

Abstract
Multiple sclerosis is an inflammatory demyelinating disease of the CNS, the aetiology of which is believed to have both genetic and environmental components. We have investigated one of the candidate viruses for the environmental component of multiple sclerosis, the neurotropic human herpesvirus 6 (HHV-6). Utilizing fluorescent in situ hybridization (FISH) techniques, we have examined human post-mortem tissues for the presence of immediate early and late viral gene expression in multiple sclerosis patient normal appearing white matter (NAWM), lesional tissue and normal control brain samples. HHV-6 gene transcription was detected in all tissue samples and was restricted to oligodendrocytes, as determined by double mRNA FISH analysis. Quantitative analysis of viral mRNA expression indicated that both NAWM and lesional multiple sclerosis samples exhibited significantly higher levels of HHV-6 expression compared with the normal control samples. Lesional samples exhibited the highest levels of viral gene expression, with NAWM exhibiting an intermediate level between lesional and control tissues. Immunofluorescence against early and late HHV-6 proteins verified active translation of HHV-6 viral mRNA in oligodendrocytes. Southern blot analysis of nested polymerase chain reactions using extracted genomic DNA and cDNA confirmed the presence of the HHV-6 genome in all individuals, with the active expression profile mirroring the FISH results. The frequent high level of HHV-6 infection in multiple sclerosis samples suggests a possible role in pathogenesis.
Keywords: multiple sclerosis, HHV-6, oligodendrocyte, mRNA FISH
Keywords: AMCA = 7-amino-4-methylcoumarin-3-acetic acid, FISH = fluorescent in situ hybridization, FITC = fluorescein isothiocyanate, GFAP = glial fibrillary acidic protein, HHV-6 = human herpesvirus 6, IE = immediate early, MOBP = myelin-associated oligodendrocytic basic protein, NAWM = normal appearing white matter, PCR = polymerase chain reaction
Introduction
Multiple sclerosis is an inflammatory demyelinating disease of the CNS with both genetic and environmental components. While immunopathogenic mechanisms appear to be important in multiple sclerosis, the trigger or triggers responsible for initiating and perpetuating the immune reaction remain undetermined. A viral trigger involved in multiple sclerosis was suggested >100 years ago (Marie, 1884), and an extensive list of candidate viruses has emerged since then. Early investigations examined differences in viral antibody titres between multiple sclerosis and normal control patients. The predominant viruses with putative positive connections were measles, mumps, varicella-zoster, Epstein–Barr virus (EBV), parainfluenza and canine distemper virus (Reed et al., 1964; Millar et al., 1971; Lehrich et al., 1974; Ito et al., 1975; Sumaya et al., 1976; Cook et al., 1979). For every implicated virus, there were also papers whose antibody titre results refuted a connection (Woyciechowska et al., 1977; Poskanzer et al., 1980; Stephenson et al., 1980). Advances in molecular biology have allowed sensitive techniques such as the polymerase chain reaction (PCR) and in situ hybridization (ISH) to identify viral nucleic acids in multiple sclerosis tissue samples. These advances have increased the number of candidate viruses to include endogenous retroviruses, coronaviruses, several new human herpesviruses and the bacterium Chlamydia pneumoniae (Murray et al., 1992; Wilborn et al., 1994; Challoner et al., 1995; Sanders et al., 1996). In spite of the new techniques, most implicated viruses sport numerous papers that refute a connection with multiple sclerosis in almost equal proportion to those that show an association (Godec et al., 1992; Hilton et al., 1994; Martin et al., 1997; Merelli et al., 1997; Tsai and Gilden, 2001; Tuke et al., 2004). No single virus so far has been successfully linked to multiple sclerosis initiation and/or exacerbation, although recent successes in identifying infectious agents involved in other chronic diseases have raised fresh hopes that the agent(s) involved in multiple sclerosis will be identified (Zimmer, 2001).
There has been much recent interest in a member of the herpesvirus family, human herpes virus 6 (HHV-6). HHV-6 was first isolated from AIDS patients (Salahuddin et al., 1986). It is a β-herpesvirus that comes in two distinct variants (A and B) (Ablashi et al., 1991; Schirmer et al., 1991) and has a linear double-stranded DNA genome of 160 kb (Gompels et al., 1995). The virus has a high seroprevalence, with most becoming infected by the age of 2 years (Okuno et al., 1989; Levy et al., 1990), and is associated with the common childhood infection exanthem subitum (Yamanishi et al., 1988). After initial infection, HHV-6 can remain in the host in a latent state, with reactivation being a particular problem after transplant surgery (Singh and Carrigan, 1996). In vitro studies have demonstrated that HHV-6 can infect neural cells (Albright et al., 1998), and primary infection with HHV-6 can cause CNS complications such as encephalitis (Yoshikawa and Asano, 2000). In addition, in vitro infection of immature dendritic cells induces functional and phenotypic alterations, suggesting an ability to affect immune function (Kakimoto et al., 2002). There is also evidence to suggest that HHV-6 infection may result in oligodendrocyte cell death (Kong et al., 2003) or a decrease in cell proliferation rate (Dietrich et al., 2004), and could initiate demyelination as the virus encodes a protein capable of generating cross-reactivity with myelin basic protein (Cirone et al., 2002; Tejada-Simon et al., 2003). The combination of all of these studies enhances the viral hypothesis and makes HHV-6 a credible virus for involvement in multiple sclerosis. The presence of HHV-6 in patient lesions was first demonstrated by Challoner et al. (1995) by PCR of DNA extracted from the lesions and immunocytochemistry (ICC) on lesion sections for viral proteins, findings that were largely confirmed by others (Friedman et al., 1999). Subsequent studies have demonstrated the presence of the HHV-6 genome in oligodendrocytes, lymphocytes and microglia of lesional tissues by performing in situ PCRs (ISPCRs) (Blumberg et al., 2000; Goodman et al., 2003). In most studies, HHV-6 has been found in normal control samples, and is frequently absent in some of the multiple sclerosis samples (Moore and Wolfson, 2002; Tyler, 2003). This is especially apparent in the majority of studies that only examine sera and/or CSF (Ablashi et al., 1998, 2000; Akhyani et al., 2000; Soldan et al., 2000; Tomsone et al., 2001; Alvarez-Lafuente et al., 2002a,b; Caselli et al., 2002; Tejada-Simon et al., 2002; Cermelli et al., 2003; Chapenko et al., 2003). This is also the case for studies where it is concluded that HHV-6 does not play a role in multiple sclerosis (Hay and Tenser, 2000; Taus et al., 2000; Al-Shammari et al., 2003).
As no studies to date have examined evidence of active viral infection through detection of RNA either in situ or in extracted nucleic acids, we reasoned that this could be important in identifying a possible connection between HHV-6 and multiple sclerosis. Experiments were therefore designed which examined expression of both an immediate early (IE) gene (U94) responsible for maintenance of latency (Yoshikawa et al., 2002) and a late gene (expression of which indicates active viral infection). We have utilized mRNA fluorescent in situ hybridization (FISH) and PCR/reverse transcription (RT)–PCR, in our search for latency-associated U94 and late gene U58 expression. By comparing normal control tissue with multiple sclerosis normal appearing white matter (NAWM) and lesional tissues, we hoped to establish whether there were any differences in viral activity between these three tissue types.
Material and methods
Samples
Tissue samples were provided by the multiple sclerosis tissue bank and consist of normal control white matter (three samples), multiple sclerosis NAWM (seven fixed samples) and multiple sclerosis lesional (nine fixed samples) white matter (see Table 2). Tissues were either fixed frozen or snap frozen, and were used for FISH and making genomic DNA and cDNA for nested PCR/Southern blot, respectively.
Table 2.
Raw data and information for each patient sample processed
| Sample ID |
Total no. of oligodendrocytes |
Percentage HHV-6 positive |
Age and sex |
Multiple sclerosis type and duration |
Post-mortem interval |
Days in fixation |
|---|---|---|---|---|---|---|
| MS 77 LES 1 | 188 (9/view) | 84% | F 57 | Multiple sclerosis 31 years | 28 h | 8 |
| MS 77 LES 2 | 4358 (32/view) | 97% | F 57 | Multiple sclerosis 31 years | 28 h | 8 |
| MS 80 LES | 1221 (24/view) | 23% | F 71 | Secondary progressive MS 35 years | 24 h | 12 |
| MS 88 NAWM | 645 (17/view) | 50% | F 54 | Chronic multiple sclerosis 20 years | 22 h | 10 |
| MS 88 LES | 1536 (29/view) | 77% | F 54 | Chronic multiple sclerosis 20 years | 22 h | 10 |
| MS 97 NAWM | 1640 (29/view) | 24% | M 55 | Multiple sclerosis, details N/A | 31 h | 14 |
| MS 97 LES | 3098 (58/view) | 10% | M 55 | Multiple sclerosis, details N/A | 31 h | 14 |
| MS 102 NAWM | 3773 (47/view) | 7% | M 73 | PPMS 52 years | 20 h | 26 |
| MS 102 LES | 6606 (43/view) | 19% | M 73 | PPMS 52 years | 20 h | 26 |
| MS 103 NAWM | 1107 (26/view) | 27% | F 77 | PPMS 20 years | 7 h | 24 |
| MS 103 LES | 3200 (53/view) | 7% | F 77 | PPMS 20 years | 7 h | 24 |
| MS 107 LES 1 | 2750 (28/view) | 36% | M 38 | Progressive RMS 18 years | 19 h | 22 |
| MS 107 LES 2 | 216 (14/view) | 56% | M 38 | Progressive RMS 18 years | 19 h | 22 |
| Control 14 | 570 (10/view) | 12% | F 64 | COD = myocardial infarction | 18 h | 22 |
| Control 15 | NA | 0% | M 82 | COD = schizophrenia | 21 h | 18 |
| Control 16 | NA | 0% | M 92 | COD = cardiac failure | 13 h | 10 |
NA = not applicable; N/A = not available; PPMS = primary progressive multiple sclerosis; RMS = relapsing multiple sclerosis; COD = cause of death
Two wax-embedded AIDS brain samples were kindly provided by Professor Peter Lantos, Institute of Psychiatry, London.
One mouse brain was kindly provided by Peter Humphreys, University of Glasgow Department of Neurology, Division of Clinical Neurosciences.
Extraction of RNA and DNA from snap-frozen tissues
RNA was extracted using the RNA Lipid Tissue Mini Kit from Qiagen (catalogue no. 74804). DNA was extracted using the QIAmp DNA Mini Kit from Qiagen (catalogue no. 51304).
Primers and probes
All probes and primers were used in the course of the investigation. Labelled probes were made by MWG, and PCR primers and Southern blot probes were made by Sigma Genosys.
Claudin-11 (BC013577): forward primer (F) = 239–262, reverse primer (R) = 721–744. FISH oligo cocktail = 241–270, 301–330, 361–390, 421–450, 481–510.
HHV-6 (U1102): IE (U94) nested outer F primer = 142 778–142 801, outer R primer = 142 058–142 081, nested inner F primer = 142 749–142 772, nested inner R primer = 142 384–142 407. FISH oligo cocktail = 142 296–142 325, 142 425–142 454, 142 488–142 517, 142 558–142 587, 142 660–142 689, 142 757–142 783, 141 408–141 437, 141 566–141 595, 141 726–141 755, 141 877–141 916. Southern blot probe = 142 591–142 614. Late (U58) nester outer F primer = 94 185–94 208, nested outer R primer = 95 207–95 230, nested inner F primer = 94 798–94 821, nested inner R primer = 95 089–95 112. FISH oligo cocktail = 94 153–94 182, 94 263–94 292, 94 365–94 394, 94 523–94 552, 94 808–94 837.
PCR, RT–PCR and Southern blotting
RT–reactions were performed on the RNA samples extracted from the snap-frozen tissues using either specific (HHV-6 outer nested primers) or oligo(dT)/random hexamer primers in conjunction with Durascript RT-enzyme/Durascript RT–PCR kit from Sigma (catalogue nos A4464/HSRT-20). PCRs were performed using RedTaq Ready Mix PCR mix from Sigma, 45 cycles at either 60°C (claudin-11, HHV-6 late) or 65°C (HHV-6 IE) annealing. Southern blot hybridizations were performed using DiG-labelled oligo probes (terminal transferase catalogue no. 3 333 547, digoxigenin-11-dUTP catalogue no. 1 093 088), Nylon membrane (catalogue no. 1 209 272) and DiG Nucleic Acid Detection Kit (catalogue no. 1 175 041), all from Roche. As a positive control, HHV-6 genomic DNA (Autogen Bioclear) was used.
Fluorescent in situ hybridization (FISH)
Tissue preparation, sectioning and pre-hybridization were essentially as described (Opsahl et al., 2002), with the initial dehydration step removed for unwaxed sections and a proteinase K (1 µg/ml) digestion step performed for 20 min at 37°C. Probes were cocktails of five (claudin-11, HHV-6 late) or 10 (HHV-6 IE) antisense or sense30-mer oligonucleotides based on cDNA sequences labelled with either rhodamine red (claudin-11, 3′ only) or fluorescein isothiocyanate (FITC) (HHV-6, 5′ and 3′) by the suppliers (MWG-BIOTECH AG, Germany). Sections overlaid with 40 ng/ml of the probe cocktail in hybridization buffer [40% formamide, 2× SSC, 1× Denhardt's, 10% dextran sulphate, 50 mM phosphate buffer, 50 mM dithiothreitol (DTT), 0.250 mg/ml tRNA, 0.5 mg/ml denatured salmon sperm DNA] were heated at 90°C for 6 min, cooled on ice for 1 min and hybridized overnight at 37°C. Sections were washed once for 10 min and once for 30 min at 37°C in 2× SSC, repeated at room temperature using 0.1× SSC. Secondary detection was with rat anti-FITC antibody conjugated to FITC (Serotec Ltd, UK) or mouse anti-rhodamine followed by goat anti-mouse IgG conjugated to rhodamine (both AbCam Ltd). Sections were blocked in incubation buffer (no antibody) for 15 min (in 100 mM Tris–HCl pH 7.4, 15 mM NaCl, 1% serum). Antibody incubation (1 : 100 dilution for anti-fluorescein, 1 : 300 for mouse anti-rhodamine and 1 : 500 for goat anti-mouse IgG-rhodamine) was for 30 min at room temperature followed by three 5 min washes in buffer minus serum. Sections were mounted using Vectashield Mounting medium with antifading agent [±4′,6-diamidino-2-phenylindole (DAPI). Images were captured using a Zeiss Axioplan microscope fitted with a cooled CCD camera (Pro-Series High Performance) and analysed with Image-Pro Plus software version 4.1 (Media Cybernetics).
Immunofluorescence
Immunofluorescence was performed on hybridized sections using a goat primary antibody directed against MOBP (myelin-associated oligodendrocytic basic protein; Autogen-Bioclear UK) with 7-amino-4-methylcoumarin-3-acetic acid (AMCA)-conjugated secondary rabbit anti-goat IgG (Vector Laboratories). Dilutions were 1 : 250 and 1 : 100, respectively. Astrocytes were visualized using a mouse monoclonal antibody against glial fibrillary acidic protein (GFAP; Sigma G3893, clone G-A-5, 1 : 250 dilution) with goat anti-mouse IgG conjugated to FITC (Southern Biotechnology, 1 : 100 dilution) as the secondary antibody. Mouse monoclonal antibodies raised against HHV-6 early (induced early nuclear protein, p41/38) and late (16/gp64/gp54) proteins (both diluted 1 : 250, Autogen-Bioclear) were used to confirm the RNA FISH results, using goat anti-mouse IgG conjugated to rhodamine (1 : 500 dilution, AbCam Ltd) as the secondary antibody. To differentiate between HHV-6A and B, two mouse monoclonal antibodies raised against either HHV-6A early antigen (USBiological 2034-01) or HHV-6B (USBiological 2034-17A) were used, both diluted 1 : 250. Goat anti-mouse IgG conjugated to rhodamine (1 : 500 dilution, AbCam Ltd) again was used as the secondary antibody.
Statistical methods
Data were analysed on the logit scale using an extended binomial model incorporating estimated weights to account for overdispersion (Genstat, 7th edn, Lawes Agricultural Trust, 2003). Effects for tissue and sample type (control, NAWM and lesion) were fitted.
Results
Validation of mRNA FISH on frozen fixed sections
Functioning both as an internal control for endogenous mRNA and an identifier of oligodendrocytes (the myelinating cells of the CNS), a cocktail of ten 30 base oligonucleotides specific for claudin-11 were labelled with rhodamine and used as a probe for mRNA FISH. Claudin-11 is an oligodendrocyte-specific gene expressed in both developing and mature oligodendrocytes, whose product is the third most abundant protein of the myelin sheath (Bronstein et al., 2000). The resulting hybridization did not provide a strong enough signal for direct visualization in any of the tissues, so the signal had to be amplified. Signal amplification was achieved using a primary antibody raised against the rhodamine hapten (mouse anti-rhodamine, AbCam), and a secondary antibody raised against mouse IgG containing a rhodamine hapten (goat anti-mouse IgG, AbCam). It was necessary to perform three rounds of signal amplification in order to obtain a strong positive signal.
The claudin-11 probe was validated by performing immunofluorescence on the probed sections (prior to rhodamine signal amplification) using an antibody against MOBP (goat anti-MOBP), with the secondary antibody containing an AMCA hapten (rabbit anti-goat IgG, Vector Laboratories). The specificity of the amplified claudin-11 probe signal was verified by co-location with the MOBP signal (Fig. 1A), confirming that the RNA FISH correctly identified oligodendrocytes.
Fig. 1.
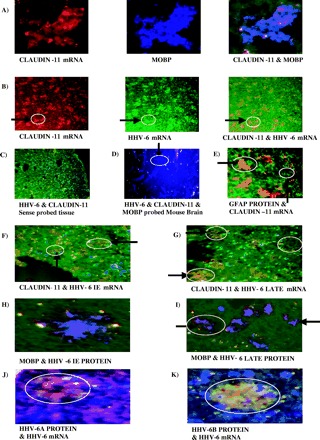
Both immediate early and late viral gene expression is seen in all samples and locates to the cytoplasm of oligodendrocytes, indicating an active infection. The number of cells that are positive for HHV-6 mRNA varies but cells tend to be seen in clusters. (A) Representative FISH of claudin-11 mRNA (rhodamine red), ISH for myelin oligodendrocyte basic protein (MOBP) using AMCA (blue)-conjugated secondary antibody with the two colour images merged to demonstrate co-localization of both signals indicating that the mRNA probe correctly identifies oligodendrocytes (magnification ×400). The image is taken from normal control tissue. (B) Representative double FISH for claudin-11 mRNA (rhodamine red), HHV-6 mRNA (fluorescein) and the two colour merged image demonstrating co-localization of both signals to oligodendrocyte cells (orange). The circle and arrow indicate two representative cells showing first claudin-11 mRNA, then HHV-6 mRNA and finally the two merged images (magnification ×200). The image is taken from multiple sclerosis NAWM tissue. (C) Normal control tissue probed with sense probes for HHV-6 (fluorescein) and claudin-11 (rhodamine red). All that can be seen are the DAPI-counterstained nuclei (magnification ×200). (D) Mouse brain section probed for HHV-6 mRNA (fluorescein), claudin-11 mRNA (rhodamine red) and MOBP protein (AMCA secondary). Only MOBP signal is present. The circle and arrow indicate a representative oligodendrocyte (magnification ×200). (E) Representative ISH for GFAP (fluorescein secondary) and claudin-11 mRNA FISH (rhodamine red). The signals do not co-localize, the circle and arrows highlighting examples of both (magnification ×200). The image is taken from normal control tissue. (F) Representative double FISH for claudin-11 and HHV-6 immediate early mRNA with both signals co-localizing (orange). The circle and arrow indicate two representative cells where the two mRNA signals co-localize (magnification ×200). (G) Representative double FISH for claudin-11 and HHV-6 late mRNA with both signals co-localizing (orange). The circle and arrow indicate two representative cells where the two mRNA signals co-localize (magnification ×200). (H) Representative double ISH for MOBP (AMCA secondary) and HHV-6 immediate early (rhodamine secondary) proteins exhibiting co-localization of the signal to oligodendrocytes (purple) confirming mRNA FISH results (magnification ×400). (I) Representative double ISH for MOBP (AMCA secondary) and HHV-6 late (rhodamine secondary) proteins exhibiting co-localization of signal to oligodendrocytes (purple) confirming mRNA FISH results. The circle and arrow indicate two representative cells where the two protein signals co-localize (magnification ×200). Images F–I are from from multiple sclerosis lesion tissue. (J) Representative ISH for HHV-6A protein (rhodamine secondary) and HHV-6 mRNA FISH (fluorescein) (magnification ×400). The image is taken from multiple sclerosis NAWM. (K) Representative ISH for HHV-6B protein (rhodamine secondary) and HHV-6 mRNA FISH (fluorescein) (magnification ×400). The image is taken from multiple sclerosis NAWM.
Double mRNA FISH on frozen sections
Having validated the technique, the tissues were probed simultaneously for claudin-11 and HHV-6 IE (U94) and/or HHV-6 late (U58) mRNA. A cocktail of ten 30 base oligonucleotides specific for the two viral genes were used as probes, in this instance labelled with FITC. As with the claudin-11 probe, the signal had to be amplified, using a rat IgM raised against FITC labelled with FITC (Serotec). Eight rounds of amplification were needed for a strong signal that could easily be captured by digital imaging (Fig. 1B). Two additional controls were the probing of DNase-treated tissue with the corresponding sense probes to the antisense ones used to pick up mRNA (Fig. 1C) and probing mouse brain sections with antisense probes against both HHV-6 and claudin-11 in conjunction with immunofluorescence against MOBP (Fig. 1D). Although the antibody against MOBP picks up both human and rodent MOBP, the sequence similarity between mouse and human claudin-11 (∼80% similarity) was sufficiently divergent that the oligo probe cocktail did not hybridize. HHV-6 probes did not give a signal in mouse tissue, nor did the sense probes in the DNase-treated human tissue (see Fig. 1C and D). As a final control, claudin-11 mRNA FISH was combined with immunofluorescence against GFAP and demonstrated clearly that the astrocyte signal (FITC) and oligodendrocyte signal (rhodamine red) did not co-localize (Fig. 1E).
The double mRNA FISH procedure provided examples of co-localization of both HHV-6 IE and late gene expression with claudin-11 in all tissues examined (Fig. 1F–I). The presence of HHV-6 late gene expression confirmed an active infection. As an additional confirmation of the mRNA results, the tissues were also probed with antibodies raised against an early and late viral gene, respectively (Autogen Bioclear). The pattern of staining revealed by these antibodies mirrored that seen for mRNA expression and co-localized with the oligodendrocyte marker signal (Fig. 1H and I). We also examined two examples of wax-embedded AIDS brain tissue, as HHV-6 reactivation/active infection is a common complication in such patients. Although the RNA in these samples was not abundant enough to enable mRNA FISH, the immunofluorescence for viral protein was successful and both samples exhibited early and late viral protein signal located to oliogodendrocytes, with virtually all oligodendrocytes expressing the viral proteins (not shown). As both multiple sclerosis and normal control tissue provided examples of active HHV-6 gene expression, no qualitative difference emerged between the tissues. The visual examination of the tissues did strongly indicate that there was a quantitative difference in the number of oligodendrocytes exhibiting active HHV-6 gene expression.
In order to determine whether there was a quantitative difference in HHV-6 gene expression between the three sets of tissues, the total number of oligodendrocytes in each section were counted and the proportion of them also harbouring HHV-6 mRNA was determined (see Table 1 and Fig. 2). Although all tissues exhibited some activity, multiple sclerosis samples averaged almost a 4-fold increase in active HHV-6 expression (or a 5-fold increase when comparing weighted averages). The weighted averages from the three sample groups (control, NAWM and lesion) were compared using a generalized linearized model with a logit link, and significant differences were found in the proportions of infected cells between tissues (P < 0.001) and between sample types (P < 0.001). Lesion tissue showed the highest proportion of infected cells, and this was significantly higher than the observed proportion infected for NAWM tissue (P < 0.05) and for control tissue (P < 0.01). Control tissue showed the lowest proportion infected, but this was not significantly lower than the proportion for NAWM (P = 0.1).
Table 1.
Mean number of oligodendrocyte cells that are positive for HHV-6 mRNA in each of the three categories of tissue
| Tissue |
Percentage of HHV-6 mRNA-positive oligodendrocytes |
Weighted averages of percentage of HHV-6-positive oligodendrocytes |
|---|---|---|
| Control | 12% | 7.5% |
| NAWM | 28.5% | 17.8% |
| Lesion | 40.9% | 37.7% |
The numbers are expressed as a percentage of the total number of oligodendrocytes counted. The second column shows the same data with weighted averages.
Fig. 2.
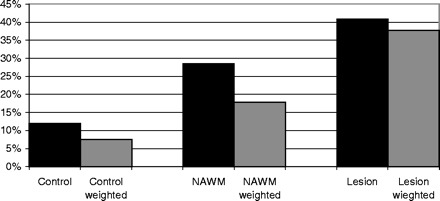
Graphical representation of the data presented in Table 1.
When the results are examined on an individual basis, it becomes apparent that individual infection levels fluctuate although most multiple sclerosis samples remain higher than the highest control level of 12% of infected cells (see Table 2 and Fig. 3). There were also differences observed in signal intensity, although it was impossible to determine whether this was the result of differential viral load or differences in RNA preservation as post-mortem intervals and fixation times varied widely between samples (see Table 2). It was also noticed that the number of oligodendrocytes varied between samples (see Table 2), but it was not possible to determine whether this could have been partly due to differences in RNA preservation between tissues. Although there appeared to be no link between post-mortem interval and mRNA preservation, we did observe that fixation time appeared to have an effect on mRNA signal. Fixation beyond 10 days appears to negatively affect mRNA signal quality, possibly due to poor probe access caused by overfixation of the tissue.
Fig. 3.
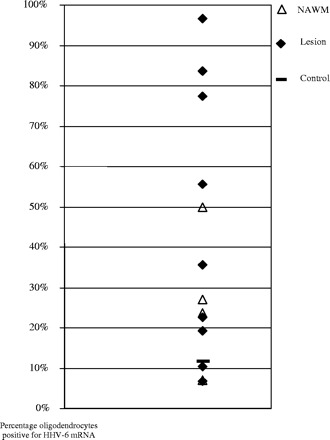
Scatterplot representing the data presented in Table 2. Each point represents an individual sample. The control sample value is represented by a broad line, lesional samples are filled diamonds and NAWM are open triangles. As two of the control samples had undetectable HHV-6 mRNA, only the third sample appears on this graph representing what has to be considered a normal level of HHV-6 infection in oligodendrocytes.
We also performed immunofluorescence using antibodies against either HHV-6A- or B-specific proteins, and found that both species were present in the three representative cases examined (multiple sclerosis NAWM and lesion tissue), with two individuals exhibiting evidence of both subspecies (see Fig. 1J and K). The tissues had also been probed for HHV-6 mRNA expression, and the protein signal (rhodamine red) co-localized with the mRNA signal (fluorescein) (see Fig. 1J and K). HHV-6 mRNA had already been established to occur exclusively in oligodendrocytes. The two subspecies were seen in both NAWM and lesion samples. There was thus no obvious species bias in multiple sclerosis tissue, nor any obvious bias between NAWM and lesional tissue in HHV-6 subspecies activity.
RT–PCR
Using the unfixed tissues, RNA and DNA were extracted and subjected to PCR analysis for the presence of the two viral genes probed for in the mRNA FISH experiment. Forty-five rounds of amplification using just a single set of primers for each viral gene failed to provide any signal except in the positive control samples. A further 45 rounds of amplification failed to improve matters, prompting the adoption of the nested PCR approach. As nested PCRs carry a higher risk of a false-positive signal, Southern blots were performed to validate the results. Southern blot of nested PCRs show positive samples for multiple sclerosis tissues for all genomic DNA samples and many of the cDNAs from mRNA (Fig. 4C and D). RT–PCR was also performed on the samples for the endogenous oligodendrocyte gene claudin-11 and the housekeeping gene β-microglobulin (see Fig. 4A and B). Although equal amounts of mRNA were used, the level of claudin-11 expression is obviously variable between the different samples, whereas the housekeeping gene remains relatively uniform (see Fig. 4A and B). This suggests that the actual number of oligodendrocytes varies between the samples.
Fig. 4.
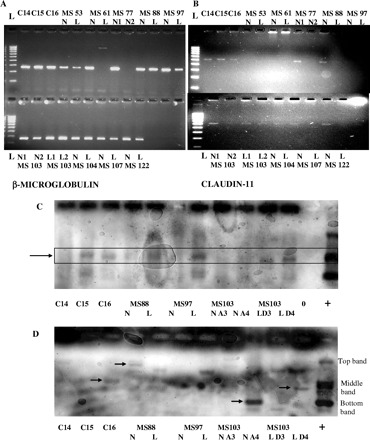
RT–PCR of snap-frozen samples for (A) the endogenous housekeeping gene β-microglobulin (250 bases) and (B) the oligodendrocyte-specific gene claudin-11 (500 bases). Note that not all samples are positive for cDNA, even for the housekeeping gene, with claudin-11 less abundant and more variable. Both are 2% agarose gels with a 1 kb ladder in increments of 100 bases. N = NAWM; L = lesion. As both fixed frozen and snap-frozen samples were not available for all multiple sclerosis patients, some of the PCR samples will not be seen with FISH data and vice versa. Southern blot of genomic DNA (C) and cDNA (D) for the HHV-6 IE gene using nested PCR demonstrates the presence of the viral genome in all samples (highlighted box in C) and transcription in most sample sets (arrows highlight the strongest bands), complementing mRNA FISH and protein data. As nested PCR was utilized, three bands are seen in the positive control lanes representing the three possible primer combinations. The main middle band is predominant in the genomic DNA samples (C), whilst examples of all three are evident in the cDNA samples (D).
Discussion
We have shown here that HHV-6 mRNA expression is found in both normal control and multiple sclerosis brain samples, with activity confined to oligodendrocytes. The difference between normal control samples and multiple sclerosis tissue is quantitative rather than qualitative, with control normal tissue exhibiting much lower levels of viral activity (our highest level being ∼12% of all oligodendrocytes surveyed, with the average level at ∼4–5%). Both NAWM and lesional multiple sclerosis tissue samples exhibited in general much higher levels of viral gene expression, with some examples of close to 100% of oligodendrocytes exhibiting active viral gene expression. Statistical analysis confirmed that the differences between the tissues were significant. The mRNA FISH results were confirmed with both immunofluorescence against viral proteins and RT–PCR. Although the presence of viral proteins indicates an active infection (from both A and B subspecies), we cannot exclude the remote possibility that there could be a post-translational block and consequent absence of virus particle production. While some individual NAWM and lesional samples had HHV-6-positive oligodendrocytes within the normal range, most had much higher levels. These results suggest that NAWM may not be ‘normal’ in multiple sclerosis patients, a notion which has been suggested previously (Peters et al., 1995). The lesional samples that exhibited lower levels of HHV-6 infection may be inactive lesions, with high levels of viral load seen only in active lesions and pre-lesional NAWM. Alternatively, it is possible that higher levels of activity are only seen immediately preceding and/or during a clinical episode. Reactivation of HHV-6 from a latent state due to an exogenous viral infection/inflammatory response is another plausible hypothesis that could explain the differences in HHV-6-positive oligodendrocytes. It is also possible that HHV-6 plays a role in multiple sclerosis pathogenesis in only a subset of patients, with other environmental agents acting in other cases (possibly another herpesvirus such as the related HHV-7 or EBV). Even if viral gene products are significantly upregulated in multiple sclerosis, the question inevitably arises as to whether this is an epiphenomenon secondary to underlying disease processes or a primary event in disease pathogenesis. Differentiation of these two scenarios is problematic and relies mainly on the interpretation of multiple relevant data. Comparing and interpreting results from different research groups is in itself difficult as a wide variety of techniques are used and there is not even a commonly adopted primer set for PCR verification of the HHV-6 genome (Enbom, 2001; Moore and Woolfson, 2002; Clark, 2004). In addition, few, if any, groups have access to the number of patient samples and controls necessary to establish a strong link between multiple sclerosis and a given candidate virus. There is also the potential pitfall that with low numbers, what appears to be a trend may only be the result of random sampling, and the problem of low patient sample numbers is also problematic.
The novel features of this study are the use of both IE and late gene probes to detect HHV-6 mRNA in white matter samples, and the use of fluorescent double labelling of two different RNA species to visualize co-localization of HHV-6 gene expression and oligodendrocytes. The detection of late gene mRNA and protein in addition to IE gene expression and translation confirmed the presence of an active infection in all the sample groups studied. The absence of any qualitative differences necessitated the adoption of a quantitative evaluation of viral activity in the samples. Multiple sclerosis samples had a predominantly higher proportion of their oligodendrocyte cells undergoing active HHV-6 infection compared with normal control levels, with lesional samples exhibiting generally higher levels of infection than NAWM. It was also interesting to note that the number of oligodendrocytes in total varied between samples, and that the RT–PCR results suggested that this was not an artefact and could be a part of the multiple sclerosis pathology where oligodendrocyte cell death is a feature. This is to our knowledge the first study to use a (semi-)quantitative approach to determine the relative viral load in tissue samples. Our results suggest that HHV-6 activity is a feature of multiple sclerosis pathology in many patients, although it is not possible from the current evidence to prove a role in initiation. It has been postulated that multiple sclerosis may be initiated and/or propagated through interactions between the immune system and one or more viruses, in a manner analogous to that documented for other diseases (Welsh and Selin, 2002; Nagai and Osame, 2003; Nowak et al., 2003). How a virus enters the brain and initiates multiple sclerosis is currently the subject of debate (Fazakerly and Walker, 2003; Tsunoda and Fujinami, 2002; Tsunoda et al., 2003). Key properties that may be expected of a candidate virus include neurotropism, latency and reactivation. In addition, some aspect of the viral infection cycle and/or genome should be able to suggest possible mechanisms in the initiation and/or propagation of the immune response that causes the demyelination typical of multiple sclerosis. In the most recent publications investigating a viral link with multiple sclerosis, herpesviruses have predominated (Simmons, 2001). One family member, EBV, was one of the early candidates and continues to be an important focus/area of current research (Ascherio et al., 2001; Levin et al., 2003; Alotaibi et al., 2004; Haahr et al., 2004). HHV-6 may be present in a latent state in oligodendrocytes and subsequently reactivated as part of multiple sclerosis pathogenesis (a raised activity preceding new lesions may explain the high levels of viral mRNA seen in morphologically NAWM of multiple sclerosis patients). It is possible that HHV-6 may even integrate into the host cell genome, staying undetected and inactive for years. Interestingly, studies have indicated that HHV-6 may have a preferred integration site located to chromosome 17p13.3 (Morris et al., 1999). Located to this site are a number of genes whose interaction with HHV-6 could theoretically contribute to multiple sclerosis pathogenesis. Inappropriate expression of CXCL16, a chemokine ligand that may be involved in T-cell migration and T- and dendritic cell interactions (Matloubian et al., 2000), could enhance the autoimmune response, whilst reduction in PITPN (phosphatidylinositol transfer protein) could result in neuronal degeneration (Hamilton et al., 1997). Activation of SERPIN/PEDF (pigment epithelium-derived factor) could also initiate neurodegeneration due to the neurotrophic properties of the protein (Simonovic et al., 2001), whilst increased expression of GPCR42 (G protein-coupled receptor 42) could render a cell sensitive to retroviral infection (Ericsson et al., 2003). Possibly the most interesting scenario is integration downstream of the TRPV 3 (transient receptor potential channel, subfamily V, member 3) promoter, which is heat activated (Smith et al., 2002; Xu et al., 2002). HHV-6 could then be transcribed/activated during febrile episodes, which would explain the frequent exacerbation of multiple sclerosis following common viral infections such as the common cold. Alternatively, HHV-6 may be constitutively active in oligodendrocytes, with infectivity kept in check in normal brains, whilst the disregulated immune system of multiple sclerosis patients is unable to control periodical flare-ups of the virus that may in turn contribute to the pathology of the disease.
The recently reported detection of HHV-6 in mesial temporal lobe epilepsy surgical brain sections (Donati et al., 2003) suggests that HHV-6 activity seen in brain samples is not necessarily linked to an inflammatory response. Consistent with these findings is the fact that we also detected viral gene expression in normal control brain samples. This does not imply that the inflammatory response fails to increase the level of viral activation, and the observation of very high levels of activation in the two AIDS brain samples does strongly suggest that this may indeed occur. In the majority of our tissue samples (multiple sclerosis and normal controls), we observed that HHV-6 gene expression tended to occur in clusters of oligodendrocytes instead of single cells evenly distributed throughout. Although we did not see active HHV-6 infection in all oligodendrocytes, we suggest that it is possible that many/most may contain the virus in a latent state (with latency transcripts below the limit of detection by our mRNA FISH) capable of subsequent reactivation (possibly as a side effect of a localized inflammatory response, or with a different virus/pathogen as co-factor in the reactivation process). Our observations complement the experimental results and theory forwarded by Rotola et al. (2004) that HHV-6 reactivation and replication may occur only in certain sites, triggered by local factors and contained within a small area.
How do our findings fit in with previous published studies on a putative link between HHV-6 and multiple sclerosis? A direct comparison is difficult to make as the majority of studies have examined peripheral blood and CSF samples, and studies on brain tissue samples have generally been limited to detection of viral DNA and/or protein. A recent review by Clark (2004) provides an up to date examination of previous publications based on research strategy, and highlights how similar studies can produce varying and often contradictory results and conclusions. The closest comparisons with our study would be published papers based on the analysis of human brain tissue. In PCR-based studies, those that examined normal control brain tissue found that HHV-6 DNA could also be detected to a varying degree, ranging from 13.5% (Friedman et al., 1999) to >70% (Challoner et al., 1995). The frequency of HHV-6 genome detection in multiple sclerosis lesion samples also varied, ranging from 36% (Friedman et al., 1999) to 78% (Challoner et al., 1995). Our own examination of genomic DNA extracted from tissue samples showed that the HHV-6 genome could be detected in all samples (see Fig. 4C) and is consistent with conclusions that the presence of the viral genome in the CNS is not implicated in the pathogenesis of multiple sclerosis (Sanders et al., 1996). Results on brain sections using immunohistochemistry may be as confusing and variable as the PCR results, although sample numbers are much lower. For studies that included non-multiple sclerosis control tissue, multiple sclerosis positivity ranged from just under 50% (Friedman et al., 1999) to >70% (Knox et al., 2000), with control tissue positivity 0 and 8%, respectively. Differences in tissue protein preservation and pre-treatment (as well as use of different antibodies) could explain the variable results. The non-uniform pattern of mRNA expression seen in our tissues also suggests that viral protein distribution may be random, increasing the chances of a negative result in tissues where viral activity is low (as we see in our control tissues as well as in some of the multiple sclerosis samples). In CSF and serum samples, detection of cell free HHV-6 DNA is considered proof of active viral infection, but the results are as variable and confusing as in the brain tissue studies. For CSF, results range from no detection of HHV-6 DNA in either multiple sclerosis or normal control samples (Taus et al., 2000) to 46% of multiple sclerosis samples and 20% of controls being positive (Tejada-Simon et al., 2002). In serum studies, the results are even more diverse, ranging from no detection in either multiple sclerosis or control samples (Wilborn et al., 1994) to 66 and 33% in multiple sclerosis and control samples, respectively (Tejada-Simon et al., 2002). The variable level of infection seen in our tissues, as quantified by the number of oligodendrocytes exhibiting active HHV-6 infection, suggests that the level of viral DNA released may vary and could easily fall below the level of detection of many of the PCR assays. The use of different primers and reaction conditions, as well as varying methods of DNA extraction, makes direct comparisons between these papers extremely difficult. Serial serum samples taken at several time points (ideally spanning a year or more) would help elucidate the extent of HHV-6 activity in the general population compared with multiple sclerosis subjects, provided a reliable sensitive PCR methodology has been established and used as routine in all laboratories. For post-mortem samples, this is of course not possible, but ideally the ‘next best thing’ would be several tissue samples from both NAWM and different lesions from the same individual.
It would be important to determine whether increased HHV-6 activity (as determined by blood markers) could be linked to multiple sclerosis exacerbations and thus be used to indicate a need for early treatment with antiviral agents specific for HHV-6 (using carrier molecules capable of passing the blood–brain barrier). It would also be important to determine whether any of the oligoclonal bands so characteristic of (Correale and Molinas, 2002), but not exclusive to, the CSF of multiple sclerosis patients contain antibodies that react with HHV-6 antigens. An unequivocal demonstration of an active HHV-6 infection in multiple sclerosis patients, but not in normal individuals, would clearly have specific therapeutic implications. There are several studies that do link multiple sclerosis exacerbations with increased HHV-6 activity (Berti et al., 2002; Chapenko et al., 2003; Alvarez-Lafuente et al., 2004). Direct comparison of the results of these studies is impossible due to the differences in techniques and PCR primers used, and may explain the divergence in results with the percentage of patients in exacerbation also exhibiting active HHV-6 infection ranging from 22% (Berti et al., 2002; Alvarez-Lafuente et al., 2004) to 76.9% (Chapenko et al., 2003). Comparison with our results is also difficult since our samples are post-mortem tissue from patients predominantly in the secondary progressive phase of the disease, whereas relapsing–remitting multiple sclerosis predominates in the serum-based studies. Before any large-scale study into treatment/prevention of HHV-6-linked exacerbations can be contemplated, establishing a commonly adopted stringent PCR-based detection assay for detection of HHV-6 in serum with maximum sensitivity is essential. Whether the global upregulation of HHV-6 gene transcription that we have detected in multiple sclerosis patients relative to controls is pathogenically significant remains an open question, but one that needs to be resolved in further studies. It is particularly important to determine whether specific cofactors exist that may render the viral infection significant enough to justify antiviral therapy.
Acknowledgments
The human tissue samples were provided by the Multiple Sclerosis Tissue Bank, London, and we thank Dr A. Vora and Professor R. Reynolds for their help with tissue acquisition. We also thank Dr A. Springbett for performing the statistical analyses. This work was funded by the Multiple Sclerosis Society (Scotland) and the project was approved by the ethics committee of South Glasgow University Hospitals NHS Trust.
References
- Ablashi DV, Balanchandran N, Josephs SF, Hung CL, Krueger GR, Kramarsky B, et al. Genomic polymorphism, growth properties and immunologic variations in human herpesvirus-6 isolates. Virology 1991; 184: 545–52. [DOI] [PubMed] [Google Scholar]
- Ablashi DV, Lapps W, Kaplan M, Whitman JE, Richert JR, Pearson GR. Human herpesvirus-6 (HHV-6) infection in multiple sclerosis: a preliminary report. Mult Scler 1998; 4: 490–6. [DOI] [PubMed] [Google Scholar]
- Ablashi DV, Eastman HB, Owen CB, Roman MM, Friedman J, Zabriskie JB, et al. Frequent HHV-6 reactivation in multiple sclerosis (MS) and chronic fatigue syndrome (CFS) patients. J Clin Virol 2000; 16: 179–91. [DOI] [PubMed] [Google Scholar]
- Akhyani N, Berti R, Brennan MB, Soldan SS, Eaton JM, McFarland, et al. Tissue distribution and variant characterization of human herpesvirus (HHV)-6: increased prevalence of HHV-6A in patients with multiple sclerosis. J Infect Dis 2000; 182: 1321–25. [DOI] [PubMed] [Google Scholar]
- Albright AV, Lavi E, Black JB, Goldberg S, O'Connor MJ, Gonzalez-Scarano F. The effect of human herpesvirus-6 (HHV-6) on cultured human neural cells: oligodendrocytes and microglia. J Neurovirol 1998; 4: 486–94. [DOI] [PubMed] [Google Scholar]
- Alotaibi S, Kennedy J, Tellier R, Stephens D, Banwell B. Epstein–Barr virus in pediatric multiple sclerosis. J Am Med Assoc 2004; 291: 1875–9. [DOI] [PubMed] [Google Scholar]
- Al Shammari S, Nelson RF, Voevodin A. HHV-6 DNAaemia in patients with multiple sclerosis in Kuwait. Acta Neurol Scand 2003; 107: 122–4. [DOI] [PubMed] [Google Scholar]
- Alvarez-Lafuente R, Martin-Estefania C, las Heras V, Castrillo C, Picazo JJ, Varela de Seijas E, et al. Active human herpesvirus 6 infection in patients with multiple sclerosis. Arch Neurol 2002; 59: 929–33. [DOI] [PubMed] [Google Scholar]
- Alvarez-Lafuente R, Martin-Estefania C, las Heras V, Castrillo C, Cour I, Picazo JJ, et al. Prevalence of herpesvirus DNA in MS patients and healthy blood donors. Acta Neurol Scand 2002; 105: 95–9. [DOI] [PubMed] [Google Scholar]
- Alvarez-Lafuente R, De las Heras V, Bartolome M, Picazo JJ, Arroyo R. Relapsing–remitting multiple sclerosis and human herpesvirus 6 active infection. Arch Neurol 2004; 61: 1523–7. [DOI] [PubMed] [Google Scholar]
- Ascherio AF, Munger KL, Lennette ET, Spiegelman D, Hernan MA, Olek MJ, et al. Epstein–Barr virus antibodies and risk of multiple sclerosis: a prospective study. J Am Med Assoc 2001; 286: 3083–8. [DOI] [PubMed] [Google Scholar]
- Berti R, Brennan MB, Soldan SS, Ohayon JM, Casareto L, McFarland HF, et al. Increased detection of serum HHV-6 DNA sequences during multiple sclerosis (MS) exacerbations and correlation with parameters of MS disease progression. J Neurovirol 2002; 8: 250–6. [DOI] [PubMed] [Google Scholar]
- Blumberg BM, Mock DJ, Powers JM, Ito M, Assouline JG, Baker JV, et al. The HHV6 paradox: ubiquitous commensal or insidious pathogen? A two-step in situ PCR approach. J Clin Virol 2000; 16: 159–78. [DOI] [PubMed] [Google Scholar]
- Bronstein JM, Chen K, Tiwari-Woodruff S, Kornblum HI. Developmental expression of OSP/claudin-11. J Neurosci Res 2000; 60: 284–90. [DOI] [PubMed] [Google Scholar]
- Caselli E, Boni M, Bracci A, Rotola A, Cermelli C, Castellazzi M, et al. Detection of antibodies directed against human herpesvirus 6 U94/REP in sera of patients affected by multiple sclerosis. J Clin Microbiol 2002; 40: 4131–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermelli C, Berti R, Soldan SS, Mayne M, D’ambrosia JM, Ludwin SK, et al. High frequency of human herpesvirus 6 DNA in multiple sclerosis plaques isolated by laser microdissection. J Infect Dis 2003; 187: 1377–87. [DOI] [PubMed] [Google Scholar]
- Challoner PB, Smith KT, Parker JD, MacLeod DL, Coulter SN, Rose TM, et al. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc Natl Acad Sci USA 1995; 92: 7440–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapenko S, Millers A, Nora Z, Logina I, Kukaine R, Murovska M. Correlation between HHV-6 reactivation and multiple sclerosis disease activity. J Med Virol 2003; 69: 111–7. [DOI] [PubMed] [Google Scholar]
- Cirone M, Cuomo L, Zompetta C, Ruggieri S, Frati L, Faggioni A, et al. Human herpesvirus 6 and multiple sclerosis: a study of T cell cross-reactivity to viral and myelin basic protein antigens. J Med Virol 2002; 68: 268–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark D. Human herpesvirus type 6 and multiple sclerosis. Herpes 2004; 11 (Suppl 2): 112A–9A. [PubMed] [Google Scholar]
- Cook SD, Dowling PC, Russell WC. Neutralizing antibodies to canine distemper virus and measles virus in multiple sclerosis. J Neurol Sci 1979; 41: 61–70. [DOI] [PubMed] [Google Scholar]
- Correale J, Molinas MDB. Oligoclonal bands and antibody responses in multiple sclerosis. J Neurol 2002; 249: 375–89. [DOI] [PubMed] [Google Scholar]
- Dietrich J, Blumberg BM, Roshal M, Baker JV, Hurley SD, Mayer-Proschel M, et al. Infection with an endemic human herpesvirus disrupts critical glial precursor cell properties. J Neurosci 2004; 24: 4875–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donati D, Akhyani N, Fogdell-Hahn A, Cermelli C, Cassiani-Ingoni R, Vortmeyer A, et al. Detection of human herpesvirus-6 in mesial temporal lobe epilepsy surgical brain resections. Neurology 2003; 61: 1405–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enbom M. Human herpesvirus 6 in the pathogenesis of multiple sclerosis. APMIS 2001; 109: 401–11. [DOI] [PubMed] [Google Scholar]
- Ericsson TA, Takeuchi Y, Templin C, Quinn G, Farhadian SF, Wood JC, et al. Identification of receptors for pig endogenous retrovirus. Proc Natl Acad Sci USA 2003; 100: 6759–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazakerley JK, Walker R. Virus demyelination. J Neurovirol 2003; 9: 148–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JE, Lyons MJ, Cu G, Ablashi DV, Whitman JE, Edgar M, et al. The association of the human herpesvirus 6 and MS. Mult Scler 1999; 5: 355–62. [DOI] [PubMed] [Google Scholar]
- Godec MS, Asher DM, Murray RS, Shin ML, Greenham LW, Gibbs CJ Jr, et al. Absence of measles, mumps, and rubella viral genomic sequences from multiple sclerosis brain tissue by polymerase chain-reaction. Ann Neurol 1992; 32: 401–4. [DOI] [PubMed] [Google Scholar]
- Gompels UA, Nicholas J, Lawrence G, Jones M, Thomson BJ, Martin ME, et al. The DNA sequence of human herpesvirus-6: structure, coding content, and genomic evolution. Virology 1995; 209: 29–51. [DOI] [PubMed] [Google Scholar]
- Goodman AD, Mock DJ, Powers JM, Baker JV, Blumberg BM. Human herpesvirus 6 genome and antigen in acute multiple sclerosis lesions. J Infect Dis 2003; 187: 1365–76. [DOI] [PubMed] [Google Scholar]
- Haahr S, Plesner AM, Vestergaard BF, Hollsberg P. A role of late Epstein–Barr virus infection in multiple sclerosis. Acta Neurol Scand 2004; 109: 270–5. [DOI] [PubMed] [Google Scholar]
- Hamilton BA, Smith DJ, Mueller KL, Kerrebrock AW, Bronson RT, van Berkel V, et al. The vibrator mutation causes neurodegeneration via reduced expression of PITP alpha: positional complementation cloning and extragenic suppression. Neuron 1997; 18: 711–22. [DOI] [PubMed] [Google Scholar]
- Hay KA, Tenser RB. Leukotropic herpesviruses in multiple sclerosis. Mult Scler 2000; 6: 66–8. [DOI] [PubMed] [Google Scholar]
- Hilton DA, Love S, Fletcher A, Pringle JH. Absence of Epstein–Barr-virus RNA in multiple sclerosis as assessed by in-situ hybridisation. J Neurol Neurosurg Psychiatry 1994; 57: 975–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Barron AL, Olszewski WA, Milgrom F. Antibody titres by mixed agglutination to varicella-zoster, herpes simplex and vaccinia viruses in patients with multiple sclerosis. Proc Soc Exp Biol Med 1975; 149: 835–9. [DOI] [PubMed] [Google Scholar]
- Kakimoto M, Hasegawa A, Fujita S, Yasukawa M. Phenotypic and functional alterations of dendritic cells induced by human herpesvirus 6 infection. J Virol 2002; 76: 10338–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox KK, Brewer JH, Henry JM, Harrington DJ, Carrigan DR. Human herpesvirus 6 and multiple sclerosis: systemic active infections in patients with early disease. Clin Infect Dis 2000; 31: 894–903. [DOI] [PubMed] [Google Scholar]
- Kong H, Baerbig Q, Duncan L, Shepel N, Mayne M. Human herpesvirus type 6 indirectly enhances oligodendrocyte cell death. J Neurovirol 2003; 9: 539–50. [DOI] [PubMed] [Google Scholar]
- Lehrich JR, Arnason BG, Fuller TC, Wray SH. Parainfluenza, histocompatibility, and multiple sclerosis. Association of parainfluenza antibodies and histocompatibility types in MS and optic neuritis. Arch Neurol 1974; 30: 327–9. [DOI] [PubMed] [Google Scholar]
- Levin LI, Munger KL, Rubertone MV, Peck C, Lenette ET, Spiegelman D, et al. Multiple sclerosis and Epstein–Barr virus. J Am Med Assoc 2003. 26; 289: 1533–6. [DOI] [PubMed] [Google Scholar]
- Levy JA, Ferro F, Greenspan D, Lennette ET. Frequent isolation of HHV-6 from saliva and high seroprevalence to the virus in the population. Lancet 1990; 335: 1047–50. [DOI] [PubMed] [Google Scholar]
- Marie P. Sclerose en plaques et maladies infectieuses. Prog Med Paris 1884; 12: 287–9. [Google Scholar]
- Martin CF, Enbom MF, Soderstrom MF, Fredrikson S, Dahl H, Lycke J, et al. Absence of seven human herpesviruses, including HHV-6, by polymerase chain reaction in CSF and blood from patients with multiple sclerosis and optic neuritis. Acta Neurol Scand 1997; 95: 280–3. [DOI] [PubMed] [Google Scholar]
- Matloubian M, David A, Engel S, Ryan JE, Cyster JG. A transmembrane CXC chemokine is a ligand for HIV-coreceptor Bonzo. Nat Immunol 2000; 1: 298–304. [DOI] [PubMed] [Google Scholar]
- Merelli E, Bedin RF, Sola PF, Barozzi P, Mancardi GL, Ficarra G, et al. Human herpesvirus 6 and human herpes virus 8 DNA sequences in brains of multiple sclerosis patients, normal adults and children. J Neurol 1997; 244: 450–4. [DOI] [PubMed] [Google Scholar]
- Millar JH, Fraser KB, Haire M, Connolly JH, Shirodaria PV, Hadden DS. Immunoglobulin M specific for measles and mumps in multiple sclerosis. Br Med J 1971; 758: 378–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore FGA, Wolfson C. Human herpesvirus 6 and multiple sclerosis. Acta Neurol Scand 2002; 106: 63–83. [DOI] [PubMed] [Google Scholar]
- Morris C, Luppi M, McDonald M, Barozzi P, Torelli G. Fine mapping of an apparently targeted latent human herpesvirus type 6 integration site in chromosome band 17p13.3. J Med Virol 1999; 58: 69–75. [DOI] [PubMed] [Google Scholar]
- Murray RS, Brown B, Brian D, Cabirac GF. Detection of coronavirus RNA and antigen in multiple sclerosis brain. Ann Neurol 1992; 31: 525–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai M, Osame M. Human T-cell lymphotropic virus type I and neurological diseases. J Neurovirol 2003; 9: 228–35. [DOI] [PubMed] [Google Scholar]
- Nowak J, Januszkiewicz D, Pernak M, Liwen I, Zawada M, Rembowska J, et al. Multiple sclerosis-associated virus-related pol sequences found both in multiple sclerosis and healthy donors are more frequently expressed in multiple sclerosis patients. J Neurovirol 2003; 9: 112–7. [DOI] [PubMed] [Google Scholar]
- Okuno T, Takahashi K, Balachandra K, Shiraki K, Yamanishi K, Takahashi M, et al. Seroepidemiology of human herpesvirus-6 infection in normal children and adults. J Clin Microbiol 1989; 27: 651–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opsahl ML, McClenaghan M, Springbett A, Ried S, Lathe R, Colman A, et al. Multiple effects of genetic background on variegated transgene expression in mice. Genetics 2002; 160: 1107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters AR, Geelen JA, den Boer JA Prevo RL, Minderhoud JM, Gravenmade EJ. A study of multiple sclerosis patients with magnetic resonance spectroscopic imaging. Mult Scler 1995; 1: 25–31. [DOI] [PubMed] [Google Scholar]
- Poskanzer DC, Sever JL, Sheridan JL, Prenney LB. Multiple sclerosis in the Orkney and Shetland islands. IV: viral antibody titres and viral infections. J Epidemiol Community Health 1980; 34: 258–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed S, Sever J, Kurtzke J, Kurland L. Measles antibody in patients with multiple sclerosis. Arch Neurol 1964; 10: 402–10. [DOI] [PubMed] [Google Scholar]
- Rotola A, Merlotti I, Caniatti L, Caselli E, Granieri E, Tola MR, et al. Human herpesvirus 6 infects the central nervous system of multiple sclerosis patients in the early stages of the disease. Mult Scler 2004; 10: 348–54. [DOI] [PubMed] [Google Scholar]
- Salahuddin SZ, Ablashi DV, Markham PD, Josephs SF, Sturzenegger S, Kaplan M, et al. Isolation of a new virus, HBLV, in patients with lymphoproliferative disorders. Science 1986; 234: 596–601. [DOI] [PubMed] [Google Scholar]
- Sanders VJ, Waddell AE, Felisan SL, Li XF, Conrad AJ, Tourtellotte WW. Herpes simplex virus in postmortem multiple sclerosis brain tissue. Arch Neurol 1996; 53: 125–33. [DOI] [PubMed] [Google Scholar]
- Schirmer EC, Wyatt LS, Yamanishi K, Rodriguez WJ, Frenkel N. Differentiation between 2 distinct classed of viruses now classified as human herpesvirus-6. Proc Natl Acad Sci USA 1991; 88: 5922–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons A. Herpesvirus and multiple sclerosis. Herpes 2001; 8: 60–3. [PubMed] [Google Scholar]
- Simonovic M, Gettins PGW, Volz K. Crystal structure of human PEDF, a potent antiangiogenic and neurite growth-promoting factor. Proc Natl Acad Sci USA 2001; 98: 11131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Carrigan DR. Human herpesvirus-6 in transplantation: an emerging pathogen. Ann Intern Med 1996; 124: 1065–71. [DOI] [PubMed] [Google Scholar]
- Smith GD, Gunthorpe J, Kelsell RE, Hayes PD, Reilly P, Facer P, et al. TRPV3 is a temperature-sensitive vanilloid receptor-like protein. Nature 2002; 418: 186–90. [DOI] [PubMed] [Google Scholar]
- Soldan SS, Leist TP, Juhng KN, McFarland HF, Jacobson S. Increased lymphoproliferative response to human herpesvirus type 6A variant in multiple sclerosis patients. Ann Neurol 2000; 47: 306–13. [PubMed] [Google Scholar]
- Stephenson JR, Ter Meulen V, Kiessing W. Search for canine distemper virus antibodies in multiple sclerosis. A detailed virological evaluation. Lancet 1980; 8198: 772–5. [DOI] [PubMed] [Google Scholar]
- Sumaya CV, Myers L, Ellison GW. Epstein–Barr virus antibodies in multiple sclerosis. Trans Am Neurol Assoc 1976; 101: 300–2. [PubMed] [Google Scholar]
- Taus C, Pucci E, Cartechini E, Fie A, Guiliani G, Clementi M, et al. Absence of HHV-6 and HHV-7 in cerebrospinal fluid in relapsing–remitting multiple sclerosis. Acta Neurol Scand 2000; 101: 224–8. [DOI] [PubMed] [Google Scholar]
- Tejada-Simon MV, Zang YCQ, Hong J, Rivera VM, Killian JM, Zhang JWZ. Detection of viral DNA and immune responses to the human herpesvirus-6 101-kilodalton virion protein in patients with multiple sclerosis and in controls. J Virol 2002; 76: 6147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejada-Simon MV, Zang YCQ, Hong J, Rivera VM, Zhang JWZ. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann Neurol 2003; 53: 189–97. [DOI] [PubMed] [Google Scholar]
- Tomsone V, Logina I, Millers A, Chapenko S, Kozireva S, Murovska M. Association of human herpesvirus 6 and human herpesvirus 7 with demyelinating diseases of the nervous system. J Neurovirol 2001; 7: 564–9. [DOI] [PubMed] [Google Scholar]
- Tsai JC, Gilden DH. Chlamydia pneumoniae and multiple sclerosis: no significant association. Trends Microbiol 2001; 9: 152–4. [DOI] [PubMed] [Google Scholar]
- Tsunoda I, Fujinami RS. Inside-out versus outside-in models for virus induced demyelination: axonal damage triggering demyelination. Springer Semin Immunopathol 2002; 24: 105–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda I, Kuang LQ, Libbey JE, Fujinami RS. Axonal injury heralds virus-induced demyelination. Am J Pathol 2003; 162: 1259–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuke PW, Hawke S, Griffiths PD, Clark DA. Distribution and quantification of human herpesvirus 6 in multiple sclerosis and control brains. Mult Scler 2004; 10: 355–9. [DOI] [PubMed] [Google Scholar]
- Tyler KL. Human herpesvirus 6 and multiple sclerosis: the continuing conundrum. J Infect Dis 2003; 187: 1360–4. [DOI] [PubMed] [Google Scholar]
- Welsh RM, Selin LK. No one is naive: the significance of heterologous T-cell immunity. Nat Rev Immunol 2002; 2: 417–26. [DOI] [PubMed] [Google Scholar]
- Wilborn FF, Schmidt CA, Brinkmann VF, Jendroska KF, Oettle HF, Siegert W. A potential role for human herpesvirus type 6 in nervous system disease. J Neuroimmunol 1994; 49: 213–4. [DOI] [PubMed] [Google Scholar]
- Woyciechowska JL, Madden DL, Sever JL. Absence of measles virus antigen in jejunum of multiple sclerosis patients. Lancet 1977; 8047: 1046–9. [DOI] [PubMed] [Google Scholar]
- Xu HX, Ramsey IS, Kotecha SA, Moran MM, Chong JA, Lawson D, et al. TRPV3 is a calcium-permeable temperature-sensitive cation channel. Nature 2002; 418: 181–6. [DOI] [PubMed] [Google Scholar]
- Yamanishi K, Okuno T, Shiraki K, Takahashi M, Kondo T, Asano Y, et al. Identification of human herpesvirus-6 as a causal agent for exanthem subitum. Lancet 1988; 1: 1065–7. [DOI] [PubMed] [Google Scholar]
- Yoshikawa T, Asano Y. Central nervous system complications in human herpesvirus-6 infection. Brain Dev 2000; 22: 307–14. [DOI] [PubMed] [Google Scholar]
- Yoshikawa T, Asano Y, Akimoto S, Ozaki T, Iwasaki T, Kurata T, et al. Latent infection of human herpesvirus 6 in astrocytoma cell line and alteration of cytokine synthesis. J Med Virol 2002; 66: 497–505. [PubMed] [Google Scholar]
- Zimmer C. Microbiology. Do chronic diseases have an infectious root? Science 2001; 293: 1974–7. [DOI] [PubMed] [Google Scholar]