

Abstract
Objectives
Some semi-synthetic derivatives of glycopeptide antibiotics have been shown to exert in vitro antiviral activity against HIV and coronaviruses. Here we report and characterize the in vitro anti-hepatitis C virus (HCV) activity of several semi-synthetic derivatives of teicoplanin aglycone.
Methods
Anti-HCV activity was analysed in: (i) three different subgenomic HCV replicon systems using a luciferase or quantitative RT–PCR (qRT–PCR) assay; and (ii) an infectious HCV cell culture system by means of qRT–PCR and immunofluorescence assays.
Results
Several teicoplanin aglycone derivatives elicited selective anti-HCV activity in replicons as well as infectious cell culture systems, with LCTA-949 being the most potent derivative. LCTA-949 proved, in contrast to several directly acting antivirals for HCV, efficient in clearing cells of their replicons. When LCTA-949 was combined with HCV protease or polymerase inhibitors an overall additive effect was observed. Likewise, LCTA-949 was equipotent against wild-type replicons as well as against replicons resistant to polymerase and protease inhibitors. Following up to 4 months of selective pressure, no drug-resistant replicons were selected. When combined with the HCV NS3 protease inhibitor VX-950, LCTA-949 prevented the development of VX-950-resistant variants.
Conclusions
Semi-synthetic derivatives of teicoplanin aglycone constitute a novel class of HCV replication inhibitors that are not cross-resistant with various HCV protease and polymerase inhibitors and in particular are potent in clearing hepatoma cells of their replicons. This class of molecules also provides a good tool to obtain novel insights into the replication cycle of HCV and into cellular factors/processes that are crucial for viral replication.
Keywords: HCV, teicoplanin, directly acting antivirals, combination therapy, resistance
Introduction
The hepatitis C virus (HCV) is an enveloped single-stranded (+) RNA virus and is the only member of the genus Hepacivirus in the family Flaviviridae.1 Worldwide, more than 170 million people are chronically infected with HCV and are thus at increased risk of developing serious life-threatening liver disease.2 Current standard therapy for chronic hepatitis C consists of the combination of pegylated interferon (IFN)-α with ribavirin. Unfortunately, a sustained virological response to this therapy is limited to about 60% of treated patients.3–5 There is thus an urgent need for new treatment strategies. One promising approach is the development of small molecules that target specific enzymatic activities associated with the viral non-structural (NS) proteins, or inhibit specific interactions between viral proteins and viral or cellular co-factors.6,7 Several selective inhibitors, including NS3 protease and NS5B polymerase inhibitors, are now in clinical development.6,8–10 For most directly acting antivirals for HCV, rapid emergence of (many) resistant variants has been reported (both in vitro and in the clinical setting).11 The most successful approach to limiting drug resistance is the implementation of rationally designed drug combinations.12 A prerequisite for this type of therapy is the availability of multiple selective drugs acting on different targets.
Glycopeptide antibiotics (i.e. teicoplanin and vancomycin) are used for the treatment of Gram-positive bacterial infections.13,14 The antibacterial activity of glycopeptide antibiotics results from the inhibition of one or both mechanisms involved in the synthesis of the bacterial cell wall, i.e. inhibition of the membrane proteins involved in transglycosylation or interaction with the d-Ala-d-Ala moiety to prevent transpeptidation. Natural glycopeptide antibiotics have not been reported to exert any antiviral activity, but some of their semi-synthetic derivatives proved to be endowed with antiviral activity against retro- or coronaviruses.15–17 For HIV, it was shown that semi-synthetic aglycone glycopeptides potentially interfere with the viral entry process.17 These compounds also inhibited coronavirus replication by interfering with early step(s) of the viral life cycle.16 Here we report on the in vitro anti-HCV activity of some aglycone derivatives of teicoplanin.
Materials and methods
Compounds
Novel semi-synthetic derivatives of teicoplanin aglycone LCTA-1701, -1704 to -1707, were synthesized by the methods described18 and dissolved in DMSO. The mass spectral data of electrospray mass spectrometry are presented in Figure 1. The mass spectral data for the compounds LCTA-563, 877, 901, 914, 949 and 1703 have been published.15 Recombinant IFN-α 2b (Intron®A) was purchased from Schering Plough (Heist-op-den-Berg, Belgium). HCV NS3 protease inhibitor VX-950 and the nucleoside NS5B polymerase inhibitors 4′-azidocytidine, 2′-C-methylcytidine and the non-nucleoside NS5B polymerase inhibitor benzofuran HCV-796 were as described before.19
Figure 1.
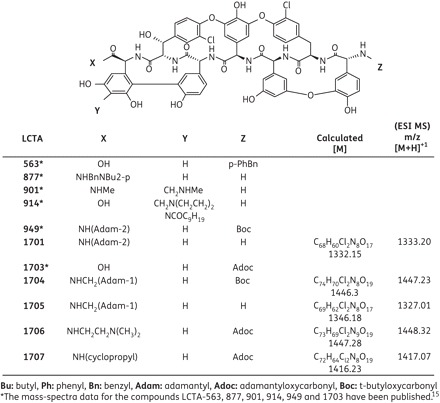
Structural formulae of teicoplanin aglycone derivatives.
Cells and viruses
The highly permissive cell line Huh 7.5.1 was kindly provided by Dr F. V. Chisari (The Scripps Research Institute, La Jolla), and cells carrying HCV replicons I389/hygro-ubi-NS3-3′/5.1 (Huh Mono), I377NS3-3′/wt (Huh 9-13) and I389luc-ubi-neo/NS3-3′/5.1 (Huh 5-2) were kindly provided by Professor R. Bartenschlager (University of Heidelberg, Germany). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, Merelbeke, Belgium) supplemented with 10% heat-inactivated fetal bovine serum (Integro, Zaandam, The Netherlands), 1× non-essential amino acids, 100 IU/mL penicillin (Gibco), 100 μg/mL streptomycin (Gibco), 25 μg/mL hygromycin (Gibco) for Huh Mono, 1000 μg/mL G418 for Huh 9-13 and 250 μg/mL G418 for Huh 5-2 cells. Cell cultures were maintained at 37°C with 5% CO2. Replicons resistant to a panel of HCV selective inhibitors were generated as previously described.20,21
Antiviral assays in HCV replicon cells
Antiviral assays were performed as described.20,21 Briefly, cells were seeded at a density of 5 × 103 cells per well in 96-well cell culture plates in DMEM containing 1000 μg/mL G418, 250 μg/mL G418 or 25 μg/mL hygromycin for Huh 9-13, Huh 5-2 and Huh Mono cells, respectively, at 37°C (5% CO2). After 24 h of incubation, medium was replaced with fresh DMEM (without G418) and serial dilutions of the test compounds. Replicon RNA levels were determined by reverse transcription quantitative PCR (qRT–PCR). For Huh 5-2 cells, HCV replication was quantified by measuring firefly luciferase activity in 96-well cell culture plates (Safire, Tecan, Austria).
Antiviral assay with infectious HCV cell culture system
The highly infectious HCV (JFH-1/CS-N6) described by Delgrange et al.22 was used for the antiviral assays. A total of 5 × 103 Huh 7.5.1 cells per well of a 96-well cell culture plate were incubated with the virus at specific infectivity of about 1:400 (specific infectivity is the ratio between HCV RNA copy number and focus-forming units per mL)23 and at the same time with serial dilutions of compounds. Following 3 days of incubation, medium was removed and cells were washed once and lysed to extract the intracellular RNA. HCV RNA was quantified by means of qRT–PCR.
qRT–PCR for infectious HCV
The 25 μL qRT–PCR reaction mixture contained 6.25 μL of 2× reaction buffer (Eurogentec, Seraing, Belgium), 12.65 μL of H2O, 5 μL of total cellular RNA extract, 200 nmol/L SF-JFH86 (5′-TGG CGT TAG TAT GAG TGT CGT ACA GCC TCC A-3′), 200 nmol/L SR-JFH194 (5′-AAA GGA CCC AGT CTT CCC GGC AAT T-3′) and 6 pmol/L probe (5′-FAM-TGG TCT GCG GAA CCG GTG AGT ACA CC-TAMRA-3′). The RT step was performed at 50°C for 30 min, 10 min at 95°C and PCR amplification comprised 40 cycles of denaturation at 94°C for 15 s and annealing and extension at 52°C for 70 s in an ABI 7500 Fast Real-Time PCR System.
Cytostatic assay
Cells were seeded at a density of 5 × 103 cells per well in 96-well cell culture plates in DMEM (with G418 or hygromycin for replicon-containing cells). Following overnight incubation at 37°C (5% CO2), medium was replaced with fresh DMEM (without G418 or hygromycin) containing serial dilutions of the test compounds. After 3 days of incubation at 37°C, cell viability was determined by means of 3-(4,5-dimethylthiazol-2yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium/phenazine-methosulphate (MTS/PMS) (Promega).
Immunofluorescence assay
Naive Huh 7.5.1 cells were seeded in eight-well chamber slides (Lab-Tek II; Nunc, Germany) at a density of 2 × 104 cells/well. Next day, cultures were inoculated with a viral stock that had a specific infectivity of about 400. Following 5–6 h incubation at 37°C, the inoculum was removed; cells were washed three times and further incubated for 72 h. Cells were stained with the rabbit polyclonal anti-HCV NS5A antibody (kindly provided by Professor Bartenschlager, University of Heidelberg, Germany) and the secondary antibody Alexa Fluor® 488 (Invitrogen). Following 4′,6-diamidino-2-phenylindole (DAPI) staining, the cultures were visualized using a confocal laser scanning microscope (LCSM; Leica Microsystems, Germany).
Drug combination studies
The effects of drug combinations, namely LCTA-949 together with other HCV protease or polymerase inhibitors, were evaluated in a chequerboard format using the method of Prichard and Shipman.24 The theoretical additive effect was calculated from the dose–response curves of individual compounds by the equation Z = X + Y(1 − X), where X represents the inhibition produced by LCTA-949 alone and Y the inhibition by the second compound alone. Z represents the effect produced by the combination of LCTA-949 with the second compound. The theoretical additive surface is subtracted from the actual experimental surface, resulting in a horizontal surface that equals the zero plane when the combination is additive. A surface that lies above the zero plane indicates a synergistic effect of the combination, and a surface below the zero plane indicates an antagonism. For each combination, three independent experiments were carried out to measure the dose–response curves of each individual compound and their combinations.
Clearance–rebound replicon assay
The assay was carried out as described before.20,21,25 Briefly, Huh 9-13 replicons (which carry the neomycin gene) were cultured for consecutive passages in the presence or absence of antiviral drugs without addition of G418 to the culture medium (clearance phase). Next, cells were further cultured in the absence of the antiviral compounds, but in the presence of G418 (rebound phase). In principle, cells that lose their replicons during the clearance phase will die in the rebound phase, whereas cells from which the replicon has not been completely cleared will be able to survive in the presence of G418.
Colony formation assay
Huh 9-13 cells were seeded at a density of 4 × 104 cells per well in 12-well cell culture plates in complete DMEM with 1 mg/mL G418 and in the presence of LCTA-949 or LCTA-1707 (10 or 20 μM) or VX-950 (1.25 or 2.5 μM) or a combination of VX-950 with either LCTA-949 or LCTA-1707. Before reaching confluency, cells were trypsinized and seeded in new 12-well cell culture plates under the same conditions. Following seven passages of selection, half of the cultures were fixed and stained with Giemsa and the remaining the cultures were stored at −80°C until further use.
Results
Teicoplanin aglycone derivatives inhibit subgenomic HCV replication
A diverse library of ∼7000 small molecules, containing semi-synthetic glycopeptide antibiotics [i.e. semi-synthetic derivatives of teicoplanin (Figure 1), eremomycin and vancomycin], was screened in the subgenomic HCV system (genotype 1b). None of the vancomycin derivatives inhibited subgenomic HCV replication, whereas one eremomycin derivative (LCTA-837) did [50% effective concentration (EC50) 6.0 ± 3.9 μM and 50% cytostatic concentration (CC50) >60 μM] (data not shown). In particular, several teicoplanin aglycone analogues were found to inhibit HCV replicon replication (in three different subgenomic replicon systems) at non-toxic concentrations (Tables 1 and 2). The most potent analogue in the first series of compounds evaluated (Table 1) was LCTA-949 (EC50 values ranging between 4 and 7 μM). Since teicoplanin is known to have high binding affinity for serum proteins, we evaluated the effect of human serum on the anti-HCV activity of LCTA-949. In the presence of 5% or 10% human serum albumin, an increase of ∼4-fold in the EC50 value was observed (data not shown).
Table 1.
Effect of selected teicoplanin aglycone analogues on the replication of HCV replicons
| Huh Mono |
Huh 9-13 |
|||
|---|---|---|---|---|
| LCTA | EC50 | CC50 | EC50 | CC50 |
| 563 | 5.1 ± 0.7 | >60 | 15 ± 2.6 | >60 |
| 877 | 14 ± 2.6 | >60 | 10 ± 8.9 | >60 |
| 901 | 29 ± 1.0 | >60 | 54 ± 12 | >60 |
| 914 | 4.8 ± 2.1 | >60 | 17 ± 2.2 | >60 |
| 949 | 6.9 ± 2.1 | >60 | 3.9 ± 0.9 | >60 |
| VX-950 | 1.0 ± 0.3 | >33 | 0.4 ± 0.1 | 24 ± 4 |
Data are expressed in μM and are means ± SD for three or four independent experiments.
The NS3 protease inhibitor (VX-950) is given as a reference HCV inhibitor.
Table 2.
Effect of selected teicoplanin aglycone derivatives on HCV reporter Huh 5-2 replicon and infectious system HCVcc replication
| Huh 5-2 |
HCVcc |
|||
|---|---|---|---|---|
| LCTA | EC50 | CC50 | EC50 | CC50 |
| 949 | 4.9 ± 0.9 | >60 | 5.2 ± 0.1 | >60 |
| 1701 | 15 ± 2.8 | 60 | >10 | >60 |
| 1703 | 5.7 ± 0.5 | >60 | 4.6 ± 3.7 | >60 |
| 1704 | 3.6 ± 1.2 | >60 | 3.4 ± 2.2 | >60 |
| 1705 | 2.9 ± 0.7 | >60 | >10 | >60 |
| 1706 | 11 ± 1.2 | >60 | >10 | >60 |
| 1707 | 7 ± 0.1 | >60 | 3.5 ± 0.5 | >60 |
| VX-950 | 1.0 ± 0.7 | >33 | 0.2 ± 0.1 | >10 |
HCVcc, infectious HCV in cell culture system.
Data are expressed in μM and are means ± SD for three to four independent experiments.
Specific anti-HCV activity of LCTA-949 in an infectious HCV system
LCTA-949 and a panel of structurally closely related derivatives (LCTA-170x series, Figure 1) were next evaluated for their effect on HCV JFH-1/CS-N6 replication. All molecules inhibited HCV replication in a concentration-dependent manner, with LCTA-1707 (among others) being slightly more efficacious than LCTA-949 (EC50 3.5 ± 0.5 and 5.2 ± 0.1 μM, respectively) (Figure 2 and Table 2). The antiviral activity was selective and was not caused by an adverse effect on the host cell. At a concentration of 20 μM, HCV replication was inhibited by 90%–95% and at 60 μM by 98%, whereas host cell metabolism was not, or only slightly, affected at these concentrations. This inhibitory effect was further corroborated by monitoring the effect on the expression of viral proteins (HCV NS5A) (Figure 3). LCTA-949 also inhibited the in vitro replication of yellow fever virus and dengue virus (type 2) at low micromolar concentrations, but not the replication of bovine viral diarrhoea virus, another member of the family Flaviviridae.
Figure 2.
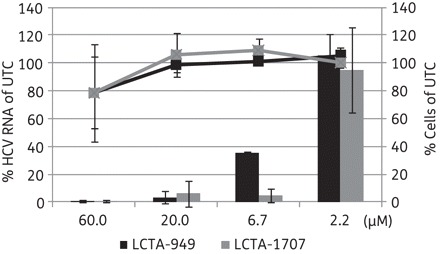
Effects of LCTA-949 and LCTA-1707 on HCV replication in the infectious cell culture system as measured by quantification of intracellular HCV RNA levels (bars) and the proliferation of exponentially growing cells (lines). Data are expressed as percentages of untreated controls (UTC) in μM and are means ± SD for two to four independent experiments.
Figure 3.
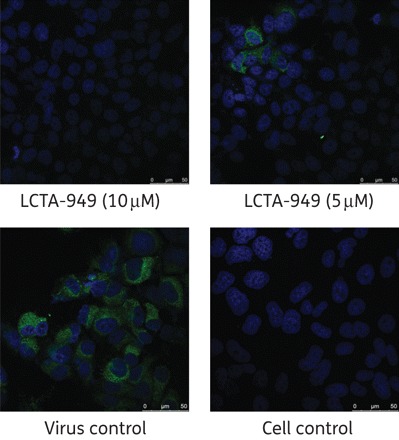
Effect of LCTA-949 on NS5A protein expression of HCV JFH-1/CS-N6 (72 h post-infection and post-treatment) in Huh 7.5.1 cells. NS5A stained in green; nuclei stained with DAPI (blue). This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
LCTA-949 when combined with interferon, protease or polymerase inhibitors results in an additive antiviral effect
The antiviral efficacy of LCTA-949 when combined with IFN, the NS3 protease inhibitor (VX-950) or the polymerase inhibitor (4′-azidocytidine) was evaluated in a 3 day antiviral assay in Huh 5-2 cells (in chequerboard format). Combinations were analysed by the method of Prichard and Shipman.24 Each combination resulted in an additive antiviral effect (Figure 4).
Figure 4.
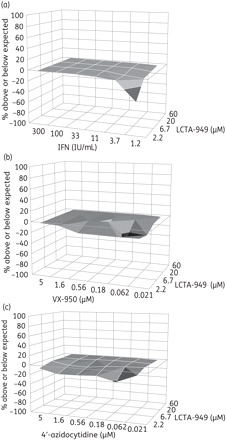
Combined effect of LCTA-949 with (a) interferon-α (IFN), (b) the NS3 protease inhibitor VX-950 or (c) the polymerase inhibitor 4′-azidocytidine on Huh 5-2 replicon replication. Values on the zero plane indicate an additive effect, values above zero represent a synergistic effect and values below zero indicate an antagonistic effect. Data are from three independent experiments.
LCTA-949 alone or in combination with HCV selective protease or polymerase inhibitors clears cells of their replicons
Experiments were set up to study whether LCTA-949 was able to clear replicon-containing cells (Huh 9-13) of their HCV replicon. Huh 9-13 cells were treated for four consecutive passages with LCTA-949, the protease inhibitor VX-950 or the polymerase inhibitors 2′-C-methylcytidine (2′CMC) or 4′-azidocytidine (each at 4× EC50), either alone or in combination. The protease and polymerase inhibitors, when used alone, reduced HCV RNA replication by 0.4–2.6 log10 (Table 3 and Figure 5), but failed to clear the cells of their replicons even following four consecutive passages. By contrast, LCTA-949 alone, but also in combination with these inhibitors, resulted in an efficient inhibition of viral replication.
Table 3.
Effect of LCTA-949 in combinations on the clearance of HCV RNA from Huh 9-13
| C1a | C2a | C2-R2b | C3a | C4a | C4–R2b | |
|---|---|---|---|---|---|---|
| LCTA-949 | 1.2 | 2.2 | 0.007 | 1.7 | UD | UD |
| LCTA-949 + VX-950 | 1.8 | >2.5 | UD | 1.8 | UD | UD |
| VX-950 | 2.6 | 2.5 | 0.003 | 1.8 | UD | 7 |
| LCTA-949 + 2′CMC | 1.7 | >2.5 | UD | 1.8 | UD | UD |
| 2′CMC | 0.4 | 0.4 | 100 | 0.1 | UD | 97 |
| LCTA-949 + 4′-azidocytidine | 1.9 | 2.9 | UD | 1.8 | UD | UD |
| 4′-azidocytidine | 2.1 | 2.1 | 80 | 1.8 | UD | 122 |
UD, undetectable by qRT–PCR; Cx, clearance phase at passage x; Rx, rebound phase at passage x; Cx–Rx, rebound x following clearance x.
aValues shown are reductions in HCV RNA (log10).
bValues shown are percentages of the untreated control.
Figure 5.
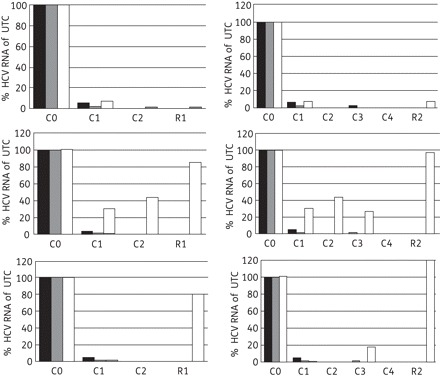
Clearance–rebound in the Huh 9-13 replicon assay. Replicon-containing cells were treated for two or four consecutive passages with LCTA-949 (black bars), white bars or VX-950 (top panels), 2′-C-methylcytidine (middle panels), 4′-azidocytidine (bottom panels) or the combination of these selective HCV inhibitors with LCTA-949 (grey bars; each compound was at a concentration of 4× EC50). In the clearance phase (C), cells were incubated in the compound's presence in the absence of G418. After this clearance phase, cells were further cultured twice in fresh medium containing 1 mg/mL G418 without antiviral agents (rebound phase, R). Intracellular HCV RNA in the surviving cells was quantified by qRT–PCR. UTC, untreated controls (Cx, clearance phase at passage x; Rx, rebound phase at passage x; Cx-Rx, rebound following clearance x).
Replicons resistant to HCV protease or polymerase inhibitors remain susceptible to LCTA-949
We studied whether replicons that are resistant to HCV protease or polymerase inhibitors remain susceptible to LCTA-949. As shown in Table 4, LCTA-949 proved equipotent against wild-type replicons as well as against a panel of replicons that are resistant to the protease inhibitor and VX-950 and the nucleoside polymerase inhibitor 2′-C-methylcytidine, and against the non-nucleoside polymerase inhibitor HCV-796-resistant replicon.
Table 4.
Effect of LCTA-949 on the replication of wild-type and several HCV protease- or polymerase-resistant HCV replicons
| Wild-type | VX-950res | HCV-796res | 2′-C-MeCytres | |
|---|---|---|---|---|
| LCTA-949 | 3.9 ± 0.9 (1.0) | 5.8 ± 0.8 (1.5) | 3.5 ± 1.1 (0.9) | 3.2 ± 1.2 (0.8) |
| VX-950 | 0.2 ± 0.1 (1.0) | 2.0 ± 0.1 (10) | 0.2 ± 0.1 (1.0) | 0.6 ± 0.1 (3) |
| HCV-796 | 0.003 ± 0.002 (1.0) | 0.003 ± 0.001 (1.0) | 7.9 ± 0.3 (2633) | 0.005 ± 0.002 (1.7) |
| 2′-C-MeCyt | 0.7 ± 0.1 (1.0) | 0.6 ± 0.2 (0.9) | 1.0 ± 0.9 (1.4) | 67.9 ± 0.8 (97) |
Data are expressed in μM and are means ± SD for at least two independent experiments.
Values in brackets indicate fold change relating to wild-type, bold indicates clear resistance.
Teicoplanin aglycone analogues prevent the development of HCV replicon resistance to the protease inhibitor VX-950
We studied whether LCTA-949 and LCTA-1707 are able to delay or even prevent resistance development of HCV replicons against the protease inhibitor VX-950. Huh 9-13 replicon-containing cells were cultured for seven consecutive passages in the presence of either 1.25 or 2.5 μM VX-950 alone. Drug-resistant replicons were readily selected (as assessed at passage 7). However, when VX-950 was combined with 10 μM LCTA-949 or LCTA-1707, no colonies developed (Table 5). This was not the result of an adverse effect on the host cell since no changes in morphology or density were observed in parallel control cultures (treated with the antiviral molecules in the absence of G418 pressure). In none of the VX-950 cultures that had also been treated with 20 μM of LCTA-949 or LCTA-1707 did colonies develop (at passage 4). These combinations can thus completely clear the cells of their replicons and suppress the emergence of drug-resistant variants.
Table 5.
Frequency of colony development following combination selection
| Compound | VX-950 (0 μM) | VX-950 (1.25 μM) | VX-950 (2.5 μM) |
|---|---|---|---|
| LCTA-949 (0 μM) | 100 | 90 | 20 |
| LCTA-949 (10 μM) | 100 | 20 | 0.0006 |
| LCTA-949 (20 μM) | 0.0002 | 0 | 0 |
| LCTA-1707 (10 μM) | 100 | 15 | 0.0004 |
| LCTA-1707 (20 μM) | 0.0002 | 0 | 0 |
Frequency was determined as number of colonies following 2 weeks of selection (third passage)/initial number of cells × 100.
Data are mean values for two to four independent experiments.
Discussion
Glycopeptide antibiotics such as vancomycin or teicoplanin are widely used for the (often last-resort) treatment of Gram-positive bacterial infections.13,14 They inhibit bacterial cell wall biosynthesis by forming non-covalent complexes with bacterial peptidoglycan precursors.26,27 The common structure shared among all glycopeptides, i.e. the heptapeptide core, is crucial for their antibacterial activity.28 The sheer size and complexity of this structure facilitates numerous chemical and biochemical modifications, although these are not always straightforward.29 Further synthesis of analogues and pharmacophore analysis should reveal which substructure is implicated in the anti-HCV activity. This may reduce the lead to a smaller molecule or delineate a substructure within the glycopeptide that may serve as a focal point for chemical modification. Most of the analogues in the LCTA-949 series (LCTA-1701 to -1707, Figure 1) were almost as active as the parent compound (Table 2 and Figure 2), arguing for a major role of the teicoplanin peptide scaffold. Chemical modification may also help to optimize the pharmacological properties of this class of molecules. Chemical modification of vancomycin resulted, for example, in the development of oritavancin, a molecule with a very long half-life (393 h) that is distributed, for example, to the liver.28 The limited oral availability of glycopeptides has been overcome by formulating, for example, vancomycin in liposomes or solid-state emulsions.30
Since neither viruses nor their mammalian host cells produce peptidoglycans, the mechanism of action of antibiotics with inhibitory activity against viruses certainly differs by definition from their mechanism against bacteria. Aglycone glycopeptide antibiotics target the entry of HIV15,17 and the early steps of the replication cycle of coronaviruses.16 The fact that these molecules inhibit the entry of these viruses does not necessarily mean that their mechanism against HCV must be similar. Geneticin, for example, has been shown to inhibit pestivirus assembly31 or release and the replication or translation of flaviviruses.32 Even though these viruses belong to the same family (Flaviviridae), they are inhibited by two different mechanisms. We demonstrate here that, unlike the case for HIV and coronaviruses, LCTA-949 inhibits HCV replication at a post-entry event. Indeed, the molecule inhibits the replication of HCV subgenomic replicons (which do not encode structural proteins) and the activity in replicons is comparable to that in the infectious HCV system. Furthermore, LCTA-949 is still active when added to HCV-infected cultures. Whether inhibition of intracellular replication is the result of a direct interaction with viral proteins or is mediated by interference with host cell factors remains to be elucidated. So far, we have not been able to select drug-resistant variants despite several months of attempts. Interestingly, oritavancin has been shown to interfere with intracellular cholesterol and phospholipid metabolism33 and membrane organization and permeability.34 Both host cell lipid metabolism35 and membrane organization36 are important during HCV replication. It is thus possible that LCTA-949 might (i) target cellular factor(s) that is (are) essential for HCV replication or (ii) influence the cell's lipid metabolism in such a way that it counteracts HCV replication or disturbs membrane organization, which is important in the HCV replication cycle.
In the subgenomic replicon model, LCTA-949 was more efficient in clearing cells of their HCV replicons than NS3 protease or NS5B polymerase inhibitors at the concentration tested (Figure 5). Future HCV therapy will consist of a combination of two or more selective inhibitors.37 In line with this, we aimed to study the combined effect of LCTA-949 with either IFN or other HCV inhibitors (protease or polymerase inhibitors). Each of the combinations resulted in an overall additive antiviral effect (Figure 4). Hence, these compounds probably do not interfere with the mechanism of action of these selective inhibitors. This is further supported by the observation that LCTA-949 was equipotent against wild-type replicons and replicons resistant to various inhibitors of NS3 protease or nucleoside or non-nucleoside NS5B polymerase. LCTA-949 delayed or prevented resistance development (of replicons) to the protease inhibitor VX-950. More importantly, even after 4 months of continuous culture in the presence of a suboptimal concentration of LCTA-949, no HCV replicons with reduced susceptibility to LCTA-949 emerged. The barrier to resistance of this class of compounds might therefore be high, assuming that the HCV quasispecies is equally diverse in vitro and in vivo.
In conclusion, an aglycone analogue(s) of teicoplanin inhibit(s) in vitro HCV replication and in particular is (are) potent in clearing hepatoma cells of their replicons. Further exploration of the structure–activity relationship may lead to the development of highly potent inhibitors of HCV replication. Moreover, this class of molecules also provides a good tool to obtain novel insights into the replication cycle of HCV and the cellular factors/processes that are crucial for viral replication.
Funding
This work was supported by a post-doctoral fellowship from the Research Foundation Flanders-FWO to J. P. and by grant G.0728.09N of the Research Foundation Flanders-FWO. The chemical part of the work was supported by a Russian fundamental research grant, # 10-03-00210-а.
Transparency declarations
None to declare.
Author contributions
S. O. generated all of the experimental data and drafted the manuscript, J. N. is principal investigator and promoter of S. O., J. P. is co-promoter of S. O. and coordinated this study. S. S. P., E. N. O. and M. N. P. synthesized and provided the compounds tested. K. D. analysed data and helped to draft the manuscript. D. D. and F. Z. provided the HCV infectious cell culture system and the necessary expertise to implement this system in the experimental set-up. All authors were involved in the interpretation of the data, reviewed and revised the manuscript for intellectual content, and approved the final version for submission.
Acknowledgements
We thank Katrien Geerts and Stijn Delmotte for excellent technical assistance, Dominique Brabants for dedicated editorial help and Els Vanstreels for her assistance with the confocal laser scanning microscope. We thank Professor Jan Balzarini for critical review of the manuscript prior to submission.
References
- 1.Lindenbach BD, Rice CM. Flaviviridae: the viruses and their replication. In: Knipe DM, Howley PM, Griffin DE, et al., editors. Fields Virology. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 991–1041. [Google Scholar]
- 2.Houghton M. Discovery of the hepatitis C virus. Liver Int. 2009;29(Suppl 1):82–8. doi: 10.1111/j.1478-3231.2008.01925.x. [DOI] [PubMed] [Google Scholar]
- 3.Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–82. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 4.Craxi A, Licata A. Clinical trial results of peginterferons in combination with ribavirin. Semin Liver Dis. 2003;23(Suppl 1):35–46. doi: 10.1055/s-2003-41633. [DOI] [PubMed] [Google Scholar]
- 5.Craxi A. PEG IFN alfa-2a vs. alfa-2b: and the winner is …? J Hepatol. 2010;52:133–5. doi: 10.1016/j.jhep.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 6.Coelmont L, Kaptein S, Paeshuyse J, et al. Debio 025, a cyclophilin binding molecule, is highly efficient in clearing hepatitis C virus (HCV) replicon-containing cells when used alone or in combination with specifically targeted antiviral therapy for HCV (STAT-C) inhibitors. Antimicrob Agents Chemother. 2009;53:967–76. doi: 10.1128/AAC.00939-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schinazi RF, Bassit L, Gavegnano C. HCV drug discovery aimed at viral eradication. J Viral Hepat. 2010;17:77–90. doi: 10.1111/j.1365-2893.2009.01246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delang L, Coelmont L, Neyts J. Antiviral therapy for hepatitis C virus: beyond the standard of care. Viruses. 2010;2:826–66. doi: 10.3390/v2040826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burton JR, Jr, Everson GT. HCV NS5B polymerase inhibitors. Clin Liver Dis. 2009;13:453–65. doi: 10.1016/j.cld.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Lange CM, Sarrazin C, Zeuzem S. Specifically targeted anti-viral therapy for hepatitis C – a new era in therapy. Aliment Pharmacol Ther. 2010;32:14–28. doi: 10.1111/j.1365-2036.2010.04317.x. [DOI] [PubMed] [Google Scholar]
- 11.Thompson AJ, McHutchison JG. Antiviral resistance and specifically targeted therapy for HCV (STAT-C) J Viral Hepat. 2009;16:377–87. doi: 10.1111/j.1365-2893.2009.01124.x. [DOI] [PubMed] [Google Scholar]
- 12.Kieffer TL, Kwong AD, Picchio GR. Viral resistance to specifically targeted antiviral therapies for hepatitis C (STAT-Cs) J Antimicrob Chemother. 2010;65:202–12. doi: 10.1093/jac/dkp388. [DOI] [PubMed] [Google Scholar]
- 13.Pea F, Brollo L, Viale P, et al. Teicoplanin therapeutic drug monitoring in critically ill patients: a retrospective study emphasizing the importance of a loading dose. J Antimicrob Chemother. 2003;51:971–5. doi: 10.1093/jac/dkg147. [DOI] [PubMed] [Google Scholar]
- 14.Sancar AA, Yegenoglu S, de Vries R, et al. Vancomycin vs teicoplanin in the treatment of Gram-positive infections: a pharmacoeconomic analysis in a Turkish University Hospital. Pharm World Sci. 2008;30:916–23. doi: 10.1007/s11096-008-9251-2. [DOI] [PubMed] [Google Scholar]
- 15.Balzarini J, Pannecouque C, De Clercq E, et al. Antiretroviral activity of semisynthetic derivatives of glycopeptide antibiotics. J Med Chem. 2003;46:2755–64. doi: 10.1021/jm0300882. [DOI] [PubMed] [Google Scholar]
- 16.Balzarini J, Keyaerts E, Vijgen L, et al. Inhibition of feline (FIPV) and human (SARS) coronavirus by semisynthetic derivatives of glycopeptide antibiotics. Antiviral Res. 2006;72:20–33. doi: 10.1016/j.antiviral.2006.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Preobrazhenskaya MN, Olsufyeva EN. Polycyclic peptide and glycopeptide antibiotics and their derivatives as inhibitors of HIV entry. Antiviral Res. 2006;71:227–36. doi: 10.1016/j.antiviral.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Printsevskaya SS, Solovieva SE, Olsufyeva EN, et al. Structure-activity relationship studies of a series of antiviral and antibacterial aglycon derivatives of the glycopeptide antibiotics vancomycin, eremomycin, and dechloroeremomycin. J Med Chem. 2005;48:3885–90. doi: 10.1021/jm0500774. [DOI] [PubMed] [Google Scholar]
- 19.Paeshuyse J, Vliegen I, Coelmont L, et al. Comparative in vitro anti-hepatitis C virus activities of a selected series of polymerase, protease, and helicase inhibitors. Antimicrob Agents Chemother. 2008;52:3433–7. doi: 10.1128/AAC.01534-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vliegen I, Paeshuyse J, De Burghgraeve T, et al. Substituted imidazopyridines as potent inhibitors of HCV replication. J Hepatol. 2009;50:999–1009. doi: 10.1016/j.jhep.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delang L, Paeshuyse J, Vliegen I, et al. Statins potentiate the in vitro anti-hepatitis C virus activity of selective hepatitis C virus inhibitors and delay or prevent resistance development. Hepatology. 2009;50:6–16. doi: 10.1002/hep.22916. [DOI] [PubMed] [Google Scholar]
- 22.Delgrange D, Pillez A, Castelain S, et al. Robust production of infectious viral particles in Huh-7 cells by introducing mutations in hepatitis C virus structural proteins. J Gen Virol. 2007;88:2495–503. doi: 10.1099/vir.0.82872-0. [DOI] [PubMed] [Google Scholar]
- 23.Zhong J, Gastaminza P, Chung J, et al. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J Virol. 2006;80:11082–93. doi: 10.1128/JVI.01307-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prichard MN, Shipman C., Jr Analysis of combinations of antiviral drugs and design of effective multidrug therapies. Antivir Ther. 1996;1:9–20. [PubMed] [Google Scholar]
- 25.Paeshuyse J, Kaul A, De CE, et al. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology. 2006;43:761–70. doi: 10.1002/hep.21102. [DOI] [PubMed] [Google Scholar]
- 26.Walsh CT. Vancomycin resistance: decoding the molecular logic. Science. 1993;261:308–9. doi: 10.1126/science.8392747. [DOI] [PubMed] [Google Scholar]
- 27.Malabarba A, Ciabatti R. Glycopeptide derivatives. Curr Med Chem. 2001;8:1759–73. doi: 10.2174/0929867013371716. [DOI] [PubMed] [Google Scholar]
- 28.Zhanel GG, Calic D, Schweizer F, et al. New lipoglycopeptides: a comparative review of dalbavancin, oritavancin and telavancin. Drugs. 2010;70:859–86. doi: 10.2165/11534440-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 29.Boger DL. Vancomycin, teicoplanin, and ramoplanin: synthetic and mechanistic studies. Med Res Rev. 2001;21:356–81. doi: 10.1002/med.1014. [DOI] [PubMed] [Google Scholar]
- 30.Anderson KE, Eliot LA, Stevenson BR, et al. Formulation and evaluation of a folic acid receptor-targeted oral vancomycin liposomal dosage form. Pharm Res. 2001;18:316–22. doi: 10.1023/A:1011002913601. [DOI] [PubMed] [Google Scholar]
- 31.Birk AV, Dubovi EJ, Zhang X, et al. Antiviral activity of geneticin against bovine viral diarrhoea virus. Antivir Chem Chemother. 2008;19:33–40. doi: 10.1177/095632020801900105. [DOI] [PubMed] [Google Scholar]
- 32.Zhang XG, Mason PW, Dubovi EJ, et al. Antiviral activity of geneticin against dengue virus. Antiviral Res. 2009;83:21–7. doi: 10.1016/j.antiviral.2009.02.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van BF, Saffran J, Mingeot-Leclercq MP, et al. Mixed-lipid storage disorder induced in macrophages and fibroblasts by oritavancin (LY333328), a new glycopeptide antibiotic with exceptional cellular accumulation. Antimicrob Agents Chemother. 2005;49:1695–700. doi: 10.1128/AAC.49.5.1695-1700.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Domenech O, Francius G, Tulkens PM, et al. Interactions of oritavancin, a new lipoglycopeptide derived from vancomycin, with phospholipid bilayers: effect on membrane permeability and nanoscale lipid membrane organization. Biochim Biophys Acta. 2009;1788:1832–40. doi: 10.1016/j.bbamem.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 35.Syed GH, Amako Y, Siddiqui A. Hepatitis C virus hijacks host lipid metabolism. Trends Endocrinol Metab. 2010;21:33–40. doi: 10.1016/j.tem.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moradpour D, Gosert R, Egger D, et al. Membrane association of hepatitis C virus nonstructural proteins and identification of the membrane alteration that harbors the viral replication complex. Antiviral Res. 2003;60:103–9. doi: 10.1016/j.antiviral.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 37.McHutchison JG, Everson GT, Gordon SC, et al. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med. 2009;360:1827–38. doi: 10.1056/NEJMoa0806104. [DOI] [PubMed] [Google Scholar]