

Abstract
Nonalcoholic fatty liver disease (NAFLD) is a heterogeneous group of liver diseases characterized by the accumulation of fat in the liver. The heterogeneity of NAFLD is reflected in a clinical and histologic spectrum where some patients develop isolated steatosis of the liver, termed nonalcoholic fatty liver, whereas others develop hepatocyte injury, ballooning, inflammation, and consequent fibrosis, termed nonalcoholic steatohepatitis (NASH). Systemic insulin resistance is a major driver of hepatic steatosis in NAFLD. Lipotoxicity of accumulated lipids along with activation of the innate immune system are major drivers of NASH. Lipid‐induced sublethal and lethal stress culminates in the activation of inflammatory processes, such as the release of proinflammatory extracellular vesicles and cell death. Innate and adaptive immune mechanisms involving macrophages, dendritic cells, and lymphocytes are central drivers of inflammation that recognize damage‐ and pathogen‐associated molecular patterns and contribute to the progression of the inflammatory cascade. While the activation of the innate immune system and the recruitment of proinflammatory monocytes into the liver in NASH are well known, the exact signals that lead to this remain less well defined. Further, the contribution of other immune cell types, such as neutrophils and B cells, is an area of intense research. Many host factors, such as the microbiome and gut–liver axis, modify individual susceptibility to NASH. In this review, we discuss lipotoxicity, inflammation, and the contribution of interorgan crosstalk in NASH pathogenesis.
Lipotoxicity and inflammation are major pathogenic drivers of nonalcoholic steatohepatitis. Here we review the sublethal and lethal lipotoxic pathways activated in nonalcoholic steatohepatitis and the contribution of the immune system to liver inflammation.
Abbreviations
- CCL
C‐C motif chemokine ligand
- CD
cluster of differentiation
- CHOP
C/EBP homologous protein
- CoA
coenzyme A
- DAMP
damage‐associated molecular pattern
- DC
dendritic cell
- DNL
de novo lipogenesis
- ER
endoplasmic reticulum
- EV
extracellular vesicle
- FGF
fibroblast growth factor
- FXR
farnesoid X receptor
- HCC
hepatocellular carcinoma
- IFN
interferon
- IL
interleukin
- IR
insulin resistance
- JNK
c‐jun N‐terminal kinase
- KC
Kuppfer cell
- LPC
lysophosphatidyl choline
- LPS
lipopolysaccharide
- MPO
myeloperoxidase
- NAFL
nonalcoholic fatty liver
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NEFA
nonesterified fatty acid
- NET
neutrophil extracellular trap
- NLRP3
Nod‐like receptor protein 3
- PAMP
pathogen‐associated molecular pattern
- PPAR
peroxisome proliferator‐activated receptor
- PUFA
polyunsaturated fatty acid
- PUMA
p53 up‐regulated modulator of apoptosis
- TG
triglyceride
- Th
T helper
- TLR
Toll‐like receptor
- TRAIL
tumor necrosis factor–related apoptosis‐inducing ligand
- TRAIL‐R2
tumor necrosis factor–related apoptosis‐inducing ligand receptor 2
Nonalcoholic fatty liver disease (NAFLD), the most common chronic liver disease in the United States, is a heterogeneous disorder.1 Based on histology, pathogenesis, and natural history, the NAFLD disease spectrum is characterized by excess fat deposition in the liver that is unassociated with injury or inflammation (isolated steatosis or nonalcoholic fatty liver [NAFL]) on one end and hepatocyte ballooning, liver injury, inflammation, and varying degrees of fibrosis (nonalcoholic steatohepatitis [NASH]), ultimately leading to cirrhosis and the associated risks of end‐stage liver disease and hepatocellular carcinoma (HCC), on the other end.2 Fibrosis has been reported in some subjects with NAFL, although NAFL is generally considered nonprogressive. Isolated steatosis is characterized by predominantly macrovesicular lipid accumulation in 5% or more hepatocytes, typically beginning around central veins. Hepatocellular ballooning, Mallory‐Denk bodies, and inflammation are observed additionally in NASH. Chronic inflammation is associated with fibrosis, which initially is pericellular and can progress to bridging fibrosis and cirrhosis. Thus, the two components of histologic assessment are disease activity (scored on steatosis, ballooning, and lobular inflammation) and fibrosis stage.3 Subject to the caveat that there is significant collinearity between the NAFLD activity score (NAS) and fibrosis, fibrosis is the only histologic factor associated with mortality.4 Modern multiomics approaches confirm the relevance of histologic observations by demonstrating a correlation between genetic predictors of progression and histologic assessment of the NAS.5 Here, we discuss the key molecular and cellular mechanisms that form the underpinnings of the observed histologic changes and global transcriptomics changes in NAFLD.
Steatosis and Lipotoxicity
The pathogenesis of NAFLD is multifactorial, and several systemic alterations have been implicated.2 The primary insult of lipid excess is followed by variable contributions from pathogenic drivers, such as lipotoxicity and immune system activation, and modifiers, such as genetic susceptibilities, alcohol, and dysbiosis. However, there is considerable heterogeneity in NAFLD progression and NASH development, and only a subset of NAFLD develops NASH. Potential explanations for this variability include differences in etiopathogenic drivers,2 dynamic multiphasic progression,5 or that they represent distinct diseases. Alcohol is a well‐recognized disease modifier. Recognizing the arbitrary cutoffs that define the level of intake, even modest levels of alcohol consumption have effects on NASH progression, including a worse histology and a risk for fibrosis progression.6 Biologic sex modulates NAFLD pathobiology both in experimental models and humans,7, 8 with women being relatively protected from disease.
Hepatic Steatosis
One key concept is the presence of a perturbed systemic energy balance state, characterized by substrate surplus, predominantly carbohydrates and fatty acids.9, 10 The major sources of nonesterified fatty acid (NEFA) delivery to the liver are increased release from adipocytes (accounting for approximately 60%), conversion from carbohydrates within the liver (de novo lipogenesis, 26%), and excess dietary intake (14%)9 (Fig. 1). Insulin resistance (IR) and NAFLD are crucially linked2, 11; IR leads to reduced glucose uptake in adipocytes and muscles, and hepatocytes can secrete dipeptidyl peptidase 4, which promotes adipose tissue inflammation and IR.12
Figure 1.
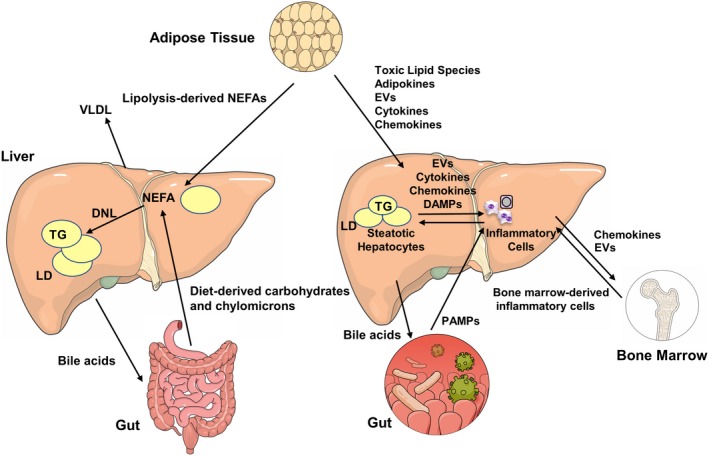
Metabolic interorgan crosstalk in NAFLD. This illustration depicts interorgan crosstalk in NAFL on the left and NASH on the right. Hepatic NEFAs are predominantly derived from three sources: lipolysis in adipose tissue, dietary lipid absorption, and DNL from carbohydrates in the liver. These NEFAs are stored in the liver as TG‐rich lipid droplets leading to hepatic steatosis or may be exported out of the liver as very low‐density lipoprotein to adipose tissue. Bile acids from the liver are key regulators of the gut–liver axis. Several mediators orchestrate the inflammatory milieu in the liver that results in NASH and fibrosis. Lipotoxic lipid species lead to hepatic stress and subsequent release of EVs, cytokines, chemokines, and DAMPs from liver cells. This results in recruitment of immune cells from the bone marrow. Bile acids from the liver, PAMPs from the gut, and adipokines from adipose tissues also influence various steps in this process. Abbreviations: LD, lipid droplet; VLDL, very low‐density lipoprotein.
At the adipocyte level, metabolic dysregulation due to impaired insulin postreceptor signaling leads to excess lipolysis of triglycerides (TGs) and NEFA release into the circulation. Albumin‐bound NEFAs are delivered to the liver. Hepatocyte NEFA uptake is mediated by fatty acid transport proteins, cluster of differentiation 36 (CD36), caveolins, and to a lesser extent passive diffusion.13 Additionally, de novo lipogenesis (DNL) from glucose and fructose occurs in the hepatocytes and is increased in subjects with NAFLD.9, 14 Unlike glucose, entry of fructose metabolites into the DNL pathway is not regulated by glycolysis.15 Fructose also induces the carbohydrate response element binding protein independent of insulin and promotes hepatic steatosis. The predominant fate of NEFAs in the liver is to either undergo mitochondrial beta‐oxidation or be esterified to form TGs. Partitioning of NEFAs into other lipid classes, such as phospholipids and ceramides, is also increased by enhanced NEFA influx into the liver.16, 17 Formation of TGs, a relatively inert storage form, appears to be an adaptive mechanism to protect the liver from toxic lipids. TGs can be exported as very low‐density lipoprotein particles or stored as lipid droplets. Lipolysis of these droplets releases NEFA back into the hepatic pool, and the regulation of this step is important in the pathogenesis of NASH.
The most strongly associated genetic variant with NASH is a single‐nucleotide polymorphism (I148M) in the patatin‐like phospholipase domain‐containing protein 3 (PNPLA3) gene,18 which encodes a lipid droplet protein and is involved at this lipolytic step. I148M variant PNPLA3 is degradation resistant, accumulates on lipid droplets, and is sufficient to induce steatosis.19 Hydroxysteroid 17β‐dehydrogenase 13 (HSD17B13), another lipid droplet protein, is up‐regulated in NASH.20 Variants of HSD17B13 are associated with increased steatosis but decreased inflammation and lower alanine aminotransferase in NAFLD. Thus, genetic studies point to an important role for lipid droplet proteins in regulating features of NAFLD; this is an area that needs further mechanistic studies to understand how steatosis can be protective against liver injury yet lipid droplet proteins may be deleterious.
Apart from quantity, the type of NEFAs that accumulate in NAFLD are also altered, with significantly more saturated fatty acids than monounsaturated and polyunsaturated fatty acids (MUFAs and PUFAs, respectively). The 16‐carbon palmitate and 18‐carbon stearate are major saturated fatty acids that accumulate and are associated with disease progression.21 Other implicated lipotoxic species include diacylglycerols, ceramides, lysophophatidyl choline (LPC), and free cholesterol.21, 22 Given the key role in the pathobiology of NAFLD and NASH, a strategy to decrease substrate delivery to the liver or to promote disposal of NEFAs from the liver represents an attractive therapeutic target (Table 1). This could be achieved, for example, by increasing fatty acid oxidation (peroxisome proliferator‐activated receptor [PPAR]α/δ agonists; fibroblast growth factor [FGF]21 agonists; thyromimetics), inhibition of DNL (acetyl‐coenzyme A [CoA] carboxylase inhibitor), increasing fatty acid desaturation (stearoyl‐CoA desaturase inhibitor), or improving IR (PPARγ and glucagon‐like peptide 1 [GLP‐1] agonists). However, worsening dyslipidemia has been observed with several pharmacologic agents that directly or indirectly target NEFA flux, lending some caution to this approach. Altered lipid flux may also impact accumulation of toxic lipids, which is discussed more in the next section.
Table 1.
Therapeutic Strategies Targeting Lipotoxicity in NAFLD
| Target | Mechanism | Example Drug |
|---|---|---|
| FXR | Agonist: improve insulin sensitivity; anti‐inflammatory and antifibrotic | Obeticholic acid |
| Acetyl‐CoA carboxylase 1/2 | Antagonist: decrease DNL | PF‐05221304 |
| FGF19/21 | Agonist: decrease bile acid synthesis; anti‐inflammatory and antifibrotic | NGM282 |
| PPARα/γ/δ | Agonist: increase fatty acid oxidation; improve IR; anti‐inflammatory | Elafibrinor |
| Steroyl CoA‐desaturase 1 | Antagonist: decrease DNL | Aramchol |
| Thyroid hormone receptor β | Agonist: decrease circulating lipids | MGL‐3196 |
| Niacin‐R | Agonist: decrease lipolysis in adipose tissue; decrease TG synthesis and increase fatty acid oxidation | Niacin |
| Sirutin‐1 | Agonist: decrease DNL; increase fatty acid oxidation; anti‐inflammatory | Resveratrol |
| Ketohexokinase | Antagonist: decrease DNL | PF‐06835919 |
Lipotoxic Hepatocellular Stress
In addition to the histologic or imaging‐based recognition of fat accumulation in the liver, there has been a rapid increase in the understanding of the deleterious role of lipotoxicity in NAFLD since the first description of lipotoxicity by Roger Unger in 1994 and the earlier recognition that many lipid species are bioactive. Concomitantly, there has been an expansion of hepatic and plasma lipidomics in NAFLD.23, 24, 25 Lipidomics analyses have typically been stochastic rather than paired and kinetic over the natural history of NAFLD. This has limited their interpretation to correlations; although in vitro observations and mouse models have elucidated the signaling pathways triggered by toxic lipids.
Lipotoxic Lipid Classes
Mechanistic studies in isolated cells and animal models have elucidated the role of several lipid classes in hepatocellular toxicity, liver injury, and inflammation. Among these, saturated NEFAs, predominantly palmitate, the glycerophospholipid LPC, free cholesterol, sphingolipids (including ceramides), and sphingosine 1‐phosphate (S1P) are well studied, although other classes of lipid, and their biosynthetic pathways are also deranged in NASH.22 While not the focus of this review, it is interesting to briefly note an increase in the ratio of n‐6 to n‐3 PUFAs and their inflammation‐regulating derivatives in NASH, especially an increase in linoleic acid and its oxidized products26 as well as an increase in oxysterols, which may have proinflammatory roles through activation of innate immune cells.27 Toxic lipids accumulate and provoke injury in hepatocytes as well as in nonparenchymal liver cells. Several extrahepatic factors, such as intestinal dysbiosis and adipokines, modulate lipotoxic exposure to the liver and subsequent injury and inflammation, with variable contributions across individuals and different stages of disease pathology.2 At a molecular level, lipotoxicity leads to endoplasmic reticulum (ER) stress, lysosomal dysfunction, inflammasome activation, cell death, and activation of inflammatory responses due to lethal and sublethal hepatocellular injury.28
Molecular Mechanisms of NEFA‐Induced Lipotoxic Cell Death
Palmitate is elevated in plasma and accumulates in its esterified form in the liver in NASH, as demonstrated by a preponderance of palmitate‐containing TGs in mouse models and humans with NASH.29 The molecular pathways that mediate the toxicity of palmitate have been elegantly elucidated in cultured hepatocytes.21, 22 Palmitate can activate both the intrinsic‐ and extrinsic‐mediated (death receptor) apoptotic machinery in hepatocytes.21 The intracellular balance of proapoptotic versus antiapoptotic proteins is shifted in palmitate‐treated hepatocytes toward apoptosis (Fig. 2). This includes the activation of intracellular stress‐activated kinase c‐jun N‐terminal kinase (JNK),30 up‐regulation of the proapoptotic proteins Bim and p53 up‐regulated modulator of apoptosis (PUMA),31, 32 degradation of the antiapoptotic proteins B‐cell lymphoma‐extra large (Bcl‐XL) and myeloid cell leukemia 1 (Mcl‐1),33 and inhibitor of apoptosis proteins.34 Many of these perturbations occur downstream of the stress kinase JNK and organelle dysfunction, such as lysosomal permeabilization and ER stress, leading to transcriptional up‐regulation of proapoptotic proteins. Posttranscriptional up‐regulation of PUMA has been described by palmitate‐induced repression of microRNA‐296‐5p.35 Palmitate also sensitizes cells to tumor necrosis factor (TNF)–related apoptosis‐inducing ligand (TRAIL)‐induced cell death by transcriptional up‐regulation of TRAIL receptor 2 (TRAIL‐R2) expression, which can lead to ligand‐independent activation of the extrinsic apoptotic pathway.36 Both the extrinsic and intrinsic pathways converge on Bax‐induced mitochondrial permeabilization, release of cytochrome c, and activation of effector caspases. Palmitate toxicity may in part be mediated by LPC, which accumulated intracellularly in palmitate‐treated hepatocytes and activated the intracellular proapoptotic pathways previously defined for palmitate, including JNK, C/EBP homologous protein (CHOP), and PUMA.37
Figure 2.
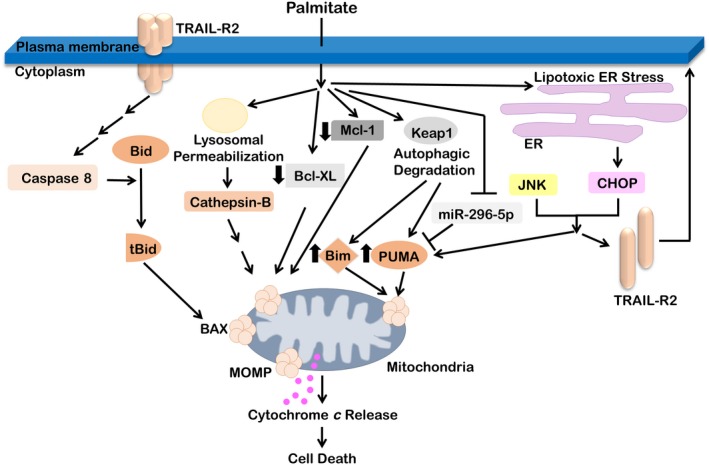
Molecular pathways of palmitate‐induced lipotoxicity in hepatocytes. Palmitate activates the extrinsic death receptor‐mediated pathway of apoptosis and also activates the intrinsic pathway of apoptosis. Lysosomal permeabilization leads to the release of the protease cathepsin B. Lipotoxic ER stress leads to up‐regulation of the proapoptotic transcription factor CHOP. The stress‐induced kinase JNK and CHOP induce the death receptor TRAIL‐R2 and the proapoptotic Bcl‐2 family proteins PUMA and Bim. PUMA and Bim are also up‐regulated by palmitate‐induced autophagic degradation of Keap1. Palmitate decreases the expression of antiapoptotic proteins Mcl‐1 and Bcl‐XL. TRAIL‐R2 can undergo ligand‐independent oligomerization, cleavage‐induced activation of caspase 8, Bid cleavage to tBid, and activation of Bax. Ologomeric Bax results in mitochondrial outer membrane permeabilization, release of cytochrome c, activation of effector caspases, and apoptosis. Abbreviations: BAX, B‐cell lymphoma 2‐like protein 4; Bcl‐XL, B‐cell lymphoma‐extra large; Bim, B‐cell lymphoma 2‐like protein 11; Keap1, Kelch‐like ECH‐associated protein 1; Mcl‐1, induced myeloid leukemia cell differentiation protein; MOMP, major outer membrane protein; tBid, truncated p15 BID.
Protective Lipid Classes
In addition to toxic classes of lipids, two broad categories of protective lipids are associated with obesity‐associated NAFLD.22 The MUFAs palmitoleate and oleate can reduce the toxicity of palmitate in cultured hepatocytes, although they promote TG formation, suggesting that the sequestration of palmitate into neutral TG is cytoprotective.22, 38 In a mouse model, adipose tissue‐derived palmitoleate suppressed hepatic steatosis and improved muscle IR, suggesting a different mechanism for its protective effect.39 The second class of lipids of interest is a set of PUFA‐derived specialized proresolving mediators (SPMs), which, through receptor‐mediated effects on immune cells, limit inflammation. These consist of n‐3 PUFA‐derived lipoxins, resolvins, maresins, and protectins.22 Exogenous resolvin D1 administration promoted the resolution of inflammation in a mouse dietary withdrawal model of NASH resolution, suggesting a therapeutic role for SPMs in NASH.40
Organelle Stress
Subcellular organelle stress, including lysosomal permeabilization and ER stress, occurs in palmitate‐treated hepatocytes and eventually contributes to cell death by up‐regulation of proapoptotic signaling (Fig. 2). Lysosomal permeabilization was mediated by translocation of Bax to the lysosomal membrane.41 Palmitate‐induced ER stress occurs due to an increase in saturated acyl chains and is referred to as lipotoxic or lipid bilayer ER stress.42 Indeed, palmitate‐induced apoptosis was partially dependent on the ER stress‐induced proapoptotic transcription factor CHOP, which in turn transcriptionally up‐regulated TRAIL‐R2 and PUMA expression.43 A recent study has linked ER stress to inflammatory responses elicited by steatotic hepatocytes through the release of proinflammatory extracellular vesicles (EVs).44 ER stress has been implicated in obesity‐associated IR and also in inflammation in various models.45 The contribution of ER stress to inflammation in NASH needs further exploration.
Relevance of Cell Death in NASH Models
A comprehensive multiomics study identified hepatocyte apoptosis as a key early signaling event in high‐fat diet‐induced NASH in mice.46 In keeping with this observation, mice deficient in apoptosis are protected from NASH.47 TRAIL receptor knockout mice are resistant to high‐fat diet‐induced obesity and diminished macrophage inflammatory responses, pointing toward a broader role for death receptor‐induced inflammatory signaling in obesity‐associated inflammation. Besides apoptosis, other forms of hepatocyte cell death have been described in NASH models. Pyroptosis, an inflammatory form of cell death that occurs due to the activation of inflammasomes and caspase 1 leading to cleavage‐induced activation of gasdermin D, is implicated in animal models of NASH and in liver biopsies of patients with NASH48; however, there are no clear links between palmitate and pyroptosis in hepatocytes. Free cholesterol, on the other hand, is known to activate the inflammasome.49 Free cholesterol accumulation in hepatocytes and cholesterol crystal formation in hepatocyte lipid droplets in subjects with NASH and not isolated steatosis is associated with aggregation of Kupffer cells (KCs) and fibrosis, suggesting that perhaps inflammasome activation may play a role in NASH.50, 51 Similarly, ferroptosis, a form of cell death dependent on iron and oxygenated phosphatidyl ethanolamine (PE), is also reported in NASH.52 Oxygenated lipids, such as PE, may be a mediator or a correlate of lipotoxicity due to other lipids in these models. Further, although necroptosis in hepatocytes with intact caspase signaling remains controversial, the receptor‐interacting protein kinases are implicated in NASH.53 This may be due to their roles in inflammatory signaling and cell death secondary to immune cell activation.
Mechanistically diverse types of hepatocellular death are observed in NASH in both animal models and human specimens. In these models, hepatocellular death is linked to inflammation, and these two processes may form a feed‐forward loop such that cell death triggers inflammatory signaling and immune cells secrete mediators, such as TRAIL or Fas, which can initiate apoptotic signaling. Although the question of primacy in these processes has not been answered, experimental data show that interruption of this loop mitigates NASH.54 Furthermore, as discussed above, rather than apoptosis alone, several other forms of cell death may play a role in activation of the immune response. Death receptor activation also leads to the release of chemokines, including interleukin (IL)‐6, IL‐8, C‐X‐C motif chemokine ligand 1 (CXCL1), and C‐C motif chemokine ligand 2 (CCL2), which can promote macrophage chemotaxis toward dying cells.55
Mechanisms of Inflammation
Inflammation and Immune Dysregulation
Activation of Immune Receptors
Inflammasomes are intracellular pattern recognition receptors (PRRs) that trigger the maturation of proinflammatory cytokines, such as IL‐1β or IL‐18.56 While the expression of the Nod‐like receptor protein 3 (NLRP3) inflammasome components is very low in healthy hepatocytes, during NASH, the expression of NLRP3 components is increased in animal models57 and humans.58 Selective pharmacologic inhibition57 or genetic deletion58 of the NLRP3 inflammasome or its components results in improved NASH pathology, including hepatocyte inflammation and fibrosis. The NLRP3 inflammasome is predominantly expressed in injured hepatocytes, KCs, and liver sinusoidal endothelial cells.59 It is also present in hepatic stellate cells where it is required for the development of fibrosis, at least in experimental mouse models.60 The NLRP3 inflammasome can be activated by multiple ligands, including pathogen‐associated molecular patterns (PAMPs) and danger associated molecular patterns (DAMPs). In NASH, potential triggers of its activation include lipopolysaccharide (LPS) and danger signals released from hepatocytes undergoing palmitate‐induced apoptosis, mitochondrial DNA released following fatty acid stimulation,61 cholesterol, and microvesicles released from fat‐laden cells undergoing lipotoxicity.62, 63, 64
Toll‐like receptors (TLRs) are transmembrane PRRs that sense invading pathogen or endogenous damage signals.65 Several studies have shown that TLR4 expression, especially in macrophages, is increased in patients with NASH66 and mouse models of disease.67 Translocation of TLR4 agonists, such as LPS and bacterial components from the gut, activate TLR signaling and has been shown to drive the progression of NASH.68 The expression of TLR9, a TLR that mainly recognizes bacterial DNA, increases in multiple mouse models of NASH, and its genetic deficiency leads to improved steatosis, inflammation, and liver fibrosis, suggesting that TLR9 promotes the progression of NASH.69
Immune Cell‐Mediated Inflammation
Crosstalk between immune cells in metabolic tissues dictates the overall inflammatory tone and systemic metabolic homeostasis. Recent evidence indicates that immunologic imbalances in the liver support the maintenance and progression of inflammation in NAFLD. In general, NASH is characterized by a robust recruitment of immune cells into the liver where they become activated and have the capacity to release molecules that cause inflammation (Fig. 3; Table 2). While a dysregulated immune response can lead to disease, the inflammatory response early during liver injury may also be important for healing and tissue repair.70 Although innate immune mechanisms are considered a major contributing factor to the inflammatory process in NASH, recent evidence indicates that adaptive immunity has an important role in the progression of this disease.71 Here, we highlight the major immune cell types involved in the progression of NASH.
Figure 3.
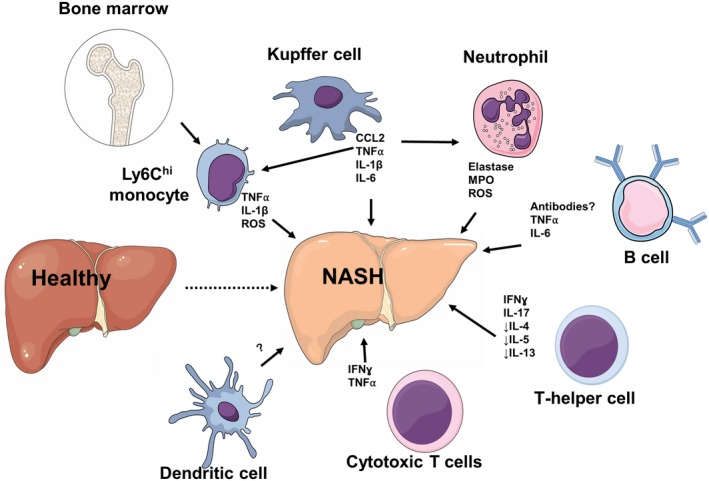
Immune dysregulation in the pathogenesis of NASH. Activation of liver‐resident KCs results in the release of CCL2 and other proinflammatory cytokines, such as TNFα, IL‐1, and IL‐6, leading to the recruitment of bone marrow‐derived Ly6Chi monocytes and neutrophils that further contribute to the inflammatory response. Activated neutrophils promote NASH by releasing elastase, MPO, and ROS. B cells can accumulate during NASH and produce TNFα and IL‐6. NASH is characterized by excessive Th1‐ and Th17‐derived IFNγ and IL‐17, respectively, and a deficiency in Th2‐derived IL‐4, IL‐5, and IL‐13. Cytotoxic CD8+ T cells are supported by type I IFN responses and lead to the production of IFNγ and TNFα. The role of DCs and their subsets in NASH is unclear as animal studies show contradictory results and it has not been investigated in humans. Abbreviation: ROS, reactive oxygen species.
Table 2.
Immune Cells Involved in Hepatic Inflammation During NASH
| Cell Type | Activator/Recruiting Factor | Role in NASH |
|---|---|---|
| Kupffer cells | NLRP3 inflammasome, TLRs, IFN genes | Promote NASH in early phase. Produce increased TNFα and CCL2 |
| Bone marrow‐derived monocytes | CCL2/CCR2 axis | Promote inflammation and fibrosis in NASH |
| Dendritic cells | Unclear. Increased intracellular lipid and proinflammatory phenotype | |
| Neutrophils | Promote NASH through antimicrobial secretion and NETs | |
| Th1 and Th17 cells | Promote NASH through increased production of IFNγ and IL‐17 | |
| Th2 cells | Decreased production of the anti‐inflammatory cytokines IL‐4, IL‐5, and IL‐13 | |
| Cytotoxic T cells | Type I IFNs | Promote NASH and HCC. Produce increased amounts of IFNγ and TNFα |
| B cells | Unclear, promote fibrosis in mouse models of hepatic injury |
Macrophages
Liver macrophages are a heterogeneous population consisting of yolk sac‐derived tissue‐resident macrophages or KCs and bone marrow monocyte‐derived macrophages. In the healthy liver, KCs exist within the hepatic sinusoids where they scavenge bacteria and microbial products from the intestine while mature monocytes show a patrolling behavior. In mouse models of NASH, KCs contribute to the early phase of disease through increased production of TNFα and CCL2.72 Indeed, depletion of KCs attenuates liver steatosis and hepatic IR in rats fed high‐fat or high‐sucrose diets.73 Determining the factors that trigger the activation of innate immune cells, such as KCs, and transition from isolated steatosis to NASH is one of the major goals in the field. KCs express the highest levels of inflammasome components among liver cell types, and NLRP3 activation in KCs promotes IL‐1β secretion fueling the progression of NASH.61 Activation of the NLRP3 inflammasome in KCs can be caused by mitochondria DNA release in response to NEFA61 or through the stimulator of interferon (IFN) genes, which induces inflammation through nuclear factor kappa B.74
The recruitment of bone marrow‐derived monocytes has been shown to be a critical event in the progression of NASH. KC‐derived factors facilitate a substantial infiltration of these monocytes into the liver, where together with KCs they contribute to the triggering and progression of disease through their inflammatory functions.75 In mouse models of NASH, inhibition of CCL2 or CCL2 receptor (CCR2) leads to reduced recruitment of monocyte‐derived macrophages, improved IR, hepatic inflammation, and fibrosis.76 Although it has been proposed that infiltrating monocytes differentiate into liver macrophages, single‐cell RNA sequencing of myeloid cells in mice fed a Western diet has shown that NAFLD also induces functional changes in their bone marrow precursors that remain stable throughout their migration into the liver,77 suggesting a role for liver–bone marrow crosstalk in maintaining hepatic inflammation in NASH.
Dendritic Cells
In the steady state, liver dendritic cells (DCs) function as antigen‐presenting cells (APCs) that internalize blood‐derived antigens and transport them to regional lymph nodes. Liver DCs, however, are poor APCs with less capacity to activate T cells compared with DCs from other tissues.78 While the role of DCs in human NASH is unclear, animal studies show contradictory results as depletion of hepatic DCs has resulted in both ameliorated or aggravated liver fibrosis and inflammation. These conflicting results are likely due to the heterogenicity of hepatic DCs and the low specificity of the methods used to experimentally manipulate DCs. Hepatic DCs can be divided into three subtypes with diverse functional capacity: classical type 1, classical type 2, and plasmacytoid. Despite this heterogeneity, most studies of liver DCs have focused on total CD11c+ or major histocompatibility complex II+ DCs. Regardless of their subclass, hepatic DCs that have increased intracellular lipid content show a proinflammatory phenotype in both mice and humans.
Neutrophils
The infiltration of neutrophils into the liver contributes to the progression of NASH through the secretion of cytokines and active molecules. Western diet‐fed mice develop increased expression of neutrophil elastase, and its genetic depletion results in decreased liver steatosis and inflammation.79 Similarly, myeloperoxidase (MPO) deficiency ameliorates liver inflammation and fibrosis in mice fed a high‐fat diet, suggesting a pathogenic role of neutrophils in NASH.80 Neutrophils can release extracellular traps (NETs) composed of nucleic acids and antimicrobials to entrap pathogens and limit infection. Increased production and reduced clearance of NETs has been linked to chronic sterile inflammation in autoimmune and inflammatory diseases. Markers of NETs are elevated in the serum of patients with NASH, and blockade of NET formation in mice protects mice from inflammation and NASH‐driven HCC.81 Another mechanism by which neutrophils can aggravate NASH is through human neutrophil peptides that induce cytokine and chemokine release during inflammation. Transgenic expression of human neutrophil peptide 1 in mice fed a NASH diet aggravated hepatic fibrosis through induction of hepatic stellate cell proliferation.82 Although the majority of studies suggest that neutrophils are pathogenic in the progression of NASH, they play a critical role in the reparative phase of sterile liver injury as they remove dead vasculature and create new channels for vascular regrowth. Whether neutrophils have divergent roles in NASH by promoting early inflammation but favoring later resolution, including fibrosis, remains to be investigated.
T‐Helper Cells
T‐helper (Th) cells are key players of adaptive immune responses as they assist B cells, macrophages, and cytotoxic T cells in eliminating pathogens and infected cells. After immune activation, Th cells can differentiate into Th1, Th2, and Th17 effector cells, depending on the cytokines in their environment. In general, NASH is characterized by excessive Th1‐derived cytokines, such as IFNγ, and a deficiency in Th2‐derived cytokines, including IL‐4, IL‐5, and IL‐13.83 IL‐17‐producing Th17 cells accumulate in the liver of mice and humans with NASH and have been shown to aggravate inflammation and fibrosis through effects on macrophages and hepatic stellate cells, respectively.
Cytotoxic T Cells
In mice and humans, cytotoxic CD8+ T cells accumulate in the liver during NAFLD, and their pharmacologic84 or genetic ablation85 results in decreased steatosis, IR, inflammation, and hepatic stellate cell activation. Activation of these cytotoxic CD8+ T cells is supported by type I IFN responses and leads to the production of the proinflammatory cytokines IFNγ and TNFα.85 Cytotoxic CD8+ T cells have also been shown to promote NASH development and subsequent transition to HCC in a process that requires crosstalk with natural killer T cells.
B cells
B cells have recently emerged as critical regulators of inflammation in adipose tissue during obesity.86 However, their role in the development of NASH is not well understood. In general, B cells can be divided into B1 and B2 subsets with differing immunophenotypes, functions, and cytokine secretion profiles. B2 cells accumulate in peripheral tissues, such as adipose tissue, where they contribute to IR through activation of T cells and secretion of proinflammatory cytokines. In contrast, adipose tissue B1 cells have protective roles against obesity‐related IR. In mice with NAFLD, B cells accumulate in the liver where they express increased amounts of TNFα and IL‐6 and have a higher capacity to activate T cells.87 B cells have also been shown to promote hepatic fibrosis in a mouse model of acute liver injury. In the same model, specific B‐cell deletion of the adaptor protein myeloid differentiation primary response 88 (MYD88) results in reduced infiltration of monocytes and DCs, suggesting that B cells are among the first responders during hepatic injury.88 Despite these findings, the exact mechanisms by which B cells become activated in the liver and promote NASH are unclear.
Intercellular and Interorgan Crosstalk
Intercellular and interorgan crosstalk are necessary for the development of liver injury and inflammation in NASH. Both soluble mediators and circulating EVs have been implicated in this crosstalk. Here, we review the adipose–liver axis, gut–liver axis, and the role of EVs in inflammatory crosstalk in NASH (Fig. 1).
Adipose–Liver Axis
Adipose tissue metabolic dysfunction is closely related to liver inflammation and fibrosis in humans and is a central driver of NASH development.89 During obesity, failure of the adipose tissue to expand and store excess energy leads to increased lipolysis and subsequent secretion of NEFAs. Diminished adipose tissue expandability and not obesity itself is believed to be the key factor linking positive energy balance and metabolic disease.90 Increased influx of adipose tissue‐derived NEFAs is a substantial source of substrate for the formation and storage of TGs in the liver, resulting in steatosis.91 Not only is the adipose tissue the major source of NEFA but it is also an endocrine organ secreting adipokines with systemic regulatory effects. Leptin and adiponectin produced by visceral adipocytes influence NAFLD and other components of the metabolic syndrome through regulation of food intake, body fat composition, insulin sensitivity, and inflammation.91 In addition, excessive production of proinflammatory cytokines by visceral adipose tissue macrophages is considered to be critical in obesity‐associated adipose tissue inflammation.92 Activated adipose tissue macrophages secrete cytokines and chemokines, including TNFα, IL‐1β, IL‐6, and CCL2, which cause local IR resulting in dysregulated lipid metabolism but which can also reach the circulation leading to systemic IR.93 Immune activation in adipose tissue likely precedes liver inflammation as mice with NASH have increased expression of macrophage and inflammatory genes in adipose tissue before the liver.89
Gut–Liver Axis
There is a bidirectional relationship between the gut, which represents an important port of first entry for external environmental influences, and the liver, which is the first line of receipt and processing of these factors. The gut contributes to both homeostasis in health as well as the pathogenesis of liver disease through several intermediaries that interact with one another, including the microbiota, bile acids, luminal products, immune mediators, and gut hormones. While causal links between the microbiota and NAFLD have not been fully elucidated, disruption in intestinal permeability94 and bacterial‐derived ligands (e.g., LPS) and metabolites (e.g., secondary bile acids, short chain fatty acids)95 are putative mediators of this association. Microbial‐derived PAMPs are capable of inciting an immune reaction and inflammation in the liver. Recently, the presence of a strain of bacterium (Klebsiella pneumoniae) that produces high levels of endogenous alcohol was associated with NAFLD in a human cohort.96 Bile acids are synthesized and secreted by hepatocytes and are involved in the absorption of dietary lipids. They are transported back to the liver by enterohepatic circulation and act on the nuclear farnesoid X receptor (FXR), which is also expressed on hepatocytes, to influence glucose97 and lipid metabolism.98 Further, release of FGF19 after ileal FXR activation is a feedback mechanism that reduces bile acid synthesis and also hepatic steatosis and IR.99 Bile acids, through their antimicrobial effects, also modulate the relationship between gut microbiota and chronic liver disease100 and improve glucose metabolism by activation of G‐protein coupled bile acid receptor (GPBAR1) in enterocytes.101 Therefore, targeting these mechanisms, for example, with an FXR agonist, is an attractive strategy in therapy for NAFLD.101, 102 Gut‐derived hormones, such as GLP‐1, play a crucial role in controlling nutrient intake, absorption, and metabolism and are attractive targets for metabolic disease in general as well as in the liver.2
Extracellular Vesicles
Recent studies have examined the role of EVs as a vector for cell‐to‐cell communication in NASH.103 Circulating EVs are elevated in human NASH samples as well as mouse models of NASH.44 Numerous bioactive cargoes have been defined for these EVs as well as the recipient cell responses activated by hepatocyte‐derived EVs.103 In NASH models, circulating EVs also differ in their cell of origin. EVs from adipocytes, macrophages, neutrophils, and platelets are reported to be elevated.104 Mechanistic studies have demonstrated that both microvesicles and exosomes are released by lipotoxic hepatocytes using distinct cell signaling pathways. LPC‐stimulated EV release was dependent on mixed lineage kinase 3. EVs released from palmitate‐ or LPC‐treated hepatocytes required activation of the death receptor 5 signaling axis leading to caspase‐mediated cleavage and activation of the rho‐associated coiled‐coil‐containing protein kinase 1 (ROCK1).103 In additional experiments, palmitate‐stimulated small EV release was dependent on the de novo synthesis of ceramide and preserved in the presence of caspase inhibitors, suggesting that lipotoxic EVs are heterogeneous in origin.44
Similar to the mechanism of their origin, EVs in NASH contain multiple bioactive cargoes. CXCL10 and S1P on EVs are implicated in macrophage chemotaxis, whereas TRAIL activated a proinflammatory response in macrophages.103 Vanin‐1‐enriched EVs were targeted to endothelial cells where they activated angiogenic pathways.103 Mitochondrial DNA‐enriched microparticles were also elevated in mouse models of NASH and found to activate myeloid cell TLR9‐dependent inflammation in the liver.103 Systemic injections of circulating EVs derived from high‐fat‐fed mice led to an increase in myeloid cells in circulation and hepatic accumulation of monocyte‐derived macrophages, suggesting that lipotoxic EVs can educate monocytes in the circulation to home into the liver.103 Thus, EVs may play a role in the local microenvironment of the liver, in circulation, and in interorgan crosstalk.
Conclusions
In the multifactorial pathogenesis of NASH, both hepatocyte lipotoxicity and immune‐mediated inflammation play key roles. Activation of hepatic stellate cells occurs as a consequence of signaling from stressed or apoptotic hepatocytes and macrophages, although other immune cells may play a role as well (Fig. 4). This triadic lesion forms the cornerstone of progressive NASH. However, recognizing the heterogeneity of NAFLD, it is very likely that our classification of NAFL and NASH will evolve as our understanding of the complexity of NASH pathogenesis grows. For example, future definitions may stratify the type of NASH based on key features in the pathogenesis continuum, such as ER stress‐predominant NASH versus apoptosis‐predominant NASH versus macrophage‐predominant NASH or B‐cell‐predominant NASH. Furthermore, each of these types of NASH may have variable activation of hepatic stellate cells. Our nascent understanding of this heterogeneity would also suggest that combination therapy, although logical, may uncover potentially redundant pathways in NASH pathogenesis. Regardless, mechanistic studies that answer fundamental questions in NASH pathogenesis are still needed. These include exploration of mechanistic questions that address early events in liver injury in NASH, such as the consequences of sublethal hepatocyte injury. Another key area is the definition of the triggers, kinetics, and magnitude of immune‐cell activation responses along the NAFLD spectrum and to define the individual and synergistic contribution of each type of immune cell.
Figure 4.
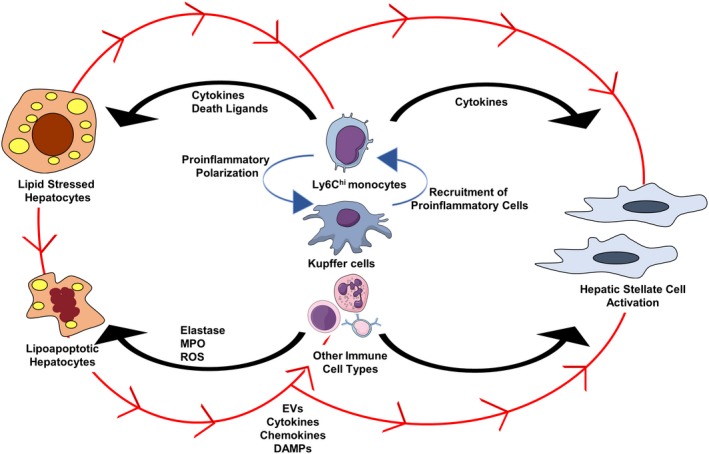
Triadic lesion in the pathogenesis of NASH. Hepatocyte injury, macrophage‐mediated inflammation, and hepatic stellate cell activation comprise the key mechanistic abnormalities in NASH. Soluble and EV signals from hepatocytes lead to proinflammatory activation of macrophages, can recruit proinflammatory monocytes into the liver, and also lead to hepatic stellate cell activation (depicted in red arrows). Other immune cells, such as neutrophils and B cells, may also respond to hepatocyte‐derived signals. Activated macrophages release cytokines and chemokines that can promote hepatocyte apoptosis, attract other immune cells into the liver, and also influence the activation of hepatic stellate cells. Abbreviation: ROS, reactive oxygen species.
Supported by the National Institutes of Health (grants DK111378 to H.M. and DK122056 to X.R.) and the Mayo Foundation (to H.M.).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Younossi ZM, Blissett D, Blissett R, Henry L, Stepanova M, Younossi Y, et al. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 2016;64:1577‐1586. [DOI] [PubMed] [Google Scholar]
- 2. Friedman SL, Neuschwander‐Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al.; Nonalcoholic Steatohepatitis Clinical Research Network . Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 4. Hagstrom H, Nasr P, Ekstedt M, Hammar U, Stal P, Hultcrantz R, et al. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy‐proven NAFLD. J Hepatol 2017;67:1265‐1273. [DOI] [PubMed] [Google Scholar]
- 5. Hoang SA, Oseini A, Feaver RE, Cole BK, Asgharpour A, Vincent R, et al. Gene expression predicts histological severity and reveals distinct molecular profiles of nonalcoholic fatty liver disease. Sci Rep 2019;9:12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chang Y, Ryu S, Kim Y, Cho YK, Sung E, Kim HN, et al. Low levels of alcohol consumption, obesity, and development of fatty liver with and without evidence of advanced fibrosis. Hepatology 2019; doi: 10.1002/hep.30867. [DOI] [PubMed] [Google Scholar]
- 7. Lonardo A, Nascimbeni F, Ballestri S, Fairweather D, Win S, Than TA, et al. Sex differences in nonalcoholic fatty liver disease: state of the art and identification of research gaps. Hepatology 2019;70:1457‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Norheim F, Hui ST, Kulahcioglu E, Mehrabian M, Cantor RM, Pan C, et al. Genetic and hormonal control of hepatic steatosis in female and male mice. J Lipid Res 2017;58:178‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005;115:1343‐1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haas JT, Francque S, Staels B. Pathophysiology and mechanisms of nonalcoholic fatty liver disease. Annu Rev Physiol 2016;78:181‐205. [DOI] [PubMed] [Google Scholar]
- 11. Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest 2016;126:12‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ghorpade DS, Ozcan L, Zheng Z, Nicoloro SM, Shen Y, Chen E, et al. Hepatocyte‐secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature 2018;555:673‐677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ipsen DH, Lykkesfeldt J, Tveden‐Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non‐alcoholic fatty liver disease. Cell Mol Life Sci 2018;75:3313‐3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lambert JE, Ramos‐Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014;146:726‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ter Horst KW, Serlie MJ. Fructose consumption, lipogenesis, and non‐alcoholic fatty liver disease. Nutrients 2017;9:pii: E981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mashek DG. Hepatic fatty acid trafficking: multiple forks in the road. Adv Nutr 2013;4:697‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Watt MJ, Barnett AC, Bruce CR, Schenk S, Horowitz JF, Hoy AJ. Regulation of plasma ceramide levels with fatty acid oversupply: evidence that the liver detects and secretes de novo synthesised ceramide. Diabetologia 2012;55:2741‐2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. BasuRay S, Wang Y, Smagris E, Cohen JC, Hobbs HH. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc Natl Acad Sci U S A 2019;116:9521‐9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ma Y, Belyaeva OV, Brown PM, Fujita K, Valles K, Karki S, et al.; Nonalcoholic Steatohepatitis Clinical Research Network . 17‐Beta hydroxysteroid dehydrogenase 13 is a hepatic retinol dehydrogenase associated with histological features of nonalcoholic fatty liver disease. Hepatology 2019;69:1504‐1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Malhi H, Gores GJ. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver Dis 2008;28:360‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Musso G, Cassader M, Paschetta E, Gambino R. Bioactive lipid species and metabolic pathways in progression and resolution of nonalcoholic steatohepatitis. Gastroenterology 2018;155:282‐302.e288. [DOI] [PubMed] [Google Scholar]
- 23. Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007;46:1081‐1090. [DOI] [PubMed] [Google Scholar]
- 24. Puri P, Wiest MM, Cheung O, Mirshahi F, Sargeant C, Min HK, et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 2009;50:1827‐1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Loomba R, Quehenberger O, Armando A, Dennis EA. Polyunsaturated fatty acid metabolites as novel lipidomic biomarkers for noninvasive diagnosis of nonalcoholic steatohepatitis. J Lipid Res 2015;56:185‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Santoro N, Caprio S, Feldstein AE. Oxidized metabolites of linoleic acid as biomarkers of liver injury in nonalcoholic steatohepatitis. Clin Lipidol 2013;8:411‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Raselli T, Hearn T, Wyss A, Atrott K, Peter A, Frey‐Wagner I, et al. Elevated oxysterol levels in human and mouse livers reflect nonalcoholic steatohepatitis. J Lipid Res 2019;60:1270‐1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ibrahim SH, Hirsova P, Gores GJ. Non‐alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut 2018;67:963‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou Y, Orešič M, Leivonen M, Gopalacharyulu P, Hyysalo J, Arola J, et al. Noninvasive detection of nonalcoholic steatohepatitis using clinical markers and circulating levels of lipids and metabolites. Clin Gastroenterol Hepatol 2016;14:1463‐1472.e1466. [DOI] [PubMed] [Google Scholar]
- 30. Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK‐dependent hepatocyte lipoapoptosis. J Biol Chem 2006;281:12093‐12101. [DOI] [PubMed] [Google Scholar]
- 31. Barreyro FJ, Kobayashi S, Bronk SF, Werneburg NW, Malhi H, Gores GJ. Transcriptional regulation of Bim by FoxO3A mediates hepatocyte lipoapoptosis. J Biol Chem 2007;282:27141‐27154. [DOI] [PubMed] [Google Scholar]
- 32. Cazanave SC, Mott JL, Elmi NA, Bronk SF, Werneburg NW, Akazawa Y, et al. JNK1‐dependent PUMA expression contributes to hepatocyte lipoapoptosis. J Biol Chem 2009;284:26591‐26602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Masuoka HC, Mott J, Bronk SF, Werneburg NW, Akazawa Y, Kaufmann SH, et al. Mcl‐1 degradation during hepatocyte lipoapoptosis. J Biol Chem 2009;284:30039‐30048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Akazawa Y, Guicciardi ME, Cazanave SC, Bronk SF, Werneburg NW, Kakisaka K, et al. Degradation of cIAPs contributes to hepatocyte lipoapoptosis. Am J Physiol Gastrointest Liver Physiol 2013;305:G611‐ G619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cazanave SC, Mott JL, Elmi NA, Bronk SF, Masuoka HC, Charlton MR, et al. A role for miR‐296 in the regulation of lipoapoptosis by targeting PUMA. J Lipid Res 2011;52:1517‐1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cazanave SC, Mott JL, Bronk SF, Werneburg NW, Fingas CD, Meng XW, et al. Death receptor 5 signaling promotes hepatocyte lipoapoptosis. J Biol Chem 2011;286:39336‐39348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kakisaka K, Cazanave SC, Fingas CD, Guicciardi ME, Bronk SF, Werneburg NW, et al. Mechanisms of lysophosphatidylcholine‐induced hepatocyte lipoapoptosis. Am J Physiol Gastrointest Liver Physiol 2012;302:G77‐G84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Akazawa Y, Cazanave S, Mott JL, Elmi N, Bronk SF, Kohno S, et al. Palmitoleate attenuates palmitate‐induced Bim and PUMA up‐regulation and hepatocyte lipoapoptosis. J Hepatol 2010;52:586‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cao H, Gerhold K, Mayers JR, Wiest MM, Watkins SM, Hotamisligil GS. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell 2008;134:933‐944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rius B, Titos E, Moran‐Salvador E, Lopez‐Vicario C, Garcia‐Alonso V, Gonzalez‐Periz A, et al. Resolvin D1 primes the resolution process initiated by calorie restriction in obesity‐induced steatohepatitis. FASEB J 2014;28:836‐848. [DOI] [PubMed] [Google Scholar]
- 41. Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF‐alpha expression via a lysosomal pathway. Hepatology 2004;40:185‐194. [DOI] [PubMed] [Google Scholar]
- 42. Volmer R, van der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci U S A 2013;110:4628‐4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cazanave SC, Elmi NA, Akazawa Y, Bronk SF, Mott JL, Gores GJ. CHOP and AP‐1 cooperatively mediate PUMA expression during lipoapoptosis. Am J Physiol Gastrointest Liver Physiol 2010;299:G236‐ G243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kakazu E, Mauer AS, Yin M, Malhi H. Hepatocytes release ceramide‐enriched pro‐inflammatory extracellular vesicles in an IRE1alpha‐dependent manner. J Lipid Res 2016;57:233‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim OK, Jun W, Lee J. Mechanism of ER stress and inflammation for hepatic insulin resistance in obesity. Ann Nutr Metab 2015;67:218‐227. [DOI] [PubMed] [Google Scholar]
- 46. Soltis AR, Kennedy NJ, Xin X, Zhou F, Ficarro SB, Yap YS, et al. Hepatic dysfunction caused by consumption of a high‐fat diet. Cell Rep 2017;21:3317‐3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Idrissova L, Malhi H, Werneburg NW, LeBrasseur NK, Bronk SF, Fingas C, et al. TRAIL receptor deletion in mice suppresses the inflammation of nutrient excess. J Hepatol 2015;62:1156‐1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu B, Jiang M, Chu Y, Wang W, Chen D, Li X, et al. Gasdermin D plays a key role as a pyroptosis executor of non‐alcoholic steatohepatitis in humans and mice. J Hepatol 2018;68:773‐782. [DOI] [PubMed] [Google Scholar]
- 49. Mehal WZ. The inflammasome in liver injury and non‐alcoholic fatty liver disease. Dig Dis 2014;32:507‐515. [DOI] [PubMed] [Google Scholar]
- 50. Ioannou GN, Haigh WG, Thorning D, Savard C. Hepatic cholesterol crystals and crown‐like structures distinguish NASH from simple steatosis. J Lipid Res 2013;54:1326‐1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ioannou GN, Landis CS, Jin G‐Y, Haigh WG, Farrell GC, Kuver R, et al. Cholesterol crystals in hepatocyte lipid droplets are strongly associated with human nonalcoholic steatohepatitis. Hepatol Commun 2019;3:776‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tsurusaki S, Tsuchiya Y, Koumura T, Nakasone M, Sakamoto T, Matsuoka M, et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis 2019;10:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, et al. A positive feedback loop between RIP3 and JNK controls non‐alcoholic steatohepatitis. EMBO Mol Med 2014;6:1062‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018;67:1270‐1283. [DOI] [PubMed] [Google Scholar]
- 55. Cullen Sean P, Henry Conor M, Kearney Conor J, Logue Susan E, Feoktistova M, Tynan Graham A, et al. Fas/CD95‐induced chemokines can serve as “find‐me” signals for apoptotic cells. Mol Cell 2013;49:1034‐1048. [DOI] [PubMed] [Google Scholar]
- 56. Schroder K, Tschopp J. The inflammasomes. Cell 2010;140:821‐832. [DOI] [PubMed] [Google Scholar]
- 57. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol 2017;66:1037‐1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wree A, McGeough MD, Pena CA, Schlattjan M, Li H, Inzaugarat ME, et al. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J Mol Med (Berl) 2014;92:1069‐1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Boaru SG, Borkham‐Kamphorst E, Tihaa L, Haas U, Weiskirchen R. Expression analysis of inflammasomes in experimental models of inflammatory and fibrotic liver disease. J Inflammation (Lond) 2012;9:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Watanabe A, Sohail MA, Gomes DA, Hashmi A, Nagata J, Sutterwala FS, et al. Inflammasome‐mediated regulation of hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol 2009;296:G1248‐G1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pan J, Ou Z, Cai C, Li P, Gong J, Ruan XZ, et al. Fatty acid activates NLRP3 inflammasomes in mouse Kupffer cells through mitochondrial DNA release. Cell Immunol 2018;332:111‐120. [DOI] [PubMed] [Google Scholar]
- 62. Garcia‐Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest 2016;126:859‐864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Povero D, Eguchi A, Niesman IR, Andronikou N, de Mollerat du Jeu X, Mulya A, et al. Lipid‐induced toxicity stimulates hepatocytes to release angiogenic microparticles that require Vanin‐1 for uptake by endothelial cells. Sci Signal 2013;6:ra88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Povero D, Eguchi A, Li H, Johnson CD, Papouchado BG, Wree A, et al. Circulating extracellular vesicles with specific proteome and liver microRNAs are potential biomarkers for liver injury in experimental fatty liver disease. PLoS One 2014;9:e113651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schuster JM, Nelson PS. Toll receptors: an expanding role in our understanding of human disease. J Leukoc Biol 2000;67:767‐773. [PubMed] [Google Scholar]
- 66. Vespasiani‐Gentilucci U, Carotti S, Perrone G, Mazzarelli C, Galati G, Onetti‐Muda A, et al. Hepatic toll‐like receptor 4 expression is associated with portal inflammation and fibrosis in patients with NAFLD. Liver Int 2015;35:569‐581. [DOI] [PubMed] [Google Scholar]
- 67. Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll‐like receptor‐4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non‐alcoholic steatohepatitis. J Hepatol 2007;47:571‐579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Henao‐Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome‐mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012;482:179‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, et al. Toll‐like receptor 9 promotes steatohepatitis by induction of interleukin‐1beta in mice. Gastroenterology 2010;139:323‐334.e327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hossain M, Kubes P. Innate immune cells orchestrate the repair of sterile injury in the liver and beyond. Eur J Immunol 2019;49:831‐841. [DOI] [PubMed] [Google Scholar]
- 71. Sutti S, Albano E. Adaptive immunity: an emerging player in the progression of NAFLD. Nat Rev Gastroenterol Hepatol 2019. 10.1038/s41575-019-0210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tosello‐Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet‐induced mouse model through tumor necrosis factor‐alpha production. J Biol Chem 2012;287:40161‐40172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ, et al. Depletion of liver Kupffer cells prevents the development of diet‐induced hepatic steatosis and insulin resistance. Diabetes 2010;59:347‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yu Y, Liu Y, An W, Song J, Zhang Y, Zhao X. STING‐mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J Clin Invest 2019;129:546‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Reid DT, Reyes JL, McDonald BA, Vo T, Reimer RA, Eksteen B. Kupffer cells undergo fundamental changes during the development of experimental NASH and are critical in initiating liver damage and inflammation. PLoS One 2016;11:e0159524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP‐1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012;61:416‐426. [DOI] [PubMed] [Google Scholar]
- 77. Krenkel O, Hundertmark J, Abdallah AT, Kohlhepp M, Puengel T, Roth T, et al. Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity‐related steatohepatitis. Gut 2019; pii: gutjnl‐2019‐318382. [DOI] [PubMed] [Google Scholar]
- 78. Pillarisetty VG, Shah AB, Miller G, Bleier JI, DeMatteo RP. Liver dendritic cells are less immunogenic than spleen dendritic cells because of differences in subtype composition. J Immunol 2004;172:1009‐1017. [DOI] [PubMed] [Google Scholar]
- 79. Chen J, Liang B, Bian D, Luo Y, Yang J, Li Z, et al. Knockout of neutrophil elastase protects against western diet induced nonalcoholic steatohepatitis in mice by regulating hepatic ceramides metabolism. Biochem Biophys Res Commun 2019;518:691‐697. [DOI] [PubMed] [Google Scholar]
- 80. Rensen SS, Bieghs V, Xanthoulea S, Arfianti E, Bakker JA, Shiri‐Sverdlov R, et al. Neutrophil‐derived myeloperoxidase aggravates non‐alcoholic steatohepatitis in low‐density lipoprotein receptor‐deficient mice. PLoS One 2012;7:e52411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. van der Windt DJ, Sud V, Zhang H, Varley PR, Goswami J, Yazdani HO, et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 2018;68:1347‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ibusuki R, Uto H, Arima S, Mawatari S, Setoguchi Y, Iwashita Y, et al. Transgenic expression of human neutrophil peptide‐1 enhances hepatic fibrosis in mice fed a choline‐deficient, l‐amino acid‐defined diet. Liver Int 2013;33:1549‐1556. [DOI] [PubMed] [Google Scholar]
- 83. Kremer M, Hines IN, Milton RJ, Wheeler MD. Favored T helper 1 response in a mouse model of hepatosteatosis is associated with enhanced T cell‐mediated hepatitis. Hepatology 2006;44:216‐227. [DOI] [PubMed] [Google Scholar]
- 84. Bhattacharjee J, Kirby M, Softic S, Miles L, Salazar‐Gonzalez RM, Shivakumar P, et al. Hepatic natural killer T‐cell and CD8+ T‐cell signatures in mice with nonalcoholic steatohepatitis. Hepatol Commun 2017;1:299‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ghazarian M, Revelo XS, Nohr MK, Luck H, Zeng K, Lei H, et al. Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci Immunol 2017;2:pii:eaai7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 2011;17:610‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhang F, Jiang WW, Li X, Qiu XY, Wu Z, Chi YJ, et al. Role of intrahepatic B cells in non‐alcoholic fatty liver disease by secreting pro‐inflammatory cytokines and regulating intrahepatic T cells. J Dig Dis 2016;17:464‐474. [DOI] [PubMed] [Google Scholar]
- 88. Thapa M, Chinnadurai R, Velazquez VM, Tedesco D, Elrod E, Han J‐H, et al. Liver fibrosis occurs through dysregulation of MyD88‐dependent innate B cell activity. Hepatology 2015;61:2067‐2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Stanton MC, Chen S‐C, Jackson JV, Rojas‐Triana A, Kinsley D, Cui L, et al. Inflammatory signals shift from adipose to liver during high fat feeding and influence the development of steatohepatitis in mice. J Inflamm (Lond) 2011;8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Virtue S, Vidal‐Puig A. Adipose tissue expandability, lipotoxicity and the metabolic syndrome‐‐an allostatic perspective. Biochim Biophys Acta 2010;1801:338‐349. [DOI] [PubMed] [Google Scholar]
- 91. Stojsavljevic S, Gomercic Palcic M, Virovic Jukic L, Smircic Duvnjak L, Duvnjak M. Adipokines and proinflammatory cytokines, the key mediators in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol 2014;20:18070‐18091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 2010;72:219‐246. [DOI] [PubMed] [Google Scholar]
- 93. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med 2012;18:363‐374. [DOI] [PubMed] [Google Scholar]
- 94. Luther J, Garber JJ, Khalili H, Dave M, Bale SS, Jindal R, et al. Hepatic injury in nonalcoholic steatohepatitis contributes to altered intestinal permeability. Cell Mol Gastroenterol Hepatol 2015;1:222‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Schubert K, Olde Damink SWM, von Bergen M, Schaap FG. Interactions between bile salts, gut microbiota, and hepatic innate immunity. Immunol Rev 2017;279:23‐35. [DOI] [PubMed] [Google Scholar]
- 96. Yuan J, Chen C, Cui J, Lu J, Yan C, Wei X, et al. Fatty liver disease caused by high‐alcohol‐producing Klebsiella pneumoniae. Cell Metab 2019;30:675‐688.e677. Erratum in: Cell Metab 2019;30:1172. [DOI] [PubMed] [Google Scholar]
- 97. Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci U S A 2006;103:1006‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP‐1c. J Clin Invest 2004;113:1408‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Alvarez‐Sola G, Uriarte I, Latasa MU, Fernandez‐Barrena MG, Urtasun R, Elizalde M, et al. Fibroblast growth factor 15/19 (FGF15/19) protects from diet‐induced hepatic steatosis: development of an FGF19‐based chimeric molecule to promote fatty liver regeneration. Gut 2017;66:1818‐1828. [DOI] [PubMed] [Google Scholar]
- 100. Parseus A, Sommer N, Sommer F, Caesar R, Molinaro A, Stahlman M, et al. Microbiota‐induced obesity requires farnesoid X receptor. Gut 2017;66:429‐437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. van Nierop FS, Scheltema MJ, Eggink HM, Pols TW, Sonne DP, Knop FK, et al. Clinical relevance of the bile acid receptor TGR5 in metabolism. Lancet Diabetes Endocrinol 2017;5:224‐233. [DOI] [PubMed] [Google Scholar]
- 102. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al.;NASH Clinical Research Network . Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet 2015;385:956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Malhi H. Emerging role of extracellular vesicles in liver diseases. Am J Physiol Gastrointest Liver Physiol 2019;317:G739‐G749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Li J, Liu H, Mauer AS, Lucien F, Raiter A, Bandla H, et al. Characterization of cellular sources and circulating levels of extracellular vesicles in a dietary murine model of nonalcoholic steatohepatitis. Hepatol Commun 2019;3:1235‐1249. [DOI] [PMC free article] [PubMed] [Google Scholar]