

Abstract
3C-like protease (3CLpro) plays pivotal roles in the life cycle of severe acute respiratory syndrome coronavirus (SARS-CoV) and only the dimeric protease is proposed as the functional form. Guided by the crystal structure and molecular dynamics simulations, we performed systematic mutation analyses to identify residues critical for 3CLpro dimerization and activity in this study. Seven residues on the dimer interface were selected for evaluating their contributions to dimer stability and catalytic activity by biophysical and biochemical methods. These residues are involved in dimerization through hydrogen bonding and broadly located in the N-terminal finger, the α-helix A′ of domain I, and the oxyanion loop near the S1 substrate-binding subsite in domain II. We revealed that all seven single mutated proteases still have the dimeric species but the monomer–dimer equilibria of these mutants vary from each other, implying that these residues might contribute differently to the dimer stability. Such a conclusion could be further verified by the results that the proteolytic activities of these mutants also decrease to varying degrees. The present study would help us better understand the dimerization-activity relationship of SARS-CoV 3CLpro and afford potential information for designing anti-viral compounds targeting the dimer interface of the protease.
Keywords: catalytic mechanism, dimerization-activity relationship, dimer interface, residue–residue interactions, site-directed mutagenesis
The disease of severe acute respiratory syndrome (SARS) broke out in China and menaced to more than 30 other countries from the end of 2002 to June 2003. SARS coronavirus (SARS-CoV) was identified as the etiological agent responsible for this infection (1, 2). SARS-CoV involves the largest viral RNA genome known to date, encompassing 29,727 nucleotides predicted to contain 14 functional open reading frames (ORFs) (3). Two large 5′-terminal ORFs, 1a and 1b, encode two overlapping polyproteins, pp1a (around 450 kDa) and pp1ab (around 750 kDa) necessary for viral RNA synthesis. Polyproteins pp1a and pp1ab are cleaved extensively by 3C-like protease (3CLpro) and a papain-like cysteine protease (PL2pro) to yield a multi-subunits protein complex called ‘viral replicase-transcriptase’ (4). Considering its functional indispensability in coronavirus life cycle, SARS-CoV 3CLpro has become an attractive target in discovering new anti-SARS agents (5).
The crystal structure revealed that SARS-CoV 3CLpro can form a dimer with the two monomers oriented perpendicular to one another (Fig. 1A and B) (6). Each monomer contains three domains: domains I (residues 8–101) and II (residues 102–184) have six-stranded antiparallel β-barrel forming a chymotrypsin fold, the substrate-binding pocket is located in a cleft between these two domains, while domain III (residues 201–303) is an antiparallel globular cluster of five α-helices connecting to domain II by a long loop region (residues 185–200). The 16-residue loop region has been implicated to mediate the substrate-binding (7). Based on the crystal structure, the N-terminal finger (residues 1–7) might play an important role in both dimerization and enzymatic activity of SARS-CoV 3CLpro. Numerous reports have proven that the N-terminal finger contributes well to dimerization of SARS-CoV 3CLpro (8–10). In addition, domain III has also been revealed to extensively involve in monomer–monomer interactions (7, 11). Furthermore, Hsu et al. (9) reported that the residue Arg4 and the last C-terminal helix (residues 293–306) are critical for stabilizing the dimer structure to maintain a correct conformation of the active site. As the crystal structures of different CoV 3CL proteases give similar dimeric structures and nearly all residues of 3CLpro involved in dimerization are conserved, it has been indicated that only the dimer is the biological functional form of SARS-CoV 3CLpro (12, 13). Tan et al.(14) revealed that the low enzymatic activity of the dissociated monomer is mainly due to the collapse of the oxyanion hole in the S1 substrate-binding subsite. Since the dissociated monomer might be inactive, the dimer interface has been suggested as a potential target for rational inhibitors design against SARS-CoV 3CLpro (7, 15). An octapeptide interface inhibitor, designed according to the sequence of the N-terminal finger, was found to bind to SARS-CoV 3CLpro specifically and competitively (16).
Fig. 1.
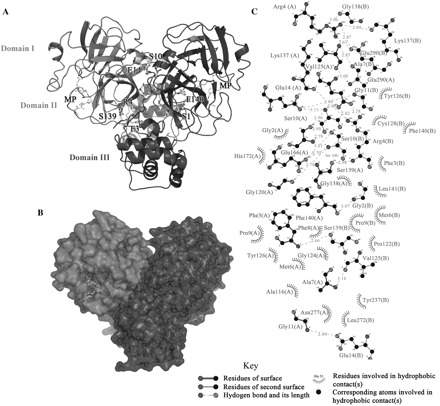
The dimeric structure of SARS-CoV 3CLpro and extensive residue–residue interactions on the dimer interface. (A) A ribbon diagram for the crystal structure of SARS-CoV 3CLpro (PDB: 1UK2). Monomer A and B are represented as black and grey, respectively and the three domains are also labelled. The residues involved in monomer–monomer interactions, which were selected for subsequent single point mutation analyses, are shown in the bond model. The binding peptide substrate (MP) is also shown as the stick model. (B) A surface model of the protease. The two monomers are in the same orientation as shown in panel A. (C) The dimer interface between monomer A and B. The bonds and residues belonging to monomer A or B are labelled respectively.
The crystal structure (6) and molecular dynamics calculations (14) revealed that the dimer interface of SARS-CoV 3CLpro mainly consists of the interactions between two helical domains III of each monomer, and the hydrogen bonding and electrostatic interactions between the N-terminal finger of one monomer and the residues near S1 substrate-binding subsite of the other monomer, in particular an oxyanion loop (residues 138–145) (Fig. 1A and C). According to the reported studies mentioned above, the structural integrity of the active site appears to be intrinsically connected with the presence of an intact dimer interface for SARS-CoV 3CLpro. To address this hypothesis, we performed the structure-guided mutagenesis analyses of the protease in this study. Totally seven residues on the dimer interface were selected, including three residues in the N-terminus (Ser1, Phe3, Arg4) (Fig. 2A), two residues in the α-helix A′ of domain I (Ser10, Glu14) (Fig. 2B), and two residues of the oxyanion loop near the S1 subsite in domain II (Ser139, Phe140) (Fig. 2A). These residues are mainly involved in dimerization of SARS-CoV 3CLpro through hydrogen bonding and highly conserved among different CoV 3CL proteases. In the following, we evaluated the effects of these residues on dimer conformational stability and catalytic activity of SARS-CoV 3CLpro using various biochemical and biophysical techniques. It was demonstrated that all seven single point mutated proteases can still form the dimer at varying concentrations, while the monomer–dimer equilibria of these mutants in solution are different from that of the wild type protease. Furthermore the proteolytic activities of these mutants decreased to varying extents compared with the wild type protease. Although the dimer formation of SARS-CoV 3CLpro could not be disrupted completely by single point mutation, individual replacement of these residues by alanine might partly disrupt the integrality of the hydrogen bonding networks on the dimer interface, which perhaps induces an altered conformation of the substrate-binding pocket, therefore, results in the decrease or loss of the enzymatic activity.
Fig. 2.
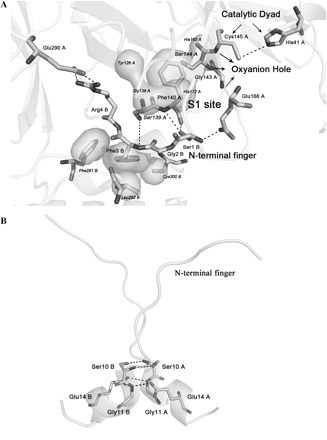
Residue–residue interactions. (A) Residue–residue interactions between the N-terminal finger and the S1 subsite in the substrate-binding pocket of SARS-CoV 3CLpro. (B) Residue–residue interactions between two α–helix A′ (residues 10–15) of domain I in SARS-CoV 3CLpro dimer. The residues belonging to monomer A or B (PDB: 1UK2) are marked respectively. The labelled residues are shown as sticks, and the rest of the proteins as cartoon. Dashes represent the hydrogen bonds formed on the dimer interface. The hydrophobic interactions between the side-chain phenyl of Phe3 or Phe140 and the neighbouring residues are also labelled as the surface model.
MATERIALS AND METHODS
Simulation System
Initial coordinates for SARS-CoV 3CLpro dimer was taken from the crystal structure (6) (PDB code: 1UK2). The missing residues were repaired using the loop search method in the Homology module of Insight II. For the simulation of SARS-CoV 3CLpro dimer in aqueous solution, the protein was first put into a suitably sized box, of which the minimal distance from the protein to the box wall was 1.5 nm. Then the box was solvated with the SPC water model (17). The protein/water system was submitted to energy minimization. Later, counterions were added to the system to provide a neutral simulation system. The whole system was subsequently minimized again.
Molecular Dynamics Simulations
Conventional molecular dynamics (CMD) simulations were carried out using the AMBER 7.0 package with NPT and periodic boundary conditions. The Amber Parm99 force field (18) was applied for the proteins. The Particle Mesh Ewald (PME) method (19) was employed to calculate the long-range electrostatics interactions. The non-bonded cutoff was set to 12.0 Å, and the non-bonded pairs were updated every 25 steps. The SHAKE method (20) was applied to constrain all covalent bonds involving hydrogen atoms. Each simulation was coupled to a 300 K thermal bath at 1.0 atm pressure by applying the algorithm of Berendsen (21). The temperature and pressure coupling parameters were set as 0.2 ps and 0.05 ps, respectively. An integration step of 2 fs was set up for the MD simulations.
Materials
The restriction and modifying enzymes in this work were purchased from NEB. The vector pQE30 and the bacterial strain M15 were from Qiagen. Isopropyl β-D-thiogalactoside (IPTG) was purchased from Promega. The Ni-chelating column and low molecular weight marker for SDS–PAGE were purchased from Amersham Pharmacia Biotech. All other chemicals were of reagent grade or ultra-pure quality, and purchased from Sigma.
Cloning, Expression and Purification of the Wild Type SARS-CoV 3CLpro
The wild type SARS-CoV 3CLpro was prepared according to our published method (22). The protease was highly pure according to SDS–PAGE and dialyzed to 20 mM Tris–HCl pH 7.5 containing 100 mM NaCl, 5 mM dithiothreitol (DTT) and 1 mM ethylene diaminetetraacetic acid (EDTA). The purified protein was further confirmed by N-terminal sequencing and mass spectrometry, and concentrated by Centriprep (Milipore). The protein concentration used in all experiments was determined from the absorbance at 280 nm (A280) using a molar extinction coefficient (ε280) for the monomer of 34,390/M cm (22, 23).
Site-directed Mutagenesis of the Residues on the Dimer Interface of SARS-CoV 3CLpro
Site-directed mutagenesis of the residues on the dimer interface of SARS-CoV 3CLpro was processed by a modified recombinant PCR method. Totally seven mutated SARS-CoV 3CLpros (Ser1Ala, Phe3 Ala, Arg4Ala, Ser10Ala. Glu14Ala, Ser139Ala and Phe140Ala) were prepared with the QuikChange site-directed mutagenesis kit (Stratagene) using pQE30-SARS-CoV 3CLpro as a template. The nucleotide sequences of the primers used for mutation were given in Table 1. The pQE30-SARS-CoV 3CLpro plasmids encoding mutated forms of SARS-CoV 3CLpro were verified by sequencing, and then Escherichia coli M15 cells were transformed by the resulting plasmids. The mutated proteins were expressed and purified in a similar procedure to that for the wild type protease. The purity and structural integrity of the mutated proteases were analysed by SDS–PAGE, N-terminal sequencing and mass spectrometry. The concentrated proteins were stored in 20 mM Tris–HCl pH 7.5, 100 mM NaCl, 5 mM DTT, 1 mM EDTA, at −20°C.
Table 1.
Nucleotide sequences of the primers used for site-directed mutagenesis of SARS-CoV 3CLpro.a
| Oligonucleotide sequence (5′→ 3′) | Polarity | Mutation introduced |
|---|---|---|
| CATCACGGATCCACCATGGCTGGTTTTAGGAAAATGGC | Forward | SARS-CoV 3CLpro Ser1Ala |
| GCCATTTTCCTAAAACCAGCCATGGTGGATCCGTGATG | Reverse | SARS-CoV 3CLpro Ser1Ala |
| CCACCATGAGTGGTGCTAGGAAAATGGCATTCCCG | Forward | SARS-CoV 3CLpro Phe3Ala |
| CGGGAATGCCATTTTCCTAGCACCACTCATGGTGG | Reverse | SARS-CoV 3CLpro Phe3Ala |
| CCACCATGAGTGGTTTTGCGAAAATGGCATTCCCGTC | Forward | SARS-CoV 3CLpro Arg4Ala |
| GACGGGAATGCCATTTTCGCAAAACCACTCATGGTGG | Reverse | SARS-CoV 3CLpro Arg4Ala |
| GGAAAATGGCATTCCCGGCAGGCAAAGTTGAAGG | Forward | SARS-CoV 3CLpro Ser10Ala |
| CCTTCAACTTTGCCTGCCGGGAATGCCATTTTCC | Reverse | SARS-CoV 3CLpro Ser10Ala |
| CGTCAGGCAAAGTTGCAGGGTGCATGGTAC | Forward | SARS-CoV 3CLpro Glu14Ala |
| GTACCATGCACCCTGCAACTTTGCCTGACG | Reverse | SARS-CoV 3CLpro Glu14Ala |
| CATACCATTAAAGGTGCTTTCCTTAATGGATCATGTGG | Forward | SARS-CoV 3CLpro Ser139Ala |
| CCACATGATCCATTAAGGAAAGCACCTTTAATGGTATG | Reverse | SARS-CoV 3CLpro Ser139Ala |
| CATACCATTAAAGGTTCTGCCCTTAATGGATCATGTGG | Forward | SARS-CoV 3CLpro Phe140Ala |
| CCACATGATCCATTAAGGGCAGAACCTTTAATGGTATG | Reverse | SARS-CoV 3CLpro Phe140Ala |
aThe mutant codons in the oligonucleotide sequences are highlighted in boldface. SARS-CoV 3CLpro amino acids are numbered continuously from the N-terminal residue, Ser1, to the C-terminal residue, Gln303.
Circular Dichroism (CD) Spectroscopy
Circular Dichroism (CD) spectra were recorded on a JASCO-810 spectropolarimeter. The protein sample was prepared in 20 mM sodium phosphate pH 7.5, 100 mM NaCl at 25°C with concentration of 10 μM. Far-UV CD spectra from 190 to 250 nm were collected with 1 nm band width using 0.1 cm path length cuvette, and normalized by subtracting the baseline recorded for the buffer. Each measurement was repeated thrice and the final result was the average of three independent scans. The Far-UV CD spectra of the mutated proteases were compared with that of the wild type SARS-CoV 3CLpro to exclude the possibility of structural misfolding caused by single point mutation.
Fluorescence Spectroscopy
The fluorescence experiments were performed on a HITACHI F-2500 fluorescence spectrophotometer. The protease sample was prepared in 20 mM Tris–HCl pH 7.5, 100 mM NaCl with concentration of 5 μM. The fluorescence emission spectra from 300 to 380 nm were collected after excitation at 280 nm, and the spectral slit width was 5 nm for excitation and emission. Fluorescence spectra of the wild type and mutated SARS-CoV 3CLpros were measured in a 1 ml quartz cuvette with 1 cm path length at 25°C. All final spectra were corrected for the buffer contribution, and were the average of three parallel measurements.
Glutaraldehyde Cross-linking SDS–PAGE
For the wild type and mutated SARS-CoV 3CLpros (final concentration from 0.2 to 5 mg/ml in 20 mM Tris–HCl pH 7.5, 100 mM NaCl, 5 mM DTT, 1 mM EDTA) an aliquot of 25% (v/v) glutaraldehyde was added to make a final concentration of 0.05 or 0.1% glutaraldehyde. The samples were incubated at 25°C for 15 min followed by quenching the reaction with the addition of 1.0 M Tris–HCl pH 8.0 (0.5% v/v). Orthophosphoric acid was thereafter added into the reaction mixture to result in precipitation of the cross-linked proteins. After centrifugation (12,000 r.p.m., 4°C), the precipitate was re-dissolved in loading buffer and heated at 100°C for 5 min. SDS–PAGE was run with 10% gels.
Size-exclusion Chromatography (SEC) Analysis
The dimer–monomer equilibria of the wild type and mutated SARS-CoV 3CLpros were analysed by size-exclusion chromatography (SEC) on a HiLoad 16/60 Superdex 75 prep grade column through an AKTA FPLC system (Amersham Biosciences). Buffer used was 20 mM Tris–HCl pH 7.5, 100 mM NaCl, 5 mM DTT and 1 mM EDTA. The buffer was degassed and the column was equilibrated with the buffer before injecting protein samples. Protein samples with a concentration of 5 mg/ml were loaded on the column and then eluted with the buffer at a flow rate of 1 ml/min by detection of absorbance at 280 nm. The integrated area values of absorbance peaks were calibrated by AKTA FPLC evaluation software. The column was calibrated using a low molecular mass gel filtration kit (Amersham Biosciences) with four marker proteins: Ribonuclease A (13.7 kDa), Chymotrypsinogen A (25.0 kDa), Ovalbumin (43.0 kDa) and Albumin (67.0 kDa).
Enzymatic Activity Assay
The catalytic activities of the wild type and mutated SARS-CoV 3CLpros were measured by FRET-based assays using a 12-amino acid fluorogenic substrate, EDANS-VNSTLQSGLRK (Dabcyl)-M, according to our published studies (23, 24). During the continuously kinetic assay, the protease (final concentration 1 μM) was pre-incubated for 30 min at 25°C with the assay buffer (20 mM Tris–HCl pH 7.5, 100 mM NaCl, 5 mM DTT and 1 mM EDTA), followed by the addition of the fluorogenic substrate (final concentration 10 μM). The fluorescence intensity was monitored on a GENios microplate reader (TECAN, Männedorf, Switzerland) and the instrument was first set to zero with the fluorogenic substrate. Cleavage of the substrate as a function of time was measured by the increase in emission fluorescence intensity upon continuous monitoring of reactions in a 96-well black microplate (BMG LABTECH, Offenburg, Germany) using wavelengths of 340 nm and 488 nm for excitation and emission, respectively. The incubation of the substrate in the assay buffer without the protease was also performed as a control. Enzymatic activity was the average of three parallel assays and the activity of the wild type SARS-CoV 3CLpro was taken as 100%.
RESULTS
Preparation of the Seven Mutated Proteases Involved in the Dimer Interface
To predict the key factors that maintain the stability of the dimer interface, 5-ns CMD simulations were firstly conducted on the dimer of SARS-CoV 3CLpro. All interactive residues between monomer A and B, as shown in Fig. 1C, were monitored for the time occupancy during the whole simulation process. The hydrogen bonds formed on the dimer interface were calculated by using HPLUS (25). Interestingly, more than 10 hydrogen bonding interactions occupy most time of simulation (Table 2), suggesting that the residues involved in these interactions might possibly make well contributions to keep the dimer conformational stability. Guided by this potential information, we selected seven residues on the dimer interface for site-directed mutagenesis (Fig. 1A). These residues are mainly involved in the dimerization of SARS-CoV 3CLpro through hydrogen bonding and hydrophobic interactions with their side-chain or main-chain groups (Fig. 2), and single Ala substitution might perturb the entirety of the hydrogen bonding networks on the dimer interface.
Table 2.
Potential residue–residue interactions on the dimer interface of SARS-CoV 3CLpro predicted by 5-ns molecular dynamics simulations.a
| Hydrogen Bond | Time occupancy (%) | ||
|---|---|---|---|
| A | B | ||
| 1 | Ser10 (OG) | Ser10 (OG) | 99.1 |
| 2 | Gly11(N) | Glu14 (OE1/OE2) | 100 |
| 3 | Glu14 (OE1/OE2) | Gly11(N) | 100 |
| 4 | Arg4(NH1/NH2) | Glu290(OE1/OE2) | 95.7 |
| 5 | Glu290(OE1/OE2) | Arg4(NH1/NH2) | 96.9 |
| 6 | Ser139(O/OG) | Gly2(N) | 99.2 |
| 7 | Gly2(N) | Ser139(O/OG) | 30.3 |
| 8 | Phe3(N) | Ser139(O/OG) | 88.2 |
| 9 | Ser139(O/OG) | Phe3(N) | 43.5 |
| 10 | Ser1(OG) | Glu166(OE1/OE2) | 98.3 |
| 11 | Glu166(OE1/OE2) | Ser1(OG) | 55.4 |
| 12 | Phe140(N) | Ser1(O/OG) | 93.0 |
| 13 | Ser1(O/OG) | Phe140(N) | 5.1 |
aA, monomer A; B, monomer B.
According to the preparation strategy previously reported in our lab (22), we expressed SARS-CoV 3CLpro as an N-terminal His-tagged protein for purification convenience. Considering the results shown in several other publications (10, 15, 26, 27) that N-terminal extra amino acids, e.g. the purification affinity tag, might interfere with dimerization of SARS-CoV 3CLpro, we also constructed the protease into a vector without affinity tag and evaluated the dimerization feature of the un-tagged protein. While there are no obvious differences observed for the dimer–monomer equilibrium in solution between the two purified proteases (data not shown), thus we performed all subsequent assays with the N-terminal His-tagged 3CLpro.
Similar with the wild type protease, all the seven single point mutants were also successfully cloned and expressed in E. coli M15 cells. The majority of the proteins could be obtained in the soluble fraction of the cell lysate. SDS–PAGE analyses indicated that all mutated proteases are highly homogeneous in solution. Although the corresponding protein bands in SDS–PAGE would shift little faster than the molar marker of 35.0 kDa, the recombinant proteins have been clearly identified as SARS-CoV 3CLpro with a molecular mass of 35.8 kDa by mass spectrometric characterization (data not shown), in agreement with the values calculated from the protein sequences and the published data from our laboratory (22).
Figure 3 shows the Far-UV CD spectra of the wild type and seven mutants of SARS-CoV 3CLpro. The spectra of the seven mutated proteases seem to be similar to that of the wild type SARS-CoV 3CLpro. All spectra give a positive peak at 196 nm and dual negative peaks at 209 and 222 nm, typical of a mixture of α-helical and β-sheet structures. These results indicated that all seven mutated proteases have well-defined secondary structures and excluded the possibility of structural misfolding caused by single residue mutation. However small changes of the CD spectra do exist as shown in Fig. 3, which might be due to minor structural changes induced by Ala mutations.
Fig. 3.
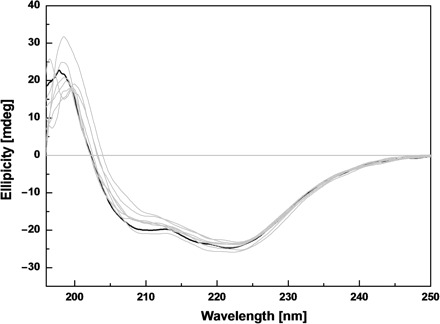
CD spectra of the wild type and site-directed mutants for SARS-CoV 3CLpro. Far-UV CD spectra of the wild type and seven mutated SARS-CoV 3CLpros at 25°C. Protein concentrations used in CD experiments were 10 μM and all protein samples were prepared in 20 mM sodium phosphate pH 7.5, 100 mM NaCl. The CD spectrum of the wild type protease is shown in black and the spectra of the mutated proteases are shown in light gray.
The fluorescence emission spectra of the wild type and seven mutants of SARS-CoV 3CLpro are also shown in Fig. 4. The emission λmax of the wild type SARS-CoV 3CLpro is 325 nm. Similar to the wild type protease, all seven mutated proteins show only minor difference on the emission λmax (varying from 324 nm to 327 nm), further demonstrating that replacement of single residue on the dimer interface by Ala has not changed the folding manner of the protease.
Fig. 4.
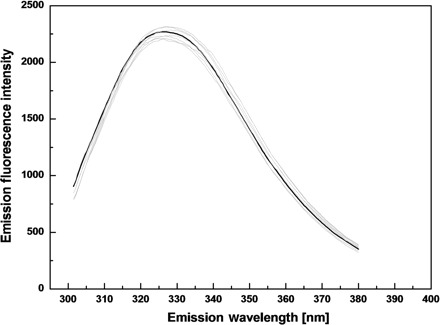
Fluorescence emission spectra of the wild type and site-directed mutants for SARS-CoV 3CLpro. Fluorescence emission spectra of the wild type and seven mutated proteases were recorded at 25°C after excitation at 280 nm. The protease samples (5 μM) were prepared in 20 mM Tris–HCl pH 7.5, 100 mM NaCl. The spectrum of the wild type protease is shown in black and those of the mutants are shown in light gray.
Chemical Cross-linking Analyses of the Seven Mutated Proteases
Similar to 3CL proteases of human coronavirus (HCoV) 229E and transmissible gastroenteritis coronavirus (TGEV) (28), SARS-CoV 3CLpro can form a dimer in the crystal structure and solution (6, 13). The dimerization features of SARS-CoV 3CLpro have been successfully characterized by various biochemical and biophysical methods (7–9, 12). According to the published method (29), we first performed the chemical cross-linking analysis of the wild type SARS-CoV 3CLpro. When incubated with 0.05% glutaraldehyde, the protease at a concentration of 0.2 mg/ml displayed a form of monomer near 35.0 kDa with the other band corresponding to the dimer (Fig. 5A, lane 6b). With protein concentration increasing, both of the dimeric and monomeric forms increased (Fig. 5A, lanes 5b–3b). A similar cross-linking pattern of the protease was observed when using a higher concentration of glutaraldehyde (0.1%), excluding the possibility of obvious artificial cross-linking effects (Fig. 5A, lanes 6a–3a). These results indicate that the wild type protease exists as a mixture of monomer and dimer at varying concentrations, which is consistent with the reported studies (7, 8).
Fig. 5.
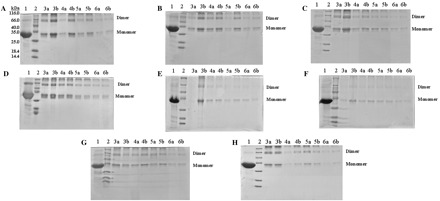
SDS–PAGE profiles of glutaraldehyde cross-linked SARS-CoV 3CLpros. (A) Cross-linking analysis of the wild type SARS-CoV 3CLpro. (B–H) Cross-linking analyses of Ser1_Ala, Phe3_Ala, Arg4_Ala, Ser10_Ala, Glu14_Ala, Ser139_Ala and Phe140_Ala mutants, respectively. Lane 1, untreated 3CLpro (5 mg/ml); lane 2, molecular weight protein standards; lane 3a, 3CLpro (5 mg/ml) cross-linked by 0.1% glutaraldehyde; lane 3b, 3CLpro (5 mg/ml) cross-linked by 0.05% glutaraldehyde; lanes 4a and 4b, 3CLpro (1 mg/ml), 0.1 and 0.05% glutaraldehyde; lanes 5a and 5b, 3CLpro (0.5 mg/ml), 0.1 and 0.05% glutaraldehyde; lanes 6a and 6b, 3CLpro (0.2 mg/ml), 0.1 and 0.05% glutaraldehyde.
To preliminarily examine the effects of Ala mutations of the selected seven residues on dimerization of SARS-CoV 3CLpro, the chemical cross-linking analyses of the mutated proteases were also performed, respectively. Conclusively all seven mutated proteases displayed similar cross-linking patterns with the wild type protease (Fig. 5B–H). The dimeric form of each mutated protease also existed within a wide range of protein concentrations, suggesting that mutation of single residue on the dimer interface could not completely abolish the dimeric structure of SARS-CoV 3CLpro in solution. However moderate differences of dimer–monomer equilibria do exist among these mutants. For the Ser10_Ala and Glu14_Ala mutants (Fig. 5E and F), the amount of the dimer was relatively low compared with the wild type and other mutated proteases, indicating that the α-helix A′ of domain I might be an important part of the dimer interface and relatively contribute more to maintain the dimer stability of SARS-CoV 3CLpro. In addition, we should note that the possibility of minor artificial cross-linking effects might still exist due to the appearance of the high-order multimers in SDS–PAGE (Fig. 5). While, considering the chemical cross-linking analyses of the wild type and mutant proteases were performed under exactly same experimental procedures, these analyses might still be convincing to preliminarily examine the effects of these mutations on dimerization of SARS-CoV 3CLpro.
SEC Analyses of the Seven Mutated Proteases
In order to more exactly evaluate the perturbation of dimer–monomer equilibrium caused by these mutations, we performed SEC analyses to further characterize the wild type and mutated SARS-CoV 3CLpros. We used a protein concentration at 5 mg/ml for each run, which represents the highest concentration used in the cross-linking experiments, and the physical states corresponding to native monomeric and dimeric protease were observed. As shown in Fig. 6A, the wild type SARS-CoV 3CLpro elutes in two peaks with the retention volumes at 44.6 and 62.1 ml. The elution profiles of four molecular mass marker proteins confirmed that the first peak might correspond to the dimer state (71.6 kDa) and the second peak would represent the monomeric species of SARS-CoV 3CLpro (35.8 kDa), in well agreement with the reported result (8). We also collected the fractions representing these two elution peaks and analysed them by SDS–PAGE, and the corresponding protein bands further indicated that both of these two peaks are SARS-CoV 3CLpro (data not shown). The amount of the dimer and monomer could be further quantified by the integrated area values of these two peaks and the dimer/monomer ratio of the wild type protease was estimated as 1.02 (Table 3). This observation thus indicates that in solution the wild type protease exhibits both forms of monomer and dimer and the amount of the monomer is almost equal to that of the dimeric form, in well agreement with the chemical cross-linking analysis and literature report (13).
Fig. 6.
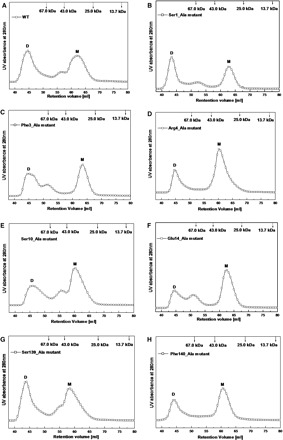
Dimer–monomer equilibria of the wild type and site-directed mutants for SARS-CoV 3CLpro analysed by SEC. (A) Elution profile of the wild type SARS-CoV 3CLpro at neutral pH (7.5) and a concentration of 5 mg/ml; (B–(H) Elution profiles of Ser1_Ala, Phe3_Ala, Arg4_Ala, Ser10_Ala, Glu14_Ala, Ser139_Ala and Phe140_Ala mutants at concentrations of 5 mg/ml, respectively. Elution profiles of four marker proteins are also shown in arrow labels. Each protein sample was loaded to a HiLoad 16/60 Superdex 75 prep grade column and then eluted at a flow rate of 1 ml/min with detection of absorbance at 280 nm.
Table 3.
Elution profiles of the wild type and site-directed mutants for SARS-CoV 3CLpro in solution by SEC analyses.
| Protein | Elution peakDimer (ml) | Elution peak monomer (ml) | Dimer (%)a | Monomer (%)a | D (%)/M (%) |
|---|---|---|---|---|---|
| WT SARS-CoV 3CLpro | 44.6 | 62.1 | 50.5 | 49.5 | 1.02 |
| SARS-CoV 3CLpro Ser1Ala | 43.4 | 62.8 | 51.9 | 48.1 | 1.08 |
| SARS-CoV 3CLpro Phe3Ala | 44.8 | 63.4 | 48.2 | 51.8 | 0.93 |
| SARS-CoV 3CLpro Arg4Ala | 44.8 | 60.4 | 31.1 | 68.9 | 0.45 |
| SARS-CoV 3CLpro Ser10Ala | 45.8 | 60.2 | 39.8 | 60.2 | 0.66 |
| SARS-CoV 3CLpro Glu14Ala | 44.4 | 62.4 | 26.5 | 73.5 | 0.36 |
| SARS-CoV 3CLpro Ser139Ala | 43.8 | 59.2 | 44.8 | 55.2 | 0.81 |
| SARS-CoV 3CLpro Phe140Ala | 44.2 | 60.6 | 38.7 | 61.3 | 0.63 |
aThe percentage of dimers (D) and monomers (M) was estimated by deconvolution of the corresponding SEC elution profiles.
For the seven mutated proteases under identical conditions, the two elution-peaks representing the dimer and monomer states were also monitored, respectively (Fig. 6B–H). The results demonstrate that dimerization of SARS-CoV 3CLpro could not be disrupted entirely by mutation of single residue on the dimer interface, further supporting the chemical cross-linking results. Compared with the wild type protease, these mutants showed minor drifts on the retention volumes of the two elution peaks (Table 3, varying from 43.4 ml to 45.8 ml and from 59.2 ml to 63.4 ml), indicative of possible subtle conformational changes of the dimer and monomer structures. Furthermore, the dimer/monomer ratios of these mutants differentiated significantly from each other (Table 3), implying that the contributions of these residues to the monomer–dimer equilibrium of SARS-CoV 3CLpro are quite different. For the Ser1_Ala, Phe3_Ala and Ser139_Ala mutants, the ratios between the dimers and monomers were 1.08, 0.93 and 0.81, respectively, which indicates that these three residues could only affect the dimer interface stability to a lesser extent. For the other mutants, especially the Arg4_Ala and Glu14_Ala mutants, the dimer/monomer ratios decreased obviously and were nearly 2 to 3-fold lower than that of the wild type protease, suggesting that the amount of the dimer has decreased and the monomer is the predominant form. Overall, Glu14, Arg4, Phe140 and Ser10 (in decreasing order) on the dimer interface are the relatively more critical residues for stabilizing the dimeric structure of SARS-CoV 3CLpro.
Enzymatic Activity Assays of the Seven Mutated Proteases
Several published results have proposed that only the dimer should be the biological functional form of SARS-CoV 3CLpro and the dissociated monomer might be enzymatic inactive (13, 30). Meanwhile, alteration of the correct conformation of the dimeric structure could also lead to a complete loss of the catalytic activity (8, 14). Although Ala replacement of single residue on the dimer interface could not completely result in the dimer dissociation in solution, the seven residues we selected still might affect the catalytic activity of SARS-CoV 3CLpro considering their contributions to stabilize the monomer–monomer interface. To verify this prediction, we determined the enzymatic activities of the wild type and seven mutated proteases by a fluorogenic substrate reported previously in our lab (23, 24). The catalytic activity of SARS-CoV 3CLpro and relevant inhibitors screening have been characterized extensively by the FRET-based assay (26, 31–33). As shown in Fig. 7, the fluorescence increase following hydrolysis of the substrate by the wild type SARS-CoV 3CLpro is significant and time-dependent, implying that the protease could hydrolyze the substrate efficiently. As expected, the fluorescence profiles of the seven mutants were obviously different from that of the wild type protease (Fig. 7), which indicates that mutation of these residues could inactivate the catalytic activity of SARS-CoV 3CLpro to varying extents. In detail, mutation of residues Ser10 and Phe140 almost produced the complete loss of the enzymatic activity and the catalytic activities of the Phe3_Ala, Glu14_Ala and Arg4_Ala mutants were also decreased to only 4–10% of that of the wild type protease (Table 4). While the mutants of Ser1_Ala and Ser139_Ala still possessed 46 and 58% of enzymatic activity, respectively. (Table 4). These results further support the conclusions derived from the SEC analyses that the extensive monomer–monomer interactions regulated by these residues could stabilize the dimeric structure at different degrees. However, it is noticeable that the influence of these mutations on the catalytic activity is more obvious than that on dimerization of SARS-CoV 3CLpro, which will be discussed below.
Fig. 7.
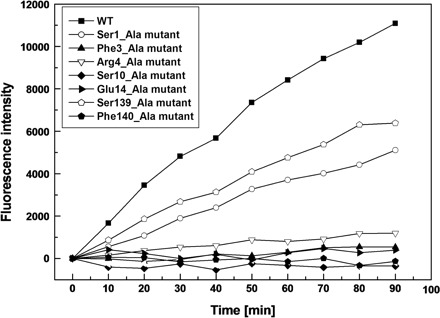
Fluorescence profiles of hydrolysis of the fluorogenic substrate by the wild type and site-directed mutants for SARS-CoV 3CLpro. The fluorogenic substrate at a concentration of 10 μM was incubated with 1 μM wild type or mutated SARS-CoV 3CLpro in 20 mM Tris–HCl pH7.5, 100 mM NaCl, 5 mM DTT, 1 mM EDTA, at 25°C. Increase of emission fluorescence intensity at 488 nm wavelength was recorded at 10 min intervals, λEX = 340 nm. The emission spectrum was recorded for 90 min and the activity of the wild type protease was taken as 100%.
Table 4.
Enzymatic activities of the wild type and site-directed mutants for SARS-CoV 3CLpro.a
| Protein | Proteolytic activity (%) |
|---|---|
| WT SARS-CoV 3CLpro | 100 |
| SARS-CoV 3CLpro Ser1Ala | 46 |
| SARS-CoV 3CLpro Phe3Ala | 5 |
| SARS-CoV 3CLpro Arg4Ala | 10 |
| SARS-CoV 3CLpro Ser10Ala | <1 |
| SARS-CoV 3CLpro Glu14Ala | 4 |
| SARS-CoV 3CLpro Ser139Ala | 58 |
| SARS-CoV 3CLpro Phe140Ala | <1 |
aEnzymatic activities were averages determined in three parallel experiments. The activity of wild type SARS-CoV 3CLpro was taken as 100%.
DISCUSSION
Proteolytic processing of the non-structural polyproteins is a vital step in the replication cycle of coronavirus, and such processing is commonly performed by virus-genome encoded proteases including 3CLpro (4, 34–36). Therefore 3CLpro has been appreciated as an attractive target in discovering anti-coronavirus agents (5). SARS-CoV 3CLpro shares high homology with the 3CLpros of other coronaviruses, and the 3D structures of different coronavirus 3CLpros are more conserved than their sequences (6, 37). SARS-CoV 3CLpro has been extensively characterized for its structural property and enzymatic activity (8, 9, 12–15, 27, 38, 39). The protease can form a homodimer in crystal and solution (Fig. 1A and B), and the dimeric structure is proposed to be indispensable for enzymatic activity. Much progress has been made for understanding the correlation between dimerization and catalytic activity of SARS-CoV 3CLpro (7–10, 12, 27, 30). Recently a systematic mutagenesis study reported an initial attempt to map the dimerization interface on the helical domain III of the protease (11).
In the present study, we focused another seven residues on the dimer interface of SARS-CoV 3CLpro for single point mutagenesis (Fig. 1A). These selected residues are predicted to involve in dimerization mainly through hydrogen bonding (Table 2 and Fig. 2). Structurally, the seven mutated proteases could be divided into three groups. The first group includes Ser1_Ala, Phe3_Ala and Arg4_Ala mutants regarding three residues on the N-terminal finger of SARS-CoV 3CLpro. The N-terminal finger of one monomer can form intensive interactions with domain II of the other monomer (6) (Fig. 2A), e.g. the NH group of Ser1 in monomer B donates hydrogen bonds to the main-chain carbonyl of Phe140 in monomer A, as well as the side-chain carboxylate of Glu166A; and the side-chain OH group of Ser1B forms a hydrogen bond with the main-chain NH group of Phe140A. In addition, the NH group of Phe3B donates a hydrogen bond to the side-chain OH group of Ser139A. This pair of hydrogen bond might be stabilized by hydrophobic interactions between the side-chain phenyl of Phe3B and the neighbouring residues, e.g. Leu282B and Phe291B. Whereas replacement of residue Ser1 or Phe3 by Ala rendered little influence on the dimer–monomer equilibrium of SARS-CoV 3CLpro (Fig. 5B, C and Table 3), indicating that these two residues might not play a vital role in dimerization. The results are in agreement with a reported study that the N-terminal residues1–3 truncated protease still exhibits a tendency to form dimer (9). The Ser1_Ala mutant maintained 46% of enzymatic activity and the activity of the Phe3_Ala mutant was nearly 10-fold lower than that of the wild type protease (Table 4), implying that in addition to dimerization, residues Ser1 and Phe3 could regulate the catalytic activity of the protease by other mechanisms. According to the crystal structure (Fig. 2A), the monomer–monomer interactions mediated by Ser1 and Phe3 might be helpful to maintain the correct catalytic conformation of the S1 subsite, and mutation of Ser1 or Phe3 possibly induces an altered uncompetitive conformation of the S1 subsite. Nevertheless, this hypothesis should be verified by the crystal structures determination of these two mutated proteases. Besides Ser1 and Phe3, another residue Arg4 was also selected for mutagenesis study. The side-chain guanidyl of Arg4 in one monomer forms a salt bridge with the side-chain of Glu290 in the other monomer (Fig. 2A), which has been reported as one of the major interactions between the two monomers (12). Here, Arg4_Ala mutant was shown to a tendency to monomer state (Table 3) and a very weak enzymatic activity (Table 4), further demonstrating the importance of the Arg4-mediated interactions in the quaternary structure and activity of the protease. Although the role of the N-terminal finger has been assessed by many investigations (9, 10), our results revealed that the residues on the N-terminal finger indeed contribute differently to the dimer stability and catalytic activity of SARS-CoV 3CLpro.
The second group of the mutants is about Ser10_Ala and Glu14_Ala, which are related to two residues on the α-helix A′ of domain I of SARS-CoV 3CLpro. The two residues are highly conserved among different coronavirus 3CL proteases and also extensively involved in monomer–monomer interactions (Fig. 1C). Residue Ser10 from each monomer can form a pair of hydrogen bond between the main-chain NH group and the side-chain OH group, and the side-chain carboxylate of Glu14 in one monomer donates a hydrogen bond with the main-chain NH group of Gly11 in the other monomer (Fig. 2B). In the present study, both of the two mutants were shown to weak dimerization (Fig. 5E–F and Table 3) and have no detectable enzyme activity either (Table 4), indicating that the α-helix A′ of domain I might also be a critical region for dimerization. Structurally, the α-helix A′ (residues Ser10-Gly15) connects to the N-terminal finger of SARS-CoV 3CLpro and might determine the correct spatial orientation of the N-terminal finger (Fig. 2B). In the dimer structure, the N-terminal finger can squeeze into the space between domain III of its parent monomer and domain II of the neighbouring monomer, which is indispensable for maintaining the correct catalytic conformation of the protease (6, 8). Mutation of Ser10 or Glu14 to Ala is possible to partly disrupt the structure of the α-helix A′ and produce a mis-oriented N-terminal finger, thus making the protease completely inactive. However, this conclusion should be further confirmed by the crystal structures of the corresponding mutated proteases (unpublished data from this laboratory).
In addition, we also performed another group of mutants including Ser139_Ala and Phe140_Ala. These two residues are located in the oxyanion loop (residues 138–145) of domain II and involved in the dimer interface by interacting with the N-terminus residues of the other monomer (Fig. 2A). The oxyanion loop is associated with the formation of the S1 subsite in the substrate-binding pocket, which determines the absolute specificity of SARS-CoV 3CLpro for Glu in the P1 position of the substrate (6, 14). The oxyanion loop is very flexible and a rearrangement of its correct conformation could induce the collapse of the oxyanion hole (Gly143-Ser144-Cys145) in the S1 subsite, therefore, inactivate the protease completely (14). The Ser139_Ala mutant showed only a minor difference in the monomer–dimer equilibrium with the wild type protease (Table 3), implying that the contribution of residue Ser139 to dimerization is not dominant. While the Ser139_Ala mutant preserved only 50% of the wild type activity (Table 4), which indicates that mutation of Ser139 might directly affect the catalysis, most probably by altering the conformation of the oxyanion loop. Although Phe140 donated hydrogen bonds to Ser1 through its main-chain groups, the Phe140_Ala mutant still had an obvious trend to the monomer state (Table 3). It is possible that the hydrophobic packing between the side-chain phenyl of Phe140 and the residues nearby (e.g. His163, His172) would also stabilize the Phe140–Ser1 interactions (Fig. 2A). Meanwhile, mutation of Phe140 completely abolished the proteolytic activity of SARS-CoV 3CLpro (Table 4), further suggesting that the interactions mediated by Phe140 might also contribute well to maintain the conformational stability of the S1 subsite (14).
In summary, our study characterized the contributions of several previously unidentified residues to the dimer stability and catalytic activity of SARS-CoV 3CLpro. Since the dimeric structure has been proved to be indispensable for enzymatic activity of SARS-CoV 3CLpro, it is easy to understand the conclusion of no dimer, no activity. In this study, some residues have been revealed to be important for both dimerization and activity. Meanwhile, SARS-CoV 3CLpro is a very flexible protein and the correct conformation state might also be vital for the protease to maintain its full activity. Thus, in addition to dimer dissociation, an altered conformation of the substrate-binding pocket possibly induced by single mutation on the dimer interface could also make the protease inactive. Our future study should be focused on determining the crystal structures of these mutated proteases, which will shed more light on understanding the dimerization-activity relationship of SARS-CoV 3CLpro.
Acknowledgments
This work was supported by the State Key Program of Basic Research of China (grants 2004CB58905 and 2006AA09Z447), the National Natural Science Foundation of China (grants 30525024 and 20472095), Shanghai Basic Research Project from the Shanghai Science and Technology Commission (grants 06JC14080, 03DZ19228) and Foundation of Chinese Academy of Sciences (grant KSCX1-YW-R-18).
Glossary
Abbreviations:
- 3Clpro
3C-like protease
- CD
circular dichroism
- Dabcyl
4-[[4-(dimethylamino) phenyl] azo] benzoic acid
- EDANS
5-[(2′-aminoethyl)-amino] naphthelenesulfonic acid
- FRET
fluorescence resonance energy transfer
- SARS-CoV
severe acute respiratory syndrome coronavirus
- SEC
size-exclusion chromatography
- WT
wild type
REFERENCES
- 1.Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W, Nicholls J, Yee WK, Yan WW, Cheung MT, Cheng VC, Chan KH, Tsang DN, Yung RW, Ng TK, Yuen KY. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TC, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus AD, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 3.Marra MA, Jones SJ, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YS, Khattra J, Asano JK, Barber SA, Chan SY, Cloutier A, Coughlin SM, Freeman D, Girn N, Griffith OL, Leach SR, Mayo M, McDonald H, Montgomery SB, Pandoh PK, Petrescu AS, Robertson AG, Schein JE, Siddiqui A, Smailus DE, Stott JM, Yang GS, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth TF, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples GA, Tyler S, Vogrig R, Ward D, Watson B, Brunham RC, Krajden M, Petric M, Skowronski DM, Upton C, Roper RL. The Genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 4.Thiel V, Ivanov KA, Putics A, Hertzig T, Schelle B, Bayer S, Weissbrich B, Snijder EJ, Rabenau H, Doerr HW, Gorbalenya AE, Ziebuhr J. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 5.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 6.Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, Sun L, Mo L, Ye S, Pang H, Gao GF, Anand K, Bartlam M, Hilgenfeld R, Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi J, Wei Z, Song J. Dissection study on the severe acute respiratory syndrome 3C-like protease reveals the critical role of the extra domain in dimerization of the enzyme: defining the extra domain as a new target for design of highly specific protease inhibitors. J. Biol. Chem. 2004;279:24765–24773. doi: 10.1074/jbc.M311744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen S, Chen L, Tan J, Chen J, Du L, Sun T, Shen J, Chen K, Jiang H, Shen X. Severe acute respiratory syndrome coronavirus 3C-like proteinase N terminus is indispensable for proteolytic activity but not for enzyme dimerization. Biochemical and thermodynamic investigation in conjunction with molecular dynamics simulations. J. Biol. Chem. 2005;280:164–173. doi: 10.1074/jbc.M408211200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu WC, Chang HC, Chou CY, Tsai PJ, Lin PI, Chang GG. Critical assessment of important regions in the subunit association and catalytic action of the severe acute respiratory syndrome coronavirus main protease. J. Biol. Chem. 2005;280:22741–22748. doi: 10.1074/jbc.M502556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei P, Fan K, Chen H, Ma L, Huang C, Tan L, Xi D, Li C, Liu Y, Cao A, Lai L. The N-terminal octapeptide acts as a dimerization inhibitor of SARS coronavirus 3C-like proteinase. Biochem. Biophys. Res. Commun. 2006;339:865–872. doi: 10.1016/j.bbrc.2005.11.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi J, Song J. The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J. 2006;273:1035–1045. doi: 10.1111/j.1742-4658.2006.05130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chou CY, Chang HC, Hsu WC, Lin TZ, Lin CH, Chang GG. Quaternary structure of the severe acute respiratory syndrome (SARS) coronavirus main protease. Biochemistry. 2004;43:14958–14970. doi: 10.1021/bi0490237. [DOI] [PubMed] [Google Scholar]
- 13.Fan K, Wei P, Feng Q, Chen S, Huang C, Ma L, Lai B, Pei J, Liu Y, Chen J, Lai L. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 2004;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan J, Verschueren KH, Anand K, Shen J, Yang M, Xu Y, Rao Z, Bigalke J, Heisen B, Mesters JR, Chen K, Shen X, Jiang H, Hilgenfeld R. pH-dependent conformational flexibility of the SARS-CoV main proteinase (Mpro) dimer: molecular dynamics simulations and multiple x-ray structure analyses. J. Mol. Biol. 2005;354:25–40. doi: 10.1016/j.jmb.2005.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu MF, Kuo CJ, Chang KT, Chang HC, Chou CC, Ko TP, Shr HL, Chang GG, Wang AH, Liang PH. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 2005;280:31257–31266. doi: 10.1074/jbc.M502577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding L, Zhang XX, Wei P, Fan K, Lai L. The interaction between severe acute respiratory syndrome coronavirus 3C-like proteinase and a dimeric inhibitor by capillary electrophoresis. Anal. Biochem. 2005;343:159–165. doi: 10.1016/j.ab.2005.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berendsen H.JC, Postma J.PM, Van Gunsteren WF, Hermans J. In: In Intermolecular Forces, Pullman B, editor. Dordrecht, The Netherlands: Reidel; 1981. pp. 331–342,. [Google Scholar]
- 18.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, David C. Spellmeyer DC, Fox D, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 19.Darden T, York D, Pedersen L. Particle Mesh Ewald: an N Log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 20.Rychaert JP, Ciccotti G, Berendsen JC. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 21.Berendsen H.JC, Postma J.PM, Van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 22.Sun H, Luo H, Yu C, Sun T, Chen J, Peng S, Qin J, Shen J, Yang Y, Xie Y, Chen K, Wang Y, Shen X, Jiang H. Molecular cloning, expression, purification, and mass spectrometric characterization of 3C-like protease of SARS coronavirus. Protein Expr. Purif. 2003;32:302–308. doi: 10.1016/j.pep.2003.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen S, Chen LL, Luo HB, Sun T, Chen J, Ye F, Cai JH, Shen JK, Shen X, Jiang HL. Enzymatic activity characterization of SARS coronavirus 3C-like protease by fluorescence resonance energy transfer technique. Acta Pharmacol. Sin. 2005;26:99–106. doi: 10.1111/j.1745-7254.2005.00010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Gui C, Luo X, Yang Q, Gunther S, Scandella E, Drosten C, Bai D, He X, Ludewig B, Chen J, Luo H, Yang Y, Yang Y, Zou J, Thiel V, Chen K, Shen J, Shen X, Jiang H. Cinanserin is an inhibitor of the 3C-like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro. J. Virol. 2005;79:7095–7103. doi: 10.1128/JVI.79.11.7095-7103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 1994;238:777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 26.Kuo CJ, Chi YH, Hsu JT, Liang PH. Characterization of SARS main protease and inhibitor assay using a fluorogenic substrate. Biochem. Biophys. Res. Commun. 2004;318:862–867. doi: 10.1016/j.bbrc.2004.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrila J, Bacha U, Freire E. Long-range cooperative interactions modulate dimerization in SARS 3CLpro. Biochemistry. 2006;45:14908–14916. doi: 10.1021/bi0616302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anand K, Palm GJ, Mesters JR, Siddell SG, Ziebuhr J, Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prakash K, Prajapati S, Ahmad A, Jain SK, Bhakuni V. Unique oligomeric intermediates of bovine liver catalase. Protein Sci. 2002;11:46–57. doi: 10.1110/ps.20102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Graziano V, McGrath WJ, DeGruccio AM, Dunn JJ, Mangel WF. Enzymatic activity of the SARS coronavirus main proteinase dimer. FEBS Lett. 2006;580:2577–2583. doi: 10.1016/j.febslet.2006.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blanchard JE, Elowe NH, Huitema C, Fortin PD, Cechetto JD, Eltis LD, Brown ED. High-throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chem. Biol. 2004;11:1445–1453. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kao RY, To AP, Ng LW, Tsui WH, Lee TS, Tsoi HW, Yuen KY. Characterization of SARS-CoV main protease and identification of biologically active small molecule inhibitors using a continuous fluorescence-based assay. FEBS Lett. 2004;576:325–330. doi: 10.1016/j.febslet.2004.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuang WF, Chow LP, Wu MH, Hwang LH. Mutational and inhibitive analysis of SARS coronavirus 3C-like protease by fluorescence resonance energy transfer-based assays. Biochem. Biophys. Res. Commun. 2005;331:1554–1559. doi: 10.1016/j.bbrc.2005.04.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dougherty WG, Semler BL. Expression of virus-encoded proteinases: functional and structural similarities with cellular enzymes. Microbiol. Rev. 1993;57:781–822. doi: 10.1128/mr.57.4.781-822.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ziebuhr J, Heusipp G, Siddell SG. Biosynthesis, purification, and characterization of the human coronavirus 229E 3C-like proteinase. J. Virol. 1997;71:3992–3997. doi: 10.1128/jvi.71.5.3992-3997.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ziebuhr J, Snijder EJ, Gorbalenya AE. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 37.Xiong B, Gui CS, Xu XY, Luo C, Chen J, Luo HB, Chen LL, Li GW, Sun T, Yu CY, Yue LD, Duan WH, Shen JK, Qin L, Shi TL, Li YX, Chen KX, Luo XM, Shen X, Shen JH, Jiang HL. A 3D model of SARS_CoV 3CL proteinase and its inhibitors design by virtual screening. Acta Pharmacol. Sin. 2003;24:497–504. [PubMed] [Google Scholar]
- 38.Chen H, Wei P, Huang C, Tan L, Liu Y, Lai L. Only one protomer is active in the dimer of SARS 3C-like proteinase. J. Biol. Chem. 2006;281:13894–13898. doi: 10.1074/jbc.M510745200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang C, Wei P, Fan K, Liu Y, Lai L. 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry. 2004;43:4568–4574. doi: 10.1021/bi036022q. [DOI] [PubMed] [Google Scholar]