

Abstract
Wiskott-Aldrich syndrome (WAS) is associated with thrombocytopenia of unclear origin. We investigated real-time cytosolic calcium dynamics, mitochondrial membrane potential and phoszphatidylserine (PS) exposure in single fibrinogen-bound platelets using confocal microscopy. The WAS platelets had higher resting calcium levels, more frequent spikes, and their mitochondria more frequently lost membrane potential followed by PS exposure (in 22.9% of platelets vs. 3.9% in controls; P<0.001) after the collapse of the last mitochondria. This phenomenon was inhibited by the mitochondrial permeability transition pore inhibitor cyclosporine A, as well by xestospongin C and lack of extracellular calcium. Thapsigargin by itself caused accelerated cell death in the WAS platelets. The number of mitochondria was predictive of PS exposure: 33% of platelets from WAS patients with fewer than five mitochondria exposed PS, while only 12% did among those that had five or more mitochondria. Interestingly, healthy donor platelets with fewer mitochondria also more readily became procoagulant upon PAR1/PAR4 stimulation. Collapse of single mitochondria led to greater cytosolic calcium increase in WAS platelets if they had one to three mitochondria compared with platelets containing higher numbers. A computer systems biology model of platelet calcium homeostasis showed that smaller platelets with fewer mitochondria could have impaired calcium homeostasis because of higher surface-to-volume ratio and greater metabolic load, respectively. There was a correlation (C=0.81, P<0.02) between the mean platelet size and platelet count in the WAS patients. We conclude that WAS platelets readily expose PS via a mitochondria-dependent necrotic mechanism caused by their smaller size, which could contribute to the development of thrombocytopenia.
Introduction
Wiskott-Aldrich syndrome (WAS) is an X-linked disorder classically characterized by thrombocytopenia, immunodeficiency and eczema.1 Its pathophysiological mechanisms relate to defective actin polymerization and abnormal signal-mediated cytoskeleton rearrangements in hematopoietic cells as a result of deficient or dysregulated activity of the WAS protein that belongs to a distinct family of proteins involved in the transduction of signals from the cell surface to the actin cytoskeleton.2 The severity of immunodeficiency varies between WAS patients, whereas the platelet defect (reduced number and size) is the universal feature of the disease, and thrombocytopenia-related bleeding contributes greatly to mortality in untransplanted patients.3 Although major platelet functions are retained in WAS platelets, there is evidence of defects in these platelets that could potentially contribute additionally to bleeding.4
The specific mechanisms of thrombocytopenia in WAS remain elusive. Studies of megakaryocytes from patients produced evidence both in favor of defects in platelet production5 and against them.6 On the other hand, platelets from WAS patients and murine WAS protein knockouts had shortened lifespan and were subject to increased phagocytosis.7–9 In particular, it was shown previously that WAS platelets have increased phosphatidylserine (PS) exposure upon storage and activation,10,11 which could be one of the mechanisms involved in their accelerated clearance by splenic macrophages and possibly contribute to thrombocytopenia. Indeed, recent evidence from diverse eukaryotic systems suggests that the actin cytoskeleton has a role in regulating apoptosis via interactions with mitochondria.12 Changes to the dynamics of the actin cytoskeleton were implicated in the release of reactive oxygen species (ROS) from mitochondria and subsequent cell death.13 Interestingly, recent studies discovered that platelets from patients with deficiency of actin filament branching regulator Arp2/3 have major phenotype features similar to those observed in WAS:14,15 microthrombocytopenia, deficiency of dense granules and spreading.
Here we investigated the mechanism underlying cell death in platelet samples from a cohort of 35 WAS patients. The main conclusion is that the platelets are prone to PS exposure upon minor stimulation, which occurs as a result of mitochondrial permeability transition pore opening. We provide evidence for the two major mechanisms responsible for this: (i) an increased surface-to-volume ratio of these micro-platelets leading to dysregulation of calcium homeostasis; (ii) a decreased number of mitochondria per platelet, which results in a dramatic increase of cytosolic calcium upon opening of the mitochondrial permeability transition pores in a single mitochondrion. Platelet size correlated with platelet count in the untreated WAS patients.
Methods
A full description of the methods and reagents is available in the Online Supplementary Material.
Patients and healthy donors
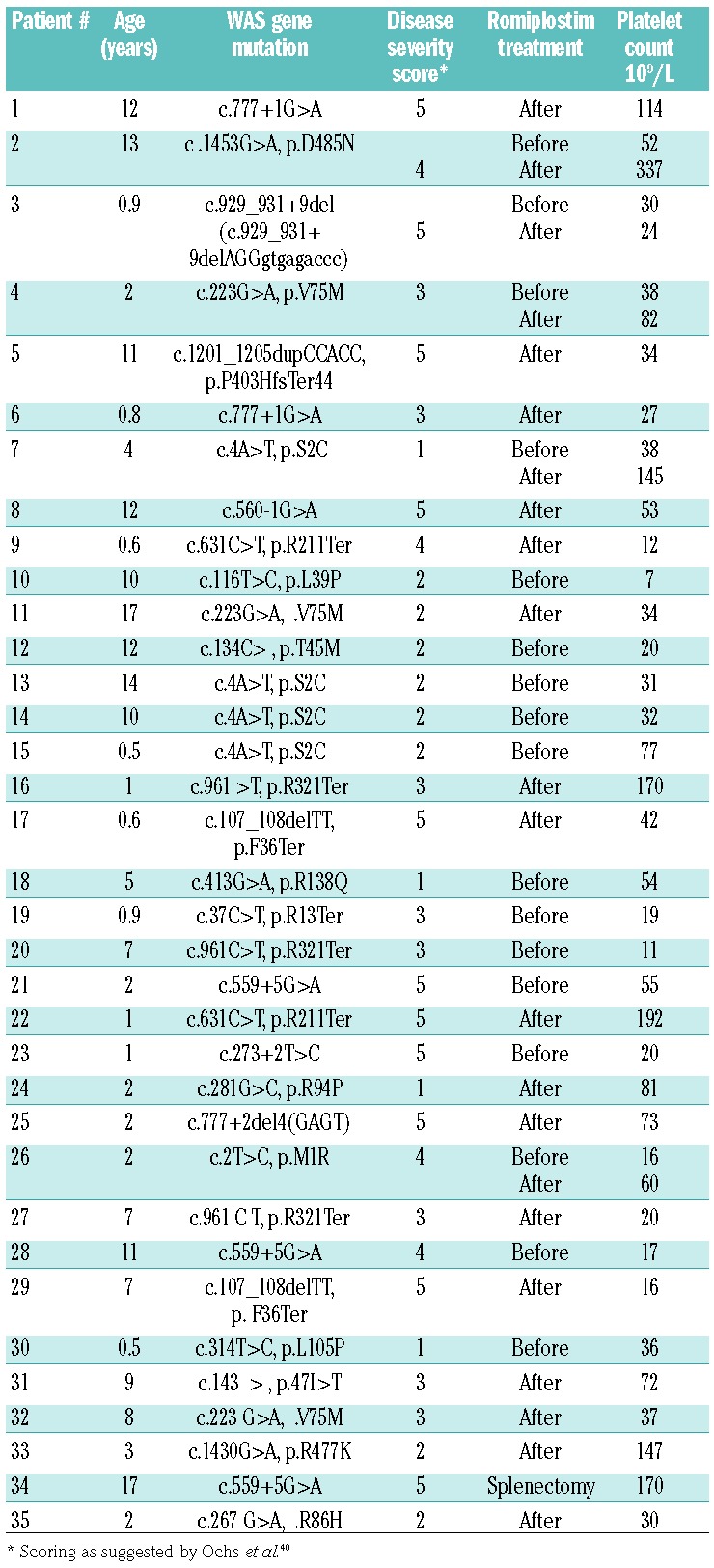
A total of 35 patients with WAS were included in the study (Table 1). The diagnosis was made according to the diagnostic criteria of the European Society for Immunodeficiencies and genetically confirmed by identification of WAS mutations. Romiplostim was administered off-label at a dose of 9 μg/kg weekly, according to an institutional protocol. Twelve of 35 patients had a Zhu score of 1 or 2. Control samples included blood from children and healthy adults, as indicated in the experimental descriptions.
Table 1.
Characteristics of the patients with Wiskott-Aldrich syndrome.
Blood collection and platelet isolation
Investigations were performed in accordance with the Declaration of Helsinki under approval of the Children’s Center for Hematology Ethical Committee, and written informed consent was obtained from all patients (or their parents) and donors. Washed platelets were prepared essentially as described previously.17
Confocal microscopy experiments: general design
Glass coverslips were cleaned and coated with 1 mg/mL fibrinogen or monafram in phosphate-buffered saline. Washed platelets were attached to the protein-coated surface by incubating them at 1.3×105/μL (or the maximal concentration attainable for WAS under conditions of thrombocytopenia) for 20 min and rinsing with buffer A with 1.5 mM CaCl2. The PS+ fraction was counted after incubation for a further 30 min.
Cytosolic calcium signaling and mitochondrial membrane potential change
The methodology for these investigations was essentially as described elsewhere.18 Calibration for ratiometric measurements was performed separately for healthy and WAS platelets. Calcium concentrations were calculated using an equation for ratiometric indicators.19 Tetramethylrhodamine methyl ester (TMRM) was used to detect mitochondrial potential dynamics.
Characterization of platelet response to TRAP-6 in platelet-rich plasma
Samples of platelet-rich plasma were diluted with platelet-poor plasma to a final concentration of 20,000/mL and buffered with HEPES at pH 7.4 (100 μM final concentration). The irreversible thrombin inhibitor Phe-Pro-Arg chloromethyl ketone (PPACK) was added to the final concentration of 100 μM in order to block spontaneous thrombin generation upon recalcification. Platelets were recalcified by addition of calcium chloride (final concentration 20 mM, which corresponds to 2 mM of free calcium20) and activated by 25 μM TRAP-6 (thrombin receptor agonist peptide-6) for 5 min at room temperature.
ATP measurement in platelets
In brief, platelets of the samples were lyzed (by addition of 90 μL dimethylsulfoxide to 10 μL platelet suspension) and analyzed by luciferase-luciferin assay as described previously21,22 while the other part was stained with CD61-FITC and annexin V-Alexa Fluor 647 and analyzed by flow cytometry.
Flow cytometry characterization of platelet functional activity
The experiments were performed essentially as described previously23,24 with minor modifications.
Transmission electron microscopy
The protocol was essentially as described elsewhere.25
Statistics
Data are presented as means ± standard deviations. The statistical significance of the differences between groups was determined with the non-parametric Mann–Whitney U-test (P) or Wilcoxon signed-rank test (P*) for paired samples. Differences were considered to be statistically significant when the P-value was <0.05.
Computational modeling of platelet calcium homeostasis
A systems biology model of platelet calcium signaling was based on one developed previously.18 In contrast to its predecessor pure calcium signaling/homeostasis models,26,27 it had several mitochondrial compartments and included equations describing dependence of ATP production and calcium pump activity on mitochondrial inner membrane potential.
Results
Phosphatidylserine exposure by fibrinogen-immobilized Wiskott-Aldrich syndrome platelets
In order to gain insight into the mechanisms of increased/accelerated PS exposure of WAS platelets, we investigated dynamics of status change by the single fibrinogen-bound cells (Figure 1A). Unexpectedly, this relatively mild method of platelet immobilization produced massive spontaneous PS exposure in the WAS platelets during 30 min without any additional stimulation compared with PS exposure on platelets from either healthy adults (n=18) or children without WAS (n=6, aged 0-7 years, median 2.5 years) (Figure 1B). The smaller number of adherent platelets in some of the WAS patients could lead to some underestimation of their PS+ platelets, so that the effect could be even greater. Taking into account that some of the patients received romiplostim, previously shown to potentially affect platelet function.28,29 it was reasonable to evaluate its effect on platelets separately.30,31 However, there was no statistically significant difference between PS externalization by platelets from untreated WAS patients and from those on romiplostim (Figure 1C): in both groups, approximately 20% of platelets, on average, exposed PS. The phenomenon was not fibrinogen-specific, as platelets attached to the αIIbβ3 antagonist monafram produced the same results (Figure 1D).
Figure 1.

Exposure of phosphatidylserine by Wiskott-Aldrich syndrome platelets upon fibrinogen binding. (A) Confocal microscopy images of healthy (left) and Wiskott-Aldrich syndrome (WAS) (right) platelets after spreading for 30 min on a fibrinogen surface in the presence of 1.5 μM Ca2+. The platelets are labeled with CD61 (green) and annexin V (red); scale bar: 10 μm. (B) Phosphatidylserine-positive (PS+) fraction of platelets from the WAS patients (27 patients, >7,500 cells), adult healthy (18 donors, >6,500 cells) and 0- to 7-year old children without WAS (6 children, age: 0, 0, 2, 3, 4, 7 years, 2,300 platelets) on a fibrinogen surface. (C) PS+ fraction of WAS platelets on the fibrinogen surface, showing a comparison of romiplostim-treated and untreated WAS patients, P=0.94. (D) Monafram-coated coverslips did not change the PS+ fraction, P=0.86, n=4, 2,500 platelets. w/o: without; FG: fibrinogen.
Functional activity of the Wiskott-Aldrich syndrome platelets
To thoroughly characterize the status of the WAS platelets involved in the study, we analyzed them by diluted whole blood flow cytometry using a comprehensive set of functional markers (Figure 2 and Online Supplementary Figure S1). Platelets were either left in the resting state or subjected to potent dual stimulation with TRAP-6 and collagen-related peptide (CRP). As a control, we used a group of 20 healthy children (9 boys and 11 girls, aged 0-13 years, median 5 years). The WAS platelets had significantly decreased forward scatter and levels of major surface glycoproteins (Figure 2A-C) reflecting their decreased size. There were two interesting exceptions: patients 5 and 18 had normal forward scatter. Patient 5 was splenectomized, which could be a plausible explanation of his larger platelets, while patient 18 had an exceptionally mild WAS phenotype. The size-independent parameter of shape change evaluated as the light scattering ratios for the resting/stimulated platelets (Online Supplementary Figure S1A, B) was significantly decreased in WAS. In order to take into account the difference in platelet size and surface area, we evaluated integrin αIIbβ3 activation and α-granule release by either the percentage of PAC1+ (Figure 2D) or CD62P+ (Figure 2E) platelets, or by normalizing the data on CD61 fluorescence intensity, as described elsewhere31 (Online Supplementary Figure S1). In both cases, the response to activation was somewhat decreased in WAS platelets compared with healthy platelets. However, there was a relative increase in baseline platelet activation in the WAS patients compared with the controls, as judged by integrin (Online Supplementary Figure S1D) and P-selectin (Online Supplementary Figure S1F) surface expression of resting platelets. Dense granule release in WAS was essentially lower than in healthy children (Figure 2F). Interestingly, the completely size-independent response of procoagulant platelet formation evaluated as a percentage of annexin V+ cells was also clearly decreased (Figure 2G). Neither of these phenomena changed in the romiplostim-treated patients compared with untreated ones. Analysis with TRAP stimulation in diluted platelet-rich plasma revealed a minor increase in PS+ platelets under resting conditions and normal PS expression upon stimulation (Online Supplementary Figure S2). In summary, WAS platelets were small and demonstrated some decreased preactivation features in the resting state but they did not appear to have any drastic functional differences from normal platelets in an activated state.
Figure 2.

Functional response of the Wiskott-Aldrich syndrome and healthy platelets. (A-G) Whole blood platelets were stimulated (designated by A) or not (designated N/A) with thrombin receptor agonist peptide-6 plus collagen-related peptide and analyzed by flow cytometry. Parameters shown for healthy children (n=21, age 0-13 years, median 5.0) and Wiskott-Aldrich patients (17 treated with romiplostim, 11 non-treated) are: platelet size, determined from the forward scatter measured by mean fluorescence intensity (MFI) (A); CD42b level, MFI (B); CD61 level, MFI (C); PAC1-positive platelets, % (D); CD62p-positive platelets, % (E); dense granule release determined by mepacrine level, MFI (F); and phosphatidylserine-positive platelet fraction, % (G). P: Mann–Whitney U-test, P*:Wilcoxon signed-rank test. FSC: forward scatter, WAS: Wiskott-Aldrich syndrome; PS+: phosphatidylserine-positive.
Signaling events in single fibrinogen-attached Wiskott-Aldrich syndrome platelets
To identify the mechanisms of the PS externalization of the WAS platelets, we simultaneously examined dynamics of calcium in the cytosol, mitochondrial membrane potential and PS exposure in WAS and control platelets (Figure 3). The mean intracellular cytosolic calcium level in the WAS platelets was 4-fold greater than that in the control platelets at the beginning, and the average difference increased with time (Figure 3A). While healthy unstimulated platelets, in line with previous reports,18,32 had only occasional calcium spikes when bound to fibrinogen (Figure 3B), unstimulated platelets from WAS patients had frequent oscillations with longer spike duration (Figure 3C, D). The mitochondria in the WAS platelets lost their membrane potential one after another and, if all of them became TMRM-negative, the cell began to bind annexin V within 10 s (Figure 3D), exactly as reported before for the PS+ platelet formation induced in healthy donors with TRAP-6 or thrombin.18,27 This is in agreement with the scenario of mitochondrial calcium-overloading-induced necrosis of procoagulant platelet formation.18,33,34
Figure 3.

Dynamics of cytoplasmic calcium, mitochondrial potentials and phosphatidylserine exposure of single platelets. Plots show dynamics of intracellular calcium concentration and annex-in V binding to single platelets during incubation on fibrinogen in the presence of 1.5 mM of extracellular calcium. (A) Averaged calcium dynamics (± standard deviation) for non-activated (N/A) platelets from patients with Wiskott-Aldrich syndrome (WAS) (4 patients, 34 platelets) and for platelets from healthy donors (HD) which were either activated with 10 μM TRAP (3 HD, 30 platelets) or not activated (3 HD, 26 platelets). (B) Dynamics for a single healthy phosphatidylserine-negative (PS−) platelet; (C) the same for a WAS PS− platelet; (D) the same for a WAS PS+ platelet. The TMRM signal is represented as a number of TMRM-positive mitochondria in the platelet (B-D). Intracellular events leading to PS exposure induced with mitochondria collapse with following cytoplasmic calcium increase and PS exposure. All three processes were almost simultaneous, lasting decades of seconds. Both WAS (E) and normal (not shown) PS+ platelets lost their mitochondrial potentials. Scale bar: 1 μm for all microscopic images. TRAP: thrombin receptor agonist peptide; TMRM: tetramethylrhodamine methyl ester.
Importantly, time lapse imaging revealed that mitochondrial collapse in WAS platelets with few mitochondria in turn led to a rapid cytosolic calcium increase (Online Supplementary Figure S3A, time point 116 s). If it was reversed, calcium concentration also decreased (Online Supplementary Figure S3A, time point 146 s). This is drastically different from healthy donors’ activated platelets, in which calcium was not so sensitive to the collapse of a single mitochondrion.18 Interestingly, WAS platelets with large numbers of mitochondria had increased background cytosolic calcium and frequent oscillations, but were not sensitive to the collapse of single mitochondria either (Online Supplementary Figure S3B).
Phosphatidylserine exposure on Wiskott-Aldrich syndrome platelets is mediated by mitochondrial permeability transition pore opening
The critical element of mitochondrially driven necrosis is mitochondrial permeability transition pore opening. To check this, we added several inhibitors of different cell death-regulating signaling pathways during platelet incubation on fibrinogen (cyclosporine A, necrostatin-1, Z-VAD-FMK) or spreading (calpeptin). The mitochondrial permeability transition pore inhibitor cyclosporine A (5 μM) significantly diminished the PS+ fraction formed by the WAS platelets on fibrinogen (Figure 4A). Other inhibitors, including necroptosis inhibitor necrostatin-1, calpain inhibitor calpeptin and pan-caspase inhibitor Z-VAD-FMK, had no significant effect on PS exposure (Figure 4B). These data strongly suggest that it is indeed the necrotic mechanism of mitochondrial permeability transition pore opening that is responsible for increased PS exposure in WAS platelets.
Figure 4.

Prevention of mitochondrial permeability transition pore opening affects spontaneous phosphatidylserine exposure of Wiskott-Aldrich syndrome platelets. (A) Phosphatidylserine (PS) exposure of fibrinogen-spread platelets incubated in the absence or presence of 5 μM cyclosporine A (3,900 platelets from 11 patients and 4,000 platelets from 6 healthy donors). (B) PS exposure of platelets incubated with DMSO or programmed cell death inhibitors calpeptin (200 μM, 20 min incubation), Nec-1 (50 μM, 50 min incubation) and Z-VAD-FMK (50 μM, 50 min incubation) (100-300 platelets were observed for each dot). (C) Modulation of intracellular calcium signaling in spread platelets by xestospongin C (3 μM, 50 min); thapsigargin (TG, 1 μM, 30 min); with lactadherin and without addition of 1.5 mM CaCl2 (n=5, 6,800 platelets). (D) Flow cytometry analysis of Wiskott-Aldrich syndrome (WAS) platelet PS exposure. Incubation of platelets in suspension with 1 mM TG in the presence of 1.5 mM CaCl2 for 10 min induced a PS+ platelet fraction comparable to that with fibrinogen-spreading for both WAS patients (n=7, mean ± standard deviation: 19.7%±11.8%) and healthy donors (n=11, 6.6%±8.0%). (E) Flow cytometry analysis of WAS platelet PS exposure in suspension without addition of CaCl2(n=3); (F) Analysis of the mitochondrial inhibitors in suspension at TG treatment (n=3); (G, H) ATP levels (G) versus the number of PS+ platelets (H): in healthy donors and WAS patients at TG and CCCP treatment (n=3). CsA: cyclosporine A, DMSO: dimethylsulfoxide; HD: healthy donor.
Role of calcium homeostasis, cellular energetics, and reactive oxygen species in phosphatidylserine exposure by Wiskott-Aldrich syndrome platelets
In order to get further insight into the necrosis of the surface-attached WAS platelets, they were treated with xestospongin C (an inositol trisphosphate receptor blocker), or thapsigargin (a sarco-endoplasmic reticulum Ca2+ ATPase inhibitor), or in buffer A without addition of calcium chloride (Figure 4C). Xestospongin C inhibited PS exposure suggesting involvement of inositol trisphosphate signaling, while thapsigargin potently boosted platelet necrosis. In contrast, the effects were drastically decreased in the absence of extracellular calcium.
Importantly, thapsigargin caused accelerated cell death in the WAS platelets compared with platelets from healthy controls in suspension as well without any surface attachment (Figure 4D), which suggests that the WAS platelets’ propensity to necrosis is caused by dysregulation of their calcium homeostasis. The same experiment with lactadherin and without addition of extracellular calcium did not show an increased PS+ fraction of WAS platelets (Figure 4E). For an additional check of the effect of outside-in signaling on thapsigargin-induced PS exposure in this design, we pre-treated platelets with the integrin αIIbβ3 antagonist monafram which did not affect the thapsigargin-induced PS exposure (Online Supplementary Figure S4). Pre-incubation of the WAS platelets with the mitochondrial ATPase inhibitor oligomycin or with the mitochondrial uncoupler CCCP increased the formation of PS+ platelets at thapsigargin treatment in the case of WAS platelets, while the mitochondrial respiratory chain complex I inhibitor rotenone had less effect on the thapsigargin-induced PS exposure (Figure 4F); none of these three drugs caused platelet necrosis by themselves. These data indicate that an energy deficiency could be a factor contributing to platelet necrosis but not the defining one. In line with this, although the levels of ATP in cells were decreased in parallel with the increase of the PS+ platelets upon thapsigargin treatment, the same decrease of ATP was caused by CCCP without PS exposure indicating that the observed phenomenon is not purely caused by an energy collapse (Figure 4G, H). ROS production in the WAS platelets was not essentially different from that in healthy donor platelets, and was only mildly increased upon stimulation with CRP (Online Supplementary Figure S5). The morphology of the mitochondria in WAS platelets was not apparently different from that of normal ones, as judged by transmission electron microscopy (Online Supplementary Figure S6).
Platelet necrosis correlates directly with the number of mitochondria
During examination of the images, it became apparent that the WAS platelets undergoing PS exposure and mitochondrial membrane potential loss rarely had more than two mitochondria per cell. We, therefore, performed experiments to count the number of mitochondria in each platelet and correlated this with the outcome (i.e. PS exposure) (Figure 5). For both WAS patients and healthy donors, the number of mitochondria was significantly lower in the platelets that became PS+ (Figure 5A). This number affected the fate of platelets in a dose-dependent manner: about 33% of the WAS platelets exposed PS if they had one to four mitochondria per platelet, and only about 11% if they had more than five mitochondria (Figure 5B). A similar dependence was observed for platelets from healthy donors (Figure 5B), although they exposed PS more rarely. The histogram in Figure 5C shows the distributions of mitochondria number for platelets from WAS patients and healthy donors side by side. Importantly, although the mean number of mitochondria in WAS platelets was not much lower than that in the control platelets, there was significant skewing to the left of the curve: a total of 27±12% of WAS platelets had fewer than three mitochondria, compared to only 8.7±4.4% of healthy platelets. In order to check if the number of mitochondria has a wider significance in platelet necrosis, we performed experiments with fibrinogen-attached healthy platelets stimulated with TRAP-6 or thrombin, revealing the same pattern (Figure 5D, E).
Figure 5.

Dependence of phosphatidylserine exposure on mitochondria count. Platelets that exposed phosphatidylserine (PS) during incubation on fibrinogen contained significantly fewer mitochondria than PS- cells. (A) Mean mitochondria number in platelet subpopulations per patient with Wiskott-Aldrich syndrome (WAS) or per healthy donor (HD) for non-activated (N/A) fibrinogen-bound platelets. Each dot represents one WAS patient (7 patients, 381 platelets) or HD (n=4, 567 platelets). (B) Averaged PS+ fraction ± standard deviation of the same WAS and HD platelets with different mitochondrial counts. (C) Averaged distribution of mitochondria per platelet (both subpopulations) for WAS patients (7 patients, 381 platelets) and HD (11 HD, 1,179 platelets). (D, E) Healthy activated platelets, overall 613 cells from seven HD activated with TRAP-6 (n=5, 306 cells) or thrombin (n=4, 307 cells). Platelets most likely to expose PS had fewer mitochondria. Mitochondria were counted by TMRM fluorescence using a microscope after spreading for 20 min (before activation in experiments with activated platelets from HD); subpopulation were determined after an additional 30 min incubation. Each dot represents the mean of the mitochondria count in a patient or HD (A,C). P: Mann–Whitney U-test. TRAP-6: thrombin receptor agonist peptide-6: TMRM: tetramethylrhodamine methyl ester.
Systems biology simulations reveal critical roles of mitochondrial number and surface-to-volume ratio in programmed cell death in Wiskott-Aldrich syndrome
In order to dissect the mechanisms of mitochondria-dependent necrosis in WAS, we developed a computational systems biology model of calcium signaling (Figure 6). In the model, which incorporated all compartments and major calcium signaling mechanisms, we investigated dependence of the platelet calcium response on two major variables that differ for the WAS platelets, the number of mitochondria and platelet size.
Figure 6.

Increased cytosolic calcium as a result of downsizing: computer systems biology simulation of calcium signaling in normal and Wiskott-Aldrich syndrome platelets. Wiskott-Aldrich syndrome (WAS) platelets were assumed to have the same content of signaling proteins, scaled to the respective volume of compartments. (A, B) Stochastic simulation of the activation of normal platelets containing two (A) or four (B) mitochondria with 10 nM thrombin. With the collapse of one mitochondrion the average cytosolic calcium increases 1.5-fold (A) in the case of two mitochondria or does not change (B) in the case of four mitochondria. (C-E) Stochastic and deterministic simulations of normal and WAS platelets stimulated with 1 nM thrombin.
The model demonstrated that a decrease in the number of mitochondria should make platelets more sensitive to mitochondrial collapse and result in a higher increase in calcium because the remaining mitochondria could not bear the ATP production load (Figure 6A, B), which agrees well with the experimental observations. We also simulated platelets of different sizes; when scaling them, the ratio between surface and volume molecules was naturally changed (Figure 6), as volume is proportional to the size to the third degree, while surface is proportional to the size to the second degree. Upon stimulation, the virtual platelets with a smaller size had comparable active phospholipase C per volume (Figure 6C), but more inositol trisphosphate and ultimately much more calcium (Figure 6E) because they had more inositol trisphosphate receptors per volume (as these were assumed to be proportional to the surface). This is again in line with the experimental data presented above that showed increased calcium levels in WAS even prior to mitochondrial permeability transition pore opening, and with the sensitivity of the phenomenon to xestospongin C.
The model, therefore, predicted that the size of platelets from untreated WAS patients would negatively affect the platelets’ ability to expose PS spontaneously and (if this is the mechanism underlying thrombocytopenia) positively affect the patients’ platelet count. Interestingly, there was a significant positive correlation between platelet size and platelet count among the untreated WAS patients (Online Supplementary Figure S7A). Although we did not observe significant correlations with PS exposure, probably as a result of the limited number of samples (Online Supplementary Figure S7B, C), it is interesting that patient #18 (indicated with a red arrow), who had normal-sized platelets and a mild phenotype, also had the least PS exposure upon immobilization and incidentally the highest platelet count.
Discussion
In this study, we show that the death of WAS platelets upon minor stimulation, such as fibrinogen attachment or low-dose thapsigargin treatment, follows the pathway of mitochondrial necrosis. It is a rapid process associated with opening of mitochondrial permeability transition pores, which actually precedes PS exposure at the single platelet level, extracellular calcium-dependent, and it is downregulated by cyclophilin D and inositol trispohosphate receptor antagonists, but not by apoptosis or necroptosis inhibitors. It is associated with decreased platelet ATP levels, and downregulation of the energy metabolism with CCCP, rotenone and oligomycin-promoted necrosis, but did not cause it by itself. This cell death phenomenon predominantly occurred in the platelet fraction with fewer than four mitochondria per platelet. Importantly, although beyond the scope of this WAS study, the low number of mitochondria turned out to be predictive of the agonist-induced formation of pro-coagulant platelets by healthy donors. The immediate causes of this necrosis are: (i) increased calcium concentration and spiking frequency of WAS platelets upon spreading; and (ii) increased sensitivity of calcium homeostasis to collapse of single mitochondria in platelets with fewer mitochondria. Computational systems biology analysis confirmed that both increased surface-to-volume ratio (leading to impaired calcium homeostasis) and a lower number of mitochondria (resulting in increased sensitivity of calcium to mitochondrial collapse) contribute to the tendency of the WAS platelets to undergo necrosis upon minor stimulation. Although the clinical consequences of this phenomenon are beyond the scope of this paper, this mechanism is supported by the observation of a correlation between platelet size and platelet count in WAS. In contrast to the observations for immune thrombocytope-nia,29,35 there were no statistically significant changes in WAS platelet functionality upon romiplostim treatment.
The phenomena investigated in the present study agree well with previous observations by Shcherbina et al.10,11 who reported increased, accelerated or spontaneous PS exposure by the platelets of WAS patients or WAS knockout mice associated with increased calcium levels at rest, and provide a molecular basis for the previous reports. The mechanism of this massive PS exposure in WAS platelets appears to be essentially similar to that determining agonist-induced procoagulant platelet formation in physiological potent platelet activation:18,26,33,34,36,37 an increase of cytosolic calcium followed by mitochondrial calcium overload and collapse, ultimately leading to necrotic cell death. The difference was that PS exposure in WAS was triggered by weak stimuli, such as fibrinogen attachment, which, although recognized as being an activating stimulus,38 produced negligible PS exposure in healthy donor platelets by itself. A high percentage of PS+ platelets seems to be a universal feature of the disease, irrespective of its severity and absence\presence of other WAS features.
The similarity between these phenomena (PS exposure by normal platelets via thrombin and/or collagen receptors and PS exposure by WAS platelets induced by fibrinogen attachment) goes so far that the phenomenon of predominant necrosis by platelets with fewer mitochondria was observed here for TRAP-6- or thrombin-stimulated healthy platelet activation as well. This is interesting in itself and might have implications beyond the scope of the present study: although several previous studies attempted to identify properties of platelets that predispose them to procoagulant formation such as age or resting calcium concentration,18,33,39 the effects were much less than those of the number of mitochondria, and it has been generally assumed in the field that it is unclear which platelets become necrotic and which do not.
Although the data of the present study clearly characterize the immediate molecular cause and sequence of events leading to spontaneous PS exposure in WAS platelets, they are more limited with regard to linking this phenomenon to the genetic cause of the disease or clinical consequences. While a decreased number of mitochondria in the WAS platelets (natural because of their decreased size) is likely to contribute to their tendency to undergo necrosis, this difference by itself is not great enough to have such drastic consequences. Non-stimulated WAS platelets undergo necrosis more efficiently than non-stimulated healthy platelets having the same number of mitochondria (Figure 5), so there should be additional mechanisms. The most promising one is disruption of cytosolic calcium balance (even without regard to mitochondria) simply because of the greater surface-to-volume ratio in WAS platelets: computer systems biology simulations indicated that this mechanism alone would be sufficient to explain the platelets’ necrosis. This hypothesis is supported by the sensitivity of the PS+ fraction to xestospongin C and a decrease in extracellular calcium, as well as by the ability of the calcium pump inhibitor thapsigargin to promote WAS platelet necrosis much more rapidly than that of healthy donor platelets. The inability of mitochondrial function antagonists to cause necrosis by itself is also in line with the proposed picture of events. However, we did not show a causal relationship between size and calcium in direct experiments, and cannot exclude participation of other contributing factors. Furthermore, although it is tempting to speculate that increased PS exposure may promote platelet clearance by macrophages,10,11 the statistics of the present study are not sufficient to confirm or disprove a relationship between clinical severity and the tendency of platelets to expose PS, despite showing a relationship between platelet count and platelet size. Involvement of impaired actin cytoskeleton dynamics due to WAS protein mutations in the programmed cell death of WAS platelets cannot be excluded either, and requires additional research.
Acknowledgments
We thank Prof. A.V. Mazurov and Prof. R.W. Farndale for their kind gifts of monafram and CRP. The project was supported by a grant from the endowment foundation "Doctors, Innovations, Science for Children", and by the Russian Foundation for Basic Research grants 17-00-00141 (17-00-00138/17-00-00139/17-00-00140), 17-04-01309 and 18-34-20026. Electron microscopy was supported by the Russian Science Foundation grant 17-15-01290. Computer modeling was supported by Russian Science Foundation grant 17-74-20045.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/105/4/1095
References
- 1.Candotti F. Clinical manifestations and pathophysiological mechanisms of the Wiskott-Aldrich syndrome. J Clin Immunol. 2018;38(1):13–27. [DOI] [PubMed] [Google Scholar]
- 2.Rivers E, Thrasher AJ. Wiskott-Aldrich syndrome protein: emerging mechanisms in immunity. Eur J Immunol. 2017;47(11):1857–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sereni L, Castiello MC, Villa A. Platelets in Wiskott-Aldrich syndrome: victims or executioners¿ J Leukoc Biol. 2018;103(3):577–590. [DOI] [PubMed] [Google Scholar]
- 4.Poulter NS, Pollitt AY, Davies A, et al. Platelet actin nodules are podosome-like structures dependent on Wiskott-Aldrich syndrome protein and ARP2/3 complex. Nat Commun. 2015;6:7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kajiwara M, Nonoyama S, Eguchi M, et al. WASP is involved in proliferation and differentiation of human haemopoietic progenitors in vitro. Br J Haematol. 1999;107(2):254–262. [DOI] [PubMed] [Google Scholar]
- 6.Haddad E, Cramer E, Riviere C, et al. The thrombocytopenia of Wiskott Aldrich syndrome is not related to a defect in proplatelet formation. Blood. 1999;94(2):509–518. [PubMed] [Google Scholar]
- 7.Prislovsky A, Zeng X, Sokolic RA, et al. Platelets from WAS patients show an increased susceptibility to ex vivo phagocytosis. Platelets. 2013;24(4):288–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prislovsky A, Marathe B, Hosni A, et al. Rapid platelet turnover in WASP(-) mice correlates with increased ex vivo phagocytosis of opsonized WASP(-) platelets. Exp Hematol. 2008;36(5):609–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sereni L, Castiello MC, Marangoni F, et al. Autonomous role of Wiskott-Aldrich syndrome platelet deficiency in inducing autoimmunity and inflammation. J Allergy Clin Immunol. 2018;142(4):1272–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shcherbina A, Rosen FS, Remold-O’Donnell E. Pathological events in platelets of Wiskott-Aldrich syndrome patients. Br J Haematol. 1999;106(4):875–883. [DOI] [PubMed] [Google Scholar]
- 11.Shcherbina A, Cooley J, Lutskiy MI, Benarafa C, Gilbert GE, Remold-O’Donnell E. WASP plays a novel role in regulating platelet responses dependent on alphaIIbbeta3 integrin outside-in signalling. Br J Haematol. 2010;148(3):416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takano K, Sato K, Negishi Y, Aramaki Y. Involvement of actin cytoskeleton in macrophage apoptosis induced by cationic liposomes. Arch Biochem Biophys. 2012;518(1):89–94. [DOI] [PubMed] [Google Scholar]
- 13.Mack TG, Kreis P, Eickholt BJ. Defective actin dynamics in dendritic spines: cause or consequence of age-induced cognitive decline? Biol Chem. 2016;397(3):223–229. [DOI] [PubMed] [Google Scholar]
- 14.Kahr WH, Pluthero FG, Elkadri A, et al. Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease. Nat Commun. 2017;8:14816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paul DS, Casari C, Wu C, et al. Deletion of the Arp2/3 complex in megakaryocytes leads to microthrombocytopenia in mice. Blood Adv. 2017;1(18):1398–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mazurov AV, Pevzner DV, Antonova OA, et al. Safety, inhibition of platelet aggregation and pharmacokinetics of Fab’2 fragments of the anti-glycoprotein IIb-IIIa monoclonal antibody FRaMon in high-risk coronary angioplasty. Platelets. 2002;13(8):465–477. [DOI] [PubMed] [Google Scholar]
- 17.Topalov NN, Yakimenko AO, Canault M, et al. Two types of procoagulant platelets are formed upon physiological activation and are controlled by integrin alpha(IIb)beta(3). Arterioscler Thromb Vasc Biol. 2012;32(10): 2475–2483. [DOI] [PubMed] [Google Scholar]
- 18.Obydennyy SI, Sveshnikova AN, Ataullakhanov FI, Panteleev MA. Dynamics of calcium spiking, mitochondrial collapse and phosphatidylserine exposure in platelet subpopulations during activation. J Thromb Haemost. 2016;14(9):1867–1881. [DOI] [PubMed] [Google Scholar]
- 19.Takahashi A, Camacho P, Lechleiter JD, Herman B. Measurement of intracellular calcium. Physiol Rev. 1999;79(4):1089–1125. [DOI] [PubMed] [Google Scholar]
- 20.Dashkevich NM, Vuimo TA, Ovsepyan RA, et al. Effect of pre-analytical conditions on the thrombodynamics assay. Thromb Res. 2014;133(3):472–476. [DOI] [PubMed] [Google Scholar]
- 21.Ugarova NN, Lomakina GY, Modestova Y, et al. A simplified ATP method for the rapid control of cell viability in a freeze-dried BCG vaccine. J Microbiol Methods. 2016;130:48–53. [DOI] [PubMed] [Google Scholar]
- 22.Lomakina GY, Modestova YA, Ugarova NN. Bioluminescence assay for cell viability. Biochemistry (Mosc). 2015;80(6):701–713. [DOI] [PubMed] [Google Scholar]
- 23.Ignatova AA, Karpova OV, Trakhtman PE, Rumiantsev SA, Panteleev MA. Functional characteristics and clinical effectiveness of platelet concentrates treated with riboflavin and ultraviolet light in plasma and in platelet additive solution. Vox Sang. 2016;110(3): 244–252. [DOI] [PubMed] [Google Scholar]
- 24.Poletaev AV, Koltsova EM, Ignatova AA, et al. Alterations in the parameters of classic, global, and innovative assays of hemostasis caused by sample transportation via pneumatic tube system. Thromb Res. 2018;170: 156–164. [DOI] [PubMed] [Google Scholar]
- 25.Podoplelova NA, Sveshnikova AN, Kotova YN, et al. Coagulation factors bound to pro-coagulant platelets concentrate in cap structures to promote clotting. Blood. 2016;128(13):1745–1755. [DOI] [PubMed] [Google Scholar]
- 26.Shakhidzhanov SS, Shaturny VI, Panteleev MA, Sveshnikova AN. Modulation and pre-amplification of PAR1 signaling by ADP acting via the P2Y12 receptor during platelet subpopulation formation. Biochim Biophys Acta. 2015;1850(12):2518–2529. [DOI] [PubMed] [Google Scholar]
- 27.Sveshnikova AN, Balatskiy AV, Demianova AS, et al. Systems biology insights into the meaning of the platelet’s dual-receptor thrombin signaling. J Thromb Haemost. 2016;14(10):2045–2057. [DOI] [PubMed] [Google Scholar]
- 28.Suntsova EV, Demina IM, Ignatova AA, et al. Bleeding tendency and platelet function during treatment with romiplostim in children with severe immune thrombocytopenic purpura. Int J Hematol. 2017;105(6):841–848. [DOI] [PubMed] [Google Scholar]
- 29.Ignatova AA, Ponomarenko EA, Polokhov DM, et al. Flow cytometry for pediatric platelets. Platelets. 2019;30(4):428–437. [DOI] [PubMed] [Google Scholar]
- 30.Rodeghiero F, Pecci A, Balduini CL. Thrombopoietin receptor agonists in hereditary thrombocytopenias. J Thromb Haemost. 2018;16(9):1700–1710. [DOI] [PubMed] [Google Scholar]
- 31.Gerrits AJ, Leven EA, Frelinger AL, 3rd, et al. Effects of eltrombopag on platelet count and platelet activation in Wiskott-Aldrich syndrome/X-linked thrombocytopenia. Blood. 2015;126(11):1367–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heemskerk JW, Hoyland J, Mason WT, Sage SO. Spiking in cytosolic calcium concentration in single fibrinogen-bound fura-2-loaded human platelets. Biochem J. 1992;283(Pt 2):379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sveshnikova AN, Ataullakhanov FI, Panteleev MA. Compartmentalized calcium signaling triggers subpopulation formation upon platelet activation through PAR1. Mol Biosyst. 2015;11(4):1052–1060. [DOI] [PubMed] [Google Scholar]
- 34.Kholmukhamedov A, Janecke R, Choo HJ, Jobe SM. The mitochondrial calcium uniporter regulates procoagulant platelet formation. J Thromb Haemost. 2018;16(11):2315–2321. [DOI] [PubMed] [Google Scholar]
- 35.Ignatova A, Suntsova E, Zharkov P, et al. Evolution of platelet function and bleeding in children and adults with chronic immune trombocytopenia on romiplostim treatment. Blood. 2017;130(Suppl 1):3636. [Google Scholar]
- 36.Jobe SM, Wilson KM, Leo L, et al. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood. 2008;111(3): 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alberio L, Ravanat C, Hechler B, Mangin PH, Lanza F, Gachet C. Delayed-onset of procoagulant signalling revealed by kinetic analysis of COAT platelet formation. Thromb Haemost. 2017;117(6):1101–1114. [DOI] [PubMed] [Google Scholar]
- 38.Mangin PH, Onselaer MB, Receveur N, et al. Immobilized fibrinogen activates human platelets through glycoprotein VI. Haematologica. 2018;103(5):898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alberio L, Safa O, Clemetson KJ, Esmon CT, Dale GL. Surface expression and functional characterization of alpha-granule factor V in human platelets: effects of ionophore A23187, thrombin, collagen, and convulxin. Blood. 2000;95(5):1694–1702. [PubMed] [Google Scholar]
- 40.Ochs HD, Filipovich AH, Veys P, Cowan MJ, Kapoor N. Wiskott-Aldrich syndrome: diagnosis, clinical and laboratory manifestations, and treatment. Biol Blood Marrow Transplant. 2009;15(1 Suppl):84–90. [DOI] [PubMed] [Google Scholar]