 from 0.52 Mb to 1.26 Mb were screened, in which the region of Chr 4 at ~8.2 Mb was associated with the highest peak (Table 1 and Supplementary Fig. S1).
Identification of the gene associated with red/green color
The results of the association analysis showed that the main associated regions were located on Chr 4, which contains more than one thousand genes (Table 1, Fig. 3, and Supplementary Fig. S1). Therefore, it is hard to define which gene is truly associated with the characteristic of red/green color. Because “Zaosu Red” is a red mutant of “Zaosu”, we further identified the different SNPs/InDels of Chr 4 between “Zaosu Red” and “Zaosu” and cross-referenced that with the data between “Zaosu Red” and “Kuala Pear”. The results showed that there were 252 and 150 mutually different SNPs and InDels between “Zaosu Red” vs. “Zaosu” and “Zaosu Red” vs. “Kuala Pear”, respectively, in the candidate-associated regions of Chr 4. Of these, five SNPs and eight InDels were heterozygous in both “Zaosu Red” and the red pool and homozygous in “Kuala Pear”, “Zaosu”, and the green pool, respectively (Supplementary Table S8).
Of the remaining five SNPs and eight InDels, only one deletion of 14 nucleotides within a gene produced a coding frame shift, and the gene was annotated as a CO-like (COL) domain-class transcription factor and subsequently named PpBBX24, while all the other SNPs and InDels were located in intergenic regions, unknown regions, or in upstream or downstream genes, which did not alter the CDS (Table 2). In addition, this deletion was located at 18,388,296 bp of Chr 4, which was near the highest associated peak region (17.3 Mb) calculated by the ED method. Thus, the PpBBX24 gene, which contains the 14 nucleotide deletion, was selected as the final candidate gene associated with red/green color for further analysis.
Table 2.
Location, annotation, and nucleotide types of selected SNPs/InDels in different samples
| Variant type | Position | Reference nucleotides | Alternate nucleotides | Zaosu Red | Zaosu | Kuala Pear | Green pool | Red pool | Effect | Gene ID | Nr annotation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SNPs | 9,115,802 | T | A | 0.5 | N | 7.0 | 11.0 | 7.6 | Upstream | Pdr4g010210 | Adenylate kinase 1, chloroplastic-like |
| 9,212,167 | C | T | 0.1 | N | 2.0 | 6.0 | 5.4 | Upstream | Pdr4g010260 | Zinc finger CCCH domain-containing protein 11-like | |
| 9,212,608 | G | T | 0.8 | N | 7.0 | 15.0 | 10.7 | Upstream | Pdr4g010260 | ||
| 9,214,082 | T | C | 0.3 | N | 8.0 | 9.0 | 12.1 | Upstream | Pdr4g010260 | ||
| 9,214,271 | A | G | 0.4 | N | 8.0 | 11.0 | 12.2 | Upstream | Pdr4g010260 | ||
| InDels | 8,553,091 | CTT | C | 2.4 | 0.3 | 0.2 | 0.6 | 6.7 | Intergenic | ||
| 9,121,649 | GT | G | 5.4 | 5.0 | 17.0 | 28.0 | 19.4 | Intergenic | |||
| 9,122,408 | G | GA | 11.4 | 8.0 | 7.0 | 20.0 | 20.8 | Intergenic | |||
| 9,178,944 | TAGA | T | 2.5 | 2.0 | 3.0 | 5.0 | 6.1 | Unknown | |||
| 9,222,351 | T | TA | 11.4 | 8.0 | 11.0 | 27.0 | 14.5 | Downstream | Pdr4g010260 | Zinc finger CCCH domain-containing protein 11-like | |
| 9,222,379 | G | GT | 12,4 | 11,0 | 12,0 | 28,0 | 17,6 | Downstream | Pdr4g010260 | ||
| 18,388,296 | AGCAGCTGAAGTTCC | A | 10,5 | 14,0 | 20,0 | 39,0 | 24,9 | Frame shift | Pdr4g016570 | COL domain-class transcription factor | |
| 16,860,274 | AG | A | 18,9 | 9,0 | 7,0 | 13,0 | 17,7 | Downstream | Pdr4g015130 | COP9 signalosome complex subunit 7-like |
The numbers in the columns represent the supported read depths of reference nucleotides. Alternate nucleotides are separated by commas, and N represents no read support.
Variant verification, cloning, and bioinformatic analysis of the PpBBX24 gene
The PCR amplification results by the primer pair F-2655 and R-2794, which encompassed the deleted region of the PpBBX24 gene (Fig. 4a), showed that two bands with a size discrepancy of 14 bp were amplified from the DNA of “Zaosu Red” and 50 red F1 plants, and only the larger band was amplified from the DNA of “Zaosu”, “Kuala Pear”, and 50 green F1 plants (Fig. 4a). This revealed that the deletion exists only in the red plants.
Fig. 4. PCR amplification results of the PpBBX24 gene fragment using the primer pair F-2655 and R-2794 in different DNA samples.

a PCR amplification results of the leaves of “Zaosu Red”, “Zaosu”, “Kuala Pear”, and green and red F1 plants. The maps were generated using LabChip GX Reviewer v5.4 (Perkin-Elmer, USA). M represents “Marker”, and the numbers 1, 2, 3, 4–53, and 54–103 above the bands represent “Zaosu Red”, “Zaosu”, “Kuala Pear”, green F1 plants, and red F1 plants, respectively. b PCR amplification results of the leaves of 18 different cultivars/strains. The band maps were generated using LabChip GX Reviewer v5.4. The numbers below the bands from 1 to 17 represent “Zaosu Red”, “Zaosu”, “Kuala Pear”, “N3-1”, “Red Anjou”, “Red Comice”, “Zhongaihongli”, “Bayuehong”, “Wenshanghongli”, “Pingguoli”, “Jinxiang”, “Zaojinxiang”, “4X Yali”, “Huangguan”, “Dangshansu”, “Jinfeng”, and “E1-2”, respectively. The leaves and fruits of these cultivars/strains are shown above the corresponding bands. Among these fruits, 1 and 4–6 are fully red, 7–9 are partly red, 3 and 10–11 are reddish, and 2 and 12–17 are fully green
The same PpBBX24 gene fragments were also amplified using the DNA of other cultivars/strains whose young leaves and young fruits are of different colors, but the small band of 126 bp appeared only for the DNA of “Zaosu Red” (Fig. 4b). This revealed that the color mechanism of “Zaosu Red” was different from that of other red pears. Although both the young leaves and young fruits of “Zaosu Red”, “Red Anjou”, and “N3-1” are red, not all the tested cultivars/strains have the same color of young leaves and fruits (Fig. 4b). This further revealed that their color mechanisms are different.
The full CDSs of the candidate PpBBX24 gene both with and without the deletion were amplified from “Zaosu”, “Kuala Pear”, and “Zaosu Red”. The sequencing results showed that the full CDS without the deletion was 720 bp and encoded 239 aa. When 14 bp from 568 bp to 581 bp (from 2736 bp to 2749 bp in the gene sequence) were deleted, a coding frame shift and early termination occurred, and only 205 aa were translated (Fig. 5a–c). Aside from the 14 bp deletion, there were four other SNPs among the different types sequences of these three cultivars, which caused one amino acid alteration (Fig. 5b).
Fig. 5. Bioinformatic analysis of the gene and protein sequences of PpBBX24 and its mutant.

a Gene structure and deletion and primer locations of PpBBX24. The gene structure schematic was generated using GSDS 2.0 (http://gsds.cbi.pku.edu.cn/). The letters F and R represent the forward and reverse primers, respectively, and the numbers behind them represent the gene sequence order of the first nucleotide at the 5ʹ terminus of each primer. b CDS alignment and translation of PpBBX24 and Ppbbx24-del in the three cultivars. c Protein sequence alignment of PpBBX24 and Ppbbx24-del in “Zaosu Red”. d Schematic of the conserved domains of PpBBX24, generated using SMART (http://smart.embl-heidelberg.de/). e Alignment of the VP domains of PpBB24, HY5 (GenBank: BAA21327.1), THY5 (GenBank: AJ011914.1), STO (GenBank: X95572), STH (GenBank: AF323666), and MdCOL4 (GenBank: ADL36673.1).
As Fig. 5a shows, this candidate gene has three exons and two introns, and the deletion is located in the last exon. The protein sequence contains two B-box domains at the N-terminus (Fig. 5d) belonging to the BBX family, also known as the COL transcription factor family, and was shown to have the highest homology with AtBBX24 (STO, AT1G06040.1) by BLASTP against the Arabidopsis thaliana Araport11 protein sequence database (https://www.arabidopsis.org/) and by phylogenetic analysis (Supplementary Fig. S2). Therefore, we named this gene and its mutant PpBBX24 and Ppbbx24-del, respectively. Because of the early termination codon, a VP domain and a nuclear localization sequence (NLS) at the end of PpBBX24 are lacking in Ppbbx24-del (Fig. 5c). The VP domains of PpBBX24 are exactly the same as those of MdCOL4 and are homologous with those of HY5, THY5, STO, and STH (Fig. 5e).
Transcription-level analysis of anthocyanin biosynthesis-related genes
Using RNA-seq, we obtained a total of 176 Gb clean data (590 M clean reads) from 24 pear samples (Supplementary Table S9), which have been archived at the NCBI SRA under accession SRP225824. According to the fragments per kilobase of transcript per million fragments mapped (FPKM) data, the transcript levels of genes encoding seven key anthocyanin synthesis enzymes, four important regulatory genes, and the PpBBX24 gene were compared in the young leaves and peels of young fruits of “Zaosu Red”, “Zaosu”, and “Kuala Pear” and in the leaves of the red pool and green pool. The results showed that the transcript levels of the seven anthocyanin synthesis enzyme-encoding genes in the young leaves and peels of young fruits of “Zaosu Red” were not significantly higher than those of “Zaosu” and “Kuala Pear”, but the transcript levels of PpPAL, PpCHS, PpCHI, PpF3H, and PpANS in the young leaves of the red pool were significantly higher than those of the green pool. The transcript levels of the PpMYB10 and PpHY5 genes in all the tested red samples were significantly higher than those in the green samples. The PpBBX24 gene presented slightly lower transcript levels in the red samples than in the green samples (Fig. 6).
Fig. 6. FPKM values of anthocyanin synthesis- and regulation-associated genes in the leaves and fruit peels of different pear cultivars according to RNA-seq.

RL, ZL, and KL and RF, ZF, and KF represent young leaves and peels of young fruits of “Zaosu Red”, “Zaosu”, and “Kuala Pear”, respectively; RP and GP represent mixed leaves of the red pool and green pool, which were randomly collected from ten red and ten green hybrid progeny of “Kuala Pear” × “Zaosu Red” (three biological replicates of each pool were collected from 30 different plants), respectively. All the data are the means of three biological replications, with the error bars indicating ±SDs. The different lowercase letters above the bars are significantly different at P ≤ 0.05 (Duncan’s multiple range test). The column color is consistent with the sample color
Discussion
Differentially sourced red-colored pear mutants have various types of mutations
Mutation selection is one of the most efficient approaches to obtain red-skinned pear cultivars, and many red-skinned pear cultivars have been successfully selected in practice, such as “Red Anjou”28, “Red Comice”, “Royal Red Hardy”9, “Max Red Bartlett”10, “Bon Rouge”29, “Nanhong Li”30, and “Red Zaosu”24. However, which loci are mutated in the genome causing a green pear cultivar to turn red is rarely reported. In this study, we identified a 14 bp deletion in chromosome 4 of the red pear mutant “Zaosu Red” by combining BSA with whole-genome sequencing (Table 2). Using PCR amplification and sequencing, we determined that the deletion was present only in “Zaosu Red” and in its red F1 plants; thus, the deletion is closely linked to the red characteristic of the red pear mutant. However, no such deletion was found in the other tested pears, regardless of their phenotype, even despite “Red Anjou” also being red mutant and having the same phenotype as “Zaosu Red” (Fig. 4). This implies that the color mechanism is complex and varies among different red pear types.
Generally, compared with red plants or red young leaves, red fruit color is the characteristic we most expect. According to the phenotypes of the tested pear cultivars/accessions in this study, it was shown that a pear that produces red young leaves is not always able to produce red fruits. For example, “Huangguan” and “E1-2”, produce red young leaves but green fruits; other cultivars, such as “Red Comice” and “Zhongaihongli”, produce red fruits but have green leaves (Fig. 4b). The “Zaosu Red” pear we used for DNA-seq in this study not only produces red fruits and red young leaves but also produces red flowers, red stems, and red F1 plants (Fig. 1). Because the F1 plants we used in this study were small seedlings without fruits, we did not know if the red plants could also produce red fruits. According to the results of our anthocyanin content analysis (Fig. 2), the anthocyanin components between the young leaves and peels of young fruits of “Zaosu Red” were nearly the same, but the proportions of the main components were different, which revealed that the color mechanisms in the leaves and fruits may have both similarities and differences. However, whether the red color of both the plants and fruits is caused by the 14 bp deletion of the PpBBX24 gene needs further study.
Structure and function of the candidate PpBBX24 gene
In this study, the candidate PpBBX24 associated with the red characteristic of “Zaosu Red” is a B-box protein and belongs to a class of zinc finger transcription factors that contain one or more B-box domains at the N-terminus involved in mediating protein–protein interactions, of which 32 members have been identified in Arabidopsis31,32. It has been verified that several plant BBXs play important regulatory roles in photomorphogenesis and anthocyanin accumulation, among which AtBBX2033, MdBBX2034, AtBBX2135,36, PpBBX166, PpBBX1825, OsBBX1437,38, and MdBBX2239,40 act as positive regulators, whereas AtBBX1941, PpBBX2123, AtBBX3242, MdBBX3743, MdCOL4 (BBX24)44, AtBBX2445, and AtBBX2535,46 act as negative regulators.
PpBBX24 shares high homology with MdCOL444 and AtBBX2445 (96.23% and 62.00%, respectively). The function of MdCOL4 and AtBBX24 would be a valuable clue for PpBBX24. In apple, MdCOL4 interacts with MdHY5 via the B-box2 domain of MdCOL4 and the bZIP domain of MdHY5 to synergistically inhibit the expression of MdMYB1. MdCOL4 also directly binds to the three G-box motifs in the promoters of MdANS and MdUFGT to suppress their expression44. AtBBX24 attenuates UV-B-induced HY5 accumulation and suppresses its transcriptional activation activity45, which is similar to that which occurs for MdCOL4. Compared with PpBBX24, Ppbbx24-del has 48 fewer aa at the C-terminus (Fig. 5b, c), so we predicted that the mutated Ppbbx24-del may cause its repressive function on the transcriptional activation activity of HY5 and the reduced expression of MYB1/10. Furthermore, premature Ppbbx24-del may lose its binding activity with the promoters of the ANS and UFGT genes and decrease the repression of gene expression. In this study, the PpMYB10 and PpHY5 genes were significantly upregulated in all the tested red samples compared with the green samples (Fig. 6), while the expression of the normal PpBBX24 gene (full type) was reversed, which would essentially support the above assumptions. In addition, studying whether PpBBX24 could regulate the transcript level of the PpHY5 gene directly would be an interesting future topic.
Previous studies have shown that AtBBX24 (STO) and AtBBX25 (STH) contain a reserved VP domain (Fig. 5e), which is necessary to interact with the WD40 domain of AtCOP147,48. AtCOP1 is an E3 ubiquitin ligase that is mainly localized to the nucleus, and its physical interaction with AtBBX24 is required for the ubiquitination and degradation of BBX24 in the light48. Due to the 14 nucleotide deletion, two motifs, the NLS and VP domain, were lost from Ppbbx24-del (Fig. 5c), which may cause Ppbbx24-del to fail to localize to the nucleus and interact with COP1 and to not be subsequently ubiquitinated and degraded, resulting in the opposite activity of that of MYB1/10 and HY5 by the newly generated amino acids. However, all of the above hypotheses need to be studied further.
The transcript level of the PpBBX24 gene in red samples is relatively low
In this study, the PpBBX24 gene also presented a lower transcript level in the red samples than in the green samples, but compared with the green samples, not all the red samples reached a significant level in terms of their PpBBX24 transcript level. The PpBBX24 gene in the red samples comprised both full and del types, while in the reference genome, the green samples represent only the full type. When the FPKM value was calculated, some of the del-type fragments failed to contribute to it, so the transcript levels (FPKM values) of the PpBBX24 gene in the red samples here were slightly lower than those of the real sum of full and del types. We also amplified the PpBBX24 gene using the cDNA of young leaves from “Zaosu Red” by PCR and sequenced the products at random. Among the 68 sequenced clones, 29 (42.65%) were full type, and 39 (57.35%) were del type. This implied that the transcript levels of the normal PpBBX24 gene in the red samples were significantly lower than those in the green samples, which may greatly reduce the negative regulatory effect of this gene on photomorphogenesis and anthocyanin accumulation. This may be the key season why the color of “Zaosu Red” becomes red. However, whether the expression differences of the MYB10 and HY5 transcriptional factors were caused by the mutated Ppbbx24-del and its underlying regulatory mechanism need to be studied further.
The combination of BSA and whole-genome sequencing is effective for the identification of causal genes
Along with the development of sequencing technology and the successful assembly of high-quality genomes, the combination of BSA and whole-genome sequencing has become a very quick and efficient approach for identifying causal genes, of which many have been successfully identified in many plant species49–51. To obtain ideal results, single plants of mixed pools are generally selected from F2 or backcross populations. Nevertheless, many fruit trees species, such as pear and apple, are limited by a long juvenile phase or self-incompatibility, making it hard to obtain F2 or backcross populations. Therefore, a few F1 populations were also used for BSA, but the target trait in the dominant parent must be heterozygous. It is also hard to obtain an accurate associated locus corresponding to the target trait. For example, Lu et al.52 located a dominant gene (temperature-sensitive semidwarf, Tssd) at an interval of 500 kb and obtained 69 candidate genes using an F1 population of peach. Xue et al.27 used an F1 pear population of “PremP109” × “Red Zaosu” to map the red foliage and fruit skin traits in a 368.6 kb region of LG4. In this study, we used an F1 pear population of “Kuala Pear” × “Zaosu Red” to identify the variants associated with the red color trait. The red parent has the same genetic background as that of the genotype that Xue et al.27 used. We also obtained a long associated region in chromosome 4; this region is located on the same chromosome and in the same neighboring region as that reported by Xue et al.27 and contains hundreds of predicted protein-coding genes (Table 1). It is fortunate that the red parent is a mutant of a green cultivar. We combined the associated analysis with the differential analysis of the red parent and its original type and ultimately identified the key associated locus and gene.
In this study, the final candidate variant was located at 18,388,296 bp of Chr 4 and was closer to the highest associated peak region (17.3 Mb) calculated by the ED method than by the SNP index method (8.2 Mb), which means that the ED method may be more suitable for highly heterozygous species such as pear in the same associated analysis. However, if the target characteristic is not sourced from a mutation, it would be very hard work or even impossible to find the associated locus accurately by this method. Therefore, we strongly suggest that when an F1 population is used for BSA, including the mutant as one of the parents is the best choice.
In conclusion, a deletion in the coding region of PpBBX24 was identified from the red skin of “Zaosu Red” pear and may be the key factor that causes “Zaosu Red” to turn red. This study will provide new perspectives for the study of plant anthocyanin regulation and red pear breeding.
Materials and methods
Plant materials
A cross of “Kuala Pear” (male parent, a local cultivar of P. sinkiangensis grown in China, Fig. 1g, j) and “Zaosu Red” (male parent, Fig. 1f, i) was performed in 2016, and more than 1000 F1 plants were obtained in 2017, out of which red and green plants were produced at ~1:1 (Fig. 1k). In this study, the young leaves of 50 red F1 plants and 50 green F1 plants selected at random from the above cross were collected to construct red and green gene pools for BSA, the young leaves of 30 red F1 plants and 30 green F1 plants (10 of which were mixed for biological replication) were collected for RNA-seq, and last, the young leaves and peels of young fruits of “Kuala Pear”, “Zaosu Red”, and “Zaosu” were collected for anthocyanin content measurement, DNA-seq, RNA-seq, and gene cloning. Three biological replications were included for anthocyanin content measurement and RNA-seq. All the trial plants were grown in the orchard of the Horticultural Crops Research Institute, Xinjiang Academy of Agricultural Sciences (Luntai, China, 41°47ʹ4ʺ N, 84°14ʹ10ʺ E) and sampled from 2017 to 2019.
Extraction and measurement of anthocyanins
The extraction of anthocyanins was performed according to the protocol described by Yao et al.23 but was slightly modified as follows. Approximately 0.2 g of pear peels was ground in liquid nitrogen and suspended in methanol consisting of 0.1% HCl in the dark for 24 h. Afterward, 2 ml of the supernatant was then pipetted into a new tube and evaporated using a rotary evaporator (R-210, Buch, Switzerland) under vacuum at 40 °C. The residue was resuspended in acidified water (1.18 mM HCl) and then extracted with a preconditioned C18 solid-phase column (Oasis HLB, 30 μm, Waters, Eschborn, Germany). The anthocyanins were ultimately eluted with 1 ml of methanol and filtered through a 0.22 μm Millipore membrane (Millipore, Merck, USA). The anthocyanins were subsequently analyzed using a Waters Acquity UPLC system coupled to an Acquity PDA eλ diode array detector and a Xevo TQD mass spectrometer (Waters, MA, USA) equipped with an Acquity UPLC HSS T3 column (2.1 mm × 150 mm, 1.8 μm particle size, Waters, Eschborn, Germany). In brief, 2 μl of the filtered sample was injected and run at 40 °C (column temperature) and 0.3 ml/min (flow rate) using mobile phases A (methanol:acetonitrile, 7:3, v/v) and B (formic acid:water, 5:95, v/v). The linear gradient of phase A was 10% for the first 2 min, after which it increased from 10 to 25% from 2 min to 30 min, increased from 25 to 40% from 30 to 60 min, increased from 40 to 80% from 60 to 61 min, and then decreased from 80 to 10% from 61 to 62 min. Finally, an isocratic elution with 10% phase A was maintained for 13 min. The MS/MS parameters were as follows: ESI source of positive ion mode, MRM model scanning, source temperature of 150 °C, probe temperature of 500 °C, desolvation gas flow of 800 L/h, cone gas flow of 50 L/h, and collision gas (high-purity argon) flow of 0.13 ml/min. The PDA detector wavelength was set to 520 nm. The standard samples of cyanidin-3-O-glucoside, cyanidin-3-O-arabinoside, and peonidin-3-O-galactoside were obtained from Sigma-Aldrich (MO, USA), and C-Ga was obtained from PhytoLab (Vestenbergsgreuth, Germany).
DNA extraction and whole-genome sequencing
Genomic DNA was extracted from fresh leaves of 50 red F1 plants and 50 green F1 plants as well as their two parents and “Zaosu” using the cetyl-trimethylammonium bromide method53 and subsequently quantified using a NanoDrop 2000 spectrophotometer (Thermo Scientific, MA, USA). Equal amounts of DNA from 50 red plants and 50 green plants were then mixed to construct the red pools and green pools. The DNA of the two parents, two pools, and “Zaosu” was sonicated to generate 350 bp fragments for the construction of DNA sequencing libraries using a TruSeq DNA Sample Preparation Kit (Illumina, CA, USA) following the manufacturer’s recommendations. The qualified libraries were sequenced at Biomarker Technologies Corporation (Beijing, China) on an Illumina HiSeq X-Ten platform (Illumina, CA, USA).
Read alignment, SNP/InDel variant detection, and annotation
The raw data were qualified, and the adapter sequences and low-quality reads (proportion of noncalled bases >5%) were removed. High-quality clean reads were aligned to the pear genome (GCA_008932095.1)54 using BWA software with the default parameters55. According to the location results of the clean reads in the reference genome, the duplicated reads were filtered using Picard (http://broadinstitute.github.io/picard/). The SNP/InDel variants were then detected and filtered using the Genome Analysis Toolkit56 (GATK, https://software.broadinstitute.org/gatk/) and annotated using SnpEff57, which included determining their locations in the genome and their effects on genes.
Identification and analysis of trait-associated SNPs/InDels
The SNPs/InDels between the two parents and between the two pools were extracted from their vcf files. Two methods, the ED58, and the SNP index59, were used to assess the association of these SNPs/InDels with the target characteristics based on their read depth information in the red pool and green pool. The ED value was calculated as follows:
in which each A, G, C, and T letter represents the frequency of its corresponding DNA nucleotide in the red pool and green pool, and the higher the ED value is, the stronger the variant associated with the target characteristic. The SNP index value was calculated as follows:
in which aa and ab represent the green pool and red pool, respectively, and M and P represent the frequency of the variant from the female and male parents, respectively; the closer to 1 the ΔSNP index value is, the stronger the variant associated with the target characteristic. To eliminate background noise and false-positive sites, the fifth power of the original ED value was taken as the association value58. The fifth power of the ED and the ΔSNP index values from the same chromosome were then fitted using a sliding window approach with 2 Mb windows sliding in 10 kb steps. Finally, the regions whose fitted ED and ΔSNP index values were over the theoretical thresholds were selected as candidate regions.
The SNPs/InDels between “Zaosu” and “Zaosu Red” in the candidate regions were also extracted, and the mutual SNPs/InDels between the two parents, the two pools, “Zaosu”, and “Zaosu Red” in the candidate regions were further obtained. The mutual SNPs/InDels in the candidate regions, which were homozygous in the green parent and pool and heterozygous in the red parent and pool, were chosen as the final variants. According to the annotated information, the genes that contain or were near the final variants were chosen as candidate genes associated with the mutated trait for further analysis.
Genotypic identification
To verify the association of a selected InDel with the red trait of “Zaosu Red”, we designed a pair of primers that encompasses the deleted region (F-2655: 5ʹ-GATTCGCTTGAGTTTGGAGA-3ʹ and R-2794: 5ʹ-GGTCTGTAGGAGGTAAGATTGC-3ʹ) according to the sequence information. PCR amplification was then performed using this pair of primers on the DNA samples of the two parents, “Zaosu”, 50 red F1 plants, 50 green F1 single plants, and other cultivars/accessions. The PCR products were examined using a DNA 1K Chip (PN 760517) & Reagent Kit (PN CLS 760673) on a LabChip GX Touch HT Nucleic Acid Analyzer (Perkin-Elmer, Connecticut, USA).
Cloning and bioinformatic analysis of the PpBBX24 gene
The total RNA of “Zaosu”, “Zaosu Red”, and “Kuala Pear” was extracted from young leaves using an EasySpin Plus Plant RNA Kit (RN38, Aidlab, Beijing, China), and first-strand cDNA was synthesized from 2 μg of DNA-free RNA using a PrimeScript First-Strand cDNA Synthesis Kit (6110A, TaKaRa, Ohtsu, Japan) following the manufacturer’s instructions. The cDNA was diluted fivefold for use in gene cloning. The CDSs of the PpBBX24 (with the deleted region) and Ppbbx24-del (without the deleted region) genes were amplified using PrimeSTAR GXL DNA Polymerase (R050Q, TaKaRa, Ohtsu, Japan) with the primer pair ColF (5ʹ-ATGAAGATTCAGTGTGATGTGTGC-3ʹ) and ColR (5ʹ-CTAAAATCTGCCGAGGTCAGG-3ʹ) and then sequenced by Taihe Biotechnology Co., Ltd (Beijing, China). The cloned sequences were subsequently translated and aligned using DNAMAN 6 software. The conserved domains were searched using the online software SMART (http://smart.embl-heidelberg.de/), and the gene structure was analyzed using the Gene Structure Display Server 2.0 (http://gsds.cbi.pku.edu.cn/).
RNA isolation, library preparation, and RNA-seq
Total RNA was isolated using TRIzol reagent (Invitrogen, San Diego, USA) following the manufacturer’s protocol and then purified using oligo (dT) magnetic beads. The integrity of the RNA was verified with an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, USA). The paired-end library was prepared using a TruSeq RNA Sample Preparation kit (Illumina, San Diego, USA), and the libraries were sequenced by Biomarker Technology Company (Beijing, China) on an Illumina HiSeq X-Ten platform (Illumina, CA, USA).
Gene transcription analysis
After filtering, the clean reads were aligned against the reference genome (GCA_008932095.1)53 using HISAT260 and StringTie61 software. The FPKM value62 of every gene was then calculated to estimate the transcript levels of every sample using the following formula:
Supplementary information
Acknowledgements
We thank Prof. Rumei Chen and her team (Biotechnology Research Institute, Chinese Academy of Agricultural Sciences) for guidance on this manuscript. We also thank the National Key Research and Development Program of China (2018YFD1000102) and the Agricultural Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences (CAAS-ASTIP-2016-RIP) programs for funding this work.
Author contributions
Z.Z, S.J., J.W., C.O., and X.Z. conceived and designed the study; C.O, X.Z., F.W., and Y.Z. prepared the materials and conducted the experiments; and C.O., X.Z., L.Z., M.F., and J.W. wrote the manuscript. C.O. and X.Z. contributed equally to this paper. All authors read and approved the final manuscript.
Data availability
The data associated with this study have been deposited in NCBI under accession number PRJNA577323. The raw data from this project have been deposited in the NCBI SRA database under accession number SRP225824.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
These authors contributed equally: Chunqing Ou, Xiaoli Zhang
Contributor Information
Jixun Wang, Email: ee_wjx@163.com.
Shuling Jiang, Email: jshling@163.com.
Zhihong Zhang, Email: zhangz@syau.edu.cn.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41438-020-0259-7).
References
- 1.Huang C, Yu B, Teng Y, Su J, Shu Q. Effects of fruit bagging on coloring and related physiology, and qualities of red Chinese sand pears during fruit maturation. Sci. Hortic. 2009;121:149–158. doi: 10.1016/j.scienta.2009.01.031. [DOI] [Google Scholar]
- 2.Sun Y, et al. Postharvest pigmentation in red Chinese sand pears (Pyrus pyrifolia Nakai) in response to optimum light and temperature. Postharvest Biol. Tec. 2014;91:64–71. doi: 10.1016/j.postharvbio.2013.12.015. [DOI] [Google Scholar]
- 3.Gamble J, Jaeger SR, Harker FR. Preferences in pear appearance and response to novelty among Australian and New Zealand consumers. Postharvest Biol. Technol. 2006;41:38–47. doi: 10.1016/j.postharvbio.2006.01.019. [DOI] [Google Scholar]
- 4.Bao L, Chen KS, Zhang D, Li X, Teng YW. An assessment of genetic variability and relationships within Asian Pears based on AFLP (amplified fragment length polymorphism) markers. Sci. Hortic. 2008;116:374–380. doi: 10.1016/j.scienta.2008.02.008. [DOI] [Google Scholar]
- 5.Bai SL, et al. Transcriptome analysis of bagging-treated red Chinese sand pear peels reveals light-responsive pathway functions in anthocyanin accumulation. Sci. Rep. 2017;7:63. doi: 10.1038/s41598-017-00069-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bai S, et al. BBX 16, a B‐box protein, positively regulates light‐induced anthocyanin accumulation by activating MYB 10 in red pear. Plant Biotechnol. J. 2019;17:1985–1997. doi: 10.1111/pbi.13114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang YN, et al. Expression differences of anthocyanin biosynthesis genes reveal regulation patterns for red pear coloration. Plant Cell Rep. 2015;34:89–198. doi: 10.1007/s00299-014-1698-0. [DOI] [PubMed] [Google Scholar]
- 8.Qian M, et al. Analysis of different pigmentation patterns in ‘Mantianhong’ (Pyrus pyrifolia Nakai) and ‘Cascade’ (Pyrus communis L.) under bagging treatment and postharvest UV-B/visible irradiation conditions. Sci. Hortic. 2013;151:75–82. doi: 10.1016/j.scienta.2012.12.020. [DOI] [Google Scholar]
- 9.Lespinasse Y, Guérif P. Inheritance of red leaf colour from pear red sports of ‘Doyenne du Comice’, ‘Bartlett’ and ‘Beurre Hardy’. Acta Hortic. 2011;909:97–102. doi: 10.17660/ActaHortic.2011.909.9. [DOI] [Google Scholar]
- 10.Dondini L, et al. The inheritance of the red colour character in European pear (Pyrus communis) and its map position in the mutated cultivar ‘Max Red Bartlett’. Plant Breed. 2008;127:524–526. doi: 10.1111/j.1439-0523.2008.01500.x. [DOI] [Google Scholar]
- 11.Steyn WJ, Wand SJE, Holcroft DM, Jacobs G. Red colour development and loss in pears. Acta Hortic. 2005;671:79–85. doi: 10.17660/ActaHortic.2005.671.9. [DOI] [Google Scholar]
- 12.Zhang X, et al. Differential gene expression analysis of Yunnan red pear, Pyrus pyrifolia, during fruit skin coloration. Plant Mol. Biol. Rep. 2011;29:305–314. doi: 10.1007/s11105-010-0231-z. [DOI] [Google Scholar]
- 13.Zhang, D., Qian, M., Yu, B. & Teng, Y. Effect of fruit maturity on UV-B-induced post-harvest anthocyanin accumulation in red Chinese sand pear. Acta Physiol. Plant35, 2857–2866 (2013).
- 14.Ni J, et al. Ethylene response factors Pp4ERF24 and Pp12ERF96 regulate blue light-induced anthocyanin biosynthesis in ‘Red Zaosu’pear fruits by interacting with MYB114. Plant Mol. Biol. 2019;99:67–78. doi: 10.1007/s11103-018-0802-1. [DOI] [PubMed] [Google Scholar]
- 15.Ngo T, Zhao Y. Stabilization of anthocyanins on thermally processed red D’Anjou pears through complexation and polymerization. LWT-Food Sci. Technol. 2009;42:1144–1152. doi: 10.1016/j.lwt.2009.02.013. [DOI] [Google Scholar]
- 16.Winkel-Shirley B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 2001;126:485–493. doi: 10.1104/pp.126.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu B, et al. Isolation of anthocyanin biosynthetic genes in red Chinese sand pear (Pyrus pyrifolia Nakai) and their expression as affected by organ/tissue, cultivar, bagging and fruit side. Sci. Hortic. 2012;136:29–37. doi: 10.1016/j.scienta.2011.12.026. [DOI] [Google Scholar]
- 18.Yang YN, et al. Molecular cloning and gene expression differences of the anthocyanin biosynthesis-related genes in the red/green skin color mutant of pear (Pyrus communis L.) Tree Genet. Genomes. 2013;9:1351–1360. doi: 10.1007/s11295-013-0644-6. [DOI] [Google Scholar]
- 19.Ban Y, et al. Isolation and functional analysis of a MYB transcription factor gene that is a key regulator for the development of red coloration in apple skin. Plant Cell Physiol. 2007;48:958–970. doi: 10.1093/pcp/pcm066. [DOI] [PubMed] [Google Scholar]
- 20.Xu WJ, Dubos C, Lepiniec L. Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 2015;20:176–185. doi: 10.1016/j.tplants.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 21.Albert NW, et al. Members of an R2R3-MYB transcription factor family in Petunia are developmentally and environmentally regulated to control complex floral and vegetative pigmentation patterning. Plant J. 2011;65:771–784. doi: 10.1111/j.1365-313X.2010.04465.x. [DOI] [PubMed] [Google Scholar]
- 22.Feng SQ, Wang YL, Yang S, Xu YT, Chen XS. Anthocyanin biosynthesis in pears is regulated by a R2R3-MYB transcription factor PyMYB10. Planta. 2010;232:245–255. doi: 10.1007/s00425-010-1170-5. [DOI] [PubMed] [Google Scholar]
- 23.Yao GF, et al. Map-based cloning of the pear gene MYB114 identifies an interaction with other transcription factors to coordinately regulate fruit anthocyanin biosynthesis. Plant J. 2017;92:437–451. doi: 10.1111/tpj.13666. [DOI] [PubMed] [Google Scholar]
- 24.Tao R, et al. The blue light signal transduction pathway is involved in anthocyanin accumulation in ‘Red Zaosu’ pear. Planta. 2018;248:37–48. doi: 10.1007/s00425-018-2877-y. [DOI] [PubMed] [Google Scholar]
- 25.Bai S, et al. Two B‐box proteins, PpBBX18 and PpBBX21, antagonistically regulate anthocyanin biosynthesis via competitive association with PpHY5 in the peel of pear fruit. Plant J. 2019;100:1208–1223. doi: 10.1111/tpj.14510. [DOI] [PubMed] [Google Scholar]
- 26.Wu J, et al. Identification of differentially expressed genes related to coloration in red/green mutant pear (Pyrus communis L.) Tree Genet. Genomes. 2013;9:75–83. doi: 10.1007/s11295-012-0534-3. [DOI] [Google Scholar]
- 27.Xue H, et al. The genetic locus underlying red foliage and fruit skin traits is mapped to the same location in the two pear bud mutants ‘Red Zaosu’ and ‘Max Red Bartlett’. Hereditas. 2018;155:25. doi: 10.1186/s41065-018-0063-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li P, Zhang Y, Einhorn TC, Cheng L. Comparison of phenolic metabolism and primary metabolism between green ‘Anjou’ pear and its bud mutation, red ‘Anjou’. Physiol. Plant. 2014;150:339–354. doi: 10.1111/ppl.12105. [DOI] [PubMed] [Google Scholar]
- 29.Booi S, van Dyk MM, du Preez MG, Rees DJG, Labuschagné I. Molecular typing of red and green phenotypes of ‘Bon Rouge’ pear trees, with the use of microsatellites. Acta Hort. 2005;671:293–297. doi: 10.17660/ActaHortic.2005.671.42. [DOI] [Google Scholar]
- 30.Li J, et al. A new red pear cultivar ‘Nanhong Li’. Acta Hort. Sin. 2011;38:1821–1822. [Google Scholar]
- 31.Khanna R, et al. The Arabidopsis B-box zinc finger family. Plant Cell. 2009;21:3416–3420. doi: 10.1105/tpc.109.069088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gangappa SN, Botto JF. The BBX family of plant transcription factors. Trends Plant Sci. 2014;19:460–470. doi: 10.1016/j.tplants.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 33.Wei C, et al. The Arabidopsis B-box protein BZS1/BBX20 interacts with HY5 and mediates strigolactone regulation of photomorphogenesis. J. Genet. Genom. 2016;43:555–563. doi: 10.1016/j.jgg.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fang H, et al. The B-box zinc finger protein MdBBX20 integrates anthocyanin accumulation in response to ultraviolet radiation and low temperature. Plant Cell Environ. 2019;42:2090–2104. doi: 10.1111/pce.13499. [DOI] [PubMed] [Google Scholar]
- 35.Job N, Yadukrishnan P, Bursch K, Datta S, Johansson H. Two B-box proteins regulate photomorphogenesis by oppositely modulating HY5 through their diverse C-terminal domains. Plant Physiol. 2018;176:2963–2976. doi: 10.1104/pp.17.00856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu D, et al. BBX21, an Arabidopsis B-box protein, directly activates HY5 and is targeted by COP1 for 26S proteasome-mediated degradation. Proc. Natl. Acad. Sci. U. S. A. 2016;113:7655–7660. doi: 10.1073/pnas.1607687113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim DH, et al. A rice B-Box protein, OsBBX14, finely regulates anthocyanin biosynthesis in rice. Int. J. Mol. Sci. 2018;19:2190. doi: 10.3390/ijms19082190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bai B, et al. OsBBX14 promotes photomorphogenesis in rice by activating OsHY5L1 expression under blue light conditions. Plant Sci. 2019;284:192–202. doi: 10.1016/j.plantsci.2019.04.017. [DOI] [PubMed] [Google Scholar]
- 39.Bai S, et al. An apple B-box protein, MdCOL11, is involved in UV-B-and temperature-induced anthocyanin biosynthesis. Planta. 2014;240:1051–1062. doi: 10.1007/s00425-014-2129-8. [DOI] [PubMed] [Google Scholar]
- 40.An JP, et al. MdBBX22 regulates UV‐B‐induced anthocyanin biosynthesis through regulating the function of MdHY5 and is targeted by MdBT2 for 26S proteasome‐mediated degradation. Plant Biotechnol. J. 2019;17:2231–2233. doi: 10.1111/pbi.13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang CQ, Sarmast MK, Jiang J, Dehesh K. The transcriptional regulator BBX19 promotes hypocotyl growth by facilitating COP1-mediated EARLY FLOWERING3 degradation in Arabidopsis. Plant Cell. 2015;27:1128–1139. doi: 10.1105/tpc.15.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holtan HE, et al. BBX32, an Arabidopsis B-Box protein, functions in light signaling by suppressing HY5-regulated gene expression and interacting with STH2/BBX21. Plant Physiol. 2011;156:2109–2123. doi: 10.1104/pp.111.177139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.An JP, et al. An apple B-Box protein MdBBX37 modulates anthocyanin biosynthesis and hypocotyl elongation synergistically with MdMYBs and MdHY5. Plant Cell Physiol. 2019;61:130–143. doi: 10.1093/pcp/pcz185. [DOI] [PubMed] [Google Scholar]
- 44.Fang H, et al. MdCOL4 interaction mediates crosstalk between UV-B and high temperature to control fruit coloration in apple. Plant Cell Physiol. 2019;60:1055–1066. doi: 10.1093/pcp/pcz023. [DOI] [PubMed] [Google Scholar]
- 45.Jiang L, et al. Arabidopsis STO/BBX24 negatively regulates UV-B signaling by interacting with COP1 and repressing HY5 transcriptional activity. Cell Res. 2012;22:1046. doi: 10.1038/cr.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gangappa SN, Holm M, Botto JF. Molecular interactions of BBX24 and BBX25 with HYH, HY5 HOMOLOG, to modulate Arabidopsis seedling development. Plant Signal. Behav. 2013;8:e25208. doi: 10.4161/psb.25208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holm M, Hardtke CS, Gaudet R, Deng XW. Identification of a structural motif that confers specific interaction with the WD40 repeat domain of Arabidopsis COP1. EMBO J. 2001;20:118–127. doi: 10.1093/emboj/20.1.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan H, et al. Nuclear localization and interaction with COP1 are required for STO/BBX24 function during photomorphogenesis. Plant Physiol. 2011;156:1772–1782. doi: 10.1104/pp.111.180208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dong W, Wu D, Li G, Wu D, Wang Z. Next-generation sequencing from bulked segregant analysis identifies a dwarfism gene in watermelon. Sci. Rep. 2018;8:2908. doi: 10.1038/s41598-018-21293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klein H, et al. Bulked-segregant analysis coupled to whole genome sequencing (BSA-Seq) for rapid gene cloning in Maize. G3 Genes Genom. Genet. 2018;8:3583–3592. doi: 10.1534/g3.118.200499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li F, et al. Identification of a locus for seed shattering in rice (Oryza sativa L.) by combining bulked segregant analysis with whole-genome sequencing. Mol. Breed. 2019;39:36. doi: 10.1007/s11032-019-0941-3. [DOI] [Google Scholar]
- 52.Lu Z, et al. Fine mapping of the temperature-sensitive semi-dwarf (Tssd) locus regulating the internode length in peach (Prunus persica) Mol. Breed. 2016;36:20. doi: 10.1007/s11032-016-0442-6. [DOI] [Google Scholar]
- 53.Doyle JJ, Doyle JL. Isolation of plant DNA from fresh tissue. Focus. 1990;12:39–40. [Google Scholar]
- 54.Ou C, et al. A de novo genome assembly of the dwarfing pear rootstock Zhongai 1. Sci. Data. 2019;6:281. doi: 10.1038/s41597-019-0291-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reumers J, et al. Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat. Biotechnol. 2012;30:61–68. doi: 10.1038/nbt.2053. [DOI] [PubMed] [Google Scholar]
- 57.Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hill JT, et al. MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq. Genome Res. 2013;23:687–697. doi: 10.1101/gr.146936.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fekih R, et al. MutMap+: genetic mapping and mutant identification without crossing in rice. PLoS One. 2013;8:e68529. doi: 10.1371/journal.pone.0068529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pertea M, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Florea L, Song L, Salzberg SL. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000 Res. 2013;2:188. doi: 10.12688/f1000research.2-188.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data associated with this study have been deposited in NCBI under accession number PRJNA577323. The raw data from this project have been deposited in the NCBI SRA database under accession number SRP225824.