

Abstract
The transforming growth factor-β (TGF-β) family controls embryogenesis, stem cell differentiation, and tissue homeostasis. However, how post-translation modifications contribute to fine-tuning of TGF-β family signaling responses is not well understood. Inhibitory (I)-Smads can antagonize TGF-β/Smad signaling by recruiting Smurf E3 ubiquitin ligases to target the active TGF-β receptor for proteasomal degradation. A proteomic interaction screen identified Vpr binding protein (VprBP) as novel binding partner of Smad7. Mis-expression studies revealed that VprBP negatively controls Smad2 phosphorylation, Smad2–Smad4 interaction, as well as TGF-β target gene expression. VprBP was found to promote Smad7–Smurf1–TβRI complex formation and induce proteasomal degradation of TGF-β type I receptor (TβRI). Moreover, VprBP appears to stabilize Smurf1 by suppressing Smurf1 poly-ubiquitination. In multiple adult and mouse embryonic stem cells, depletion of VprBP promotes TGF-β or Activin-induced responses. In the mouse embryo VprBP expression negatively correlates with mesoderm marker expression, and VprBP attenuated mesoderm induction during zebrafish embryogenesis. Our findings thereby uncover a novel regulatory mechanism by which Smurf1 controls the TGF-β and Activin cascade and identify VprBP as a critical determinant of embryonic mesoderm induction.
Keywords: Smurf1, TGF-β type I receptor, ubiquitination, Activin, mesoderm induction
Introduction
The transforming growth factor-β (TGF-β) signaling pathway is critical for multiple biological processes in vertebrates including embryonic development, immunity, and organ homeostasis (Moustakas and Heldin, 2009). Aberrant TGF-β signaling may cause developmental defects, vascular disorders, kidney, lung and liver fibrosis, as well as cancer (Akhurst and Hata, 2012). To initiate signal transduction, TGF-β binds to the transmembrane serine/threonine kinase type I (TβRI) and type II (TβRII) receptors. Receptor complex formation allows TβRI to be phosphorylated by the TβRII kinase and this induces TβRI kinase-mediated phosphorylation of the downstream R-Smad proteins. R-Smads then bind to Smad4 and translocate into the nucleus, where these heteromeric complexes regulate downstream target gene expression (Feng and Derynck, 2005). All TGF-β family members signal via structurally and functionally related receptors. Whereas TGF-βs, Activins and Nodal signal mainly via Smad2 and Smad3, BMPs activate Smad1, Smad5, and Smad8 (Feng and Derynck, 2005).
Various non-Smad pathways can also be induced by activated TGF-β family receptor complexes, resulting in stimulation of e.g. MAP kinase signaling, Rho-like GTPase pathways, and AKT signaling (Zhang, 2009). Upon activation of TGF-β family signaling, important negative feedback mechanisms operate to attenuate and/or terminate the signaling cascade(s) (Itoh and ten Dijke, 2007). One way by which this is achieved is through transcriptional induction of inhibitory (I)‐Smad7 (Nakao et al., 1997). Smad7 recruits the E3 ligases Smurf1 and Smurf2 to TβRI resulting in poly-ubiquitination and degradation of this receptor (Kavsak et al., 2000; Ebisawa et al., 2001).
Smurf1 and Smurf2 are HECT E3 ubiquitin ligases that belong to the Nedd4 family (Miyazono, 2002). The Smurf proteins are comprised of an N-terminal C2 domain for substrate selection and subcellular location, a carboxyl-terminal HECT domain for catalytic activity, and several WW domains (named after the presence of two conserved tryptophans) mediating protein–protein interactions in between C2 and HECT (Zhu et al., 1999; Lu et al., 2011). Identified as modifier of TGF-β/Smad signaling, Smurf2 was found to interact with Smad2 and TβRI and mediate ubiquitin-mediated degradation of these signaling components (Zhang et al., 2001). Smurf1 was reported to selectively induce proteasomal degradation of BMPR-Smad1/5 and TGF-β/BMP type I receptors (Ebisawa et al., 2001; Murakami et al., 2003). Although Smurf1 has a high sequence similarity with Smurf2, it targets distinct substrates, including RUNX2/3, MEKK2, JunB, RhoA, and TRAF proteins (Wang et al., 2003; Yamashita et al., 2005; Shen et al., 2006; Li et al., 2010a; Zhao et al., 2010). In addition, Smurf1 and Smurf2 use distinct self-regulatory mechanisms to control their catalytic activity. Intramolecular interaction between the Smurf2 C2 and HECT domains leads to auto-inhibition and results in inhibition of ubiquitin thioester formation (Wiesner et al., 2007). In contrast, Smurf1 displays a high level of auto-ubiquitination due to the absence of one WW domain (Lu et al., 2008, 2011). Multiple functions of Smurf1 remain to be investigated including how its activity is regulated, and how the selection of substrates occurs.
Vpr binding protein (VprBP), an adaptor protein also identified as DCAF1, can act as a substrate recognition adaptor of CRL4 or Dyrk2 protein complexes and modulate protein ubiquitination, cell-cycle progression, and tumorigenic potential in human cancers (Jin et al., 2006; Maddika and Chen, 2009). VprBP has also been reported to contain a functional kinase domain that phosphorylates histone H2A and controls histone modification (Kim et al., 2013). In addition, recent studies demonstrated that the CRL4–DDB1–VprBP complex is crucial for activation of TET methylcytosine dioxygenases in oocytes and regulates oocyte reprogramming (Yu et al., 2013). These notions indicate that VprBP is functionally involved in protein modification and activation and plays an important role in multiple biological processes. Here, we identify VprBP as a novel binding partner of Smad7 and Smurfs, and highlight its relevance in controlling Smurf1 activity and defining TGF-β and Activin-induced responses in adult and embryonic cells and tissues.
Results
VprBP interacts with both Smad7 and Smurfs
To investigate mechanisms of TGF-β signaling fine-tuning, we previously performed a tandem affinity purification (TAP) assay of the whole proteome to identify interacting proteins of I-Smads (Zhang et al., 2013b). In this assay, HEK293T cells were transfected with TAP-tagged Smad7, followed by immunoprecipitation, SDS–PAGE, and Coomassie staining. Several distinct bands were enriched at different molecular weights when using Smad7 as the bait (Figure 1A). We analysed the enriched putative interactors by mass spectrometry(MS)-based proteomics and identified VprBP, as a novel candidate Smad7 binding protein (Figure 1B; Supplementary Table S1). Several known binding partners of Smad7 were confirmed in our screen, including ITCH, WWP2, USP11, and UBE20 (Soond and Chantry, 2011; Al-Salihi et al., 2012; Zhang et al., 2013b; Park et al., 2015). VprBP is a multifunctional protein that has a potential regulating role in cancer, development, protein degradation, and cell signaling (Nakagawa et al., 2013). We therefore tested the interaction of VprBP with Smad7 and Smad7-associated TGF-β pathway components by co-immunoprecipitation assays. HEK293T cells were transfected with Flag-tagged Smad2, Smad7, TβRI, Smurf1, or Smurf2 (both wild-type (WT) and catalytically inactive (CA) mutants), and immunoprecipitated with Flag antibodies to analyse for binding of endogenous VprBP. VprBP robustly interacted with Smad7, but not with Smad2 and TβRI (ALK5) (Figure 1C). In addition, we also noticed the moderate association between VprBP and Smurf1/2 (Figure 1D). We did not observe interaction between VprBP with other I-Smad modifiers such as ITCH (Supplementary Figure S1A). Endogenous interactions between VprBP and Smurf1 were detected as well (Figure 1E). These findings indicate that VprBP interacts with both Smad7 and Smurf1, and may thereby play a role in TGF-β signaling.
Figure 1.

VprBP interacts with Smad7 and Smurf1/2. (A) Proteomic screening of Smad7-interacting proteins. HEK293T cells were transfected with Smad7-TAP and lysed for immunoprecipitation. The bound complexes were separated by SDS–PAGE and stained with Coomassie Brilliant Blue. Arrow indicates Smad7-TAP at 53 kDa. (B) Network graph of Smad7-binding partners identified by mass spectrometry-based proteomics, such that more significant interactors will tend to be closer to SMAD7. Green dots indicated for known Smad7 interacting proteins. (C) HEK293T cells were transfected with Flag-tagged Smad2 and Smad7 or TβRI (ALK5) expression constructs and their vector control, in untreated or treated with TGF-β (5 ng/ml) for 1 h and subjected to immunoprecipitate with Flag antibody followed by blotting with VprBP and Flag antibodies. Total cell lysates were also blotted for VprBP. (D) HEK293T cells were transfected with Flag-Smurf constructs (WT and C699A variants) and vector control and subjected to immunoprecipitation with Flag antibody followed by VprBP and Flag immunoblotting. (E) Endogenous interaction between VprBP with Smurf1 in HEK293T cell lysates as identified by immunoprecipitation of Smad7 or Smurf1 followed by immunoblotting with VprBP antibody.
VprBP negatively regulates TGF-β signaling
To further evaluate the function of VprBP on the TGF-β pathway, we detected the effect of its mis-expression onTGF-β/Smad transcriptional activity using the Smad-dependent CAGA-luciferase transcriptional reporter. As shown in Figure 2A, ectopic expression of VprBP significantly inhibited the activity of this reporter in HEK293T cell, whereas depletion of VprBP promoted the response. However, VprBP had no significant effect on BMP-induced BMP response element (BRE)-luciferase transcriptional reporter activity and Smad1 phosphorylation (Supplementary Figure S1B and C), demonstrating selectivity of VprBP in regulating TGF-β signaling. Since VprBP can function as a substrate recognition adaptor in the CRL4 E3 ligase complex, we next examined whether CUL4A also has an effect on TGF-β signaling. However, in contrast to VprBP, ectopic expression of CUL4A did not significantly decrease TGF-β-induced reporter activity, also not when co-expressed with VprBP (Supplementary Figure S1D), indicating that the inhibitory effect of VprBP is independent of its CRL4 adaptor function.
Figure 2.

VprBP inhibits TGF-β signaling. (A) Effect of VprBP mis-expression on CAGA-Luc TGF-β Smad-dependent transcriptional reporter activity in HEK293T cells, treated with or without 2.5 ng/ml TGF-β for 16 h. Left, VprBP ectopic expression; right, VprBP knockdown using two short hairpin RNA (shRNA) constructs (#1 and #2). (B) Effect of shRNA-mediated VprBP knockdown on TGF-β-induced Smad2 phosphorylation (p-Smad2), measured by immunoblot analysis. Total Smad2, VprBP, and actin are also shown. HaCaT cells were treated with 2.5 ng/ml TGF-β for the indicated hours. (C) Effect of VprBP knockdown on TGF-β-induced Smad2/Smad4 complex formation, measured by immunoprecipitation followed by immunoblotting. HaCaT cells were treated with 2.5 ng/ml TGF-β for 1 h. (D) TGF-β (2.5 ng/ml)-induced gene expression in control and VprBP-depleted HaCaT cells, measured by real-time quantitative PCR (qPCR). GAPDH was used for normalization; mean ± SD of triplicates. (E) qPCR analysis of mRNA levels of TGF-β-induced EMT markers in control or VprBP knockdown HaCaT cells upon 48 h TGF-β (2.5 ng/ml) treatment. Normalized to GAPDH expression, mean ± SD of triplicates. (F) Immunoblotting of p-Smad2, Smad2, and VprBP in control and VprBP-depleted MDA-MB-231 cells. (G) Immunoblotting of p-Smad2, VprBP, Smad2 (lower gel 55 kDa), and actin (lower gel 40 kDa) in MDA-MB-231 cells ectopically expressing VprBP vs. control cells. (H and I) Migration assay of control vector-infected MDA-MB-231 cells and cells ectopically expressing VprBP. Cells were treated with or without 5 ng/ml TGF-β for 36 h. (H) Representative images of scratch and migration. (I) Fold changes in distance of cell migration of the experiment shown in H. Mean ± SD, 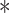
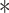
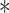 P < 0.001.
P < 0.001.
To investigate the effect of VprBP on TGF-β signaling more directly, we stably knocked down VprBP in the human keratinocyte cell line HaCaT and found that TGF-β-induced Smad2 phosphorylation was potentiated (Figure 2B). Moreover, compared with control vector-infected HaCaT cells, the endogenous interaction between Smad2 and Smad4 was increased upon VprBP knockdown, while the total protein levels of Smad2 and Smad4 were unchanged (Figure 2C). In line with these results, the TGF-β-induced messenger RNA (mRNA) levels of CTGF, p15, and p21, PAI-1, Smad7, and SnoN were all significantly enhanced after depletion of VprBP in HaCaT cells (Figure 2D).
As TGF-β can act as a promoter of epithelial–mesenchymal transition (EMT) and cell migration, we next examined the impact of VprBP on these TGF-β-induced phenotypes (Lamouille et al., 2014). First of all, we found the mRNA levels of the TGF-β-induced mesenchymal markers N-Cadherin, Slug, and Twist to be enhanced in HaCaT cells upon VprBP knockdown (Figure 2E). Additionally, in MDA-MB-231 breast cancer cells, we also detected increased levels of TGF-β-induced phosphorylated Smad2 after VprBP depletion whereas Smad2 phosphorylation was inhibited in these cells when VprBP was ectopically expressed (Figure 2F and G). Ectopic expression of VprBP decreased the TGF-β-induced expression of CTGF, IL-11, and pTHrP, genes associated with breast cancer aggressiveness in MDA-MB-231 cells (Supplementary Figure S1E). Moreover, VprBP strongly inhibited the TGF-β-induced migration of MDA-MB-231 cells (Figure 2H and I). Collectively, these data show that VprBP acts as suppressor of TGF-β/Smad signal transduction and target gene activation.
VprBP promotes Smad7–Smurf1–TβRI complex formation and degradation of TβRI
As an E3 ligase adaptor for substrate recognition, VprBP might block TGF-β/Smad signaling by promoting degradation of critical pathway components via its association to Smad7 and Smurf1. We therefore examined the effect of VprBP on the ubiquitination of Smad7, various other Smad members, and TβRI. For this, stable HA-tagged ubiquitin-expressing HEK293T cells were co-transfected with Myc-tagged VprBP and Flag-tagged Smad expression constructs, treated with the proteasome inhibitor MG132, precipitated with anti-Flag M2 affinity gel, and immunoblotted. Under these conditions, we did not observe any alterations in the ubiquitination of Smad1, Smad2, Smad3, Smad5, and Smad7 (Supplementary Figure S2A–C). However, poly-ubiquitination of TβRI (Activin receptor-like kinase 5, ALK5 was increased upon co-expression of VprBP and addition of MG132 in HEK293T cells (Figure 3A). Moreover, we observed increased protein levels of TβRI in VprBP-depleted HaCaT cells (Figure 3B), whereas the total protein levels of Smad2 and Smad4 were not affected (Figure 2B and C). To confirm that VprBP affects TGF-β signaling via TβRI, we analysed the levels of biotin-labeled TβRI at the cell membrane. As shown in Figure 3C, silencing endogenous VprBP in HaCaT cells increased the amount of cell-surface TβRI and delayed TGF-β-linduced TβRI turnover. VprBP had no impact on the mRNA levels of the analysed TGF-β components (Supplementary Figure S2D). Together, these results indicate that VprBP promotes the degradation of TβRI and not of Smads.
Figure 3.

VprBP promotes Smurf1–Smad7 complex formation and degradation of TβRI. (A) Stable HA-ubiquitin expressing HEK293T cells were transfected with TβRI-Flag and/or Myc-VprBP constructs, treated with the proteasome inhibitor MG132 (5 μM) for 5 h, immunoprecipitated with Flag antibodies, and analysed by immunoblotting for HA, Flag, and Myc. (B) The effect of VprBP depletion on TβRI protein level. (C) Immunoblotting and quantification of biotinylated TβRI in control or VprBP knockdown HaCaT cells treated with TGF-β (2.5 ng/ml) for the indicated time. Mean ± SD, 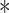
 P < 0.001. (D) Co-immunoprecipitation (co-IP) of ectopically expressed Flag-Smad7 and endogenous Smurf1 in control or VprBP-depleted HEK293T cells treated for 5 h with MG132 (5 μM). Co-IP and input samples were analysed by immunoblotting. (E) Co-IP of endogenous TβRI and ectopically expressed Flag-Smurf1 in control or VprBP-depleted HEK293T cells after 5 h MG132 (5 μM) treatment.
P < 0.001. (D) Co-immunoprecipitation (co-IP) of ectopically expressed Flag-Smad7 and endogenous Smurf1 in control or VprBP-depleted HEK293T cells treated for 5 h with MG132 (5 μM). Co-IP and input samples were analysed by immunoblotting. (E) Co-IP of endogenous TβRI and ectopically expressed Flag-Smurf1 in control or VprBP-depleted HEK293T cells after 5 h MG132 (5 μM) treatment.
Interestingly, we originally had detected direct interaction of VprBP with Smad7 and Smurf1, which cooperate in poly-ubiquitination and degradation of type I receptors (Ebisawa et al., 2001), but no interaction between VprBP and TβRI itself (Figure 1). This suggested that VprBP might induce degradation of TβRI by increasing the binding of Smurf1 to Smad7. We therefore examined the effect of VprBP on Smurf1–Smad7 complex formation. HEK293T cells transfected with Flag-tagged Smad7 were treated with MG132 and analysed for co-immunoprecipitation of endogenous Smurf1 protein. As shown in Figure 3D, the interaction between Smad7 and Smurf1 decreased when VprBP was depleted. In addition, the interaction between endogenous TβRI and ectopically expressed Flag-tagged Smurf1 was strongly reduced in MG132-treated HEK293T cells upon VprBP knockdown (Figure 3E). In contrast, ectopic expression of VprBP increased Smurf1–TβRI complex formation (Supplementary Figure S2E). Depletion of VprBP did not interfere with Smurf2–Smad7 complex formation (Supplementary Figure S2F). Together, these data indicate that VprBP mediates TβRI degradation by promoting Smurf1–Smad7–TβRI complex formation.
VprBP inhibits Smurf1 poly-ubiquitination
We next focused on the mechanism by which VprBP regulates Smad7–Smurf1 complex formation. Interestingly, knockdown of VprBP in HaCaT cells significantly decreased the total levels of endogenous Smurf1, but not of Smurf2 (Figure 4A). In contrast to the defect on Smurf1 protein expression, VprBP had no impact on both Smurf1 and Smurf2 mRNA level (Figure 4B). This indicates that VprBP might control the stabilization of Smurf1. To get more clues, we studied the interaction between VprBP and Smurf1 in more detail. Domain analysis revealed that the N-terminal part of VprBP (including the Armadillo-like domain) could bind to Smurf1, suggesting that VprBP may be involved in Smurf1 substrate selection and ubiquitination (Supplementary Figure S3A). We therefore examined the ubiquitination of ectopically expressed WT and catalytically inactive (C699A) Smurf1 in the absence and presence of VprBP overexpression. VprBP strongly decreased ubiquitination of WT Smurf1, but had no significant effect on the E3 ligase-inactive C699A mutant in MG132-treated HEK293T cells (Figure 4C). However, Smurf2 poly-ubiquitination was not affected by VprBP (Supplementary Figure S3B). To confirm that VprBP is specifically targeting Smurf1, we also checked the impact of VprBP on Smurf2 auto-inhibition, by analysing the Smurf2 HECT-C2 domain interaction (Wiesner et al., 2007). Indeed, the interaction between Smurf2 and its HECT domain was unchanged upon VprBP overexpression (Supplementary Figure S3C). To further verify the effect of VprBP on Smurf1 poly-ubiquitination, we analysed the ubiquitination of Smurf1 in VprBP-depleted HEK293T cells. In line with the previous experiments, the poly-ubiquitination of Smurf1 in MG132-treated cells was enhanced when VprBP was absent (Figure 4D). Similar results were obtained when we analysed the poly-ubiquitination of Smurf1 by His-ubiquitin nickel pull-down assays (Figure 4E). Ubiquitination of endogenous Smurf1 was also remarkably interrupted by VprBP overexpression in HEK293T cells (Figure 4F). Finally, in a pulse chase assay using the protein synthesis inhibitor cycloheximide, VprBP knockdown significantly accelerated Smurf1 protein degradation (Figure 4G).
Figure 4.
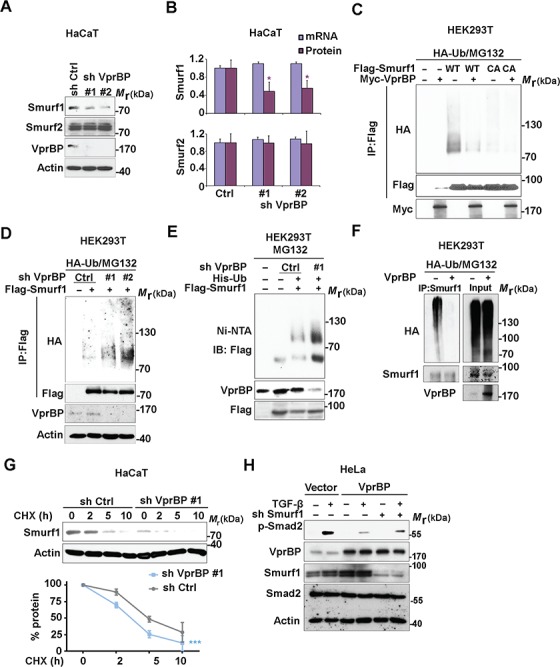
VprBP decreases Smurf1 poly-ubiquitination. (A) Effect of VprBP knockdown on Smurf1 and Smurf2 protein expression in HaCaT cells, examined by immunoblotting with the indicated antibodies. Actin served as control for protein loading. (B) Comparison of Smurf1/2 mRNA and protein expression in control and VprBP knockdown HaCaT cells. Mean ± SD,  P < 0.05. (C) Stable HA-ubiquitin-expressing HEK293T cells were co-transfected with Flag-tagged Smurf1 (WT or C699A mutant) and Myc-tagged VprBP and treated with the proteasome inhibitor MG132 (5 μM) for 5 h. Cell lysates were immunoprecipitated with Flag antibody followed by immunoblotting with HA and Flag antibodies. Input samples were analysed with Myc antibody. (D) Effect of VprBP knockdown on Smurf1 ubiquitination, examined by immunoblotting of input lysates and immunoprecipitates from HA-ubiquitin-expressing HEK293T cells with or without VprBP depletion, transfected with Flag-Smurf1, and treated with MG132 (5 μM). (E) HEK293T cells were infected with lentiviruses expressing VprBP shRNA, His-ubiquitin (His-ub), and/or control virus, transfected with Flag-Smurf1 plasmid, treated with MG132 (5 μM) for 5 h, and lysed for Ni-NTA pull down assay followed by immunoblotting. (F) VprBP controls endogenous Smurf1 ubiquitination. Stable HA-ubiquitin-expressing HEK293T cells with or without VprBP depletion were treated with MG132 (5 μM) for 5 h and lysed for ubiquitination assay followed by immunoblotting. (G) Immunoblotting and quantification of Smurf1 expression in control or VprBP knockdown HaCaT cells treated with CHX (20 μg/ml) for the indicated time. Mean ± SD,
P < 0.05. (C) Stable HA-ubiquitin-expressing HEK293T cells were co-transfected with Flag-tagged Smurf1 (WT or C699A mutant) and Myc-tagged VprBP and treated with the proteasome inhibitor MG132 (5 μM) for 5 h. Cell lysates were immunoprecipitated with Flag antibody followed by immunoblotting with HA and Flag antibodies. Input samples were analysed with Myc antibody. (D) Effect of VprBP knockdown on Smurf1 ubiquitination, examined by immunoblotting of input lysates and immunoprecipitates from HA-ubiquitin-expressing HEK293T cells with or without VprBP depletion, transfected with Flag-Smurf1, and treated with MG132 (5 μM). (E) HEK293T cells were infected with lentiviruses expressing VprBP shRNA, His-ubiquitin (His-ub), and/or control virus, transfected with Flag-Smurf1 plasmid, treated with MG132 (5 μM) for 5 h, and lysed for Ni-NTA pull down assay followed by immunoblotting. (F) VprBP controls endogenous Smurf1 ubiquitination. Stable HA-ubiquitin-expressing HEK293T cells with or without VprBP depletion were treated with MG132 (5 μM) for 5 h and lysed for ubiquitination assay followed by immunoblotting. (G) Immunoblotting and quantification of Smurf1 expression in control or VprBP knockdown HaCaT cells treated with CHX (20 μg/ml) for the indicated time. Mean ± SD, 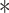
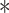
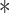 P < 0.001. (H) Immunoblot detection of p-Smad2, VprBP, Smurf1, Smad2, and actin protein levels in HeLa cells infected with lentivirus plasmids as indicated.
P < 0.001. (H) Immunoblot detection of p-Smad2, VprBP, Smurf1, Smad2, and actin protein levels in HeLa cells infected with lentivirus plasmids as indicated.
To confirm that Smurf1 is required for the inhibition ofTGF-β/Smad signaling by VprBP, we overexpressed VprBP in Smurf1-depleted HeLa cells. In line with the previous results, VprBP overexpression enhanced endogenous Smurf1 expression and decreased TGF-β-induced Smad2 phosphorylation, whereas Smurf1 knockdown partly restored the Smad2 activation (Figure 4H). Together, these results show that VprBP can protect Smurf1 against poly-ubiquitination and degradation, and thereby enhances the inhibitory function of Smurf1 on TGF-β signaling.
VprBP stabilizes Smurf1 by modification of lysine 667 and serine 672
Our next aim was to identify post-translational modification(s) in Smurf1 that are regulated by VprBP, including the poly-ubiquitination attachment site. For this, we overexpressed Flag-tagged Smurf1 with or without Myc-tagged VprBP in HEK293T cells, followed by Flag antibody immunoprecipitation and mass spectrometry (MS) analysis. When compared with overexpressed Smurf1 alone, VprBP co-expression reduced Smurf1 ubiquitination at lysine 667 and Smurf1 phosphorylation at serine 672. Both sites are located in the Smurf1 HECT domain and highly conserved between species (Supplementary Table S2; Figure 5A). Consistent with the MS analysis, we observed that the poly-ubiquitination of the Smurf1 HECT domain was strongly decreased by VprBP co-expression (Figure 5B).
Figure 5.

Lysine 667 and serine 672 of Smurf1 are targeted by VprBP. (A) Top, schematic diagram of Smurf1 ubiquitination on lysine 667 and phosphorylation on serine 672. Bottom, alignment of K667 and S672 across species. (B) Stable HA-ubiquitin-expressing HEK293T cells transfected with control vector or Flag-tagged Smurf1 deletions (C2, WW, HECT) and with or without Myc-VprBP were treated with MG132 (5 μM) for 5 h and lysed for the ubiquitination assay followed by immunoblotting. (C) Stable HA-ubiquitin expressing HEK293T cells transfected with control vectors and Smurf1 WT or mutants together with or without Myc-VprBP were treated with MG132 (5 μM) for 5 h and lysed for ubiquitination assay followed by immunoblotting. (D) Validation of TGF-β-induced CAGA transcriptional response in HEK293T cells infected with control vector or Smurf1 mutants. Mean ± SD, 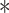 P < 0.05,
P < 0.05, 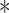
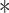 P < 0.01. (E) Immunoblotting of phosphorylated Smad2, total Smad2, Smurf1, and actin in HaCaT cells infected with control vector or Smurf1 mutants. (F) Immunoblotting of biotinylated cell surface TβRI, phosphorylated Smad2, total Smad2, Flag, and actin in COS7 cells transfected with empty vector (Vector), Flag-tagged Smurf1-WT, K667R, or S672A plasmids and treated with TGF-β (5 ng/ml) for the indicated time.
P < 0.01. (E) Immunoblotting of phosphorylated Smad2, total Smad2, Smurf1, and actin in HaCaT cells infected with control vector or Smurf1 mutants. (F) Immunoblotting of biotinylated cell surface TβRI, phosphorylated Smad2, total Smad2, Flag, and actin in COS7 cells transfected with empty vector (Vector), Flag-tagged Smurf1-WT, K667R, or S672A plasmids and treated with TGF-β (5 ng/ml) for the indicated time.
To further validate the role of these modified Smurf1-HECT residues, we constructed and analysed K667R and S672A Smurf1 substitution mutants. Whereas VprBP strongly inhibited poly-ubiquitination of WT Smurf1, poly-ubiquitination of both the K667R and S672A mutants were severely compromised (Figure 5C). To investigate the effects of these Smurf1 mutations on TGF-β signal transduction, we established stable cell lines expressing either WT Smurf1 or the Smurf1 mutants. Compared with WT Smurf1, both the K667R and S672A mutants showed a stronger inhibition of CAGA-luciferase reporter activity in HEK293T cells (Figure 5D). Consistently, the protein levels of the K667R and S672A mutants were somewhat increased in HaCaT cells, and they had a stronger inhibitory effect onTGF-β-induced Smad2 phosphorylation compared to ectopically expressed WT Smurf1 (Figure 5E). Moreover, Smurf1 K667R and S672A mutants showed stronger capacity than Smurf1-WT to induce the turnover of TβRI at the cell surface in COS7 cells (Figure 5F). These findings suggest that VprBP inhibits Smurf1 poly-ubiquitination at lysine 667 directly and/or indirectly by interfering with the phosphorylation at serine 672.
VprBP antagonizes Activin signaling in mouse embryonic stem cells
Activin and Nodal regulate differentiation of embryonic stem cells (ESCs) by activating Smad2, whereas Smad7 exerts inhibitory feedback on this process (Ogawa et al., 2007; Fei et al., 2010). Interestingly, VprBP overexpression decreased Activin-induced activation of the Activin response element (ARE)-Luc transcriptional reporter in HepG2 cells (Figure 6A). Moreover, depletion of endogenous VprBP in HepG2 cells by shRNA promoted Activin-induced Smad2 phosphorylation and downstream target expression (Figure 6B; Supplementary Figure S4A). In addition, we found that in MG132-treated HEK293T cells, poly-ubiquitination of the Activin/Nodal type I receptor ALK4 to be enhanced upon co-expression of VprBP (Figure 6C). These data suggest that the function of VprBP is conserved between the TGF-β and Activin/Nodal pathways.
Figure 6.

The function of VprBP in Activin signaling and Activin-mediated mESCs differentiation. (A) Ectopic expression of VprBP suppresses ARE-Luc Activin reporter activity in HepG2 cells with or without 25 ng/ml Activin A treatment for 16 h. Mean ± SD, 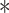
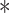
 P < 0.001. (B) Detection of VprBP knockdown in HepG2 cells on Activin (25 ng/ml)-induced Smad2 phosphorylation, Smad2, VprBP, and actin, measured by immunoblotting. (C) Stable HA-ubiquitin-expressing HEK293T cells were transfected with control vector, ALK4-Flag, or Myc-VprBP, treated with MG132 (5 M) for 5 h, and lysed for ubiquitination assay followed by immunoblotting. (D) mESCs were transfected with control or VprBP siRNA, treated with or without 25 ng/ml Activin A for 2 h, and followed by immunoblotting. (E) Immunoblotting of protein expression in VprBP knockdown mESCs. (F) qPCR analysis of Lefty1, Lefty2, Nanog, and Oct4 in control and VprBP knockdown mESCs. Normalized to GAPDH expression, mean ± SD of triplicates. (G) Schematic representation of mESCs differentiation assay. (H) Detection of VprBP depletion on Activin A (25 ng/ml)-induced mesendoderm markers by qPCR analysis. Normalized to GAPDH expression, mean ± SD of triplicates. (I) Detection of VprBP depletion on Activin-inhibited neural markers by qPCR analysis. Normalized to GAPDH expression, mean ± SD of triplicates. (J) Scatter plots of differential gene expression (compare with control) displayed positive correlation between VprBP depletion and Activin A response in mESCs. si VprBP (top) or si VprBP + Activin A (bottom) vs. si Ctrl + Activin A (25 ng/ml) after 4 days of mESCs differentiation. Gray dots indicate mRNA expressions not changed. (K) Venn diagram analysis of genes significantly altered upon 25 ng/ml Activin A treatment in both control and VprBP knockdown mESCs. (L) Gene expression profile shows that VprBP depletion in mESCs displayed stronger response to Activin A treatment. Differentially expressed genes were presented as heatmap in the indicated groups.
P < 0.001. (B) Detection of VprBP knockdown in HepG2 cells on Activin (25 ng/ml)-induced Smad2 phosphorylation, Smad2, VprBP, and actin, measured by immunoblotting. (C) Stable HA-ubiquitin-expressing HEK293T cells were transfected with control vector, ALK4-Flag, or Myc-VprBP, treated with MG132 (5 M) for 5 h, and lysed for ubiquitination assay followed by immunoblotting. (D) mESCs were transfected with control or VprBP siRNA, treated with or without 25 ng/ml Activin A for 2 h, and followed by immunoblotting. (E) Immunoblotting of protein expression in VprBP knockdown mESCs. (F) qPCR analysis of Lefty1, Lefty2, Nanog, and Oct4 in control and VprBP knockdown mESCs. Normalized to GAPDH expression, mean ± SD of triplicates. (G) Schematic representation of mESCs differentiation assay. (H) Detection of VprBP depletion on Activin A (25 ng/ml)-induced mesendoderm markers by qPCR analysis. Normalized to GAPDH expression, mean ± SD of triplicates. (I) Detection of VprBP depletion on Activin-inhibited neural markers by qPCR analysis. Normalized to GAPDH expression, mean ± SD of triplicates. (J) Scatter plots of differential gene expression (compare with control) displayed positive correlation between VprBP depletion and Activin A response in mESCs. si VprBP (top) or si VprBP + Activin A (bottom) vs. si Ctrl + Activin A (25 ng/ml) after 4 days of mESCs differentiation. Gray dots indicate mRNA expressions not changed. (K) Venn diagram analysis of genes significantly altered upon 25 ng/ml Activin A treatment in both control and VprBP knockdown mESCs. (L) Gene expression profile shows that VprBP depletion in mESCs displayed stronger response to Activin A treatment. Differentially expressed genes were presented as heatmap in the indicated groups.
In line with the effects of VprBP on TGF-β signaling, in undifferentiated mouse (m) ESCs, small interference RNA (siRNA)-mediated knockdown of VprBP enhanced Activin-induced Smad2 activation and decreased Smurf1 protein expression (Figure 6D and E). In addition, the mRNA levels of the Smad2 target genes lefty1 and lefty2 were increased upon VprBP depletion, whereas the stem cell self-renewal markers Nanog and Oct4 remained unchanged (Figure 6F).
To investigate whether VprBP is critical for Activin-mediated mESCs differentiation, we examined the effect of its mis-expression in the stem cell differentiation assay (Figure 6G; Ying and Smith, 2003). After 3 days, VprBP depletion dramatically increased the expression of Activin-induced mesendoderm markers such as Gsc, Lhx-1, Foxa2, and T (Figure 6H), whereas after 6 days, the VprBP-depleted mESCs displayed a stronger suppression of the neuronal markers Pax6 and Sox3 (Figure 6I). shRNA-mediated VprBP knockdown alone caused similar effects on mESC differentiation markers (Supplementary Figure S4B and C).
To further address the importance of VprBP on Activin-dependent transcriptional responses in mESCs, we analysed the genome-wide expression profiles of control and VprBP knockdown mESCs with or without Activin treatment or TGF-β/Activin type I receptor kinase inhibitor SB431542 challenge for 4 days (Supplementary Table S3). As shown in Figure 6J, VprBP knockdown positively correlated with the Activin-induced response regarding differentially expressed genes in comparison to control mESCs. Of note, 775 genes were found significantly changed in both Activin A-treated control (si Ctrl + Activin) and VprBP knockdown mESCs (si VprBP + Activin A) (Figure 6K). The heatmap also demonstrates that a large population of Activin-dependent genes with significant differential expression were more enhanced or repressed by VprBP knockdown mESCs plus Activin treatment (si VprBP + Activin), which were oppositely regulated by the SB431542 treatment (Figure 6L). These findings demonstrate that VprBP modulates mESCs differentiation by restricting Activin-induced mesendodermal marker gene expression and is a critical determinant in the Activin-dependent transcriptional output.
VprBP expression negatively correlates with genes associated with Activin/Nodal activation during embryogenesis
The negative role of VprBP on Activin-mediated mESCs differentiation brought us to evaluate its function during mesoderm induction in vivo. Based on a published 3D spatial transcriptome data set of the mouse embryo (GSE65924), we first analysed the gene expression profile of the mid-streak stage (E7.0) (Peng et al., 2016). By analysing a 3D signal and fate map of mouse embryo from this data set, VprBP mRNA expression is highly expressed in anterior epiblast and lower expressed in the posterior epiblast, which is opposite to the expression of genes that are associated with Nodal signal activation and development fate of mesoderm progenitors (Figure 7A). In addition, we observed significant negative correlations between expression of VprBP and the expression of mesoderm marker genes such as T, Snail, Twist, and Flk1 (Figure 7B). Moreover, we noted positive correlations between the VprBP mRNA and neural markers like Sox2, Sox3, Pou3f1, and Tubb3 in the same data set (Figure 7B). Thus, VprBP is potentially involved in mesoderm induction during mouse embryogenesis.
Figure 7.
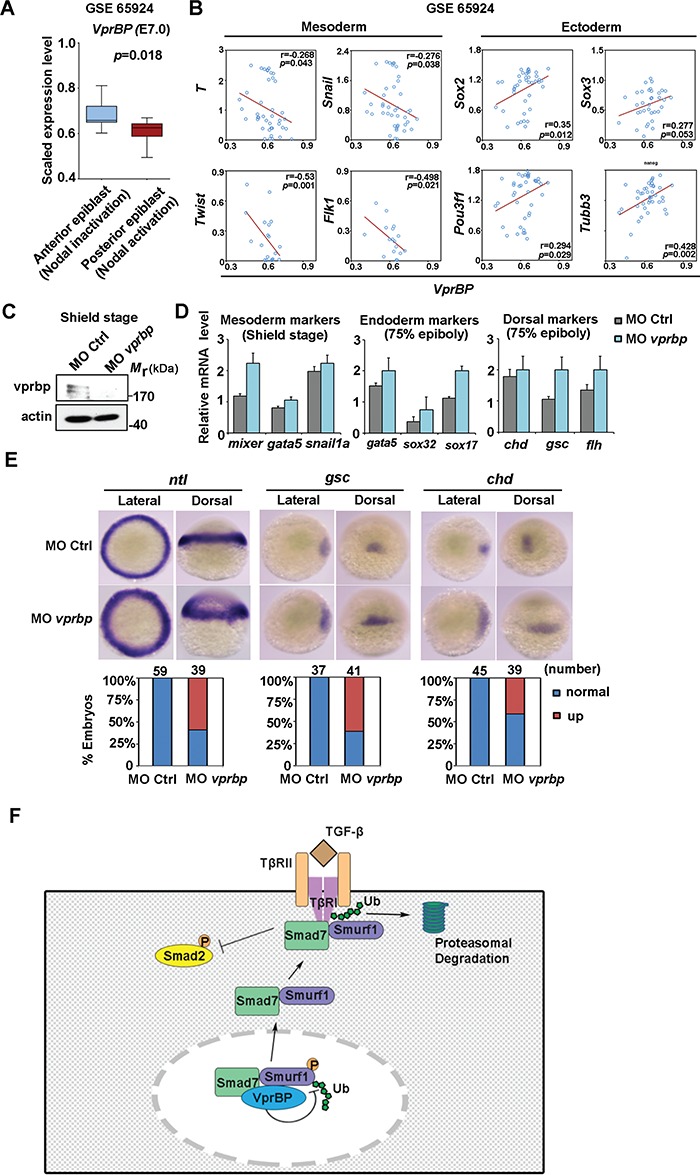
VprBP controls mesoderm induction during embryogenesis. (A) mRNA expression plot of VprBP mRNA of anterior and posterior epiblast at mouse mid-gastrulation (E7.0) in published data set (GSE 65924). (B) Correlations between VprBP mRNA expression and that of the indicated mesoderm or ectoderm markers in the same data set. (C) Immunoblotting of vprbp and actin in control zebrafish embryos and vprbp morphants at shield stage. (D) qPCR analysis of mRNA expression of mesoderm, endoderm, and dorsal markers as indicated in control embryos and vprbp morphants. (E) Expression patterns of developmental markers ntl, gsc, and chd in control embryos and vprbp morphants. Representative embryos are shown for each marker from both lateral and dorsal view, with the number and percentage of affected embryos shown at the bottom. (F) Schematic working model of VprBP action in controlling TGF-β receptor signaling.
VprBP inhibits mesoderm induction during zebrafish embryogenesis
To determine the regulation of VprBP on mid-gastrulation, we examined the vprbp expression in zebrafish embryos. Cloning of zebrafish vprbp complementary DNA (cDNA) revealed that the encoded protein displays 73% identity to the human protein (Supplementary Figure S5). The region that was used to generate antibody against human Vprbp is similar to that in zebrafish (Supplementary Figure S5). This antibody cross-reacts with zebrafish vprbp as a protein band with the expected molecular weight that is decreased upon morpholino-mediated vprbp knockdown at the shield stage (Figure 7C). Consistent with the negative correlation of VprBP to the mesoderm markers in mouse embryo, the expression of zebrafish mesoderm markers mixer, gata5, and snail1a were upregulated in vprbp morphants at shield stage; similarly, vprbp knockdown enhanced mRNA expression of endodermal markers including gata5, sox32, and sox17, as well as dorsal markers chd, gsc, and flh at 75% epiboly (Figure 7D). We then assessed the expression pattern of certain developmental markers by in situ hybridization. Compared with embryos injected with control morpholino (MO Ctrl), vprbp morphants displayed an expansion of the expression of mesoderm marker ntl and dorsal markers gsc and chd (Figure 7E). These results are consistent with the notion that vprbp limits mesoderm induction and dorsal development during early zebrafish embryonic development.
Discussion
In this study, we identified VprBP as a Smad7 and Smurf1 binding partner that negatively modulates TGF-β/Smad signaling by promoting Smad7–Smurf1–TβRI complex formation and inducing proteasomal degradation of TβRI. This effect of VprBP appears to be mediated, at least in part, via interference with Smurf1 poly-ubiquitination and degradation, and results in reduced TGF-β-mediated Smad2 phosphorylation and TGF-β/Smad target gene expression (Figure 7F). Furthermore, VprBP restricts Activin/Nodal-mediated mESCs differentiation and is a critical determinant for controlling mesoderm induction during embryogenesis.
Interestingly, Itch and other known Smad7 binding proteins were also identified in our TAP-Smad7 pull-down. However, in contrast to VprBP, Itch has been identified as a positive regulator of the TGF-β pathway that can increase Smad2 activation and induce EMT by enhancing Smad7 ubiquitination (Bai et al., 2004). Since VprBP has a negative effect on TGF-β/SMAD signaling, did not affect Smad7 ubiquitination (Supplementary Figure S2A), and did not interact with Itch (Supplementary Figure S1A), VprBP is unlikely to regulateTGF-β/Smad signaling via Itch.
Although various regulators of TGF-β signal transduction have been identified, it remains relatively unknown how these signaling regulators are controlled themselves. The activity state of the TGF-β cascade is diminished when Smad7 translocates to the plasma membrane with the help of Smurf1 (Itoh et al., 1998; Suzuki et al., 2002). We demonstrate here that VprBP can inhibit poly-ubiquitination of Smurf1, resulting in increased Smurf1 protein levels and enhanced Smad7–Smurf1 complex formation. Previous studies showed that the Smurf1 E3 ligase is regulated by multiple proteins, including kinases and other enzymes, and that specific conditions can control its activation and substrate specificity. For example, the E3 ligase complex SCFFBXL15 mediates poly-ubiquitination and degradation of Smurf1 and selectively enhances BMP signaling (Cui et al., 2011). Conversely, the deubiquitinase FAM/USP9X inhibits Smurf1 poly-ubiquitination and stabilizes its E3 ligase activity (Xie et al., 2013). Moreover, Smurf1 can be phosphorylated by protein kinase A (PKA) at Thr306, resulting in the stabilization of polarity protein Par6 and increased ubiquitination of RhoA (Cheng et al., 2011). Furthermore, neddylation at Cys426 in the Smurf1 HECT domain has been observed (Xie et al., 2014). Smurf1 physically forms a complex with Nedd8 and the E2 ligase Ubc12, catalyzing neddylation on itself and stimulation of E3 activity. However, the mechanism by which Smurf1 mediates substrate selection is unknown. Our study indicates that VprBP enables Smurf1-dependent degradation of the TGF-β type I receptor and points to a regulatory role of VprBP in Smurf1 poly-ubiquitination.
In epithelial cells, TGF-β can induce EMT and growth inhibition (Massague, 2012). VprBP strongly limited TGF-β-mediated EMT signatures and migration. We previously reported that TRAF4 promotes TGF-β signaling by inducing poly-ubiquitination of Smurf2, suggesting that ubiquitination of Smurfs is critical for TGF-β signal activation (Zhang et al., 2013a). Unlike Smurf2, an E3 ligase specific for the TGF-β pathway, Smurf1 also mitigates BMP signaling (Zhu et al., 1999). Here, we highlight that VprBP targets TGF-β, while having no impact on the BMP/Smad-induced transcriptional response. Notably, VprBP mediated stabilization of Smurf1, but not of Smurf2, and prevented TGF-β–Smad signaling and functionally inhibited TGF-β target genes.
Recent studies have shown that CRL4VprBP could activate Hippo–Yap signaling by inducing proteasomal degradation of Lats1 and Lats2 (Li et al., 2014). This finding shows that VprBP also contributes to regulation of cellular signaling depending on the Merlin–CRL4 context. However, the CRL4-independent role of VprBP in cell signaling is still unclear. Our data show that Smurf1 interacts with the N-terminal Armadillo-like domain of VprBP, which are different from the DBB1 or Merlin-binding motifs on VprBP (Li et al., 2010b; Gerard et al., 2014). Additionally, VprBP can associate with the scaffold protein DYRK2 and the HECT E3 ligase EDD/UBR5(Maddika and Chen, 2009). An open question is whether VprBP interacts with HECT-type E3 ligases. In this study, we did not observe interactions between Smurf1 and DYRK2 or EDD/UBR5 (data not shown). By MS analysis, we observed that VprBP controls site-specific ubiquitination of Smurf1 directly and/or indirectly by inhibiting phosphorylation in the HECT domain of Smurf1. The underlying mechanism by which VprBP interferes with this phosphorylation remains to be elucidated. Smurf1 regulation by other E3 ligases, kinases, and phosphatases may participate in this context.
By analysing Activin-dependent transcriptional output in mESCs, we demonstrated that VprBP inhibition is associated with enhanced Activin signaling. In addition, high VprBP expression possibly associated to Activin/Nodal signal suppression and neural commitment in early stage of mouse embryonic development. Moreover, vprbp expression is critical to control mesoderm induction at gastrulation in the zebrafish embryos. Our data indicate that dysregulation of VprBP contributes to the fine tuning of TGF-β/Activin/Nodal controlled mesoderm induction.
Materials and methods
Tandem affinity purification
TAP assay was performed as previous described (Zhang et al., 2013b). Briefly, I-Smads and Smurfs were N- and C-terminally TAP-tagged and used for affinity purification. Cells were harvested and immunoprecipitated by TAP. Then purified protein complexes were separated by one-dimensional SDS–PAGE and stained by colloidal Coomassie Brilliant Blue. Gel lanes were cut into slices across the full separation range and subjected to in-gel tryptic digestion, peptide extracts were analysed by liquid chromatography-tandem MS using an Eksigent 1D+ HPLC system coupled to an LTQ Orbitrap mass spectrometer (Thermo Scientific). Peptide mass and fragmentation data were used to query an in-house curated version of the International Protein Index (IPI) database using Mascot (Matrix Science).
Candidate lists were generated as follows. For this paper, we focus on Smad7 interactors. Proteins that were observed more frequently than expected are shown in Supplementary Table S1. Non-ribosomal proteins (ribosomal proteins are also found in many other ‘bait proteins’ interactome) with P-values ≤ 0.001 are represented as a graph in Figure 1A. All IPI identifiers were mapped to Entrez Gene symbols prior to the Fisher Exact calculation.
Cell culture and reagents
The HEK293T, HaCaT, and MDA-MB-231 cells were cultured in DMEM high glucose containing L-glutamine, 10% FBS, and 100 U/ml Pen/Strep (GIBCO, Invitrogen). All the cell lines were maintained at 37°C with 5% CO2. The HEK293T and HaCaT cell lines were obtained from the American Tissue Culture Collection (ATCC) and MDA-MB-231 cell line was previously described (Deckers et al., 2006).
Nickel pull-down
The HEK293T cells were lysed by buffer containing 8 M urea, 0.1 M Na2HPO4, 0.1 M NaH2PO4, 10 mM Tris, 10 mM imidazole, and β-mercaptoethanol for 10 min. The lysate supernatants were obtained by a 10 min centrifugation (4000 rpm) and incubated with Ni-agarose for 2 h. Then, bound proteins were eluted with sample buffer at 100°C for 5 min and analysed by western blotting.
In vivo ubiquitination assay
The HEK293T cells were lysed using 140 μl pellet buffer with 1% SDS, 10 mM N-Ethylmaleimide (NEM), and protease inhibitors. After sonication, lysates were boiled at 100°C for 5 min and centrifuged at 12000 rpm for 15 min. The supernatant was diluted using RIPA buffer and incubated with specific antibodies and protein A-Sepharose for 3 h at 4°C. After three RIPA buffer washings, the immunoprecipitated proteins were eluted with sample buffer at 100°C for 5 min and analysed by western blotting.
Statistical analysis
Statistical analyses were conducted using GraphPad (Prism 4). Results are expressed as mean ± standard deviation (SD). Student’s t-test was used for statistically significant. Analysis of variance (ANOVA) was performed to analyse the quantification curves of protein degradation and turnover. The correlation between genes expression were analysed by Pearson’s coefficient tests.
Supplementary Material
Acknowledgements
We thank Midory Thorikay and Martijn Rabelink for excellent technical assistance and all other members in ten Dijke’s laboratory for their kind help. We thank Drs Susana M. Chuva de Sousa Lopes and Qiang Wang for discussion. We thank Drs Éric Cohen, Hongrui Wang, and Jun Huang for constructs.
Funding
This research was supported by Cancer Genomics Centre Netherlands and a grant from the National Natural Science Foundation of China (31471315).
Conflict of interest: A.B. is employed by Novartis Institutes for Biomedical Research. All other authors do not have any competing interests.
Author contributions: Y.L. designed and performed experiments, analysed and interpreted data, and wrote the manuscript; C.C. performed zebrafish experiments and data analysis. F.X. and A.B. contributed to MS experiments and data analysis. H.M. and S.K. assisted for bioinformatics analysis; M.v.D. assisted for gene cloning; H.v.D. and L.Z. contributed in writing, review, and revision of the manuscript. P.t.D. directed the research, interpreted the data, and wrote the manuscript.
References
- Akhurst R.J., and Hata A. (2012). Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 11, 790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Salihi M.A., Herhaus L., Macartney T., et al. (2012). USP11 augments TGFβ signalling by deubiquitylating ALK5. Open Biol. 2, 120063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y., Yang C., Hu K., et al. (2004). Itch E3 ligase-mediated regulation of TGF-β signaling by modulating Smad2 phosphorylation. Mol. Cell 15, 825–831. [DOI] [PubMed] [Google Scholar]
- Cheng P.L., Lu H., Shelly M., et al. (2011). Phosphorylation of E3 ligase Smurf1 switches its substrate preference in support of axon development. Neuron 69, 231–243. [DOI] [PubMed] [Google Scholar]
- Cui Y., He S., Xing C., et al. (2011). SCFFBXL15 regulates BMP signalling by directing the degradation of HECT-type ubiquitin ligase Smurf1. EMBO J. 30, 2675–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckers M., van Dinther M., Buijs J., et al. (2006). The tumor suppressor Smad4 is required for transforming growth factor β-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res. 66, 2202–2209. [DOI] [PubMed] [Google Scholar]
- Ebisawa T., Fukuchi M., Murakami G., et al. (2001). Smurf1 interacts with transforming growth factor-β type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 276, 12477–12480. [DOI] [PubMed] [Google Scholar]
- Fei T., Zhu S., Xia K., et al. (2010). Smad2 mediates Activin/Nodal signaling in mesendoderm differentiation of mouse embryonic stem cells. Cell Res. 20, 1306–1318. [DOI] [PubMed] [Google Scholar]
- Feng X.H., and Derynck R. (2005). Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 21, 659–693. [DOI] [PubMed] [Google Scholar]
- Gerard F.C., Yang R., Romani B., et al. (2014). Defining the interactions and role of DCAF1/VPRBP in the DDB1-cullin4A E3 ubiquitin ligase complex engaged by HIV-1 Vpr to induce a G2 cell cycle arrest. PLoS One 9, e89195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh S., Landstrom M., Hermansson A., et al. (1998). Transforming growth factor β1 induces nuclear export of inhibitory Smad7. J. Biol. Chem. 273, 29195–29201. [DOI] [PubMed] [Google Scholar]
- Itoh S., and ten Dijke P. (2007). Negative regulation of TGF-β receptor/Smad signal transduction. Curr. Opin. Cell Biol. 19, 176–184. [DOI] [PubMed] [Google Scholar]
- Jin J., Arias E.E., Chen J., et al. (2006). A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 23, 709–721. [DOI] [PubMed] [Google Scholar]
- Kavsak P., Rasmussen R.K., Causing C.G., et al. (2000). Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGFβ receptor for degradation. Mol. Cell 6, 1365–1375. [DOI] [PubMed] [Google Scholar]
- Kim K., Kim J.M., Kim J.S., et al. (2013). VprBP has intrinsic kinase activity targeting histone H2A and represses gene transcription. Mol. Cell 52, 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S., Xu J., and Derynck R. (2014). Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Lu K., Wang J., et al. (2010a). Ubiquitin ligase Smurf1 targets TRAF family proteins for ubiquitination and degradation. Mol. Cell. Biochem. 338, 11–17. [DOI] [PubMed] [Google Scholar]
- Li W., Cooper J., Zhou L., et al. (2014). Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell 26, 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., You L., Cooper J., et al. (2010b). Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell 140, 477–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K., Li P., Zhang M., et al. (2011). Pivotal role of the C2 domain of the Smurf1 ubiquitin ligase in substrate selection. J. Biol. Chem. 286, 16861–16870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K., Yin X., Weng T., et al. (2008). Targeting WW domains linker of HECT-type ubiquitin ligase Smurf1 for activation by CKIP-1. Nat. Cell Biol. 10, 994–1002. [DOI] [PubMed] [Google Scholar]
- Maddika S., and Chen J. (2009). Protein kinase DYRK2 is a scaffold that facilitates assembly of an E3 ligase. Nat. Cell Biol. 11, 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. (2012). TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazono K. (2002). A new partner for inhibitory Smads. Cytokine Growth Factor Rev. 13, 7–9. [DOI] [PubMed] [Google Scholar]
- Moustakas A., and Heldin C.H. (2009). The regulation of TGFβ signal transduction. Development 136, 3699–3714. [DOI] [PubMed] [Google Scholar]
- Murakami G., Watabe T., Takaoka K., et al. (2003). Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol. Biol. Cell 14, 2809–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T., Mondal K., and Swanson P.C. (2013). VprBP (DCAF1): a promiscuous substrate recognition subunit that incorporates into both RING-family CRL4 and HECT-family EDD/UBR5 E3 ubiquitin ligases. BMC Mol. Biol. 14, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao A., Afrakhte M., Moren A., et al. (1997). Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature 389, 631–635. [DOI] [PubMed] [Google Scholar]
- Ogawa K., Saito A., Matsui H., et al. (2007). Activin–Nodal signaling is involved in propagation of mouse embryonic stem cells. J. Cell Sci. 120, 55–65. [DOI] [PubMed] [Google Scholar]
- Park S.H., Jung E.H., Kim G.Y., et al. (2015). Itch E3 ubiquitin ligase positively regulates TGF-β signaling to EMT via Smad7 ubiquitination. Mol. Cells 38, 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng G., Suo S., Chen J., et al. (2016). Spatial transcriptome for the molecular annotation of lineage fates and cell identity in mid-gastrula mouse embryo. Dev. Cell 36, 681–697. [DOI] [PubMed] [Google Scholar]
- Shen R., Chen M., Wang Y.J., et al. (2006). Smad6 interacts with Runx2 and mediates Smad ubiquitin regulatory factor 1-induced Runx2 degradation. J. Biol. Chem. 281, 3569–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soond S.M., and Chantry A. (2011). Selective targeting of activating and inhibitory Smads by distinct WWP2 ubiquitin ligase isoforms differentially modulates TGFβ signalling and EMT. Oncogene 30, 2451–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki C., Murakami G., Fukuchi M., et al. (2002). Smurf1 regulates the inhibitory activity of Smad7 by targeting Smad7 to the plasma membrane. J. Biol. Chem. 277, 39919–39925. [DOI] [PubMed] [Google Scholar]
- Wang H.R., Zhang Y., Ozdamar B., et al. (2003). Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science 302, 1775–1779. [DOI] [PubMed] [Google Scholar]
- Wiesner S., Ogunjimi A.A., Wang H.R., et al. (2007). Autoinhibition of the HECT-type ubiquitin ligase Smurf2 through its C2 domain. Cell 130, 651–662. [DOI] [PubMed] [Google Scholar]
- Xie P., Zhang M., He S., et al. (2014). The covalent modifier Nedd8 is critical for the activation of Smurf1 ubiquitin ligase in tumorigenesis. Nat. Commun. 5, 3733. [DOI] [PubMed] [Google Scholar]
- Xie Y., Avello M., Schirle M., et al. (2013). Deubiquitinase FAM/USP9X interacts with the E3 ubiquitin ligase SMURF1 protein and protects it from ligase activity-dependent self-degradation. J. Biol. Chem. 288, 2976–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M., Ying S.X., Zhang G.M., et al. (2005). Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell 121, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Q.L., and Smith A.G. (2003). Defined conditions for neural commitment and differentiation. Methods Enzymol. 365, 327–341. [DOI] [PubMed] [Google Scholar]
- Yu C., Zhang Y.L., Pan W.W., et al. (2013). CRL4 complex regulates mammalian oocyte survival and reprogramming by activation of TET proteins. Science 342, 1518–1521. [DOI] [PubMed] [Google Scholar]
- Zhang L., Zhou F., Garcia de Vinuesa A., et al. (2013a). TRAF4 promotes TGF-β receptor signaling and drives breast cancer metastasis. Mol. Cell 51, 559–572. [DOI] [PubMed] [Google Scholar]
- Zhang X., Zhang J., Bauer A., et al. (2013b). Fine-tuning BMP7 signalling in adipogenesis by UBE2O/E2-230K-mediated monoubiquitination of SMAD6. EMBO J. 32, 996–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Chang C., Gehling D.J., et al. (2001). Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc. Natl Acad. Sci. USA 98, 974–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.E. (2009). Non-Smad pathways in TGF-β signaling. Cell Res. 19, 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L., Huang J., Guo R., et al. (2010). Smurf1 inhibits mesenchymal stem cell proliferation and differentiation into osteoblasts through JunB degradation. J. Bone Miner. Res. 25, 1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H., Kavsak P., Abdollah S., et al. (1999). A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 400, 687–693. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.