

Abstract
Motivation: Many secretory proteins are synthesized as inactive precursors that must undergo post-translational proteolysis in order to mature and become active. In the current study, we address the challenge of sequence-based discovery of proteolytic sites in secreted proteins using machine learning.
Results: The results revealed that only half of the extracellular proteolytic sites are currently annotated, leaving over 3600 unannotated ones. Furthermore, we have found that only 6% of the unannotated sites are similar to known proteolytic sites, whereas the remaining 94% do not share significant similarity with any annotated proteolytic site. The computational challenges in these two cases are very different. While the precision in detecting the former group is close to perfect, only a mere 22% of the latter group were detected with a precision of 80%. The applicability of the classifier is demonstrated through members of the FGF family, in which we verified the conservation of physiologically-relevant proteolytic sites in homologous proteins.
Contact: kliger@compugen.co.il; yossef.kliger@gmail.com
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Many secretory proteins and peptides are initially synthesized as larger precursors, usually in the form of pre-pro-proteins. Such precursor proteins undergo post-translational proteolysis: the N-terminal pre-region, known as signal peptide, is cleaved by a well-characterized signal peptidase [reviewed in (Paetzel et al., 2002)], while various proteases liberate the active proteins from the pro-proteins. The following examples demonstrate the importance of the latter process and its regulation: (i) The envelope (Env) glycoprotein of HIV-1 is synthesized as a precursor polypeptide. In the trans-Golgi network, Env is cleaved by the cellular protease furin into two functional subunits. Cleavage of Env occurs at a conserved sequence. Mutagenesis of this sequence produces non-infectious HIV-1 particles containing unprocessed Env (Earl et al., 1991; Kowalski et al., 1987; McCune et al., 1988). This finding establishes the importance of furin-mediated processing for virus-infectivity. Accordingly, inhibitors of the host protease furin impede HIV-1 replication by interfering with the proteolytic processing of Env, suggesting they are useful for combating HIV-1 (Bahbouhi et al., 2002; Hallenberger et al., 1992; Kibler et al., 2004). Furthermore, inhibiting the production of peptides involved in various diseases by blocking the activity of the proteolytic enzymes is a promising approach (Basak, 2005; Bergeron et al., 2005; de Haan et al., 2004). (ii) The release of peptide hormones is subject to a complex and finely tuned regulation system. Post-translational proteolysis plays a key role by specifically converting the pro-hormone precursor into biologically active products. Examples of peptide hormones, whose proteolytic processing regulates their activities, are: insulin, somatostatin, parathyroid hormone, glucagon and GLP-1. Many of these are used as therapeutic peptides for treating various disorders.
The importance of identifying mature proteins fuels both experimental and computational approaches aimed at discovering and predicting proteolytic sites. Experimental attempts to unveil the human plasma proteome using proteomics methods fail to detect most cytokines and protein hormones, presumably due to their low abundance [summarized in (Anderson et al., 2004)]. Currently, most computational approaches are protease-oriented and rely on proteolytic site data of specific enzymes (Blom et al., 1996; Cai et al., 1998; Kiemer et al., 2004; Yang and Berry, 2004). However, while proteolytic sites in a protein can be experimentally identified, for example, by N-terminal sequencing of the processed protein fragments, it is much harder to find out the catalyzing protease involved. Hence, only a limited number of experimentally verified proteolytic sites can be associated with a specific proteolytic enzyme, and therefore the data available as training sets for these methods is relatively limited.
Many of the proteolytic sites whose catalyzing enzymes are known are processed by members of one family of serine proteases, called pro-hormone convertases (PCs) (Seidah et al., 1998). All known proteolytic sites of mammalian PCs have an arginine or a lysine at the first position N-terminal to the proteolytic sites. Furthermore, no other enzyme that catalyzes the processing of proteins in the secretory pathway is known to cleave immediately after these basic amino acid residues. It is therefore reasonable to assume that proteolysis after a basic residue is catalyzed by a member of the PC family. This allows data extraction of sequences of proteins, which are processed by a PC member, from databases of precursor proteins and proteolytic sites. Such extracted data, together with the evolutionary relatedness between the members of the PC family, suggests that it might be possible to construct a classifier that will discriminate between PC proteolytic sites, regardless of the specific PC member, and other sites. Such an approach was taken by Blom and colleagues (Duckert et al., 2004), who extracted PC proteolytic sites based on Swiss-Prot (version 40) annotation. Herein, we describe an improved data extraction process, which considered more proteolytic sites. The extracted data was used for training classifiers, which are based on two different classification algorithms—Random Forest and Support Vector Machines. The best classifier was used to provide a comprehensive list of predicted proteolytic sites in the mammalian secretome. Several interesting predictions of proteolytic sites are discussed.
2 METHODS
2.1 Data preparation
All eukaryotic proteins were downloaded from the Swiss-Prot knowledgebase version 47.4 (Boeckmann et al., 2003). Proteins whose first residue is not methionine were discarded, as they might not contain the full-length sequence of the precursor protein. The same holds for Swiss-Prot entries that include the phrase ‘PROTEIN SEQUENCE’, but do not include ‘NUCLEOTIDE SEQUENCE’ in their RP annotation lines, as these entries might contain sequences of processed proteins, rather than the full-length precursor proteins. Data of proteolytic sites were extracted from the post-translational modifications annotation lines (FT) of the Swiss-Prot knowledgebase (Farriol-Mathis et al., 2004).
2.2 Classifiers
Two types of classifiers were tested: Random Forest (RF) (Breiman, 2001) and Support Vector Machines (SVM) (Vapnik and Cortes, 1995). For the SVM classifier, we used Joachims’ SVMlight package (Joachims, 1999).
2.3 Signal peptide prediction
Predicting whether a protein has an N-terminal signal sequence, was performed using the SignalP 3.0 prediction tool (Bendtsen et al., 2004).
2.4 Multiple sequence alignment
Multiple sequence alignments were computed with ProbCons (Do et al., 2005) and were edited using Jalview (Clamp et al., 2004).
3 RESULTS
3.1 Proteolytic site data extraction
Since the aim of the classifier was to model proteolytic processes taking place in the secretory pathway, only secreted proteins and extracellular parts of membranal proteins (secretome) were considered. Thus, only proteins annotated as containing a signal peptide or a transmembrane domain in the feature table (FT) lines of the Swiss-Prot annotation record, or annotated as being secreted or extracellular in the comment (CC) lines of the Swiss-Prot annotation record were selected.
In the case of integral membrane proteins, cytoplasmic domains were not considered. The membrane topology information, i.e. the location of the membrane-spanning regions and their orientation, was extracted from the topology annotation lines of the Swiss-Prot entry (FT TOPO_DOM and FT TRANSMEM). When these lines do not span the full length of the protein, we completed the full topology of the protein according to the annotated signal peptide, transmembrane domains, extracellular domains and cytoplasmic domains. This process was performed twice: once by starting from the most N-terminal topology annotation, and once by starting from the most C-terminal topology annotation. Whenever discrepancies between the two completion processes were found, the Swiss-Prot entry was discarded. Such discrepancies point to mistakes in the topology annotation of multi-span proteins. Ideally, the extracted proteolytic sites should be divided into sites that are catalyzed by enzymes working in the secretory pathway, the extracellular matrix, the cytoplasm, the digestive system or in extracellular fluids. When available, annotation of the identity of the proteolytic enzyme was extracted from the FT annotation lines (following the phrase ‘Removed by’ in the description of PROPEPs lines, or following ‘by’ in the description of ‘SITE…CLEAVAGE’ lines). As the aim of this study is to model the processes that take place in the secretory pathways, proteolysis processed by enzymes that are known to act outside the secretory pathway were discarded. The list of enzymes known to act outside the secretory pathway that appear in the annotation of Swiss-Prot entries of the proteins they cleave includes: adam17, aggrecanase, alpha-secretase, beta-secretase, caspase-6, cathepsin G, arginine-specific endoprotease, C3 convertase, chymosin, collagenase, dipeptidase, dipeptidylpeptidase, DPP4, easter, elastase, kallikrein and kallikrein-like serine protease, MMPs (2, 3, and 9), coagulation factors (I, VIIa, IXa, Xa and XIa), plasmin, procollagen C-endopeptidase, procollagen N-endopeptidase, rennin, thrombin, trypsin and u-PA.
Blom and colleagues (Duckert et al., 2004) extracted PC proteolytic sites based on Swiss-Prot annotation. They screened for precursor proteins that are annotated to have a signal peptide, followed by a PROPEP that ends with an arginine or a lysine, and then followed by a PEPTIDE or a CHAIN. They were then able to construct an artificial neural network-based classifier for predicting proteolytic sites catalyzed by members of the pro-hormone convertase family of proteases (Duckert et al., 2004). However, this procedure is too strict for part of the proteolytic sites. For example, human insulin (Swiss-Prot ID: INS_HUMAN) is composed of a signal peptide, followed by a PEPTIDE, a PROPEP and then another PEPTIDE. These two well-characterized proteolytic sites were ignored by the conservative extraction, because insulin has no PROPEP immediately after the signal peptide. Therefore, due to the scarcity of data, we used a less strict data extraction procedure as described below.
This study focuses on proteolytic sites of enzymes that cut immediately after lysines or arginines. Such enzymes are often classified as members of the pro-hormone convertase family. Therefore, only sites with a lysine or arginine at the first position N-terminal to the proteolytic site were considered. We extracted all 30-mers of the secretome, arranged symmetrically around a potential proteolytic site after a basic residue, and designated them as follows: (i) Experimentally-validated proteolytic sites, which are annotated by a Swiss-Prot FT annotation line according to the word template ‘SITE…CLEAVAGE’, were marked VALIDATED. (ii) Experimentally-validated proteolytic sites, whose existence is indicated by the annotation of the two protein segments right before and immediately after the proteolytic site, were also marked VALIDATED. The annotation for protein segments is in the form of Swiss-Prot FT annotation lines having the word template ‘PEPTIDE (or PROPEPTIDE or CHAIN) [first residue] [last residue]’, and the two segments of the protein should be consecutive, i.e. the first residue of the second segment immediately follows the last residue of the first segment. We do allow for a short linker section in between the two segments, provided that it is likely to be removed by exopeptidase E after the processing of the protein precursor by a pro-hormone convertase (Day et al., 1998; Friis-Hansen et al., 2001). We consider linker sections consisting of K, R, KK, KR, RK, RR, or successive Ks and/or Rs followed by a classical furin proteolytic site (RXKR or RXRR, where X is any natural amino acid) as likely to be cut by exopeptidase E. We also allow for a glycine to immediately upstream of the basic residue/s at the C-terminus of the first PEPTIDE, PROPEPTIDE or CHAIN, as it is likely that these peptides are substrates for C-terminal alpha-amidating enzymes that convert the peptides to the corresponding desglycine peptide amide, where glycine is the amide donor (Bradbury et al., 1982). The ambiguous sites (after each of the residues located in-between the two annotation lines) are marked AMBG. (iii) When only one PEPTIDE, PROPEPTIDE or CHAIN annotation line suggests the existence of a proteolytic site, our confidence in the proteolysis site is reduced and the site is marked POTENTIAL. (iv) When comments like ‘PROBABLE’, ‘BY SIMILARITY’ or ‘POTENTIAL’ (Farriol-Mathis et al., 2004; Junker et al., 1999) appear in the description of the FT lines in the cases described in (i) and (ii), the proteolytic site is designated as POTENTIAL. (v) When the distance between two proteolytic sites does not exceed four residues, the reliability of both sites is reduced. Such proteolytic sites are marked POTENTIAL unless there is strong support for their reliability. Strong support for one or both of the two proteolytic sites is considered if a proteolytic site is marked VALIDATED according to the criterion in (i). Strong support for one or both of the two proteolytic sites is also considered if a proteolytic site is marked POTENTIAL according to the SITE…CLEAVAGE annotation line, and also marked VALIDATED according to the criterion in (ii). (vi) All other positions were marked NON (Table SI).
3.2 Training, validation, and test sets
Ideally, data would be separated into distinct training, test and validation sets. However, the relative scarcity of cleavage sites, and their different levels of reliability, present a challenge when preparing datasets for classification, and necessitate a different approach. A validation set consisting of a random quarter of the data was held out and used for parameter optimization. The rest of the data were used, once optimal parameters were chosen, in cross-validation to evaluate performance. When training, only the most reliable proteolytic sites, namely, sites that were marked VALIDATED, were used as positive examples, while a subset of the sites marked NON was used as negative examples. For the purpose of performance evaluation, on the other hand, it is important to use a set representative of all data. Thus, in the parts of the data used for testing, proteolytic sites that were marked VALIDATED or POTENTIAL were labeled positive, while those marked NON or AMBG were labeled negative.
3.3 Classifier construction
Homologous sequences raise special difficulties due to the relationship between redundancy and information. It is therefore essential to handle them with care. One approach is to discard some of the protein sequences, in a way that maximizes coverage and minimizes redundancy (Hobohm et al., 1992). The weakness of this approach is that it prevents learning from the subtle changes that exist between very similar sequences. For this reason, and due to the scarcity of annotated data, others and we decided to use all available data. This approach requires special precautions in order to minimize the risk of overestimating the predictive performance owing to training set and test set similarities. One way to avoid training and testing on homologous data is to divide the data into several partitions based on a phylogenetic tree, and then calculate the performance by cross-validation (Duckert et al., 2004). We used a different approach, which is described in what follows.
We argue that the task of classifying a site is naturally divided into two cases, depending on whether or not this site is similar (to a degree, homologous) to a known proteolytic site, i.e. a proteolytic site present in the training set. Classifying ‘seen before’ sites and ‘new’ sites are tasks that are different in nature, and have a different level of difficulty. This implies the need for two methods of classification, and, more important, for separate performance evaluation for the two tasks. In order to discriminate between the classification tasks, we analyzed 18-mers, arranged symmetrically around a potential proteolytic site, which were marked as VALIDATED or POTENTIAL. Each 18-mer was compared to its most similar known proteolytic site, and the number of identical residues was counted. Our analysis confirmed that 18-mer sites that share more than nine residues with a known proteolytic site are most likely to be proteolytic sites themselves (Figure S1).
We chose this threshold for dividing the data into ‘new’ and ‘seen before’ sites. The number of identical residues to the closest known proteolytic site was also used as an additional input feature for the classifier. This feature improves the classification results (Figure S2).
Figure 1 reveals that, as expected, the tasks of classifying ‘seen before’ sites and classifying ‘new’ sites, are different in nature, and confirms the need for two separate performance evaluations. In addition, a classifier trained to identify ‘new’ sites was more successful at identifying ‘new’ sites than a classifier trained to identify ‘seen before’ sites (Figure 1B).
Fig. 1.

The effect of creating two specialized classifiers. It is clear that the performance of classifiers for ‘seen before’ and ‘new’ sites should be evaluated separately. Furthermore, the figure shows that it is worth training specialized classifiers: (A) Identification of ‘seen before’ sites. The classifier trained to identify ‘seen before’ sites is somewhat better at identifying such sites than the classifier trained to identify ‘new’ sites. (B) Identification of ‘new’ sites. The classifier trained to identify ‘new’ sites performs better than the classifier trained to identify ‘seen before’ sites at identifying ‘new’ sites.
3.3.1 Parameter tuning
A quarter of the data was picked out at random to serve only for tuning parameters, while the rest was used at the tuning stage for training. The held out set was divided into ‘seen before’ and ‘new’ sites, based on the maximal similarity to known sites in the training set. The two classifiers, for ‘seen before’ sites and for ‘new’ sites, were then, separately, optimized by evaluating precision vs. recall graphs based on the raw score output of Random Forest (RF). The inputs to the classifier were (i) a symmetrical window around the site, and (ii) the maximal identity to a known cleavage site, divided by the window size. For the classifier specialized in ‘seen before’ sites, we used a symmetrical window of 20 residues surrounding each site, a negative set 50 times larger than the positive set, and the internal weighting mechanism of RF was set to give a weight of 50 to the positive set, and 1 to the negative set. Mtry was set to 5, and 200 trees were found to be sufficient. For the classifier aimed at identifying ‘new’ proteolytic sites, we used a symmetrical window of 12 residues around each site, a negative set 50 times larger than the positive set, and the internal weighting was set to 2 for the positive set and 1 for the negative set. Mtry was set to 2 and 200 trees were again found to be sufficient. For the SVM classifier, we tried different polynomial kernels. The best degrees were found to be 4 and 6 for the ‘seen before’ and ‘new’ classifiers, respectively. The vectors fed to the SVMs were in sparse representation (Qian and Sejnowski, 1988). The maximal identity value was used with the SVM the same way as with the RF classifier.
3.3.2 Classifier construction and performance evaluation
The data that was not used as testing data in the parameter optimization step (three quarters of the data) was used for 10-fold stratified cross-validation. Specifically, at each step of the cross-validation, nine-tenths of the data were used for training. The remaining tenth was used for testing after being divided into ‘seen before’ and ‘new’ sets with respect to the current training set. By ‘stratified’ we mean that each tenth part of the data contained the same proportion of VALIDATED, POTENTIAL, etc. sites. The parameters used were those found to be optimal in the parameter tuning step.
3.3.3 Performance evaluation correction
As explained above, all the data that was not used for parameter tuning was used for testing, in order to reflect the heterogeneity of the data as much as possible. However, there is uncertainty as to the label of any data that is not VALIDATED. To a large degree, we trust sites designated POTENTIAL to be real proteolytic sites. Manual reviewing of many of the POTENTIAL sites suggests that this assumption is reasonable. We assume that most AMBG and NON sites are not proteolytic sites. Still, it is expected that yet undiscovered proteolytic sites are hidden among the sites marked NON or AMBG. The sheer volume of NON sites raises the suspicion that there are even more unknown proteolytic sites labeled NON than known proteolytic sites. This may distort performance evaluation statistics. We present below a calculation that attempts to tackle this problem.
| (1) |
| (2) |
| (3) |
| (4) |
Where TPi denotes instances in the positive set, correctly classified as positive, TPo represents mislabeled instances in the negative set, correctly classified as positive, Ti denotes instances in the positive set, To represents mislabeled instances in the negative set, and Po denotes instances in the negative set, classified as positive. It is now easy to note that calculated precision evaluations are always underestimated. The reason is that while the denominator in Equation (2) is the same as in Equation (4), the numerator does not include TPo, which may be even larger than TPi.
We now proceed under the assumption that negative data is a mixture of two statistical types of data—mislabeled positives (a fraction α of the negative data) and real negatives. Mislabeled positives are assumed to have the same statistical nature as positive data. Let Fi (Fo) be the cumulative distribution function of the score for positive (negative) data. Let Ni (No) be the number of positive (negative) instances. Let t be a threshold for the score.
| (5) |
Note that the real recall is independent of α, and is therefore equal to the ordinary recall calculated without assuming any mislabeling.
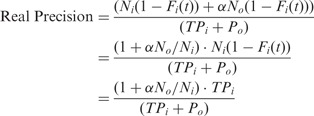 |
(6) |
The real precision is the ordinary precision multiplied by a correction factor: (1 + α No/Ni). Therefore, for α = 0 we recover the ordinary precision.
To summarize, mislabeling leaves the recall unchanged, while the precision is enhanced by a factor (1 + α No/Ni) = 1 + To/Ti.
For furin proteolysis, we can obtain a reasonable estimate of this factor, because furin sites have an easily detectable consensus (Nakayama, 1997). We extrapolate from furin to proteolytic sites of other members of the pro-hormone convertase family, in an attempt to reflect the curation level of proteolysis annotation in the Swiss-Prot knowledgebase. We look for the furin proteolysis consensus site, after RXKR or after RXRR, in the positive and negative sets. The instances in the positive set are real positives, whereas the ones in the negative set are a mixture of proteolytic and non-proteolytic sites. There is evidence that a lysine located two positions after the putative proteolytic site prevents cleavage, so such instances were excluded.
In addition, we observed which residues are most frequent immediately after the proteolytic site in the positive set. Our method for finding the ratio To/Ti was to look for the same subfamily of sites in both positive and negative sets: instances of a furin consensus followed by one of the 3 most frequent residues (as found in the positive set), excluding lysine in the second post-cleavage position. The calculated furin correction factor was found to be 1.11 for the ‘seen before’ classifier, and 3.04 for the ‘new’ classifier. Note that because of the inaccuracy of this correction procedure, corrected precision values may exceed 1. It must be emphasized that the furin correction factor is based on the assumptions that the ratio of annotated proteolytic sites to unannotated sites is equal for furin and other PC sites, and that classifier score distributions are mixtures as described above. Both these assumptions are very rough approximations. Still, we believe this correction gives a better evaluation of classifier performance. A comparison between the performance of RF and SVM classifiers specialized in ‘new’ sites is shown in Figure 2. The RF classifier performs better in the high precision/low recall area, while SVM performs better in the high recall/low precision area. Figure 2 also shows the effect of the furin correction factor on the raw score output of the RF and SVM classifiers. The performance of both the RF and SVM ‘seen before’ classifiers is almost perfect (Figure S3), as expected, and becomes perfect when applying correction (data not shown).
Fig. 2.

Comparison between RF and SVM classifiers specialized in ‘new’ sites, and the effect of the furin correction factor. VALIDATED and POTENTIAL data are treated as positive for testing, the rest as negative. The furin correction is a way to compensate for the fact that some of the data we treated as negative for cleavage is actually mislabeled (unknown proteolytic sites). (A) Raw score output of the RF and SVM classifiers; (B) Precision is multiplied by 3.04, which is the calculated furin correction factor. It should be remarked that because of the imperfection of the correction procedure, corrected precision values may exceed 1. Precision values that exceed 1 are set to 1.
3.4 Proteolytic site prediction
The classification procedure described above was repeated, but this time, no holdout set was removed, and 10-fold stratified cross-validation was applied to the whole eukaryotic secretome. For each classifier, scores were replaced by their corresponding precision values. Each site was given a single score: a ‘seen before’ site was given its score according to the ‘seen before’ classifier, and a ‘new’ site was given its score according to the ‘new’ classifier.
For ‘new’ sites, there are 1663 VALIDATED and POTENTIAL sites, and 569 820 NON and AMBG sites, and the furin correction factor is 3.04. For ‘seen before’ sites, there are 2099 VALIDATED and POTENTIAL sites, and 1035 NON and AMBG sites, and the furin correction factor is 1.11. Based on our data extraction, performance evaluation, and the furin correction factor, we estimate that the eukaryotic secretome is comprised of about 7385 proteolytic sites, of which 2330 (2099 * 1.11) are ‘seen before’, i.e. quite similar to known proteolytic sites, and 5055 (1663 * 3.04) are ‘new’, i.e. do not share significant sequence similarity to any annotated proteolytic site.
The furin correction factor also allows us to estimate the fraction of unannotated proteolysis for ‘seen before’ and ‘new’ sites. Our results reveal that only 9.9% (0.11/1.11) of ‘seen before’ sites are still unannotated, while 67% (2.04/3.04) of ‘new’ sites are yet to be discovered. Furthermore, the RF classifier specialized in ‘seen before’ sites predicts apparently all 231 ‘seen before’ sites with a precision greater than 90%, while the RF classifier specialized in ‘new’ sites predicts about 33% of the 3393 unknown ‘new’ sites with a precision of 50%, and 22% with a precision of 80% (Fig. 2).
3.5 Predicted proteolytic sites in members of the fibroblast growth factor family
Swiss-Prot 47.4 does not include annotation for proteolytic sites in any of the members of the Fibroblast Growth Factor (FGF) family. Yet, our prediction method suggests several proteolytic sites in some of the proteins in this family, resulting in a classification of the FGF proteins into three groups of orthologs: FGFs that have conserved N-terminal proteolytic sites, FGFs that have conserved C-terminal proteolytic sites and all others (Table SII). A literature search confirmed some of our predictions.
Functional proteolytic sites are expected to be conserved among close species. Our classifier revealed that the proteolytic site in FGF23 is indeed conserved in all available FGF23 orthologs (Fig. 3). The C-terminal proteolytic site of FGF23 is important for normal activity of the protein. Several groups reported proteolysis in FGF23 between Arg179 and Ser180, and mutations in proximity to this site (R179W, R179Q and R176Q) were identified in patients with autosomal-dominant hypophosphatemic rickets (ADHR) (Bowe et al., 2001; Shimada et al., 2002; White et al., 2000, 2001). The authors suggested that the proteolysis causes protein inactivation, and that these mutations created a polypeptide less sensitive to proteolysis, thus leading to elevated concentrations of FGF23, and to phosphate wasting in ADHR patients. Our prediction method revealed that these mutated forms of FGF23 do not undergo C-terminal proteolysis (Fig. 3). Furthermore, our predictions of proteolytic sites in the C-terminus of the other FGF family members might also imply their deactivation by proteolysis processing.
Fig. 3.

Proteolytic site predictions for FGF23 of human, three mutant forms from ADHR patients, and three vertebrate orthologs. Sequences of FGF23 of human, mouse, rat and pufferfish were aligned together with R179W, R179Q and R176Q human FGF23 mutants (mutations are highlighted in dark grey). High score cleavage predictions were assigned to the true cleavage sites (highlighted in light grey). In normal FGF23, cleavage is known to take place between the two amino acids in light grey.
Another known case is the N-terminal proteolytic of FGF3. The amino-terminal region downstream of the signal peptide of the protein is involved in its retention in the Golgi apparatus and the regulation of its secretion (Kiefer et al., 1993). We predicted proteolytic sites in the N-terminus of human, mouse, zebrafish, chicken and xenopus FGF3. Indeed, in xenopus, proteolysis between Arg45 and Asp46 is essential for FGF3's biological activity (Antoine et al., 2000). We suggest that proteolysis of 10–27 N-terminal amino acids occurs during the maturation of other FGFs, and may be important for their biological activity. The multiple sequence alignment in Figure 4 confirms that the N-terminal proteolytic site is conserved between some FGF family members and in proximity to an upstream variable region. It is worth noting that the proteolytic site is conserved even among remote homologs. Some of these homologs possess an N-terminal signal peptide and are secreted via the classical secretory pathway, while others do not possess a signal peptide and are secreted via an alternative pathway (Nickel, 2003).
Fig. 4.

FGF3 and other FGF family members that undergo proteolysis in their N-terminal region. Proteolysis of the N-terminal region of FGF3 is important for regulating its activity. FGF11 to 14 were also assigned high score N-terminal cleavage site predictions, although they do not have a leading signal peptide. Removing the signal peptides of FGF3 members allows alignment of the N-terminal proteolytic sites. The high conservation of the proteolytic site signatures in contrast to the variability of the flanking sequences, confirms the importance of the proteolytic processing that as in FGF3 may be involved in the regulation of protein activity.
4 DISCUSSION
This study revealed a big potential for proteolytic site predictors, because most proteolytic sites are currently still unannotated. Furthermore, the furin correction factor gives an estimate of the total number of proteolytic sites. We estimate the eukaryotic secretome to comprise about 7385 (1663 × 3.04 + 2099 × 1.11) proteolytic sites, which means that about 1.3% of R/K in the secretome are proteolytic sites (7385/(1663 + 569820 + 2099 + 1035) = 0.0129). An important conclusion is that currently only about half of the proteolytic sites are annotated [(1663 + 2099)/7385 = 0.509], meaning there is a great value for predictors of proteolytic sites.
Another important issue raised in this article is performance evaluation when some of the data is mislabeled. This mislabeling is a result of missing annotation in our case, and these sites are often unknown proteolytic sites. We showed that such mislabeling leaves the recall unchanged, while the precision is reduced by a factor that can be estimated. Furthermore, by relying on a well-characterized subgroup, namely furin sites, we were able to estimate the degree of mislabeling. As mislabeling is very common in perhaps most current biological data, we believe that our calculation is relevant for performance evaluation in other biological classification problems.
Many sites are currently not annotated as proteolytic sites, but are predicted by our classifier with high precision. These include sites in currently developed therapeutic proteins, and in a few cases, the exact boundaries of peptides identified experimentally as minimal sequences required for functionality.
We demonstrate the prediction capability of the novel classifier in an analysis of members of the Fibroblast Growth Factor (FGF) family. We were able to discriminate real proteolysis sites from non-cleaving sites of mutant FGF23 proteins of ADHR patients. Additionally the predictor was able to identify cleavage sites in remote homologs, suggesting a regulatory role for the predicted cleavages by annotation transfer.
In summary, proteolysis has a great influence on the biological function of proteins, and therefore the accurate prediction of proteolytic sites is important for basic research and biotechnological applications. It allows identification of biologically active peptides from non-active precursors. In addition, it allows identification of mutations and polymorphisms that influence the generation of active peptides and proteins.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to P. Duckert, W.L. McKeehan, M. Havilio, I. Borukhov, H. Ashkenazy, I. Myslyuk, E. Schreiber, and Y. Mansour for useful comments and helpful discussions.
Conflict of Interest: none declared.
REFERENCES
- Anderson NL, et al. The human plasma proteome: a nonredundant list developed by combination of four separate sources. Mol. Cell Proteomics. 2004;3:311–326. doi: 10.1074/mcp.M300127-MCP200. [DOI] [PubMed] [Google Scholar]
- Antoine M, et al. NH2-terminal cleavage of xenopus fibroblast growth factor 3 is necessary for optimal biological activity and receptor binding. Cell Growth Differ. 2000;11:593–605. [PubMed] [Google Scholar]
- Bahbouhi B, et al. Effects of L- and D-REKR amino acid-containing peptides on HIV and SIV envelope glycoprotein precursor maturation and HIV and SIV replication. Biochem. J. 2002;366:863–872. doi: 10.1042/BJ20020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak A. Inhibitors of proprotein convertases. J. Mol. Med. 2005;83:844–855. doi: 10.1007/s00109-005-0710-0. [DOI] [PubMed] [Google Scholar]
- Bendtsen JD, et al. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Bergeron E, et al. Implication of proprotein convertases in the processing and spread of severe acute respiratory syndrome coronavirus. Biochem. Biophys. Res. Commun. 2005;326:554–563. doi: 10.1016/j.bbrc.2004.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom N, et al. Cleavage site analysis in picornaviral polyproteins: discovering cellular targets by neural networks. Protein Sci. 1996;5:2203–2216. doi: 10.1002/pro.5560051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckmann B, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowe AE, et al. FGF-23 Inhibits Renal Tubular Phosphate Transport and Is a PHEX Substrate. Biochem. Biophys. Res. Commun. 2001;284:977. doi: 10.1006/bbrc.2001.5084. [DOI] [PubMed] [Google Scholar]
- Bradbury AF, et al. Mechanism of C-terminal amide formation by pituitary enzymes. Nature. 1982;298:686–688. doi: 10.1038/298686a0. [DOI] [PubMed] [Google Scholar]
- Breiman L. Random forests. Machine Learning. 2001;45:5–32. [Google Scholar]
- Cai YD, et al. Artificial neural network method for predicting HIV protease cleavage sites in protein. J. Protein Chem. 1998;17:607–615. doi: 10.1007/BF02780962. [DOI] [PubMed] [Google Scholar]
- Clamp M, et al. The Jalview Java alignment editor. Bioinformatics. 2004;20:426–427. doi: 10.1093/bioinformatics/btg430. [DOI] [PubMed] [Google Scholar]
- Day R, et al. Prodynorphin processing by proprotein convertase 2. Cleavage at single basic residues and enhanced processing in the presence of carboxypeptidase activity. J. Biol. Chem. 1998;273:829–836. doi: 10.1074/jbc.273.2.829. [DOI] [PubMed] [Google Scholar]
- de Haan CA, et al. Cleavage inhibition of the murine coronavirus spike protein by a furin-like enzyme affects cell–cell but not virus–cell fusion. J. Virol. 2004;78:6048–6054. doi: 10.1128/JVI.78.11.6048-6054.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do CB, et al. ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Res. 2005;15:330–340. doi: 10.1101/gr.2821705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckert P, et al. Prediction of proprotein convertase cleavage sites. Protein Eng. Des. Sel. 2004;17:107–112. doi: 10.1093/protein/gzh013. [DOI] [PubMed] [Google Scholar]
- Earl PL, et al. Biological and immunological properties of human immunodeficiency virus type 1 envelope glycoprotein: analysis of proteins with truncations and deletions expressed by recombinant vaccinia viruses. J. Virol. 1991;65:31–41. doi: 10.1128/jvi.65.1.31-41.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farriol-Mathis N, et al. Annotation of post-translational modifications in the Swiss-Prot knowledge base. Proteomics. 2004;4:1537–1550. doi: 10.1002/pmic.200300764. [DOI] [PubMed] [Google Scholar]
- Friis-Hansen L, et al. Attenuated processing of proglucagon and glucagon-like peptide-1 in carboxypeptidase E-deficient mice. J. Endocrinol. 2001;169:595–602. doi: 10.1677/joe.0.1690595. [DOI] [PubMed] [Google Scholar]
- Hallenberger S, et al. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature. 1992;360:358–361. doi: 10.1038/360358a0. [DOI] [PubMed] [Google Scholar]
- Hobohm U, et al. Selection of representative protein data sets. Protein Sci. 1992;1:409–417. doi: 10.1002/pro.5560010313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joachims T. Making large-scale support vector machine learning practical. In: Schölkopf B, et al., editors. Advances in Kernel Methods – Support Vector Learning ch. 11, Cambridge, USA: MIT Press; 1999. pp. 169–184. [Google Scholar]
- Junker VL, et al. Representation of functional information in the SWISS-PROT data bank. Bioinformatics. 1999;15:1066–1067. doi: 10.1093/bioinformatics/15.12.1066. [DOI] [PubMed] [Google Scholar]
- Kibler KV, et al. Polyarginine inhibits gp160 processing by furin and suppresses productive human immunodeficiency virus type 1 infection. J. Biol. Chem. 2004;279:49055–49063. doi: 10.1074/jbc.M403394200. [DOI] [PubMed] [Google Scholar]
- Kiefer P, et al. Retention of fibroblast growth factor 3 in the Golgi complex may regulate its export from cells. Mol. Cell Biol. 1993;13:5781–5793. doi: 10.1128/mcb.13.9.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiemer L, et al. Coronavirus 3CLpro proteinase cleavage sites: possible relevance to SARS virus pathology. BMC Bioinformatics. 2004;5:72. doi: 10.1186/1471-2105-5-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski M, et al. Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science. 1987;237:1351–1355. doi: 10.1126/science.3629244. [DOI] [PubMed] [Google Scholar]
- McCune JM, et al. Endoproteolytic cleavage of gp160 is required for the activation of human immunodeficiency virus. Cell. 1988;53:55–67. doi: 10.1016/0092-8674(88)90487-4. [DOI] [PubMed] [Google Scholar]
- Nakayama K. Furin: a mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem. J. 1997;327:625–635. doi: 10.1042/bj3270625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel W. The mystery of nonclassical protein secretion. A current view on cargo proteins and potential export routes. Eur. J. Biochem. 2003;270:2109–2119. doi: 10.1046/j.1432-1033.2003.03577.x. [DOI] [PubMed] [Google Scholar]
- Paetzel M, et al. Signal peptidases. Chem. Rev. 2002;102:4549–4580. doi: 10.1021/cr010166y. [DOI] [PubMed] [Google Scholar]
- Qian N, Sejnowski TJ. Predicting the secondary structure of globular proteins using neural network models. J. Mol. Biol. 1988;202:865–884. doi: 10.1016/0022-2836(88)90564-5. [DOI] [PubMed] [Google Scholar]
- Seidah NG, et al. Precursor convertases: an evolutionary ancient, cell-specific, combinatorial mechanism yielding diverse bioactive peptides and proteins. Ann. NY Acad. Sci. 1998;839:9–24. doi: 10.1111/j.1749-6632.1998.tb10727.x. [DOI] [PubMed] [Google Scholar]
- Shimada T, et al. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology. 2002;143:3179–3182. doi: 10.1210/endo.143.8.8795. [DOI] [PubMed] [Google Scholar]
- Vapnik V, Cortes C. Support vector networks. Machine Learning. 1995;20:1–25. [Google Scholar]
- White KE, et al. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001;60:2079–2086. doi: 10.1046/j.1523-1755.2001.00064.x. [DOI] [PubMed] [Google Scholar]
- White KE, et al. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat. Genet. 2000;26:345. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- Yang ZR, Berry EA. Reduced bio-basis function neural networks for protease cleavage site prediction. J. Bioinform. Comput. Biol. 2004;2:511–531. doi: 10.1142/s0219720004000715. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.