

Abstract
Severe human disease associated with influenza A H5N1 virus was first detected in Hong Kong in 1997. Its recent reemergence in Asia and high associated mortality highlight the need to understand its pathogenesis. We investigated the roles of death receptor ligands (DRLs) in H5N1 infection. Significant up-regulation of tumor necrosis factor (TNF)–related apoptosis-inducing ligand (TRAIL) and TNF-α, but not Fas ligand (FasL) mRNA, was detected in human monocyte–derived macrophages (MDMs) infected with avian influenza viruses A/Hong Kong/483/97 (H5N1/97) or its precursor, A/Quail/Hong Kong/G1/97. H5N1/97-infected MDMs exhibited the strongest induction of apoptosis in Jurkat T cells, and it could be reduced by TRAIL–receptor 2 blocking antibody. Furthermore, influenza virus infection enhanced the sensitivity of Jurkat T cells to apoptosis induced by TNF-α, TRAIL, and FasL. Our data suggested that functional TRAIL produced by influenza virus–infected MDMs was related to their cytotoxicity and that the enhanced sensitization to DRL-induced apoptosis detected in avian influenza may contribute to disease pathogenesis
In 1997, the avian influenza virus H5N1 crossed the species barrier and caused 18 confirmed human infections in Hong Kong with a case-fatality rate of 33%. The clinical manifestations include lymphopenia and severe pneumonia progressing to the syndromes of acute respiratory distress and multiple organ dysfunction [1, 2]. The proinflammatory cytokine dysregulation detected in human H5N1 disease is thought to contribute to the severity of this influenza [3]. However, the immune response in human to the avian influenza virus remains unclear
It has been shown in mallard ducks that H5N1 virus enhances macrophage phagocytic activity but suppresses T cell function [4]. In mice, H5N1 virus depleted lymphocytes through apoptosis, which resulted in the reduction of interferon (IFN)–γ, interleukin (IL)–1β, macrophage inflammatory protein, and tissue cellularity, leading to a higher mortality rate [5]. Indeed, cross-reactive cytotoxic T lymphocyte responses have been observed in humans with no previous exposure to avian influenza virus [6], and B cell–dependent heterosubtypic cross-protection against H5N1 virus could also be induced in mice [7]. Because lymphopenia is a notable observation in H5N1 infection, the clinical severity of human disease is associated with low peripheral white blood cell and lymphocyte counts at admission [1, 2, 8, 9], the induction of lymphocyte apoptosis may contribute to disease pathogenesis
Death receptors (DRs) and their ligands play important roles in orchestrating innate and adaptive immune responses against pathogens by regulating cell death and survival [10]. Tumor necrosis factor (TNF)–related apoptosis-inducing ligand (TRAIL) is a member of the TNF superfamily and was identified by sequence homology with 2 well-characterized DR ligands (DRLs), TNF-α and Fas ligand (FasL). There are 5 receptors (R) for TRAIL, of which TRAIL-R1 and TRAIL-R2 contain the death domains. The binding of DRLs to their specific receptors leads to the activation of a cascade of specific proteolytic enzymes (termed “caspases”), leading to apoptosis [11]. Several viruses—including measles virus, HIV, cytomegalovirus, papillomavirus, and herpes simplex virus—trigger immune-cell cytotoxicity through the induction of TRAIL expression, which is involved in the killing of virus-infected cells or bystander lymphocytes and NK cells [12–16]. However, little is known about the apoptosis of immune cells mediated by DRLs, particularly TRAIL, in H5N1 infection
We have previously reported that avian influenza virus H5N1/97—but not the human strains A/Hong Kong/54/98 (H1N1) and A/Hong Kong/1174/99 (H3N2)—could induce high production of proinflammatory cytokines, most notably TNF-α, in human primary macrophages [3]. The precursor of H5N1 virus, A/Quail/Hong Kong/G1/97 (H9N2/G1), which shares 6 internal gene segments with H5N1/97, also shared the high cytokine induction phenotype [3, 17]. In the present study, we hypothesized that the innate immune responses to H5N1 result in a strong expression of DRLs that lead to lymphocyte apoptosis. Using Jurkat T cells as a model, we demonstrate the release of functional TRAIL and cytotoxicity of H5N1/97- or H9N2/G1-infected macrophages
Materials and methods
CellsHuman monocyte-derived macrophages (MDMs) were generated from buffy coats of samples from healthy blood donors as described elsewhere [3]. The research protocol was approved by the Institutional Review Board of The University of Hong Kong/Hospital Authority Hong Kong West Cluster. The purity of monocytes, as determined by flow cytometry (Coulter Epics Elite; Beckman Coulter) with anti-CD14 monoclonal antibody (BD PharMingen), was consistently >90%. The monocytes were refed by fresh medium every 2 days and allowed to differentiate for 14 days in vitro
The Jurkat T cell line (American Type Culture Collection [ATCC]) was maintained in RPMI 1640 medium (Invitrogen Life Technologies) with 10% fetal bovine serum (FBS; Invitrogen Life Technologies). MDCK cells (ATCC) and murine fibroblasts L929 (ATCC) were cultured in MEM (Invitrogen Life Technologies) supplemented with 10% FBS
Virus preparation, titration, and infectionIn view of the biosafety issues involved in handling highly pathogenic viruses, we used avian influenza virus H9N2/G1 for optimization of the experimental system. Then, findings obtained with H9N2/G1 were confirmed using H5N1/97 virus in a biohazard level 3 facility. Avian influenza virus H9N2/G1 and human influenza A virus 54/98 (H1N1/98) were grown in MDCK cells that contained 2 mg/L N-p-tosyl-L-phenylalaninechloromethyl ketone–treated (TPCK) trypsin (Sigma). H5N1/97 virus was propagated in MDCK cells without TPCK trypsin, because these highly pathogenic avian influenza viruses are not dependent on exogenously added trypsin for productive virus replication. Virus stocks were purified by adsorption to and elution from turkey red blood cells [18] and were stored at −70°C until use. The titer of virus stock was determined by daily observation for cytopathic effect in MDCK cells and confirmed by hemagglutination assay [19]. To inactivate viruses, samples were irradiated at a dose of 0.2 J/cm2 for 15 min in a UV cross-linker (Spectrolinker)
Day-14 differentiated MDMs and Jurkat T cells were infected by influenza viruses at MOIs of 2 and 1, respectively. This was set as the 0-h point of infection (POI) for the experiments described below. After virus adsorption for 1 h at 37°C, unadsorbed virus was removed by washing with PBS. Mock-treated cells were handled in parallel, except that virus was not added. To determine infectivity, cells were fixed and analyzed by immunofluorescent staining specific for influenza A virus nucleoprotein (DAKO Imagen; Dako Diagnostics)
Quantification of mRNA by real-time, quantitative reverse -transcription (RT) polymerase chain reaction (PCR)Infected MDMs (∼5×105) cultured in macrophage serum-free medium (Invitrogen Life Technologies) were harvested at the 4-, 8-, and 12-h POI for total RNA extraction by TRIzol Reagent (Invitrogen Life Technologies). RT was performed on DNase-treated total RNA. The cDNA was synthesized from mRNA with oligo(dT)12–18 primer and Superscript II reverse transcriptase (Invitrogen Life Technologies). The cDNA samples were diluted (1:10) and used as the template. Specific primers and probes (table 1) were used in a real-time PCR assay (ABI PRISM 7700 Sequence Detection System; Applied Biosystem). The standard curve was generated using serial dilutions of plasmids (∼10–107 copies) that contained the respective cloned gene targets. The results were normalized and expressed as the number of target gene copies per 104 copies of β-actin
Table 1.

Primer sequences and probes used in real-time polymerase chain reaction
Bioassay for TNF-α in the culture supernatants from virus-infected MDMsTo avoid the generation of aerosol, we used a sensitive bioassay for quantitating bioactive TNF-α in culture supernatants [20]. Some samples were being assayed by both bioassay and ELISA, and a strong correlation was observed (data not shown). Supernatants from mock- or virus-infected 106 MDMs/well in a volume of 800 μL of RPMI 1640 plus 10% FBS for 24 h were collected and stored at −70°C, thawed, and UV irradiated before use
The culture supernatants or samples diluted to 1:10 or 1:50 were added into a confluent culture of L929 cells in 96-well plate with 1 μg/mL actinomycin D (Sigma) in duplicate, and cell viability was determined by crystal violet staining. The concentration of TNF-α in the supernatants was calculated with reference to the standard curve generated by serial dilution of recombinant human TNF-α (R & D Systems)
Apoptosis of T lymphocytes by culture supernatants from virus-infected MDMsJurkat T cells (5×105/mL) were cultured in the presence of 50% UV-treated culture supernatants generated from mock- or virus-infected MDMs for 24 h. The cell death was detected by anti–active caspase–3 antibody staining (BD PharMingen), as described in our previous study [21]. The experiments were also performed in the presence of 1 μg/mL TRAIL-R2/Fc chimera (631-T2; R & D Systems) for the blocking of TRAIL or 1 μg/mL neutralizing anti–FasL antibody (clone 100419; R & D Systems) for the determination of the specific pathway of apoptosis. The optimal concentration used was determined by titration
Coculture of PKH-26–labeled Jurkat T cells with virus-infected MDMsTo distinguish between effector and target cells, Jurkat T cells were labeled with PKH-26 (Sigma) before coculture at a MDMs:Jurkat T cell ratio of 2:1 for a further 24 h. The experiments were also performed in the presence of TRAIL-R2/Fc chimera (1 μg/mL) or anti-FasL (1 μg/mL). Then, cell death was detected by active caspase–3 staining, as described above. To ensure that H1N1/98 and H9N2/G1 released by infected MDMs would not infect the Jurkat T cells, TPCK trypsin was not added to the coculture system
Statistical analysisData are expressed as means ± SEs. Statistical significance was determined by Student t test or the nonparametric equivalent, the Mann-Whitney U test, using Instat software (version 3.05; GraphPad). P<.05 was considered to be significant
Results
Up-regulation of the expression of TNF-α and TRAIL but not FasL mRNA in MDMs by avian influenza virus infection MDMs are susceptible to both human and avian influenza virus infection. As evidenced by the expression of viral nucleoprotein, >90% of MDMs were infected by the 12-h POI. As is shown in figure 1A the expression of TNF-α mRNA was rapidly induced by avian virus H9N2/G1 infection. It was detected at the 4-h POI and then gradually decreased over time. In contrast, TRAIL mRNA expression in H9N2/G1-infected MDMs progressively increased with time (figure 1B). At all time points assessed (the 4-, 8-, and 12-h POI), the expression level of TRAIL mRNA in H9N2/G1-infected MDMs (range, 60–1000 copies/104 copies of β-actin) was significantly higher than that in H1N1/98-infected MDMs (range, 10–100 copies/104 copies of β-actin; n=6; P<.01). A very low level of FasL mRNA expression (<30 copies/104 copies of β-actin) was detected in mock- or influenza virus–infected MDMs (figure 1C)
Figure 1.

mRNA expression of tumor necrosis factor (TNF)–α, TNF-related apoptosis-inducing ligand (TRAIL), and Fas ligand (FasL) in influenza virus–infected monocyte-derived macrophages (MDMs). A, B and C The kinetics of TNF-α, TRAIL, and FasL mRNA expression in H1N1/98- and H9N2/G1-infected MDMs. Total RNA was harvested at 4, 8, and 12 h after influenza virus infection. The target genes were quantified by quantitative reverse-transcription polymerase chain reaction and normalized to 1×104 copies of β-actin mRNA. Data are the mean ± SE from 6 independent experiments. D, E and F TNF-α, TRAIL, and FasL mRNA expression in MDMs treated by H5N1 and UV-irradiated H5N1 (UVH1N1) at 7 h after infection. Data are the mean ± SE from 4 independent experiments. *P<.05, **P<.01
We subsequently repeated the experiment on H5N1/97-infected MDMs in a biohazard level 3 laboratory. At the 7-h POI, both TNF-α (figure 1D) and TRAIL (figure 1E) mRNA were significantly up-regulated (P=.0289 and P=.01, respectively, vs. H1N1/98-infected MDMs; n=4), and the expression of FasL was very low in H5N1/97-infected MDMs (figure 1F). This up-regulation of TNF-α and TRAIL was dependent on viral replication. As is shown in figure 1D and 1E a significantly lower level of TNF-α and TRAIL mRNA expression was detected in MDMs treated by UV-irradiated H5N1/97. The UV-irradiated H5N1/97-induced TRAIL mRNA expression was, however, significantly higher than that in mock-treated MDMs (P=.001) (figure 1E). A similar pattern of TNF-α and TRAIL expression was found in UV-treated H9N2/G1 virus (data not shown)
Release of soluble bioactive TNF-α and TRAIL from virus-infected MDMsSimilar to that reported elsewhere [3], we detected high levels of bioactive TNF-α in culture supernatant from avian influenza virus–infected MDMs (4.43±0.38 and 5.28±1.34 ng/mL for H9N2/G1 and H5N1/97, respectively, vs. 0.53±0.28 ng/mL for H1N1/98; P<.001; n=4). In the search for a specific cell line to determine the release of bioactive TRAIL by influenza virus–infected MDMs, we tested the sensitivity of Jurkat T cells to recombinant DRLs. It was intriguing to find that Jurkat T cells are sensitive to TRAIL and FasL in a dose-dependent manner but not to TNF-α (figure 2A). This specific induction of cell death by high-dose TRAIL (100 ng/mL) and FasL (100 ng/mL) could be reversed by the addition of TRAIL-R2/Fc chimera (1 μg/mL) and anti-FasL neutralizing antibody (1 μg/mL), respectively (figure 2B)
Figure 2.

Sensitivity of Jurkat T cells to tumor necrosis factor (TNF)–related apoptosis-inducing ligand (TRAIL)– and Fas ligand (FasL)–induced apoptosis but not to TNF-α. A Jurkat T cells cultured in the presence of recombinant TNF-α, TRAIL, and FasL at concentrations of 0–100 ng/mL for 24 h. Cell death was determined by active caspase–3 staining. B Jurkat T cell death, induced by 100 ng/mL recombinant human (rh) TRAIL and 100 ng/mL FasL and inhibited by specific antibodies at a final concentration of 1 μg/mL. Data are representative of 3 independent experiments. TRAIL-R2, TRAIL receptor 2
By incubating Jurkat T cells in the presence of 50% UV-irradiated supernatant, we demonstrated that supernatants from H1N1/98-, H9N2/G1-, and H5N1/97-infected MDMs induced the apoptosis of Jurkat T cells, but supernatants from mock-treated MDMs did not (figure 3A–3D). The cell death could be significantly reduced by the addition of TRAIL-R2/Fc chimera but not by anti-FasL. The combination of both antibodies did not reduce the level of apoptosis further
Figure 3.
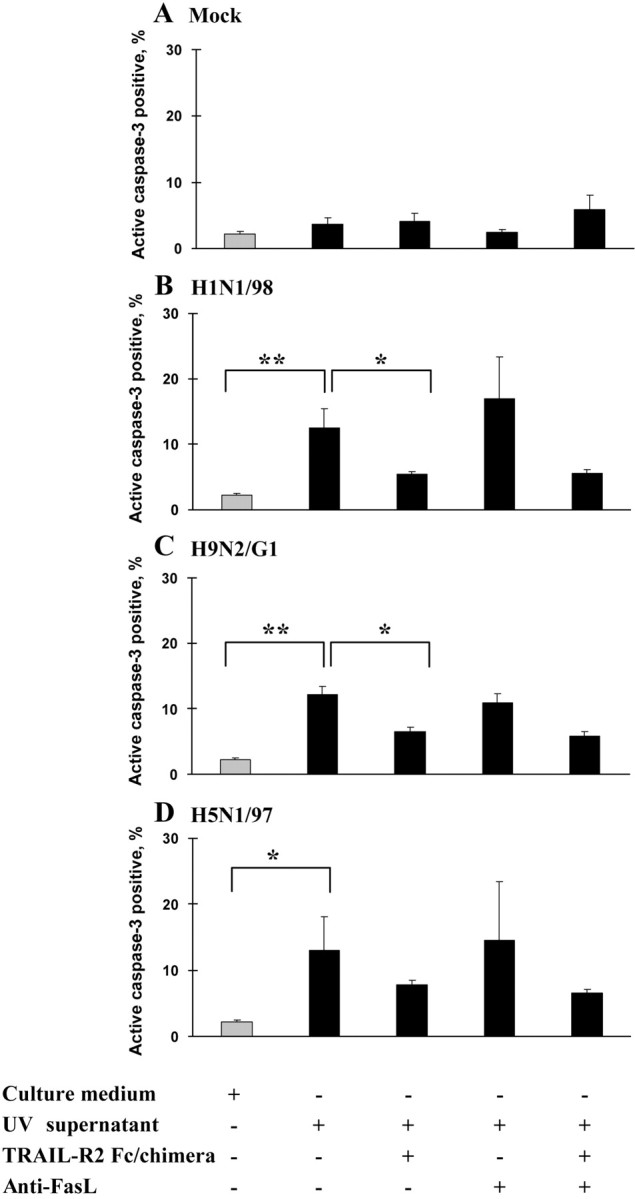
Release of functional soluble tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) by influenza virus–infected monocyte-dervied macrophates (MDMs) that was cytotoxic to Jurkat T cells. UV-irradiated supernatants from mock-treated (A) and influenza virus-infected (B–D) MDMs were tested for cytotoxic activity against Jurkat T cells. Jurkat T cells were incubated for 24 h with culture medium alone or with 50% supernatant from mock- or H1N1/98-, H9N2/G1-, and H5N1/97-infected MDMs, with or without TRAIL–receptor 2 (R2)/Fc chimera (1 μg/mL) or anti–Fas ligand (FasL; 1 μg/mL) (n=4, 4, 4, and 3, respectively). Cell death was determined by active caspase–3 staining. *P<.05, **P<.01)
Stronger apoptosis of Jurkat T cells infected by H1N1/98, compared with H5N1/97 and H9N2/G1To test the direct cytopathic effects of influenza virus on Jurkat T cells, we infected Jurkat T cells with the 3 virus strains at MOIs of 1. As assessed by immunofluorescence staining of viral nucleoprotein at the 24-h POI, >70% of Jurkat T cells were infected by either human or avian influenza viruses. Marked apoptosis was detected in H1N1/98-infected Jurkat T cells, with the percentage of active caspase–3–positive cells at 82.50%±1.1% (n=3; figure 4). Fewer than 30% of apoptotic cells were detected in H9N2/G1- and H5N1-infected Jurkat T cells (12.2% ± 2.89% and 22.2%±7.36%, respectively; n=3) (figure 4)
Figure 4.

Marked apoptosis of Jurkat T cells directly infected by human influenza virus H1N1/98 but not by avian influenza viruses H9N2/G1 and H5N1/97. Jurkat T cells were infected with H1N1/98, H5N1/97, and H9N2/G1 at an MOI of 1 for 1 h; then, unadsorbed virus was washed off and cells were cultured for further 24 h. Cell death was determined by active caspase–3 staining. A high level of apoptosis was observed in H1N1-infected Jurkat T cells. Data are the mean ± SE from 3 independent experiments
Direct induction of Jurkat T cells apoptosis by virus-infected MDMsIt has been reported that the expression of TRAIL on the surface of virus-infected cells is also important in inducing apoptosis [14–16]. Hence, we tested the direct cytotoxicity of virus-infected MDMs by coculturing Jurkat T cells with virus-infected MDMs for 24 h. No TPCK trypsin was added, to ensure no infection of Jurkat T cells by H1N1/98 or H9N2/G1 released from infected MDMs. The infection of cells by H5N1/97 was, however, independent of the presence of trypsin. Similar to that observed in MDM supernatant, increased apoptosis was detected in Jurkat T cells cocultured with virus-infected MDMs (figure 5). In contrast to H1N1/98- and H9N2/G1-infected MDMs, H5N1/97-infected MDMs exhibited the strongest induction of apoptosis in Jurkat T cells (figure 5C), with the percentage of active caspase–3–positive cells at 55.30% ± 13.52% (vs. H9N2/G1 at 20.75%±2.12% and H1N1/98 at 20.96%±3.78%; P=.0144 and P=.0485, respectively). The cytotoxic effects of H1N1/98- and H9N2/G1-infected MDMs on Jurkat T cells could be significantly inhibited by TRAIL-R2/Fc chimera but not by anti-FasL alone (figure 5A and 5B). Partial inhibition of apoptosis in Jurkat T cells cocultured with H5N1/97-infected MDMs by TRAIL-R2/Fc chimera was observed, but the difference did not yield statistical significance (figure 5C)
Figure 5.

Influenza virus-infected monocyte-dervied macrophates (MDMs) becoming cytotoxic to Jurkat T cells. Mock- or virus-infected MDMs were coincubated with PKH-26–labeled Jurkat T cells at a ratio of 2:1. Cell death was determined by active caspase–3 staining (A and B) The cytotoxicity of H1N1/98-infected MDMs (n=9) and H9N2/G1-infected MDMs (n=14) was significantly inhibited by tumor necrosis factor–related apoptosis-inducing ligand (TRAIL)–receptor 2 (R2)/Fc chimera (1 μg/mL) but not anti–Fas ligand (FasL; 1 μg/mL). C Strong cytotoxicity in avian influenza virus H5N1/97–infected MDMs and Jurkat T cell coculture. The apoptosis could only be reduced partially by TRAIL-R2 Fc/chimera. Data are the mean ± SE from 3 independent experiments. *P<.05, **P<.01
Sensitization of Jurkat T cells by avian influenza viruses to apoptosis induced by death receptor ligandsThe stronger induction of Jurkat T cells apoptosis by H5N1/97-infected MDMs (figure 5C), compared with the corresponding UV-treated supernatants (figure 3D), suggested that the effects of virus itself may be involved. According to the virus titer measured in H5N1 coculture systems (105 log10 TCID50/mL), the MOI in the coculture was only 0.2. Even when Jurkat T cells were directly infected with H5N1 at an MOI of 1, the percentage of active caspase–3–positive cells was not very high (figure 4). Hence, there may be an increased sensitivity of Jurkat T cells in the coculture system. To test the sensitivity of infected cells to DRL-induced apoptosis, exogenous TNF-α (500 ng/mL), TRAIL (10 ng/mL), or FasL (10 ng/mL) was added into H9N2/G1- or H5N1/97-infected Jurkat T cells, and they were incubated for 24 h. As shown in figure 6, mock-infected Jurkat T cells were resistant to the apoptosis induction effect of TNF-α, even at a high dose (500 ng/mL), and only a moderate increase in apoptosis was observed with recombinant human (rh) TRAIL and rhFasL. In contrast, a significantly enhanced level of apoptosis was detected in H5N1/97- and H9N2/G1-infected Jurkat T cells in the presence of TNF-α. A similar phenomenon was also observed with low doses of TRAIL and FasL (figure 6)
Figure 6.

H9N2/G1 and H5N1/97 infection sensitizing Jurkat T cells to induction of apoptosis by tumor necrosis factor (TNF)–α, TNF-related apoptosis-inducing ligand (TRAIL), and Fas ligand (FasL). After 1 h of virus adsorption, H9N2/G1- and H5N1/97-infected Jurkat T cells were cultured in presence of exogenous TNF-α (500 ng/mL), TRAIL (10 ng/mL), or FasL (10 ng/mL), respectively, for another 24-h culture. A significantly higher percentage of apoptotic cells was observed (P<.05). Data are the mean ± SE from 3 independent experiments
Discussion
Accelerated macrophage accumulation was detected in the lungs of patients with fatal H5N1 disease [22] and in primates [23], but their roles were not defined. Here, we used human MDMs as an in vitro model and showed that influenza virus–infected MDMs may exert a cytotoxic effect on lymphocytes. In particular, we demonstrated that T cell apoptosis was mediated by functional TRAIL
TRAIL, a member of the TNF superfamily, is involved in a number of viral infections [12–16, 24, 25]. Its expression is up-regulated directly by the binding of viral hemagglutinin (HA) and neuraminidase of Newcastle virus to the target cells [26] and by viral matrix protein of lyssavirus in the cytoplasm of infected and transfected cells [27]. Alternatively, various cytokines, including IFN-α, IFN-β, IFN-γ, and TNF-α, can modulate the expression of TRAIL [28, 29]. In the present study, we showed that avian influenza virus H9N2/G1 and H5N1/97 significantly up-regulated the expression of TNF-α and TRAIL, but not FasL, in MDMs (figure 1A–1C). Because ∼90% of MDMs were infected at the 12-h POI, the differential expression caused by avian and human influenza viruses was not due to the difference in infectivity. Our findings are supported by those of recent studies that demonstrated that TRAIL was up-regulated in avian influenza virus A/Bratislava/79 (H7N7)–infected epithelial cells [30] and in NK cells and T cells of influenza virus A/PR/8/34 (H1N1)–infected mice [31]
We also demonstrated that the induction of TNF-α and TRAIL mRNA expression is dependent on viral replication. Interestingly, UV-treated H5N1 virus could induce a low but significant expression of TRAIL but not TNF-α mRNA in MDMs (figure 1D and 1E). We have shown previously that the high production of TNF-α in H5N1/97 infection was associated, at least in part, with the viral nonstructural protein [3]. Avian influenza virus H9N2/G1 and H5N1/97 share 6 common internal genes. Their similar ability to induce DRLs suggested that ⩾1 of the internal genes or their proteins were likely to be involved
Consistent with our previous findings [3], there was abundant production of bioactive TNF-α by avian influenza virus–infected MDMs. To determine the mediator of apoptosis other than TNF-α, we selected Jurkat T cells, which are only sensitive to TRAIL- and FasL-mediated apoptosis (figure 2). We documented that the influenza virus–infected MDMs induced Jurkat T cells apoptosis was specially mediated by TRAIL but not by FasL (figures 3 and 5). It has been reported that Fas-FasL was involved in the apoptosis of lymphocytes induced by human influenza virus H1N1- and H3N2-infected monocytes through direct cell-to-cell contact [32]. However, our experiments did not indicate the involvement of FasL in the apoptosis of Jurkat T cells induced by influenza virus–infected MDMs or its supernatant (figures 3 and 5)
The influenza viruses used in the present study infected Jurkat T cells to a similar extent; however, a lower level of apoptosis was detected in avian than in human influenza infection (figure 4). Apoptosis induced by viral infection is a multifactor process in which various influenza viral proteins are involved [33]. The reduced cytopathic effects of avian viruses on Jurkat T cells suggest a possible mechanism of immune evasion for avian viruses
Although there was no significant difference in the level of Jurkat T cell apoptosis when cells were exposed to the supernatants from virus-infected MDMs (figure 3), H5N1/97-infected MDMs exhibited the strongest induction of Jurkat T cell apoptosis via cell-to-cell contact (figure 5C). This higher level of apoptosis in cell-to-cell coculture may be due to (1) direct cytopathic effects caused by the virus, (2) enhanced responses of infected cells to death signals, and/or (3) molecules other than TRAIL
The sensitization of avian influenza virus–infected Jurkat T cells to apoptosis induced by TNF-α, TRAIL, and FasL was observed in our experiments (figure 6). H5-subtype avian influenza virus has a characteristic cleavage-site sequence in the viral HA that renders HA susceptible to the effect of a wide spectrum of cellular proteases and permits H5N1 virus to become infectious to many tissues and organs [34]. Therefore, the sensitization to DRLs in the infected cells is probably common to many cell types in H5N1 infection and may contribute to the severe lung damage or lymphopenia observed clinically. Enhanced sensitization of virus-infected cells to apoptosis induced by TRAIL or FasL has been reported in HIV, reovirus, and respiratory syncytial virus [16, 35–37]. This is caused by the up-regulation of TRAIL-R2 or Fas expression in virus-infected cells that leads to enhanced TRAIL- and FasL-mediated apoptosis. Other mechanisms that are responsible for the modulation of sensitivity or resistance to DRLs include the involvement of TRAIL decoy receptors [38] and the regulation of downstream molecules such as Fas-associated death domain (FADD), caspase 8, cellular FADD-like IL-1β–converting enzyme inhibitory protein, and mitochondrial factors [39]. Further study on the underlying mechanism is needed
In addition to the induction of apoptosis [12–16, 24, 25, 40], TRAIL acts as an important immunomodulator. At an early stage of influenza virus infection, TRAIL may suppress immune responses [31]. It inhibits calcium influx and IL-2–dependent growth in T cell blasts [41, 42] and enhances the elimination of immature dendritic cells by NK cells in vivo [43]. TRAIL has also been reported to enhance the propagation of influenza virus in epithelial cells [30]
The recent episodes of human H5N1 infection—with high mortality, notable lymphopenia, and thrombocytopenia [44]—highlight the urgency to understand its pathogenesis. The production of functional TRAIL and enhanced sensitization to DRLs-induced apoptosis shown in the present study may contribute to disease pathogenesis
Acknowledgment
We thank Winsie Luk, Department of Microbiology, The University of Hong Kong, for her technical assistance
Footnotes
Presented in part: Keystone Symposium 2005 (B2), 5 February 2005, Vancouver, Canada (abstract 450)
Financial support: Research Grants Council, Hong Kong Special Administrative Region, China (project HKU 7532/05M); Outstanding Researcher Awards (to Y.L.L. and J.S.M.P.); University of Hong Kong (Postgraduate Studentship and Vice Chancellor’s Development Fund to J.Z.)
Potential conflicts of interest: none reported
References
- 1.Yuen KY, Chan PK, Peiris M, et al. Clinical features and rapid viral diagnosis of human disease associated with avian influenza A H5N1 virus. Lancet. 1998;351:467–71. doi: 10.1016/s0140-6736(98)01182-9. [DOI] [PubMed] [Google Scholar]
- 2.Tran TH, Nguyen TL, Nguyen TD, et al. Avian influenza A (H5N1) in 10 patients in Vietnam. N Engl J Med. 2004;350:1179–88. doi: 10.1056/NEJMoa040419. [DOI] [PubMed] [Google Scholar]
- 3.Cheung C, Poon L, Lau A, et al. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease. Lancet. 2002;360:1831–7. doi: 10.1016/s0140-6736(02)11772-7. [DOI] [PubMed] [Google Scholar]
- 4.Laudert EA, Sivanandan V, Halvorson DA. Effect of an H5N1 avian influenza virus infection on the immune system of mallard ducks. Avian Dis. 1993;37:845–53. [PubMed] [Google Scholar]
- 5.Tumpey TM, Lu X, Morken T, Zaki SR, Katz JM. Depletion of lymphocytes and diminished cytokine production in mice infected with a highly virulent influenza A (H5N1) virus isolated from humans. J Virol. 2000;74:6105–16. doi: 10.1128/jvi.74.13.6105-6116.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jameson J, Cruz J, Terajima M, Ennis FA. Human CD8+ and CD4+ T lymphocyte memory to influenza A viruses of swine and avian species. J Immunol. 1999;162:7578–83. [PubMed] [Google Scholar]
- 7.Tumpey TM, Renshaw M, Clements JD, Katz JM. Mucosal delivery of inactivated influenza vaccine induces B-cell-dependent heterosubtypic cross-protection against lethal influenza A H5N1 virus infection. J Virol. 2001;75:5141–50. doi: 10.1128/JVI.75.11.5141-5150.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shortridge KF, Zhou NN, Guan Y, et al. Characterization of avian H5N1 influenza viruses from poultry in Hong Kong. Virology. 1998;252:331–42. doi: 10.1006/viro.1998.9488. [DOI] [PubMed] [Google Scholar]
- 9.Shortridge KF. Poultry and the influenza H5N1 outbreak in Hong Kong, 1997: abridged chronology and virus isolation. Vaccine. 1999;17:S26–9. doi: 10.1016/s0264-410x(99)00102-4. [DOI] [PubMed] [Google Scholar]
- 10.Benedict CA, Banks TA, Ware CF. Death and survival: viral regulation of TNF signaling pathways. Curr Opin Immunol. 2003;15:59–65. doi: 10.1016/s0952-7915(02)00018-3. [DOI] [PubMed] [Google Scholar]
- 11.Sheridan JP, Marsters SA, Pitti RM, et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277:818–21. doi: 10.1126/science.277.5327.818. [DOI] [PubMed] [Google Scholar]
- 12.Vidalain PO, Azocar O, Lamouille B, et al. Measles virus induces functional TRAIL production by human dendritic cells. J Virol. 2000;74:556–9. doi: 10.1128/jvi.74.1.556-559.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raftery MJ, Schwab M, Eibert SM, Samstag Y, Walczak H, Schonrich G. Targeting the function of mature dendritic cells by human cytomegalovirus: a multilayered viral defense strategy. Immunity. 2001;15:997–1009. doi: 10.1016/s1074-7613(01)00239-4. [DOI] [PubMed] [Google Scholar]
- 14.Hubert P, Giannini SL, Vanderplasschen A, et al. Dendritic cells induce the death of human papillomavirus-transformed keratinocytes. FASEB J. 2001;15:2521–3. doi: 10.1096/fj.00-0872fje. [DOI] [PubMed] [Google Scholar]
- 15.Muller DB, Raftery MJ, Kather A, Giese T, Schonrich G. Frontline: induction of apoptosis and modulation of c-FLIPL and p53 in immature dendritic cells infected with herpes simplex virus. Eur J Immunol. 2004;34:941–51. doi: 10.1002/eji.200324509. [DOI] [PubMed] [Google Scholar]
- 16.Lichtner M, Maranon C, Vidalain PO, et al. HIV type 1-infected dendritic cells induce apoptotic death in infected and uninfected primary CD4 T lymphocytes. AIDS Res Hum Retroviruses. 2004;20:175–82. doi: 10.1089/088922204773004897. [DOI] [PubMed] [Google Scholar]
- 17.Guan Y, Shortridge KF, Krauss S, et al. H9N2 influenza viruses possessing H5N1-like internal genomes continue to circulate in poultry in southeastern China. J Virol. 2000;74:9372–80. doi: 10.1128/jvi.74.20.9372-9380.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirst GK. Adsorption of influenza hemagglutinins and virus by red blood cells. J Exp Med. 1942;76:195–209. doi: 10.1084/jem.76.2.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Payment P, Trudel M. Isolation and identification of viruses. In: Payment P, Trudel M, editors. Methods and techniques in virology. New York: Marcel Dekker; 1993. pp. 19–38. [Google Scholar]
- 20.Levesque A, Paquet A, Page M. Improved fluorescent bioassay for the detection of tumor necrosis factor activity. J Immunol Methods. 1995;178:71–6. doi: 10.1016/0022-1759(94)00243-p. [DOI] [PubMed] [Google Scholar]
- 21.Law HK, Cheung CY, Ng HY, et al. Chemokine upregulation in SARS coronavirus infected human monocyte derived dendritic cells. Blood. 2005;106:2366–74. doi: 10.1182/blood-2004-10-4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peiris JS, Yu WC, Leung CW, et al. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet. 2004;363:617–9. doi: 10.1016/S0140-6736(04)15595-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rimmelzwaan GF, Kuiken T, van Amerongen G, Besterbroer TM, Fouchier RA, Osterhaus AD. A primate model to study the pathogenesis of influenza A (H5N1) virus infection. Avian Dis. 2003;47:931–3. doi: 10.1637/0005-2086-47.s3.931. [DOI] [PubMed] [Google Scholar]
- 24.Roe MF, Bloxham DM, White DK, Ross-Russell RI, Tasker RT, O’Donnell DR. Lymphocyte apoptosis in acute respiratory syncytial virus bronchiolitis. Clin Exp Immunol. 2004;137:139–45. doi: 10.1111/j.1365-2249.2004.02512.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hensley LE, Young HA, Jahrling PB, Geisbert TW. Proinflammatory response during Ebola virus infection of primate models: possible involvement of tumor necrosis factor receptor superfamilly. Immunol Lett. 2002;80:169–79. doi: 10.1016/s0165-2478(01)00327-3. [DOI] [PubMed] [Google Scholar]
- 26.Zeng J, Fournier P, Schirrmacher V. Induction of interferon-alpha and tumor necrosis factor-related apoptosis-inducing ligand in human blood mononuclear cells by hemagglutinin-neuraminidase but not F protein of Newcastle disease virus. Virology. 2002;297:19–30. doi: 10.1006/viro.2002.1413. [DOI] [PubMed] [Google Scholar]
- 27.Kassis R, Larrous F, Estaquier J, Bourhy H. Lyssavirus matrix protein induces apoptosis by a TRAIL-dependent mechanism involving caspase-8 activation. J Virol. 2004;78:6543–55. doi: 10.1128/JVI.78.12.6543-6555.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sedger LM, Shows DM, Blanton RA, et al. IFN-gamma mediates a novel antivival activity through dynamic modulation of TRAIL and TRAIL receptor expression. J Immunol. 1999;163:920–6. [PubMed] [Google Scholar]
- 29.Kamohara H, Matsuyama W, Shimozato O, et al. Regulation of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) and TRAIL receptor expression in human neutrophils. Immunology. 2004;111:186–94. doi: 10.1111/j.0019-2805.2003.01794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wurzer WJ, Ehrhardt C, Pleschka S, et al. NF-kappaB-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas/FasL is crucial for efficient influenza virus propagation. J Biol Chem. 2004;279:30931–7. doi: 10.1074/jbc.M403258200. [DOI] [PubMed] [Google Scholar]
- 31.Ishikawa E, Nakazawa M, Yoshinari M, Minami M. Role of tumor necrosis factor-related apoptosis-inducing ligand in immune response to influenza virus infection in mice. J Virol. 2005;79:7658–63. doi: 10.1128/JVI.79.12.7658-7663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Human lymphocyte apoptosis after exposure to influenza A virus. J Virol. 2001;75:5921–9. doi: 10.1128/JVI.73.13.5921-5929.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morris SJ, Nightingale K, Smith H, Sweet C. Influenza A virus-induced apoptosis is a multifactorial process: exploiting reverse genetics to elucidate the role of influenza A virus proteins in virus-induced apoptosis. Virology. 2005;335:198–211. doi: 10.1016/j.virol.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 34.Tollis M, Di Trani L. Recent developments in avian influenza research epidemiology and immunoprophylaxis. Vet J. 2002;164:202–15. doi: 10.1053/tvjl.2002.0716. [DOI] [PubMed] [Google Scholar]
- 35.Clarke P, Meintzer SM, Gibson S, et al. Reovirus-induced apoptosis is mediated by TRAIL. J Virol. 2000;74:8135–9. doi: 10.1128/jvi.74.17.8135-8139.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Oliveira Pinto LM, Garcia S, Lecoeur H, Rapp C, Gougeon ML. Increased sensitivity of T lymphocytes to tumor necrosis factor receptor 1 (TNFR1)- and TNFR2-mediated apoptosis in HIV infection: relation to expression of Bcl-2 and active caspase-8 and caspase-3. Blood. 2002;99:1666–75. doi: 10.1182/blood.v99.5.1666. [DOI] [PubMed] [Google Scholar]
- 37.Kotelkin A, Prikhod’ko EA, Cohen JI, Collins PL, Bukreyev A. Respiratory syncytial virus infection sensitizes cells to apoptosis mediated by tumor necrosis factor-related apoptosis-inducing ligand. J Virol. 2003;77:9156–72. doi: 10.1128/JVI.77.17.9156-9172.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang SL, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22:8628–33. doi: 10.1038/sj.onc.1207232. [DOI] [PubMed] [Google Scholar]
- 39.Suliman A, Lam A, Datta R, Srivastava RK. Intracellular mechanisms of TRAIL: apoptosis through mitochondrial-dependent and independent pathways. Oncogene. 2001;20:2122–33. doi: 10.1038/sj.onc.1204282. [DOI] [PubMed] [Google Scholar]
- 40.Zheng SJ, Jiang J, Shen H, Chen YH. Apoptosis and ameliorated listeriosis in TRAIL-null mice. J Immunol. 2004;173:5652–8. doi: 10.4049/jimmunol.173.9.5652. [DOI] [PubMed] [Google Scholar]
- 41.Lunemann JD, Waiczies S, Ehrlich S, et al. Death ligand TRAIL induces no apoptosis but inhibits activation of human (auto)antigen-specific T cells. J Immunol. 2002;168:4881–8. doi: 10.4049/jimmunol.168.10.4881. [DOI] [PubMed] [Google Scholar]
- 42.Bosque A, Pardo J, Martinez-Lorenzo MJ, et al. Down-regulation of normal human T cell blast activation: roles of APO2L/TRAIL, FasL, and c-FLIP, Bim, or Bcl-x isoform expression. J Leukoc Biol. 2005;77:568–78. doi: 10.1189/jlb.0904514. [DOI] [PubMed] [Google Scholar]
- 43.Hayakawa Y, Screpanti V, Yagita H, et al. NK cell TRAIL eliminates immature dendritic cells in vivo and limits dendritic cell vaccination efficacy. J Immunol. 2004;172:123–9. doi: 10.4049/jimmunol.172.1.123. [DOI] [PubMed] [Google Scholar]
- 44.Beigel JH, Farrar J, Han AM, et al. Consultation on human influenza A/H5: avian influenza A (H5N1) infection in humans. N Engl J Med. 2005;353:1374–85. doi: 10.1056/NEJMra052211. [DOI] [PubMed] [Google Scholar]