

Abstract
Farnesoid X receptor (FXR) is the nuclear receptor of bile acids and is involved in innate immune regulation. FXR agonists have been shown to protect multiple organs from inflammatory tissue injuries. Because liver expresses high levels of FXR, we explored the potential therapeutic benefits and underlying mechanisms of pharmacologic FXR activation in a murine model of partial liver warm ischemia. Pretreatment of mice with FXR agonist 3‐(2,6‐dichlorophenyl)‐4‐(3′‐carboxy‐2‐chlorostilben‐4‐yl)oxymethyl‐5‐isopropylisoxazole (GW4064) attenuated liver ischemia/reperfusion injuries (IRIs) in wild‐type but not FXR knockout mice. Posttreatment with GW4064 facilitated liver recovery from IRI. Mechanistically, Kupffer cells (KCs) expressed much higher levels of FXR than bone marrow‐derived macrophages (BMMs). Pretreatment of KCs but not BMMs with GW4064 resulted in lower tumor necrosis factor α but higher interleukin‐10 expressions following toll‐like receptor stimulation. FXR‐targeted gene small heterodimer partner (SHP) was critical for the regulation of KC response by GW4064. In vivo, the depletion of KCs but not cluster of differentiation (CD) 11b+ cells or knockdown of SHP diminished the immune regulatory effect of GW4064 in liver IRI. Thus, FXR activation protects liver from IRI by up‐regulating SHP in KCs to inhibit the liver proinflammatory response.
Abbreviations
- ASC
apoptosis‐associated speck‐like protein
- B6 mice
C57BL/6J mice
- BMM
bone marrow‐derived macrophage
- Bsep
bile salt export pump
- CD
cluster of differentiation
- CL
clodronate‐encapsulated liposome
- Ctl
control
- Cyp7a1
cytochrome P450 family 7 subfamily A member 1
- DT
diphtheria toxin
- DTR
DT receptor
- FBS
fetal bovine serum
- FXR
farnesoid X receptor
- GW4064/GW
3‐(2,6‐dichlorophenyl)‐4‐(3′‐carboxy‐2‐chlorostilben‐4‐yl)oxymethyl‐5‐isopropylisoxazole
- HE
hematoxylin and eosin
- HPRT
hypoxanthine‐guanine phosphoribosyltransferase
- IL
interleukin
- iMՓ
infiltrating macrophage
- IR
ischemia/reperfusion
- IRI
ischemia/reperfusion injury
- KC
Kupffer cell
- KD
knockdown
- LPS
lipopolysaccharide
- MФ
macrophage
- NF‐κB
nuclear factor kappa B
- NLR
nucleotide‐binding oligomerization domain‐like receptor
- NLRP3
NLR family pyrin domain containing 3
- NPC
nonparenchymal cell
- OCA
obeticholic acid
- Ostβ
organic solute transporter beta
- qRT‐PCR
quantitative reverse‐transcription polymerase chain reaction
- sALT
serum alanine aminotransferase
- SHP
small heterodimer partner
- siRNA
small interfering RNA
- ssiRNA
scrambled small interfering RNA
- TGR5
Takeda G protein‐coupled receptor 5
- TLR
toll‐like receptor
- TNF‐α
tumor necrosis factor alpha
- WT
wild type
Ischemia/reperfusion injury (IRI) is a major complication in liver surgeries.1 In liver transplantation, despite the advances in surgical techniques and organ preservations, IRI remains responsible for approximately 10% of early graft failure2 and leads to a higher incidence of acute and chronic rejection. These adverse effects may become more significant in liver transplants with expanded criteria, including those from marginal, deceased, and nonbeating‐heart donors. Thus, development of an effective therapeutic strategy against liver IRI will help to not only improve the outcome of liver transplantation but also expand the donor pool to alleviate the severe organ shortage for liver transplantation.3
The pathophysiology of liver IRI is complicated. Hepatocellular damages result from at least two insults: primary cellular destruction due to metabolic/oxidative stress and secondary cytotoxicity sequent to tissue inflammation. Liver ischemia/reperfusion (IR) triggers an innate immune‐dominated inflammatory response mediated by the sentinel pattern recognition receptor (PRR) system.3, 4 It has been documented that a danger‐associated molecular pattern (DAMP), such as high‐mobility group box 1 (HMGB‐1), derived from necrotic/stressed cells4, 5, 6 activates PRRs, including toll‐like receptors (TLRs) and nucleotide‐binding oligomerization domain‐like receptors (NLRs),5, 6, 7, 8, 9, 10, 11 to initiate the response. Macrophages (MФs) are the major innate immune cells responding to DAMPs in liver IRI. Infiltration and activation of peripheral monocyte‐derived infiltrating MՓs (iMՓs) is the cardinal feature of IR‐induced inflammatory response. The liver is a unique organ in that it contains a large number of resident MΦs (Kupffer cells [KCs]) in the homeostatic state. KCs represent approximately 35% of liver nonparenchymal cells (NPCs) and 80% to 90% of all tissue MΦs in the body.12 Both KCs and iMՓs are involved in the pathogenesis of liver IRI.
Farnesoid X receptor (FXR) is the nuclear receptor for bile acids and a member of the nuclear hormone receptor family with transcriptional factor activities. It is highly expressed in liver, kidneys, and small intestine.13 FXR binds to promoters of its target genes in a ligand‐dependent manner and regulates transcriptions of genes involved in cholesterol/bile acid metabolism, hepatic gluconeogenesis, and lipogenesis. FXR regulates hepatic fibrosis, nonalcoholic fatty liver disease liver regeneration, and acute hepatitis.14, 15 In organ ischemia models, the roles of FXR have been controversial and seem to be organ specific. Pharmacologic inhibition or genetic ablation of FXR significantly reduced myocardial apoptosis, decreased infarct, and improved cardiac function in IR myocardium.16 FXR activation by its agonists, such as 3‐(2,6‐dichlorophenyl)‐4‐(3′‐carboxy‐2‐chlorostilben‐4‐yl)oxymethyl‐5‐isopropylisoxazole (GW4064) and 6α‐ethyl‐chenodeoxycholic acid (6α‐ECDCA), protected small intestine and kidneys from IRI and attenuated local inflammatory responses.17, 18 In liver, obeticholic acid (OCA) marginally reduced IRI in a rat model.19 In the current study, we explored the therapeutic potential of pharmacologic FXR activation and the underlying mechanism in the pathophysiology of liver IRI in a murine model of partial hepatic warm ischemia.
Materials and Methods
Animals
Male C57BL/6J (B6) and FXR‐deficient (Nr1h4tm1Gonz/J) mice (6‐8 weeks old) were purchased from the Jackson Laboratory (Bar Harbor, ME). All mice were housed in the University of California Los Angeles (UCLA) animal facility under a specific pathogen‐free condition and received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health. All animal experiments were approved by the UCLA Office of the Animal Research Oversight Committee.
Liver IRI Model
After a midline laparotomy, mice were injected with heparin (100 μg/kg), and an atraumatic clip was used to interrupt arterial/portal venous blood supply to the cephalad liver lobes. After 60 to 90 minutes of ischemia, the clip was removed to initiate liver reperfusion. Sham controls underwent the same procedure but without vascular occlusion. Mice were killed after 6 hours or 5 days of reperfusion, and liver and serum samples were collected. To test the effect on acute liver IRI, GW4064 (5 mg/kg, intraperitoneally; Tocris Bioscience) or OCA (30 mg/kg, oral gavage; Sigma‐Aldrich, St Louis, MO) was administered 1 to 2 hours before the onset of liver ischemia. To test the effect on liver recovery from IRI, GW4064 (5 mg/kg) was administered intraperitoneally at 24 hours, day 2, day 3, and day 4 after reperfusion. Levels of serum alanine aminotransferase (sALT) were measured with an autoanalyzer by IDEXX Laboratories (Westbrook, Maine). A portion of the liver specimens was fixed in 10% buffered formalin and embedded in paraffin. Liver sections (4 μm) were stained with hematoxylin and eosin (HE). The severity of liver IRI was graded blindly using Suzuki's criteria20 on a scale from 0 to 4. Liver sections were processed for immunohistochemical staining of myeloperoxidase (Novus Biologicals) to quantitate polymorphonuclear leukocyte infiltration as described.21, 22
In Vivo Depletion of KCs and Cluster of Differentiation 11b+ MФs
KCs were depleted using the Macrophage Depletion Kit (Encapsula NanoSciences, LLC, Brentwood, TN). In brief, 200 μL/mouse of clodronate‐encapsulated liposomes (CLs) or control liposomes was injected in B6 mice intravenously 48 hours before the onset of liver ischemia. In cluster of differentiation (CD) 11b‐diphtheria toxin (DT) receptor (DTR) mice, 10 μg/g DT was injected intravenously 48 hours before the onset of liver ischemia. The depletion of KCs or CD11b MФs was confirmed by fluorescence‐activated cell sorting (FACS) analysis of liver NPCs 6 hours after reperfusion. CL treatment effectively depleted KCs but spared iMՓs; DT treatment depleted CD11b cells (both iMՓs and neutrophils) but spared KCs in CD11b‐DTR mice (Supporting Fig. S1A). Furthermore, immunofluorescence staining of liver sections with anti‐F4/80‐fluorescein isothiocyanate and 4′,6‐diamidino‐2‐phenylindole revealed the depletion of KCs by CL treatment (Supporting Fig. S1B).
Quantitative Reverse‐Transcription Polymerase Chain Reaction
Total RNA (2.0 μg) was reverse transcribed into complementary DNA (cDNA) using Oligo (dT) 12‐18 Primer and M‐MLV Reverse Transcriptase kit (Invitrogen, Carlsbad, CA). Quantitative reverse‐transcription polymerase chain reaction (qRT‐PCR) was performed using the QuantStudio 3 System (Thermo Fisher Scientific, Waltham, MA). In a final reaction volume of 20 µL, we mixed 1× PCR Mix (Power SYBR Green PCR Master Mix; Life Technologies, Woolston, United Kingdom), cDNA, and 0.125 μM of each primer. The amplification condition was 50°C/2 minutes, 95°C/10 minutes, followed by 40 cycles of 95°C/15 seconds, 60°C/1 minute. Primers for Fxr and its target genes are listed in Table 1. Other primers used to amplify specific mouse gene fragments are the same as described.23
Table 1.
Primers for qRT‐PCR
| Gene | 5′ Primer | 3′ Primer |
|---|---|---|
| Fxr | GGGATGAGCTGTGTGTTGTC | GGCGTTCTTGGTAATGCTTC |
| Abcb4 | AATTGGCCTGTTGACACTGTT | GGAAGACCTGATCCATGAGC |
| Shp | CTCTGCAGGTCGTCCGACTATTCTG | CCTCGAAGGTCACAGCATCCTG |
| Ostβ | AGCCTGAAGTCCCAGGAATC | TGCACTGTGGTCCATGTTTC |
| Bsep | GGACACACCTGAGAGGACCTT | AGATCGTTGACGGATGGAAG |
| Cyp7a1 | TCAAGCAAACACCATTCCTG | GGCTGCTTTCATTGCTTCA |
Abbreviation: Abcb4, adenosine triphosphate binding cassette subfamily B member 4.
KC Isolation
Liver NPCs were isolated from normal or IR livers of B6 mice by in situ collagenase perfusion. In brief, livers were perfused through the portal vein with calcium‐ and magnesium‐free Hank's balanced salt solution (HBSS) supplemented with 2% heat‐inactivated fetal bovine serum (FBS), followed by 0.27% collagenase IV (Sigma‐Aldrich). Perfused livers were dissected and filtered through 70‐μm nylon mesh cell strainers (BD Biosciences, San Diego, CA). NPCs were separated from hepatocytes by low‐speed centrifugation (50g, 2 minutes) 3 times. NPCs were stained with fluorescence‐labeled antibodies and analyzed by FACS. To enrich KCs, NPCs were suspended in HBSS and layered onto a two‐layer 25% to 50% Percoll gradient (Sigma‐Aldrich) in a 50‐mL conical centrifuge tube and centrifuged at 1,800g at 4°C for 15 minutes. KCs in the middle layer were collected and allowed to attach to cell‐culture plates in supplemented Dulbecco's modified Eagle's medium (DMEM) with 10% FBS for 15 minutes at 37°C. Nonadherent cells were removed by replacing the culture medium. The purity of KCs in the adherent cells was approximately 80%, as determined by immunofluorescent staining with anti‐F4/80.
Bone Marrow‐Derived Macrophages
Bone marrow cells were isolated from mouse femurs and tibias. Cells were cultured in DMEM supplemented with 10% FBS and 20% L929‐conditioned medium for 7 days. Bone marrow‐derived macrophages (BMMs) were then replated and cultured overnight in new culture dishes for further experiments.
Western Blot Analysis
Cellular proteins were extracted with ice‐cold lysis buffer (1% Triton X‐100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 10% glycerol, 137 mM sodium chloride, 20 mM Tris, pH 7.4). Proteins (20 μg) were subjected to 12% SDS‐polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride nitrocellulose membrane. Antibodies against FXR (D‐3, SC25309) and β‐actin (Santa Cruz Technology) were used to probe the membrane.
Small Heterodimer Partner Knockdown
Small heterodimer partner (SHP) small interfering RNA (siRNA) with a pool of three target‐specific siRNAs (sc‐44870; Santa Cruz Biotechnology) was used to knock down SHP gene expression. In vitro, BMMs were transiently transfected with siRNA using LipoJet In Vitro Transfection Kit (SignaGen Laboratories) according to the manufacturer's protocol. In vivo, siRNAs were mixed with mannose‐conjugated polymers (Polyplus‐transfection SA) at a ratio specified by the manufacturer and administered intraperitoneally (siRNA 2 mg/kg) 3 hours before the onset of liver ischemia.
In Vitro MФ Activation
KCs (control or SHP siRNA transfected) or BMMs were stimulated with lipopolysaccharide (LPS) (100‐500 ng/mL; InvivoGen, San Diego, CA) in the absence or presence of GW4064 (10 μM; Tocris), or OCA (10 μM; MedChemExpress), or INT‐777 (3 μM; Cayman Chemical). Culture supernatants were collected at 0, 6, and 24 hours after stimulation for cytokine measurements. Cells were collected at 3 to 6 hours after stimulation for RNA/protein preparation.
Statistics
Results are shown as mean ± SD. Comparisons between two parameters were analyzed by the unpaired Student t test. Multiple group comparisons were performed using one‐way analysis of variance followed by Bonferroni's post hoc test. All analyses were performed using Stata software (version 11.0). P < 0.05 (two‐tailed) was considered statistically significant.
Results
FXR Pharmacologic Activation Protects Liver From IRI
To test whether pharmacologic activation of FXR protected livers from IRI, the selective FXR agonist GW4064 was administered in wild‐type (WT) B6 mice before the onset of liver ischemia (90 minutes). Liver FXR activation by GW4064 in vivo was confirmed by qRT‐PCR analysis of FXR target gene expressions, including up‐regulation of organic solute transporter beta (Ostβ), bile salt export pump (Bsep), and Shp and the down‐regulation of cytochrome P450 family 7 subfamily A member 1 (Cyp7a1) in sham livers of WT mice (Fig. 1A). IR inhibited liver FXR expression and its activities, as evidenced by the lower levels of expressions of FXR proteins and FXR‐targeted genes in IR livers than those in shams (Fig. 1A,B). However, FXR activation remained able to up‐regulate its targeted genes in IR livers as their expression levels were higher in GW4064‐treated livers than those in controls at 6 hours after reperfusion. Functionally, GW4064 treatment resulted in a significant protection of livers from IRI. There were lower levels of sALT and better preserved liver histologic architectures with improved Suzuki scores in the GW4064‐treated versus vehicle‐treated cohorts (Fig. 1C). The therapeutic benefit of FXR activation in liver IRI was confirmed with a second FXR agonist, OCA (INT‐747), under a milder ischemic condition (60 minutes of ischemia; Supporting Fig. S2). Liver inflammatory immune activation was also inhibited by GW4064, as indicated by lower levels of tumor necrosis factor α (TNF‐α), interleukin‐1β (IL‐1β), and IL‐6 expressions in IR livers (Fig. 1D). The liver‐protective effects of GW4064 were absent in FXR knockout (KO) hosts (Supporting Fig. S3), confirming the target specificity of the pharmacologic agent in vivo.
Figure 1.
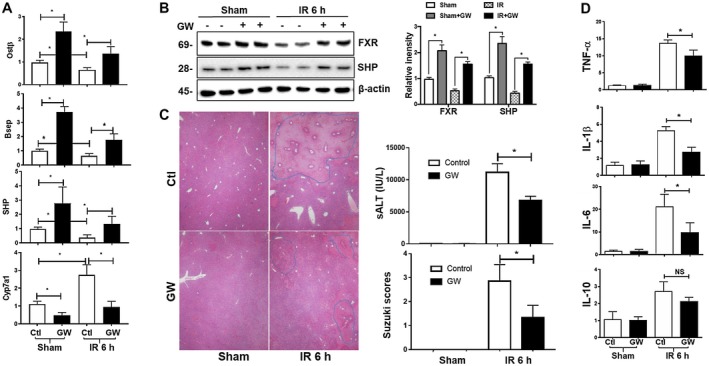
FXR activation protects livers from IRI. B6 mice pretreated with or without (control) FXR agonist GW4064 were subjected to 90 minutes of partial liver warm ischemia followed by 6 hours of reperfusion, as described in Materials and Methods. (A) Plots of average FXR target gene/HPRT ratios in livers of different experimental groups. Gene expressions in liver tissues were measured by qRT‐PCR. (B) Western blotting of FXR and SHP in liver tissues. (C) Representative liver HE staining (magnification ×100), plots of average sALT levels, and liver Suzuki scores of different experimental groups. (D) Plots of average inflammatory gene/HPRT ratios in livers. Data represent Mean ± SD, n = 4‐6/group; *P < 0.05. Abbreviations: h, hour; NS, not significant.
To further explore the therapeutic potential of pharmacologic activation of FXR in liver IRI, we determined whether postischemia treatment could improve liver recovery from IRI. In our model of liver IRI, it usually takes liver 7 days to recover from 90 minutes warm ischemia‐induced liver reperfusion injury (data not shown). GW4064 was administered daily starting at 24 hours after reperfusion. Liver recovery from IRI was evaluated at day 5 after reperfusion when the repair of hepatocellular damage was incomplete in IR livers of controls. Posttreatment with GW4064 facilitated liver recovery from IRI such that liver histologic damage in these treated mice was nearly all repaired at this time point, with significantly lower Suzuki scores compared with those in controls (Fig. 2A). The sALT levels were all at baseline at this time point of reperfusion in both groups of mice (data not shown). At the molecular level, GW4064 treatment resulted in lower TNF‐α levels but higher levels of gene expressions related to reparative MФs, such as Mer tyrosine kinase (MerTK) and T‐cell immunoglobulin mucin protein 4 (TIM‐4) (Fig. 2B). The activation of FXR in IR livers by GW4064 was confirmed by qRT‐PCR analysis of FXR target gene expressions (Fig. 2C). Thus, FXR pharmacologic activation protects liver from IRI. The pretreatment regimen reduces hepatocellular damage and inhibits liver inflammatory response against IR, and the postischemic regimen facilitates liver recovery from IRI.
Figure 2.
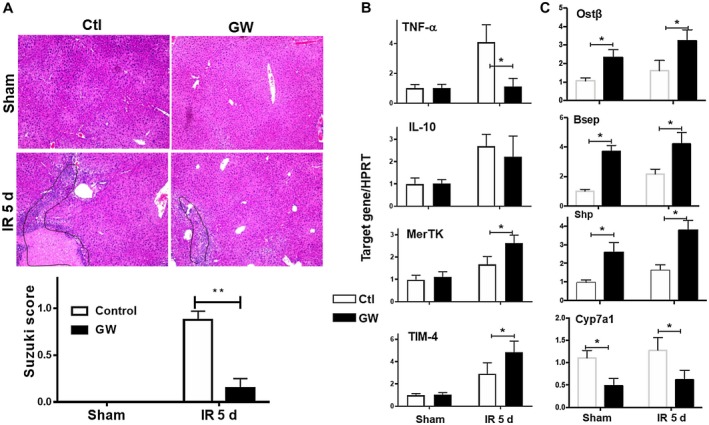
FXR activation facilitates liver recovery from IRI. B6 mice were subjected to 90 minutes of partial liver warm ischemia and divided into two groups treated with daily injection of either the vehicle control or GW4064, as described in Materials and Methods. IR liver tissues were harvested at day 5 after reperfusion. (A) Representative liver HE sections (magnification ×100) and plots of average Suzuki scores of control and GW groups. (B,C) Plots of average target gene/HPRT ratios of different experimental groups. Inflammatory and FXR‐targeted gene expressions in sham and IR livers were measured by qRT‐PCR. Representative results of two independent experiments; Data represent Mean ± SD, n = 4‐6/group; *P < 0.05, **P < 0.01. Abbreviations: d, day; MerTK, Mer tyrosine kinase; TIM‐4, T‐cell immunoglobulin mucin protein 4.
Differential Regulation of KCs and BMMs by FXR Activation
FXR is expressed in both parenchymal cells and NPCs in the liver. FXR activation‐induced liver protection could potentially result from direct cytoprotection in hepatocytes or immune regulation by immune cells. Because FXR activation has been shown to regulate hepatocyte proliferation and immune response, we focused on whether liver‐resident KCs were involved in the protection of liver from IRI. We first analyzed FXR expression in liver tissues and KCs/NPCs at resting state and during IRI as well as in BMMs in response to LPS. Liver tissues expressed the highest level of FXR. KCs/NPCs also expressed the receptor gene, whereas BMMs expressed it at minimal levels, which was nearly 1,000‐fold lower compared with KCs (Fig. 3A). IR down‐regulated FXR expression in liver and NPCs, and LPS stimulation up‐regulated FXR in BMMs. At the protein level, western blot analysis showed that KCs but not BMMs expressed FXR, which was increased by GW4064 treatment and decreased by LPS stimulation (Fig. 3B).
Figure 3.
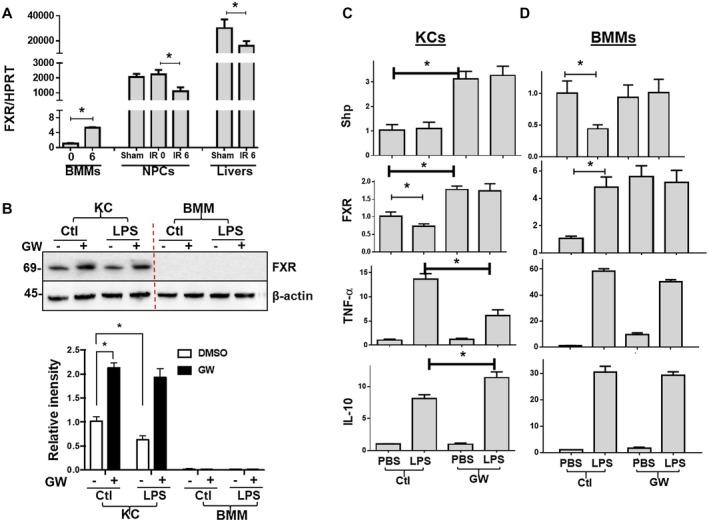
FXR expressions and functions in macrophages. (A) Plots of average FXR/HPRT gene expression ratios in livers and macrophages. FXR expressions were determined by qRT‐PCR in sham or IR livers (6 hours after reperfusion), liver NPCs (isolated from sham or IR livers after 0 or 6 hours of reperfusion), or BMMs stimulated with LPS for 0 and 6 hours in vitro. (B) Western blots of FXR expressions in KCs and BMMs. (C, D) Plots of target gene/HPRT ratios in KCs and BMMs. KCs were isolated from sham livers, and BMMs were derived from 7‐day cultures of bone marrow cells, as described in Materials and Methods. These macrophages were pretreated with either vehicle controls or GW4064 followed by 3 hours of stimulation of LPS. Gene expressions were determined by qRT‐PCR. Representative results of at least two independent experiments; Data represent Mean ± SD, n = 3‐6/group; *P < 0.05. Abbreviations: DMSO, dimethyl sulfoxide; PBS, phosphate‐buffered saline.
Because FXR was differentially expressed in KCs and BMMs, we next determined whether pharmacologic FXR activation regulated these MФs differently in their inflammatory activation in vitro. Preincubation with 10 μM GW4064 for 2 hours resulted in Shp up‐regulation in both resting and LPS‐stimulated KCs but not in BMMs, indicative of functional FXR in KCs but not BMMs (Fig. 3C). Interestingly, although LPS down‐regulated Shp in control BMMs, the SHP level was sustained in GW4064‐treated BMMs. FXR gene expression was increased by GW4064 treatment in KCs and BMMs. LPS stimulation decreased FXR in KCs but increased it in BMMs. FXR activation inhibited KC proinflammatory activation by decreased TNF‐α (~50%) but increased IL‐10 (~30%) gene expression following LPS stimulation (Fig. 3C). In BMMs, however, GW4064 only marginally inhibited LPS‐induced TNF‐α expression (~10%) without significant impact on IL‐10 (Fig. 3D). These results demonstrate distinctive expression patterns and regulatory effects of FXR in KCs and BMMs.
We also tested the potential immune regulatory effect of the FXR agonist OCA (INT‐747) and Takeda G protein‐coupled receptor 5 (TGR5) agonist INT‐777 on KCs and BMMs in vitro (Supporting Fig. S4). Similar to GW4064, OCA selectively modulated KCs but not BMMs in response to LPS stimulation. Importantly, OCA increased Shp expression in KCs in both resting and activation states. Interestingly, TGR5 activation by INT‐777 regulated both KCs and BMMs in response to LPS such that the expression of TNF‐α was decreased with simultaneous increases of IL‐10. However, INT‐777 failed to induce Shp expression in KCs. These results indicate that FXR activation selectively targets KCs whereas TGR5 activation affects both KCs and BMMs in their inflammatory response and that Shp is an FXR‐ but not a TGR5‐inducible gene.
KCs but not BMMs Are Critical for FXR‐Mediated Protection in Liver IRI
To test whether KCs mediated FXR activation‐induced liver protection against IRI, KCs were depleted by CLs and administered intravenously 48 hours before the onset of liver ischemia. Liver inflammation and injury were evaluated at 6 hours after reperfusion. Consistent with previous studies,24, 25, 26 CL‐mediated KC depletion resulted in increases in liver IRI, with higher levels of sALT and more severely damaged liver histologic architectures in CL‐treated versus blank liposome‐treated mice (Fig. 4A). Interestingly, FXR activation by GW4064 was no longer able to protect liver from IRI in KC‐depleted mice because there were no differences in sALT levels or liver histopathology between GW4064‐treated and vehicle‐treated cohorts (Fig. 4A). FXR activation also failed to inhibit liver inflammatory gene induction by IR, including TNF‐α, IL‐1β, and IL‐6, in the absence of KCs (Fig. 4B). It was noted that the GW4064‐induced alterations of FXR‐targeted genes in liver were diminished by CL treatment, indicating that KCs were responsive to FXR activation in vivo (Fig. 4C). These results indicate that KCs are required for FXR‐mediated inhibition of liver proinflammatory response and protection of liver from IRI. To address the possibility that the KC‐depleted livers were damaged too much to rescue by FXR activation, we repeated the experiment under a milder ischemia condition (60 minutes). Liver IRI at 6 hours after reperfusion was significantly less after 60 minutes of ischemia compared with that after 90 minutes of ischemia (Supporting Fig. S2), sALT levels were lower, and liver parenchymal damage was less severe. Under the 60‐minute ischemia condition, KC depletion resulted in higher liver IRI, comparable to that in control livers after 90 minutes of ischemia. Pretreatment with OCA was not able to protect those KC‐depleted livers from IRI, confirming the role of KCs in mediating the liver‐protective effect of FXR activation.
Figure 4.
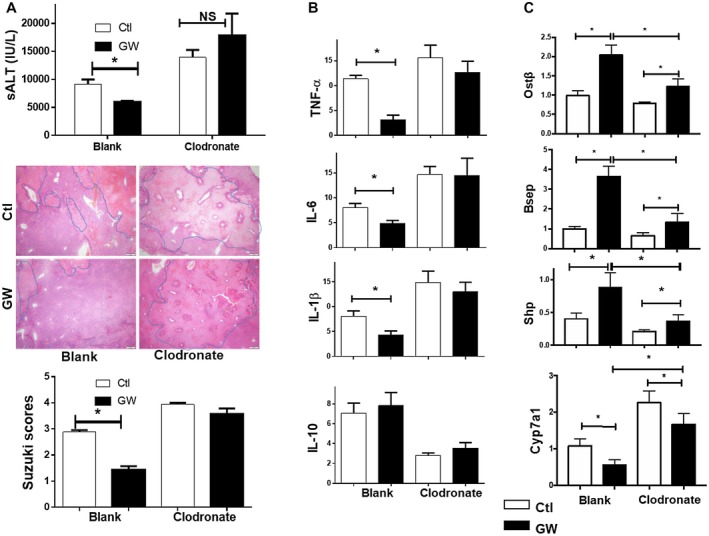
KCs mediate the immune regulatory effect of FXR activation in liver IRI. Groups of WT B6 mice were treated with either blank or clodronate liposomes 48 hours before, followed by the treatment of either vehicle controls or GW4064 2 hours before the onset of liver ischemia. Liver injuries and inflammatory immune responses were evaluated at 6 hours after reperfusion. (A) Plots of average sALT levels, representative liver histologic analysis (HE staining, magnification ×100), and plots of average Suzuki's scores in different experimental groups. (B,C) Plots of average target gene/HPRT expression ratios in different experimental groups. Liver inflammatory and FXR‐targeted gene expressions were determined by qRT‐PCR. Representative results of at least two independent experiments; Data represent Mean ± SD, n = 3‐6/group; *P < 0.05. Abbreviation: NS, not significant.
Next, we tested whether iMՓs, derived from bone marrow, were also involved in FXR activation‐induced liver protection. DT was used to deplete CD11b+ BMMs in CD11b‐DTR mice 48 hours before the onset of liver ischemia. CD11b‐depleted mice developed less severe liver IRI compared with controls. Interestingly, GW4064 treatment remained effective in ameliorating liver injury in these CD11b‐depleted cohorts, with sALT levels reduced, liver histologic architectures better preserved, and lower Suzuki scores (Fig. 5A). FXR activation remained inhibitory to the liver inflammatory response against IR in the absence of CD11b BMMs. Levels of TNF‐α, IL‐6, and IL‐1β were lower and IL‐10 was higher in IR livers treated with GW4064/DT compared with controls in these CD11b‐depleted cohorts (Fig. 5B). DT treatment did not affect the expressions of FXR target genes induced by GW4064, indicating that CD11b+ cells were not involved in responding to FXR activation in liver (Fig. 5C). Thus, BMMs are dispensable in FXR‐mediated liver protection from IRI.
Figure 5.
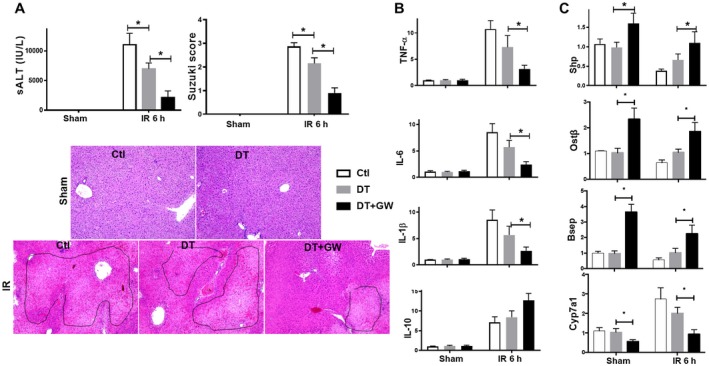
CD11b+ BMMs are dispensable for FXR‐mediated liver protection. CD11b‐DTR mice were treated with either phosphate‐buffered saline (Ctl) or DT 48 hours before, followed by the treatment of either vehicle controls or GW4064 2 hours before the onset of liver ischemia, as described in Materials and Methods. Liver injuries and inflammatory immune responses were evaluated at 6 hours after reperfusion. (A) Plots of average sALT levels and Suzuki scores in different experimental groups. Representative HE staining (magnification ×100) of IR livers. (B, C) Plots of average target gene/HPRT expression ratios in different experimental groups. Expressions of liver inflammatory and FXR‐targeted genes were determined by qRT‐PCR. Representative results of at least two independent experiments; Data represent Mean ± SD, n = 3‐6/group; *P < 0.05. Abbreviation: h, hour.
FXR Activation Induces SHP to Inhibit the Liver Inflammatory Immune Response
As a well‐known FXR target gene, SHP was demonstrated recently as a potent innate immune regulator.27, 28 We have shown in our own model that SHP induction is critical for the inhibition of the liver proinflammatory response and protection of liver from IRI in myeloid‐glycogen synthase kinase 3 (Gsk3b)‐deficient mice.29 As shown above, Shp was induced in liver in vivo and KCs in vitro by GW4064 (Figs. 1 and 3). IR inhibited liver Shp expression, which was reversed at least partially by pharmacologic FXR activation (Fig. 3). KC depletion by CLs resulted in decreased liver Shp expression in both control and GW4064‐treated hosts, indicating that KCs responded to FXR activation to up‐regulate Shp expression (Fig. 4). To test the functional significance of SHP induction in FXR‐mediated liver protection, we administered SHP siRNA carried by mannose‐conjugated polymers (selective delivery to phagocytes) in vivo before the onset of liver ischemia.30 Compared with control (scrambled [s]) siRNA, SHP‐specific siRNA prohibited Shp up‐regulation induced by GW4064 after IR, whereas other FXR target genes, such as Ostβ and Bsep, remained responsive to GW4064 treatment (Fig. 6A). Shp knockdown (KD) diminished inhibition of Cyp7a1 expression by GW4064. Most importantly, FXR activation was no longer able to protect liver from IRI or inhibit the liver proinflammatory response in SHP KD hosts. Mice treated with SHP siRNA had significantly higher levels of sALT, more severely damaged liver histologic architectures (Fig. 6B), and increased levels of TNF‐α, IL‐1β, and IL‐6 but not IL‐10 gene inductions in IR livers (Fig. 6C) compared with those treated with ssiRNA when both groups received the preoperational GW4064 treatment.
Figure 6.
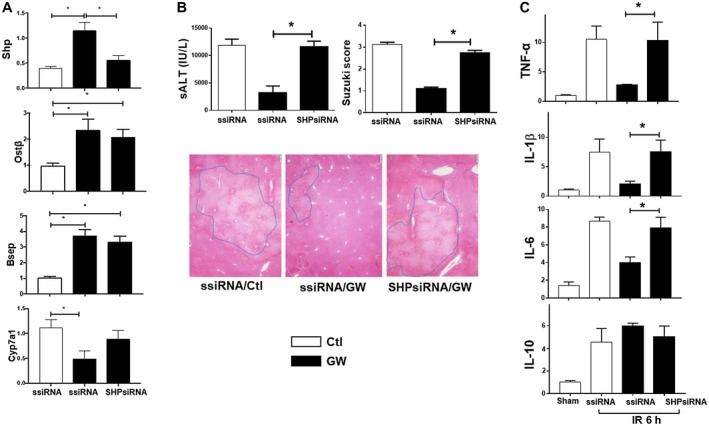
SHP is critical for FXR‐mediated liver protection. Scrambled or SHP‐specific siRNA was injected into different groups of B6 mice, followed by the administration of vehicle control or GW4064, as described in Materials and Methods. Liver IRI was evaluated at 6 hours after reperfusion. Expressions of FXR‐target genes and inflammatory genes in IR livers were measured by qRT‐PCR. (A) Plots of average ratios of target gene/HPRT expressions in different experimental groups. (B) Plots of average sALT levels and Suzuki's scores in different experimental groups. Representative liver histologic images (HE staining, magnification ×100). (C) Plots of average target gene/HPRT rations in different experimental groups. Gene expressions were determined by qRT‐PCR. Representative results of at least two independent experiments; Data represent Mean ± SD, n = 3‐6/group; *P < 0.05. Abbreviations: h, hour; ssiRNA, scrambled small interfering RNA.
In vitro, SHP KD in KCs resulted in higher levels of TNF‐α, IL‐1β, and IL‐6 but a lower level of IL‐10 gene inductions following LPS stimulation (Fig. 7). Importantly, SHP KD diminished the immune regulatory effect of pharmacologic activation of FXR in KCs such that levels of TNF‐α, IL‐1β, IL‐6, and IL‐10 remained the same in GW4064‐treated and vehicle‐treated cells following LPS stimulation. These results demonstrate that SHP induction in KCs is critical for the anti‐inflammatory and liver‐protective effects of pharmacologic activation of FXR against IRI.
Figure 7.
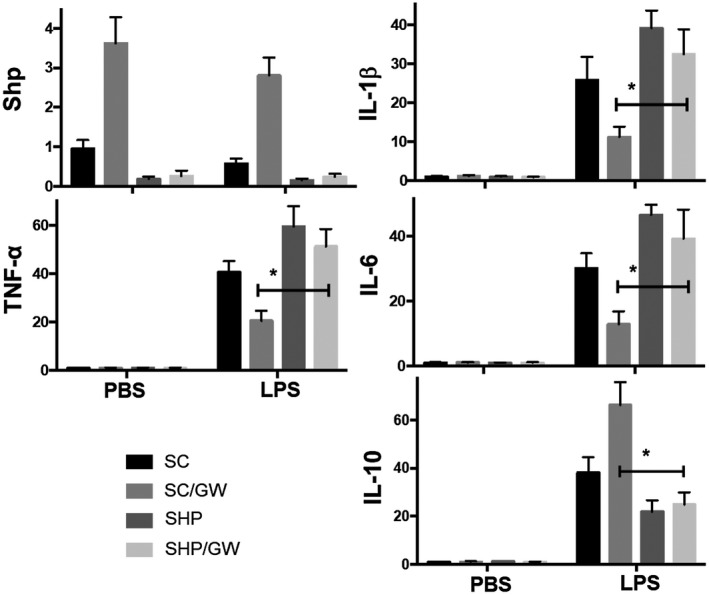
SHP is critical for FXR‐mediated immune regulation in KCs. KCs were transfected with either scrambled or SHP‐specific siRNA. Transfected cells were pretreated with either PBS or GW4064, followed by 3 hours of stimulation of LPS. Gene expressions were determined by qRT‐PCR. Average ratios of target gene/HPRT ratios in different experimental groups were plotted. Representative results of at least two independent experiments; Data represent Mean ± SD, n = 3/group; *P < 0.05. Abbreviations: PBS, phosphate‐buffered saline; SC, scrambled small interfering RNA.
Discussion
Our study demonstrates that pharmacologic activation of FXR protects liver from IRI at both acute and recovery stages. Although FXR agonists have been shown to protect multiple organs from IRI in the heart, intestine, kidney, and liver models16, 17, 18, 19 and modulate liver inflammation in non‐IRI models, cellular targets of FXR activation and immune regulatory mechanisms have not been well defined. Our results showed that the FXR‐targeted gene SHP was critical for the therapeutic effect of FXR activation in liver IRI. Liver‐resident KCs rather than infiltrating BMMs respond to FXR activation to up‐regulate SHP, which suppresses proinflammatory gene expressions downstream of TLRs.
Consistent with findings in a rat liver transplantation model,31 we showed that IR down‐regulated FXR expressions and transcriptional activities in liver and KCs. We showed that KCs isolated from ischemic liver increase proinflammatory cytokine production but decrease anti‐inflammatory IL‐10 production following TLR stimulation in vitro compared with those from sham livers.30 Because FXR regulates KC activation, its down‐regulation by IR may potentially facilitate the KC proinflammatory response. In hepatocytes, it was shown that FXR could selectively inhibit nuclear factor kappa B (NF‐κB) activation‐initiated inflammatory gene expressions, including inducible nitric oxide synthase (iNOS) and cyclooxygenase 2 (COX‐2), even in the absence of its ligands.32 FXR‐deficient mice have been shown to develop much worse liver necrosis and more severe inflammation after treatment with LPS32 or concanavalin A (ConA),33 and FXR agonists reduce not only acute inflammatory hepatocellular damage34 but also inflammatory cell infiltration and hepatic fibrosis/steatosis35, 36, 37 in chronic liver disease models. Although liver MФs have been implicated as FXR‐targeted cells in liver, evidence is derived from in vitro cell‐culture experiments.35 Our study identifies KCs as the key mediator of the therapeutic effect of FXR activation in vivo by differential analysis of KCs versus BMMs both in vitro and in vivo. Our results show that KCs express much higher levels of FXR than BMMs. FXR activation decreases TNF‐α and increases IL‐10 gene expressions in KCs in vitro by inducing SHP expression. KC depletion results in diminished expressions of FXR target genes in liver and abrogates the protection of liver from IRI by the treatment of FXR agonist GW4064 in vivo. In contrast, there are no SHP inductions or alterations in inflammatory gene expressions in BMMs following FXR activation in vitro. Depletion of BMMs in vivo did not affect the liver‐protective effect by the agonist treatment. Although CLs and DT are not specific in targeting KCs and BMMs, respectively, in vivo, our regimen was a single‐dose treatment before the onset of liver ischemia. Thus, CLs would mainly deplete tissue‐resident MФs, including KCs, without affecting circulating monocytes (low phagocytic), whereas DT would deplete monocytes without affecting KCs (CD11blow/−) in the acute phase of liver IRI. In chronic liver disease models, FXR stimulation decreased or altered infiltrations of inflammatory monocytes.35, 37 In our study, GW4064 significantly inhibited KC chemokine production, for example, monocyte chemoattractant protein 1 (MCP1) and macrophage inflammatory protein 2 (MIP2), in vitro following TLR4 stimulation (data not shown), which may lead to decreases in liver infiltration of peripheral monocytes in vivo. Hepatocytes or natural killer T cells have been shown to respond to FXR activation, leading to protection from endotoxin‐ or ConA‐induced hepatitis, respectively.32, 33 Because FXR is expressed in various types of liver cells, cellular targets of FXR therapeutics in vivo may vary depending on the pathologic mechanism of liver diseases.
In hepatocytes, FXR has been shown to suppress NF‐κB DNA binding32 and up‐regulate suppressor of cytokine signaling 3 (SOCS3).34 In MФ cell lines, nuclear receptor corepressor 1 (NCoR1) mediates the anti‐inflammatory effects of FXR.38 NCoR1 prevents direct binding of NF‐κB subunit to promoters of proinflammatory genes in unstimulated cells, which is stabilized by activated FXR. More recently, FXR was found to coprecipitate with NLR family pyrin domain containing 3 (NLRP3), caspase‐1, and possibly apoptosis‐associated speck‐like protein (ASC) in MФs to prevent NLRP3 inflammasome assembly and activation.39 Paradoxically, natural FXR ligand bile acids, alone or in synergy with LPS or adenosine triphosphate, activated NLRP3 inflammasome by promoting calcium influx.39 This indicates that FXR suppresses NLRP3 inflammasome possibly by their direct physical interactions but not by FXR ligand‐induced transcriptional activities. Because GW4064 does not inhibit NLRP3 inflammasome activation,39 this particular mechanism may not be relevant to the effect of FXR activation in our model. Interestingly, bile acids have a G protein‐coupled membrane receptor, TGR5, which has been shown to not only repress NLRP3 inflammasome40 but also inhibit NF‐κB transcriptional activities by facilitating the interaction between inhibitor of κBα (IκBα) and β‐arrestin2.41 Although molecular and cellular details of the distinctive effects of FXR and TGR5 on inflammasome activation in the absence or presence of bile acids remain to be fully elucidated, differential expressions of these receptors in different types of cells may be a key factor in determining their involvement in immune regulation. Our data show that KCs rather than BMMs express functional FXR. Therefore, pharmacologic activation of FXR selectively regulates KCs but not BMMs in their response to inflammatory stimulation. TGR5, on the other hand, is expressed in both types of MФs,42, 43 and pharmacologic activation of TGR5 therefore regulates both of them in response to TLRs. One key difference in the immune regulatory mechanisms between FXR and TGR5 activation in KCs is the induction of SHP, which turns out to be the critical mediator of the FXR therapeutic effect in liver IRI.
Our results reveal that SHP, as a direct FXR‐targeted gene, is crucial for the immune regulatory effect of FXR activation in KCs in liver IRI. SHP was identified recently as an endogenous negative regulator in inflammatory signaling downstream of TLRs and NLRs.27, 28, 44 It suppresses NF‐κB activities by physically binding to either the NF‐κBp65 subunit at resting state and/or TNF receptor‐associated factor 6 (TRAF6) following stimulation to inhibit its polyubiquitination and activation, leading to down‐regulation of TNF‐α and IL‐6 transcriptions.28 SHP also interferes with the interaction between NLRP3 and ASC to intercept the assembly of the NLRP3 inflammasome complex, resulting in down‐regulation of IL‐1β and IL‐18 production in NLRP3‐activated MФs.27 SHP could be translocated to the mitochondria with the NLRP3–ASC complex after inflammasome activation and acts to regulate mitochondrial homeostasis through the recovery of mitochondrial reactive oxygen species generation and damage.28 Most recently, we have shown in the same liver IR model that Gsk3b promotes liver inflammatory immune activation through inhibition of adenosine monophosphate‐activated protein kinase activation and SHP induction.29 SHP has been implicated in other liver disease models. FXR‐induced SHP in hepatic stellate cells has been shown to promote resolution of liver fibrosis by interacting with c‐Jun to prevent activator protein 1 (AP‐1) binding to inflammatory genes.45 SHP genetic deletion sensitizes mice to liver injuries triggered by bile duct ligation.46 Interestingly, FXR and SHP double‐KO mice are protected from hepatic steatosis due to their roles in lipid metabolism.47
In summary, we document by pharmacologic activation that FXR is a negative regulator of liver inflammatory immune response against IR. Its activation induces SHP up‐regulation in KCs, leading to decreases of proinflammatory but increases of anti‐inflammatory gene expression following TLR stimulation and protection of liver from IRI.
Supporting information
Supported by the National Institutes of Health (grants R21AI126516 and P01 AI120944‐01 to Y.Z.), the Dumont Research Foundation, Municipal Key Clinical Specialty and Science and Technology Commission of Shanghai (18441904800 to D.J.), and the National Science and Technology Major Project (2018ZX10302206 to N.X.).
Potential conflict of interest: Nothing to report.
Contributor Information
Ning Xu, Email: xuning@renji.com.
Yuan Zhai, Email: yzhai@mednet.ucla.edu.
REFERENCES
Author names in bold designate shared co‐first authorship.
- 1. Ohkohchi N. Mechanisms of preservation and ischemic/reperfusion injury in liver transplantation. Transplant Proc 2002;34:2670‐2673. [DOI] [PubMed] [Google Scholar]
- 2. Tsai YF, Liu FC, Sung WC, Lin CC, Chung PC, Lee WC, et al. Ischemic reperfusion injury‐induced oxidative stress and pro‐inflammatory mediators in liver transplantation recipients. Transplant Proc 2014;46:1082‐1086. [DOI] [PubMed] [Google Scholar]
- 3. Zhai Y, Busuttil RW, Kupiec‐Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate‐adaptive immune‐mediated tissue inflammation. Am J Transplant 2011;11:1563‐1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kaczorowski DJ, Tsung A, Billiar TR. Innate immune mechanisms in ischemia/reperfusion. Front Biosci (Elite Ed) 2009;1:91‐98. [DOI] [PubMed] [Google Scholar]
- 5. Huang H, Evankovich J, Yan W, Nace G, Zhang L, Ross M, et al. Endogenous histones function as alarmins in sterile inflammatory liver injury through Toll‐like receptor 9 in mice. Hepatology 2011;54:999‐1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, et al. HMGB1 release induced by liver ischemia involves Toll‐like receptor 4 dependent reactive oxygen species production and calcium‐mediated signaling. J Exp Med 2007;204:2913‐2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia‐reperfusion. J Exp Med 2005;201:1135‐1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhai Y, Qiao B, Shen XD, Gao F, Busuttil RW, Cheng G, et al. Evidence for the pivotal role of endogenous toll‐like receptor 4 ligands in liver ischemia and reperfusion injury. Transplantation 2008;85:1016‐1022. [DOI] [PubMed] [Google Scholar]
- 9. Zhai Y, Shen XD, O'Connell R, Gao F, Lassman C, Busuttil RW, et al. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3‐dependent MyD88‐independent pathway. J Immunol 2004;173:7115‐7119. [DOI] [PubMed] [Google Scholar]
- 10. Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP. Toll‐like receptor 9 inhibition confers protection from liver ischemia‐reperfusion injury. Hepatology 2010;51:621‐632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Inoue Y, Shirasuna K, Kimura H, Usui F, Kawashima A, Karasawa T, et al. NLRP3 regulates neutrophil functions and contributes to hepatic ischemia‐reperfusion injury independently of inflammasomes. J Immunol 2014;192:4342‐4351. [DOI] [PubMed] [Google Scholar]
- 12. Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol 2013;14:996‐1006. [DOI] [PubMed] [Google Scholar]
- 13. Zhu Y, Liu H, Zhang M, Guo GL. Fatty liver diseases, bile acids, and FXR. Acta Pharm Sin B 2016;6:409‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim SG, Kim BK, Kim K, Fang S. Bile acid nuclear receptor farnesoid X receptor: therapeutic target for nonalcoholic fatty liver disease. Endocrinol Metab (Seoul) 2016;31:500‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fiorucci S, Biagioli M, Zampella A, Distrutti E. Bile acids activated receptors regulate innate immunity. Front Immunol 2018;9:1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pu J, Yuan A, Shan P, Gao E, Wang X, Wang Y, et al. Cardiomyocyte‐expressed farnesoid‐X‐receptor is a novel apoptosis mediator and contributes to myocardial ischaemia/reperfusion injury. Eur Heart J 2013;34:1834‐1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ceulemans LJ, Verbeke L, Decuypere JP, Farre R, De Hertogh G, Lenaerts K, et al. Farnesoid X receptor activation attenuates intestinal ischemia reperfusion injury in rats. PLoS One 2017;12:e0169331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gai Z, Chu L, Xu Z, Song X, Sun D, Kullak‐Ublick GA. Farnesoid X receptor activation protects the kidney from ischemia‐reperfusion damage. Sci Rep 2017;7:9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ferrigno A, Di Pasqua LG, Berardo C, Siciliano V, Rizzo V, Adorini L, et al. The farnesoid X receptor agonist obeticholic acid upregulates biliary excretion of asymmetric dimethylarginine via MATE‐1 during hepatic ischemia/reperfusion injury. PLoS One 2018;13:e0191430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Suzuki S, Toledo‐Pereyra LH, Rodriguez FJ, Cejalvo D. Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury. Modulating effects of FK506 and cyclosporine. Transplantation 1993;55:1265‐1272. [DOI] [PubMed] [Google Scholar]
- 21. He K, Chen X, Han C, Xu L, Zhang J, Zhang M, et al. Lipopolysaccharide‐induced cross‐tolerance against renal ischemia‐reperfusion injury is mediated by hypoxia‐inducible factor‐2alpha‐regulated nitric oxide production. Kidney Int 2014;85:276‐288. [DOI] [PubMed] [Google Scholar]
- 22. Zhang S, Han CH, Chen XS, Zhang M, Xu LM, Zhang JJ, et al. Transient ureteral obstruction prevents against kidney ischemia/reperfusion injury via hypoxia‐inducible factor (HIF)‐2alpha activation. PLoS One 2012;7:e29876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhai Y, Shen XD, Gao F, Zhao A, Freitas MC, Lassman C, et al. CXCL10 regulates liver innate immune response against ischemia and reperfusion injury. Hepatology 2008;47:207‐214. [DOI] [PubMed] [Google Scholar]
- 24. Yue S, Zhou H, Wang X, Busuttil RW, Kupiec‐Weglinski JW, Zhai Y. Prolonged ischemia triggers necrotic depletion of tissue‐resident macrophages to facilitate inflammatory immune activation in liver ischemia reperfusion injury. J Immunol 2017;198:3588‐3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Devey L, Ferenbach D, Mohr E, Sangster K, Bellamy CO, Hughes J, et al. Tissue‐resident macrophages protect the liver from ischemia reperfusion injury via a heme oxygenase‐1‐dependent mechanism. Mol Ther 2009;17:65‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ellett JD, Atkinson C, Evans ZP, Amani Z, Balish E, Schmidt MG, et al. Murine Kupffer cells are protective in total hepatic ischemia/reperfusion injury with bowel congestion through IL‐10. J Immunol 2010;184:5849‐5858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang CS, Kim JJ, Kim TS, Lee PY, Kim SY, Lee HM, et al. Small heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasome. Nat Commun 2015;6:6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yuk JM, Shin DM, Lee HM, Kim JJ, Kim SW, Jin HS, et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll‐like receptors. Nat Immunol 2011;12:742‐751. [DOI] [PubMed] [Google Scholar]
- 29. Zhou H, Wang H, Ni M, Yue S, Xia Y, Busuttil RW, et al. Glycogen synthase kinase 3beta promotes liver innate immune activation by restraining AMP‐activated protein kinase activation. J Hepatol 2018;69:99‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rao J, Yue S, Fu Y, Zhu J, Wang X, Busuttil RW, et al. ATF6 mediates a pro‐inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia‐reperfusion injury. Am J Transplant 2014;14:1552‐1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cheng L, Tian F, Tian F, Tang L, Chen G, Luo Z, et al. Repression of farnesoid X receptor contributes to biliary injuries of liver grafts through disturbing cholangiocyte bile acid transport. Am J Transplant 2013;13:3094‐3102. [DOI] [PubMed] [Google Scholar]
- 32. Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008;48:1632‐1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mencarelli A, Renga B, Migliorati M, Cipriani S, Distrutti E, Santucci L, et al. The bile acid sensor farnesoid X receptor is a modulator of liver immunity in a rodent model of acute hepatitis. J Immunol 2009;183:6657‐6666. [DOI] [PubMed] [Google Scholar]
- 34. Xu Z, Huang G, Gong W, Zhou P, Zhao Y, Zhang Y, et al. FXR ligands protect against hepatocellular inflammation via SOCS3 induction. Cell Signal 2012;24:1658‐1664. [DOI] [PubMed] [Google Scholar]
- 35. McMahan RH, Wang XX, Cheng LL, Krisko T, Smith M, El Kasmi K, et al. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J Biol Chem 2013;288:11761‐11770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Verbeke L, Mannaerts I, Schierwagen R, Govaere O, Klein S, Vander Elst I, et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci Rep 2016;6:33453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang S, Wang J, Liu Q, Harnish DC. Farnesoid X receptor agonist WAY‐362450 attenuates liver inflammation and fibrosis in murine model of non‐alcoholic steatohepatitis. J Hepatol 2009;51:380‐388. [DOI] [PubMed] [Google Scholar]
- 38. Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol 2009;183:6251‐6261. [DOI] [PubMed] [Google Scholar]
- 39. Hao H, Cao L, Jiang C, Che Y, Zhang S, Takahashi S, et al. Farnesoid X receptor regulation of the NLRP3 inflammasome underlies cholestasis‐associated sepsis. Cell Metab 2017;25:856‐867.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 2016;45:802‐816. Erratum in: Immunity 2016;45:944. [DOI] [PubMed] [Google Scholar]
- 41. Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G‐protein‐coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light‐chain enhancer of activated B cells (NF‐kappaB) in mice. Hepatology 2011;54:1421‐1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Calmus Y, Poupon R. Shaping macrophages function and innate immunity by bile acids: mechanisms and implication in cholestatic liver diseases. Clin Res Hepatol Gastroenterol 2014;38:550‐556. [DOI] [PubMed] [Google Scholar]
- 43. Schubert K, Olde Damink SWM, von Bergen M, Schaap FG. Interactions between bile salts, gut microbiota, and hepatic innate immunity. Immunol Rev 2017;279:23‐35. [DOI] [PubMed] [Google Scholar]
- 44. Yuk JM, Jin HS, Jo EK. Small heterodimer partner and innate immune regulation. Endocrinol Metab (Seoul) 2016;31:17‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fiorucci S, Antonelli E, Rizzo G, Renga B, Mencarelli A, Riccardi L, et al. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology 2004;127:1497‐1512. [DOI] [PubMed] [Google Scholar]
- 46. Park YJ, Qatanani M, Chua SS, LaRey JL, Johnson SA, Watanabe M, et al. Loss of orphan receptor small heterodimer partner sensitizes mice to liver injury from obstructive cholestasis. Hepatology 2008;47:1578‐1586. [DOI] [PubMed] [Google Scholar]
- 47. Akinrotimi O, Riessen R, VanDuyne P, Park JE, Lee YK, Wong LJ, et al. Small heterodimer partner deletion prevents hepatic steatosis and when combined with farnesoid X receptor loss protects against type 2 diabetes in mice. Hepatology 2017;66:1854‐1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials