

Abstract
Myxovirus resistance A (MxA) is an antiviral protein induced by interferon α and β (IFN-α, IFN-β) that can inhibit viral replication. The minor alleles of the -88G>T and -123C>A MxA promoter single-nucleotide polymorphisms (SNPs) are associated with increased promoter activity and altered response to IFN-α and IFN-β treatment. Here, we demonstrate that the 123A minor allele provided stronger binding affinity to nuclear proteins extracted from IFN-β-untreated cells than did the wild-type allele, whereas the -88T allele showed preferential binding after IFN-β stimulation. Endogenous IFN-α and IFN-β induction can be suppressed in severe acute respiratory syndrome (SARS) coronavirus infection. In support of our in vitro findings, a large case-control genetic-association study for SARS coronavirus infection confirmed that the -123A minor-allele carriers were significantly associated with lower risk of SARS coronavirus infection, whereas the -88T minorallele carriers were insignificant after adjustment for confounding effects. This suggests that -123C>A plays a more important role in modulating basal MxA expression, thus contributing more significantly to innate immune response against viral infections that suppress endogenous IFN-α and IFN-β induction such as SARS coronavirus.
Severe acute respiratory syndrome (SARS) coronavirus infected 8422 people worldwide and caused 916 deaths [1–3]. Genetic polymorphisms have been demonstrated to be associated with the risk of SARS coronavirus infection. Although homozygosity of the extracellular neck region of the L-SIGN receptor plays a protective role in SARS coronavirus infection [4], it was reported that SARS patients who were homozygous for ICAM3 Gly143 (dbSNP: rs2304237) were associated with higher lactate dehydrogenase levels and lower total white blood cell counts [5].
Type I interferons α and β (IFN-α and IFN-β) can inhibit SARS coronavirus replication in vitro [6–10] by inducing antiviral proteins such as myxovirus resistance A (MxA; HGNC Symbol: MX1) [11–14]. The minor alleles -88T and -123A of 2 promoter singlenucleotide polymorphisms (SNPs), -88G>T (dbSNP: rs2071430) [15] and -123C>A (rs17000900) [16] of the MxA gene, have been shown with luciferase reporter assay to be associated with increased promoter activity [16, 17]. The -88G>T SNP is located in an interferonstimulated response element-like sequence; the -88T allele makes the sequence more similar to consensus interferon-stimulated response element [16, 18]. With IFN-α and IFN-β treatment, the reporter assays showed a greater increase in promoter activity for -88T but no significant effect for the -123C>A SNP [19]. In contrast, in the absence of IFN-α and IFN-β, 123A contributed to increased promoter activity, whereas the difference between -88G and -88T was small or insignificant. Consistently, on receipt of IFN-α treatment, the MxA messenger RNA (mRNA) level of healthy human peripheral blood mononuclear cells that have heterozygous -88GT or homozygous TT genotypes has been shown to be significantly higher than those that have a homozygous -88GG genotype [20]. Despite these studies, the binding preferences of the SNPs to nuclear proteins have not been reported.
Although exogenous IFN-α and IFN-β can inhibit SARS coronavirus replication in vitro [6–10], most studies report low induction of IFN-α and IFN-β in SARS coronavirus-infected cells, which suggests that the virus evades the antiviral activity of IFN-α and IFN-β [21–24]. As the -123C>A SNPs affect basal MxA expression without IFN-α and IFN-β induction, we hypothesized that this SNP is likely to be important for innate immune response against SARS, in which endogenous IFN-α and IFN-β induction is suppressed.
Because of their proximity (35 base pair [bp] apart), the -88G>T and -123C>A SNPs have been assumed to be in linkage disequilibrium (LD) [16, 17, 25], thus leaving the -123C>A SNPs less studied by comparison [16, 25]. The -88G>T SNPs have been studied in many diseases, including hepatitis B [26, 27] and hepatitis C [15, 28, 29], multiple sclerosis [19, 25], and subacute sclerosing panencephalitis [17]. For SARS susceptibility, only the -88G>T SNPs have been evaluated. Sample sizes of the studies conducted were small, and results were inconclusive [30, 31]. It would be important to clarify the contribution of both the -88G>T and -123C>A SNPs in relation to susceptibility of SARS coronavirus infection.
In this study, the possible differences in binding affinity of these 2 MxA promoter SNPs to nuclear proteins extracted from IFN-stimulated and nonstimulated cells were investigated for the first time to our knowledge. With one of the largest collections of SARS patients and exposed control subjects, this genetic-association study of the -88G>T and -123C>A SNPs for SARS susceptibility and progression was performed on 817 serology-confirmed SARS patients and 422 seronegative household members of SARS patients of the Hong Kong Chinese population.
Materials And Methods
Electrophoretic mobility shift assay. Nuclear proteins were extracted from the 293 cell line as described elsewhere [32] . Extraction of nuclear proteins from the IFN-β-stimulated 293 cells was performed after 24 h treatment with 1000 U/mL IFN-β1a (R&F Systems). The probes used in the electrophoretic mobility shift assay (EMSA) were prepared by denaturing 2 complementary single-stranded oligonucleotides (Figure 1A)at 95°C for 5 min, followed by cooling at room temperature. Each probe was radiolabeled by [γ-32P] ATP (3000 Ci/mmol/L at 10 mCi/mL; Perkin Elmer) by using T4 polynucleotide kinase (Promega), followed by purification using MicroSpin G-25 columns (GE Healthcare Life Science).
Figure 1.

Electrophoretic mobility shift assay (EMSA) for the myxovirus resistance A (MxA) -88G>T and -123C>A single-nucleotide polymorphisms (SNPs). A , The sequence of the probes used in EMSA. S, sense; A, anti-sense. The SNPs are underlined in each oligonucleotide for the MxA SNPs. B , EMSA for the -88G>T SNP, using nuclear extracts from untreated 293 cells. Comparison of lanes 3 and 4 and lanes 5 and 6 showed that the -88T probe bound slightly stronger than did the -88G probe. Comparison between lanes 9 and 10 and lanes 11 and 12 also showed that the -88T probe bound slightly stronger (also shown in the bar chart). However, the intensity of the upshifted band was low, and the competition by cold probes was weak. C , EMSA for the -123C>A SNP, using nuclear extracts from untreated 293 cells. The presence of 2 upshifted bands suggested that there might be 11 transcription factor complexes binding to the probes. The stronger band intensity of the -123A probes and the stronger competition by the cold -123A probe suggest that the binding affinity of -123A allele is stronger than that of -123C. D , EMSA for -88G>T SNP, using nuclear extracts from IFN-β-treated 293 cells. There was an upshifted band of the -88T probe, which could be competed by the cold probe. However, the -88G probe was not upshifted. E , EMSA for the -123C>A SNP, using nuclear extracts from IFN-β-treated 293 cells. The upshift pattern of the probes is similar to that with untreated 293 cells nuclear extract, indicating that the binding affinity of the probe is not significantly affected by IFNb treatment. In the figures, the arrows indicate the upshifted bands being competed away by the cold (unlabeled) probes. The relative intensities of the upshifted bands were quantified, and the results were plotted as bar charts. Asterisks indicate the intensity of the upshifted band used to normalize that of other bands in the respective EMSA assays.
EMSA reactions were performed using Gel Shift Binding 5X buffer (Promega) according to manufacturer's protocol. In brief, each reaction contained 7.5 µg of 293 cell nuclear protein extract, 0.035 µmol/L of the labeled probe, and various concentrations of unlabeled probes as appropriate and then incubated at room temperature for 30 min. Bovine serum albumin was used in place of 293 cell nuclear protein extract as a negative control. The reaction mixtures underwent electrophoresis on an 8% polyacrylamide gel (29:1 acrylamide/bisacrylamide, 1X TBE). After electrophoresis, the gel was vacuum-dried. The radioactive signal was detected by exposing the dried gel to X-ray film at 70°C overnight. Intensity of the upshifted bands was quantitatively measured with a densitometer [33]. The EMSAs were duplicated for the 2 SNPs in each experimental setting.
SARS patients and control subjects. The subjects were recruited for this study as described elsewhere [5], with approval from the respective institutional review boards of hospitals involved. Written informed consent was obtained from SARS patients who donated peripheral blood and from control subjects who donated saliva.
A total of 817 Chinese SARS patients [5] had been recruited either during the outbreak in 2003 or at the SARS follow-up outpatient clinics in 6 hospitals in Hong Kong, namely, Pamela Youde Nethersole Hospital, Princess Margaret Hospital, United Christian Hospital, Queen Mary Hospital, Alice Ho Miu Ling Nethersole Hospital, and Prince of Wales Hospital. All 817 SARS patients were confirmed to be seropositive. Their clinical data were retrospectively obtained from the Hospital Authority, Hong Kong, with permission from all attending clinicians of the respective hospitals.
Household contact control subjects were members of the households of SARS patients and who remained unaffected and seronegative at the end of the outbreak. All recruited household contact control subjects were asked to state their relationship with the SARS patient(s) and other family members in the household. In addition to the 309 control subjects (described elsewhere [5]), more control samples were collected, resulting in a total of 422 control subjects.
Genomic DNA from peripheral blood samples were extracted using conventional methods and level 3 bio-safety precautions in accordance with the World Health Organization and Center for Disease Control guidelines. The household contact control subjects donated saliva samples for extraction of buccal DNA by Oragene DNA self-collection kit (DNA Genotek). The demographic characteristic features and the clinical profile of the SARS patients and household contact control subjects who were successfully genotyped are summarized in Table 1.
Table 1.

Demographic Feature and Clinical Profile of Patients with Severe Acute Respiratory Syndrome and Control Subjects Who Were Successfully Genotyped
Genotyping. Two MxA promoter SNPs (NCBI NM_002462) were studied: -88G>T (rs2071430) and -123C>A (rs17000900). Genotyping of all subjects was performed by polymerase chain reaction (PCR) restriction fragment length polymorphism as described, except with the use of different reverse primer [28]. The described forward primer (5'-TGAAGACCCCCAATTA-CCAA-3') was used with the reverse primer designed by us (5'-GAAACTCACAGACCCTGTGCTGA-3') for PCR using AmpliTaq Gold DNA polymerase (Applied Biosystems) according to manufacturer's instruction and the following thermal cycle conditions: initial denaturation at 95°C for 7 min, 35 subsequent cycles at 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s, followed by a final extension at 72°C for 7 min. The PCR products were digested by either 1 U of HhaI or 2 U of PstI (GE Healthcare). The digested products were analyzed on 2% agarose gel (Figure 2). The size of the undigested product was 296-bp. The major -88G allele creates an HhaI restriction site (GCGC), which cleaves the 296-bp fragment into 260 bp and 36 bp, whereas the minor -88T allele is resistant to HhaI restriction. The major -123C allele creates a PstI site (CTGCAG), which cleaves the 296-bp fragment into 225 bp and 71 bp, whereas the minor -123A allele abolishes the PstI restriction site. Ten percent of all samples were duplicated to confirm the genotyping data. Control samples of known homozygous and heterozygous genotypes and no-template control were included in each run of PCR and restriction fragment length polymorphism. The calling of genotypes on the basis of electrophoresis was blindly checked by 2 individual researchers independently.
Figure 2.

Electrophoresis of the digested polymerase chain reaction (PCR) products of PCR restriction fragment length polymorphism for the myxovirus resistance A (MxA) -88G>T single-nucleotide polymorphism (SNP) and the MxA -123C>A SNP.
Genetic and risk association analyses. The genotypic and allelic distributions between the patient groups and the control groups were assessed by X2 test using SPSS for Windows software (version 13.0; SPSS Inc.) and logistic regression using STATA for Windows software (version 9.2; StataCorp LP) [34], where applicable. Odds ratios (ORs) and 95% confidence intervals (CIs) were used to measure the strength of association. Hardy-Weinberg equilibrium was assessed by a X2 test, with P<.05 considered to be significant. The D' and r2 values and the haplotype frequencies were calculated with Haploview software (version 4.0; Daly Lab) [35].
Because a portion of our patients and control subjects came from the same households, logistic regression analysis with the robust cluster method was performed to factor in the effect of genetically related subjects confounding the risk association. The subjects were clustered according to their genetic relationship. A sample subset that contained only genetically unrelated subjects was also generated for analysis. Logistic regression was also performed to adjust for the age and sex of the subjects. The status (patient or control) of the subject was used as the dependent variable; the genotypes of either one of the SNPs, age, and sex were used as independent variables.
A Bonferroni adjustment [1-(1-0.05)1/n] was applied to adjust the level of significance for multiple testing, the ordinary level of significance .05 adjusted to !.025 for n = 2, and <.0127 for n = 4(n , number of SNPs analyzed).
Analysis for association with clinical outcome measures. Twenty-three clinical outcome measures were analyzed (Table 2). The association of the SNPs with binomial clinical outcome measures was analyzed using a X2 test as described elsewhere [5]. The numeric variables were analyzed using 1-way analysis of variance (SPSS software, version 13.0).
Table 2.

List of Clinical Outcome Measures of Severe Acute Respiratory Syndrome (SARS) Patients
Results
Electrophoretic mobility shift assay. EMSA was used to investigate the binding affinity of nuclear proteins to the probes bearing the -88G>T and -123C>A SNPs (Figure 1A). Expression of MxA in the 293 cells has been reported elsewhere [36] and also confirmed by reverse-transcription PCR (RT-PCR) in this study (Figure 3A). Using quantitative PCR (qPCR), we also show induction of MxA mRNA by IFN-β treatment (Figure 3B). To investigate the differences in response to IFN-β treatment, EMSA was performed using nuclear-protein extracts from the 293 cells that were either untreated or treated with IFN-β before nuclear-protein extraction.
Figure 3.

Expression of Myxovirus resistance-A (MxA) and induction of MxA messenger RNA (mRNA) by IFN-β in the 293 cell line.
EMSA with nuclear extracts from the untreated 293 cells. The MxA -88G and -88T probes both bound with the 293 cell nuclear proteins, resulting in upshifted bands in lanes 2 and 8 (Figure 1B) when compared with lanes with no nuclear proteins (lanes 1 and 7) (Figure 1B). The binding of nuclear proteins to each hot probe was competed against a cold (unlabeled) probe of the same or the other allele in separate reactions. The upshifted band of the hot -88G probe in lanes 3 and 4 was not significantly diminished in intensity by competition with the cold -88G probe, whereas competition with the cold -88T probe decreased the band intensity in lanes 5- 6 in a dosage-dependent manner. Consistently, the upshifted band intensity of the hot -88T probe was diminished by competition with the cold -88T probe (lanes 9–10) but not by competition with the cold -88G probe (lanes 11–12). Because a stronger band intensity indicates stronger binding, these findings suggest that the binding affinity of the nuclear proteins to the -88T allele was slightly stronger than that to the -88G allele. Nevertheless, the decrease in band intensities by competition with either cold probe was weak, indicating that the binding of proteins to the probes was not specific or strong.
The MxA -123C and -123A probes also bound with the 293 cell nuclear proteins, resulting in 2 upshifted bands (lanes 2 and 8 in Figure 1C). The band intensities of the upshifted -123A probe (lane 8) was obviously stronger than that of the -123C probe (lane 2), indicating that the binding affinity of nuclear proteins to -123A was stronger. The upshifted bands of hot -123C were almost completely eliminated by competition with both the cold -123C probes (lanes 3–4) and the cold -123A probes (lanes 5–6). On the other hand, the intensity of upshifted hot -123A probe could be diminished by the cold -123A probe (lanes 9–10) but much less effectively by the cold -123C probe (lanes 11–12).
EMSA with nuclear extracts from the IFN- b -treated 293 cells. Although the difference in binding affinities of nuclear proteins to the -88G and -88T probes is weak, the interferonstimulated response element in which the -88G>T SNP resides suggests that the SNP may affect the binding affinities to nuclear proteins extracted from the IFN-β-treated 293 cells, with -88T binding more strongly. EMSA was thus repeated using nuclear proteins extracted from the IFN-β-treated 293 cells (Figure 1D). As predicted, there is an upshifted band with strong intensity observed for the hot -88T probe (lane 6) which could be competed away by the cold -88T probes in a dosage-dependent manner (lanes 7 and 8). However, such strong intensity shifted band was not observed for the hot -88G probe (lanes 2–4).
However, for -123C>A, EMSA that used nuclear proteins from the IFN-β-treated 293 cells showed a similar pattern to that of untreated cells; ie, the upshifted band of -123A was obviously stronger than that of -123C (lanes 2 and 6) (Figure 1 E), and both bands could be competed by their corresponding cold probes in a dosage-dependent manner (lanes 2–4 and lanes 6–8). These findings suggest that preferential binding of nuclear proteins to the -88T allele was enhanced after IFN-β stimulation, but binding to the -123C>A SNP was irrespective of IFN-β treatment.
Genotype analyses of the MxA SNPs. Our EMSA results suggested the MxA -88G>T and -123C>A SNPs may each have different effects in relation to IFN-mediated immune response, a known important factor in the pathogenesis of SARS. As in vivo evidence to support our in vitro observations, genetic association study was performed.
Of the 1239 subjects recruited, 1210 subjects (792 patients and 418 control subjects) were successfully genotyped for -88G>T and -123C>A SNPs. Both SNPs were in Hardy-Weinberg equilibrium for both the patients and control subjects (P>.05). Of these, 989 subjects (664 patients and 325 control subjects) were genetically unrelated (Table 3). Logistic regression was performed with robust cluster method on genotype data of all 1210 subjects to factor the impact of genetic relationships between some subjects. Results show that both -88T-positive and -123A-positive genotypes were significantly associated with decreased susceptibility to SARS coronavirus infection, P = .010 (OR, 0.72 [95%CI, 0.56–0.92]) and P = .007 (OR, 0.68 [95%CI, 0.52–0.90]), respectively, with a stronger association clearly demonstrated by the -123C>A SNP (Table 4).
Table 3.

Numbers of Household Subjects (Genetically Related or Unrelated) and Independent Subjects
Table 4.

Risk Association Analyses of the Myxovirus Resistance A Promoter -88G>T and -123C>A Single-Nucleotide Polymorphisms
Analysis of the 989 genetically unrelated subjects also showed significantly decreased risk similar to that of SARS coronavirus infection, for the -88T-positive genotype (P = .004; OR, 0.68 [95% CI, 0.52–0.88]) and the -88T allele (P = .009; OR, 0.75 [95% CI, 0.61–0.93]), as well as the -123A-positive genotype (P = .002; OR, 0.62 [95% CI, 0.46–0.85]) and the -123A allele (P = .005; OR, 0.67 [95% CI, 0.51–0.89]), again with a stronger association demonstrated by the -123C>A SNP (Table 4).
To exclude the confounding effect of age and sex, logistic regression analysis adjusted for age and sex was performed. The results confirmed the significant association remained valid for the -123C>A SNP, whereas the -88G>T SNP became marginally significant (Table 5). Previous reports had suggested that 2 SNPs of 2'01,5'01-oligoadenylate synthetase 1 (OAS-1) gene-OAS-1 +590A>G (rs1131454) and +1295A>G SNP (rs2660)—were associated with the risk of SARS coronavirus infection [30, 31]. Our genotyping of these 2 SNPs, however, showed no such association (see the Appendix, which is not available in the print edition of the Journal ; Table 6). To explore any possible confounding effect the OAS-1 SNPs may confer on the risk association of the MxA SNPs, logistic regression adjusted for the 2 OAS-1 SNPs together with age and sex was performed (Table 7). Although the OAS-1 SNPs did not significantly affect the association of the -123C>A SNP (P = .002), it rendered the association of -88G>T SNP insignificant (P p.039), compared with the adjusted significant cutoff of .0127 (Bonferroni adjustment for n = 4).
Table 5.

Risk Association Analyses of Myxovirus Resistance A (MxA) Promoter -88G>T and -123C>A Single-Nucleotide Polymorphisms Adjusted for Age and Sex in Genetically Unrelated Subjects
Table 6.

Risk Association Analyses of OAS-1 +1295A>G and OAS-1 +590A>G Single-Nucleotide Polymorphism (SNP) Using X2 Test and Logistic Regression Adjusted for Genetic
Table 7.
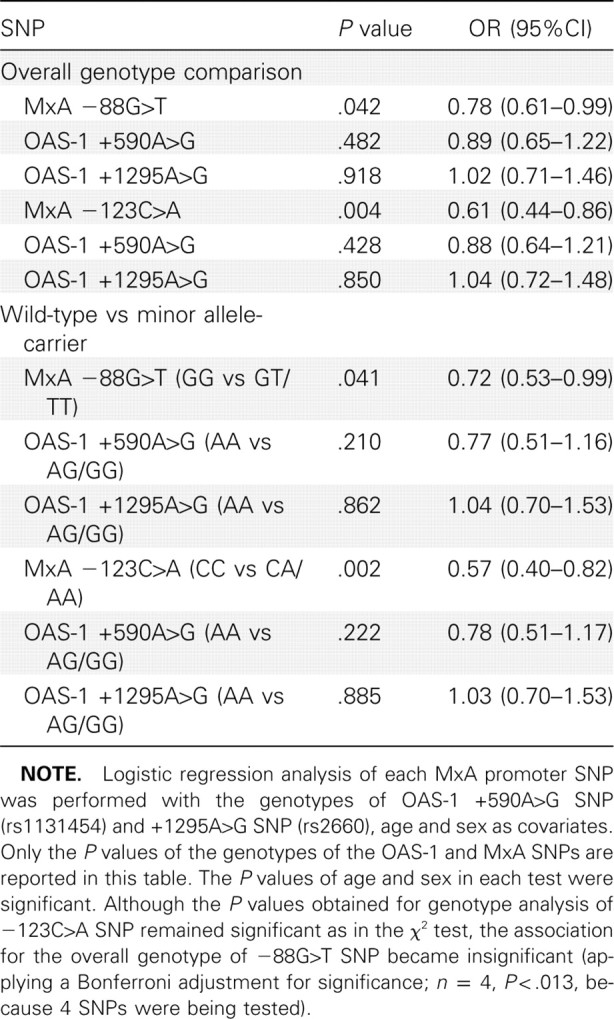
Risk Association Analyses of Myxovirus Resistance A (MxA) Promoter -88G>T and -123C>A Single-Nucleotide Polymorphisms (SNPs) and the OAS-1 SNPs Adjusted for Age and Sex in Genetically Unrelated Subjects
Linkage disequilibrium of MxA 88G>T SNP and - 123C>A SNP. The D'01 values for all subjects and the unrelated subjects were both 1.0, whereas r 2 was 0.391 and 0.384, respectively. This indicates that the SNPs are not of perfect LD. The haplotype frequency analysis between patients and control subjects (Table 8) showed that with respect to the wildtype C-123G-88 haplotype, the non-C-123G-88 haplotypes (ie, C-123T-88, A-123T-88, and A-123G-88) were associated with lower risk of SARS (P = .009; OR, 0.75 [95% CI, 0.61–0.93]) (Table 8).
Table 8.

Haplotype Risk Association Analyses of Genetically Unrelated Subjects
Analysis for association with clinical outcome. To check whether the -88G>T and -123C>A SNPs might influence clinical outcome, genotypes were analyzed against clinical outcome measures for the possible association with prognosis of SARS. The analyses of assisted ventilation and other outcome measures showed no significant association with both SNPs (data not shown).
Discussion
Our EMSA data showed that, for the -88G>T SNP, preferential binding to the minor -88T allele was enhanced after IFN-β stimulation, consistent with the -88T allele being more similar to the consensus interferon-stimulated response element; for the -123C>A SNP, there was preferential binding to the minor -123A allele irrespective of IFN-β treatment. These results are consistent with previous luciferase findings that greater increase in promoter activity was seen with IFN-β treatment for the -88T allele but with no significant effect for the -123C>A SNP, whereas the -123A allele contributed to increased promoter activity in the absence of IFN-β treatment [19]. These data suggest that in the absence of IFN induction, the-123C>A SNP may have greater effect on basal MxA levels than the -88G>T SNP. Induction of IFNs has been reported to be suppressed in SARS coronavirus-infected cells, suggesting that the virus evades the antiviral activity of IFN-α and IFN-β [21–24]. Thus the -123C>A SNP may probably play a more important role in modifying basal MxA expression, thus contributing more significantly to the innate immune response against SARS coronavirus and in the pathogenesis of SARS.
Age and sex are well-known confounding factors for SARS, because individuals aged ≥65 years and <18 years are associated with a lower risk, and the proportion of women was higher in SARS patients [2, 37, 38], as also reflected in our case patients (Table 1). When logistic regression was used to adjust for these confounding effects, the risk association of -123C>A SNP remained significant but the association of -88G>T SNP was weakened (Table 5). Thus, the association with risk of SARS coronavirus infection was clearly stronger for the -123C>A SNP.
Despite the proximity of the -88G>T and -123C>A SNPs with a high D'01 of 1.0, our genotype data showed their allelic correlation was low (r2 = 0.391). The absence of A-123G-88 haplotype in our samples is consistent with previous reports, which showed the -123A allele always coexisted with -88T allele, but never with the -88G allele [16, 19, 25], with dominance of the C-123G-88 haplotype. The fact that C-123 can coexist with T-88 indicates the LD of these SNPs is not complete. Given the differences in the contribution of the -88G>T and -123C>A SNPs to nuclear-protein binding (Figure 1) and MxA promoter activity (depending on whether there was prior stimulation by IFN-β [19]), we have also clarified an independent contribution of both these SNPs in association with SARS coronavirus infection.
Previous association studies of the -88G>T MxA SNP for SARS coronavirus infection on the basis of small sample sizes [30, 31] were inconclusive. Hamano et al [30] studied 44 SARS patients and 103 control subjects in Vietnam and found no significant association, whereas the He et al [31] Chinese Han cohort of 66 SARS patients and 64 control subjects suggested the -88GT heterozygous genotype was associated with increased susceptibility to SARS. Another IFN-inducible gene, OAS-1, was also studied on these same small population cohorts, and the +1295A>G [30, 31] and +590A>G SNPs [30] reported to be associated with SARS coronavirus infection. Our large-scale case-control analyses of the OAS-1 SNPs, however, found no significant association (Table 6). Interestingly, the confounding effect of these OAS-1 SNPs did not significantly alter the association of the -123C>A SNP, but rendered the association of -88G>T SNP insignificant (Table 7).
Our genetic association analysis of probably one of the largest case-control cohorts demonstrates for the first time, to our knowledge, the significant association of the -123A-positive genotype with lower risk of SARS coronavirus infection (Tables 4 and 5). The apparent association of -88T became insignificant after adjustment for the confounding effects of age, sex and the OAS-1 SNPs (Table 7). Because SARS coronavirus infection can suppress endogenous IFN-α and IFN-β induction [21–24], the -123C>A SNP probably exerts greater effect on basal MxA expression in innate immune response which can affect susceptibility to SARS. The importance of the -123C>A SNP was also confirmed by consistently stronger association of -123C>A SNP that was not affected after adjustment of age, sex and genetic background of the OAS-1 SNPs.
MxA can reduce influenza viral protein [39], and mice that express Mx1, the mouse homolog of MxA, have been shown to be protected against the highly lethal human H5N1 influenza virus [40, 41]. A recent report [42] showed reduced and delayed production of IFN-β in H5N1 virus-infected human bronchial epithelial cells, suggesting that the attenuation of IFN-β response in innate immune response may contribute to the virulence of H5N1 viruses. This similarity between H5N1 influenza and SARS in the evasion of IFN-α and IFN-β induction suggests that the -123C>A SNP may also be an important susceptibility factor for H5N1 infection, which warrants future study.
In conclusion, we showed that, in keeping with previously reported promoter assay findings [19], the -123A allele for the MxA gene provided stronger nuclear-protein binding than did the -123C allele in the absence of IFN stimulation, whereas the -88T allele was shown to be preferentially bound by nuclear proteins extracted from the IFN-stimulated cells. In support of these in vitro findings, our large genetic association study provided in vivo data that confirm that, although both SNPs demonstrated significantly decreased susceptibility to SARS coronavirus infection, the association was clearly stronger for the -123C>A SNP. This is in keeping with the condition of suppression of endogenous IFN induction in the innate immune response against SARS coronavirus infection. Knowledge of these differences in contribution of these 2 MxA promoter SNPs in the innate immune response to infection would be important for the assessment of one's susceptibility to infectious diseases that may emerge in the future, especially those sharing a mode of action similar to that of SARS coronavirus.
Acknowledgments
We thank Dr Vivian Wong and Ms Edwina Shung (Hospital Authority Severe Acute Respiratory Syndrome Collaborative Group) for the retrieval of clinical data of SARS patients from the central database. This work was supported by the Research Fund for the Control of Infectious Diseases, Hong Kong SAR Government (Project 04050252) and Seed Funding Programme for Basic Research, The University of Hong Kong (Project 200511159125).
Footnotes
Potential conflicts of interest: none reported.
Financial support: Research Fund for the Control of Infectious Diseases, Hong Kong (project 04050252).
References
- 1.World Health Organization [10 May 2010];Summary table of SARS cases by country, 1 November 2002 to 7 August 2003. http://www.who.int/csr/sars/country/2003_08_15/en/index.html.
- 2.Peiris JS, Yuen KY, Osterhaus AD, Stohr K. The severe acute respiratory syndrome. N Engl J Med. 2003;349:2431–2441. doi: 10.1056/NEJMra032498. [DOI] [PubMed] [Google Scholar]
- 3.Cheng VC, Lau SK, Woo PC, Yuen KY. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin Microbiol Rev. 2007;20:660–694. doi: 10.1128/CMR.00023-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan VS, Chan KY, Chen Y, et al. Homozygous L-SIGN (CLEC4M) plays a protective role in SARS coronavirus infection. Nat Genet. 2006;38:38–46. doi: 10.1038/ng1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan KY, Ching JC, Xu MS, et al. Association of ICAM3 Genetic Variant with Severe Acute Respiratory Syndrome. J Infect Dis. 2007;196:271–280. doi: 10.1086/518892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cinatl J, Morgenstern B, Bauer G, Chandra P, Rabenau H, Doerr HW. Treatment of SARS with human interferons. Lancet. 2003;362:293–294. doi: 10.1016/S0140-6736(03)13973-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stroher U, DiCaro A, Li Y, et al. Severe acute respiratory syndromerelated coronavirus is inhibited by interferon-alpha. J Infect Dis. 2004;189:1164–1167. doi: 10.1086/382597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hensley LE, Fritz LE, Jahrling PB, Karp CL, Huggins JW, Geisbert TW. Interferon-beta 1α and SARS coronavirus replication. Emerg Infect Dis. 2004;10:317–319. doi: 10.3201/eid1002.030482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan EL, Ooi EE, Lin CY, et al. Inhibition of SARS coronavirus infection in vitro with clinically approved antiviral drugs. Emerg Infect Dis. 2004;10:581–586. doi: 10.3201/eid1004.030458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sainz B, Jr., Mossel EC, Peters CJ, Garry RF, et al. Interferon-beta and interferon-gamma synergistically inhibit the replication of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) Virology. 2004;329:11–17. doi: 10.1016/j.virol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao H, De BP, Das T, Banerjee AK. Inhibition of human parainfluenza virus-3 replication by interferon and human MxA. Virology. 1996;220:330–338. doi: 10.1006/viro.1996.0321. [DOI] [PubMed] [Google Scholar]
- 12.Landis H, Simon-Jodicke A, Kloti A, et al. Human MxA protein confers resistance to Semliki Forest virus and inhibits the amplification of a Semliki Forest virus-based replicon in the absence of viral structural proteins. J Virol. 1998;72:1516–1522. doi: 10.1128/jvi.72.2.1516-1522.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chieux V, Chehadeh W, Harvey J, Haller O, Wattre P, Hober D. Inhibition of coxsackievirus B4 replication in stably transfected cells expressing human MxA protein. Virology. 2001;283:84–92. doi: 10.1006/viro.2001.0877. [DOI] [PubMed] [Google Scholar]
- 14.Gordien E, Rosmorduc O, Peltekian C, Garreau F, Brechot C, Kremsdorf D. Inhibition of hepatitis B virus replication by the interferoninducible MxA protein. J Virol. 2001;75:2684–2691. doi: 10.1128/JVI.75.6.2684-2691.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hijikata M, Ohta Y, Mishiro S. Identification of a single nucleotide polymorphism in the MxA gene promoter (G/T at nt-88) correlated with the response of hepatitis C patients to interferon. Intervirology. 2000;43:124–127. doi: 10.1159/000025035. [DOI] [PubMed] [Google Scholar]
- 16.Hijikata M, Mishiro S, Miyamoto C, Furuichi Y, Hashimoto M, Ohta Y. Genetic polymorphism of the MxA gene promoter and interferon responsiveness of hepatitis C patients: revisited by analyzing two SNP sites (-123 and -88) in vivo and in vitro. Intervirology. 2001;44:379–382. doi: 10.1159/000050075. [DOI] [PubMed] [Google Scholar]
- 17.Torisu H, usuhara K, Kira R, et al. Functional MxA promoter polymorphism associated with subacute sclerosing panencephalitis. Neurology. 2004;62:457–460. doi: 10.1212/01.wnl.0000106940.95749.8e. [DOI] [PubMed] [Google Scholar]
- 18.Nakade K, anda H, Nagata K. Promoter structure of the MxA gene that confers resistance to influenza virus. FEBS Lett. 1997;418:315–318. doi: 10.1016/s0014-5793(97)01372-0. [DOI] [PubMed] [Google Scholar]
- 19.Furuyama H, Chiba S, Okabayashi T, et al. Single nucleotide polymorphisms and functional analysis of MxA promoter region in multiple sclerosis. J Neurol Sci. 2006;249:153–157. doi: 10.1016/j.jns.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 20.Fernandez-Arcas N, Blanco A, Gaitan MJ, Nyqvist M, lonso A, Reyes-Engel A. Differential transcriptional expresion of the polymorphic myxovirus resistance protein A in response to interferon-alpha treatment. Pharmacogenetics. 2004;14:189–193. doi: 10.1097/00008571-200403000-00007. [DOI] [PubMed] [Google Scholar]
- 21.Cheung CY, Poon LL, Ng IH, et al. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J Virol. 2005;79:7819–7826. doi: 10.1128/JVI.79.12.7819-7826.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Law HK, Cheung CY, Ng HY, et al. Chemokine up-regulation in SARScoronavirus-infected, monocyte-derived human dendritic cells. Blood. 2005;106:2366–2374. doi: 10.1182/blood-2004-10-4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.eghunathan R, Jayapal M, Hsu LY, et al. Expression profile of immune response genes in patients with Severe Acute Respiratory Syndrome. BMC Immunol. 2005;6:2. doi: 10.1186/1471-2172-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spiegel M, Pichlmair A, Martinez-Sobrido L, et al. Inhibition of beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. JVirol. 2005;79:2079–2086. doi: 10.1128/JVI.79.4.2079-2086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weinstock-Guttman B, Tamano-Blanco M, Bhasi K, Zivadinov R, Ramanathan M. Pharmacogenetics of MXA SNPs in interferon-beta treated multiple sclerosis patients. J Neuroimmunol. 2007;182:236–239. doi: 10.1016/j.jneuroim.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 26.Peng XM, Lei RX, Gu L, Ma HH, Xie QF, Gao ZL. Influences of MxA gene -88 G/T and IFN-gamma +874 A/T on the natural history of hepatitis B virus infection in an endemic area. Int J Immunogenet. 2007;34:341–346. doi: 10.1111/j.1744-313X.2007.00696.x. [DOI] [PubMed] [Google Scholar]
- 27.Kong XF, Zhang XX, Gong QM, et al. MxA induction may predict sustained virologic responses of chronic hepatitis B patients with IFNalpha treatment. J Interferon Cytokine Res. 2007;27:809–818. doi: 10.1089/jir.2006.0163. [DOI] [PubMed] [Google Scholar]
- 28.Knapp S, Yee LJ, Frodsham AJ, et al. Polymorphisms in interferoninduced genes and the outcome of hepatitis C virus infection: roles of MxA, OAS-1 and PKR. Genes Immun. 2003;4:411–419. doi: 10.1038/sj.gene.6363984. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki F, Arase Y, Suzuki Y, et al. Single nucleotide polymorphism of the MxA gene promoter influences the response to interferon monotherapy in patients with hepatitis C viral infection. J Viral Hepat. 2004;11:271–276. doi: 10.1111/j.1365-2893.2004.00509.x. [DOI] [PubMed] [Google Scholar]
- 30.Hamano E, Hijikata M, Itoyama S, et al. Polymorphisms of interferoninducible genes OAS-1 and MxA associated with SARS in the Vietnamese population. Biochem Biophys Res Commun. 2005;329:1234–1239. doi: 10.1016/j.bbrc.2005.02.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He J, Feng D, de Vlas SJ, et al. Association of SARS susceptibility with single nucleic acid polymorphisms of OAS-1 and MxA genes: a casecontrol study. BMC Infect Dis. 2006;6:106. doi: 10.1186/1471-2334-6-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan KY, Liu W, Long JR, et al. Functional polymorphisms in the promoter of BRCA1 influence transcription and are associated with decreased risk for breast cancer in Chinese women. J Med Genet. 2009;46(1):32–39. doi: 10.1136/jmg.2007.057174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan KY, Liu W, Long JR, et al. Functional polymorphisms in the BRCA1 promoter influence transcription and are associated with decreased risk for breast cancer in Chinese women. J Med Genet. 2009;46:32–39. doi: 10.1136/jmg.2007.057174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boston RC, Sumner AE. STATA: a statistical analysis system for examining biomedical data. Adv Exp Med Biol. 2003;537:353–369. doi: 10.1007/978-1-4419-9019-8_23. [DOI] [PubMed] [Google Scholar]
- 35.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 36.Lussier MP, Cayouette S, Lepage PK, et al. MxA, a member of the dynamin superfamily, interacts with the ankyrin-like repeat domain of TRPC. JBiolChem. 2005;280:19393–19400. doi: 10.1074/jbc.M500391200. [DOI] [PubMed] [Google Scholar]
- 37.Leung GM, Rainer TH, Lau FL, et al. A clinical prediction rule for diagnosing severe acute respiratory syndrome in the emergency department. Ann Intern Med. 2004;141:333–342. doi: 10.7326/0003-4819-141-5-200409070-00106. [DOI] [PubMed] [Google Scholar]
- 38.Leung GM, Hedley AJ, Ho LM, et al. The epidemiology of severe acute respiratory syndrome in the 2003 Hong Kong epidemic: an analysis of all 1755 patients. Ann Intern Med. 2004;141:662–673. doi: 10.7326/0003-4819-141-9-200411020-00006. [DOI] [PubMed] [Google Scholar]
- 39.ibayashi M, Nakad K, Nagata K. Promoted cell death of cells expressing human MxA by influenza virus infection. Microbiol Immunol. 2002;46:29–36. doi: 10.1111/j.1348-0421.2002.tb02673.x. [DOI] [PubMed] [Google Scholar]
- 40.Salomon R, Staeheli P, Kochs G, et al. Mx1 gene protects mice against the highly lethal human H5N1 influenza virus. Cell Cycle. 2007;6:2417–2421. doi: 10.4161/cc.6.19.4779. [DOI] [PubMed] [Google Scholar]
- 41.Tumpey TM, Szretter KJ, Van Hoeven N, et al. The Mx1 gene protects mice against the pandemic 1918 and highly lethal human H5N1 influenza viruses. J Virol. 2007;81:10818–10821. doi: 10.1128/JVI.01116-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeng H, Goldsmith C, Thawatsupha P, et al. Highly pathogenic avian influenza H5N1 viruses elicit an attenuated type i interferon response in polarized human bronchial epithelial cells. J Virol. 2007;81:12439–12449. doi: 10.1128/JVI.01134-07. [DOI] [PMC free article] [PubMed] [Google Scholar]